
Ionic Liquids as Carbene Catalyst Precursors in the One-Pot Four-Component Assembly of Oxo Triphenylhexanoates (OTHOs)


Abstract
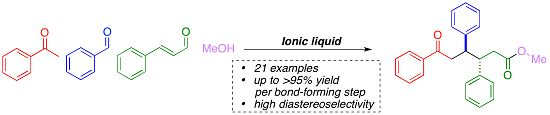
: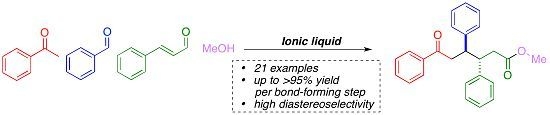
1. Introduction
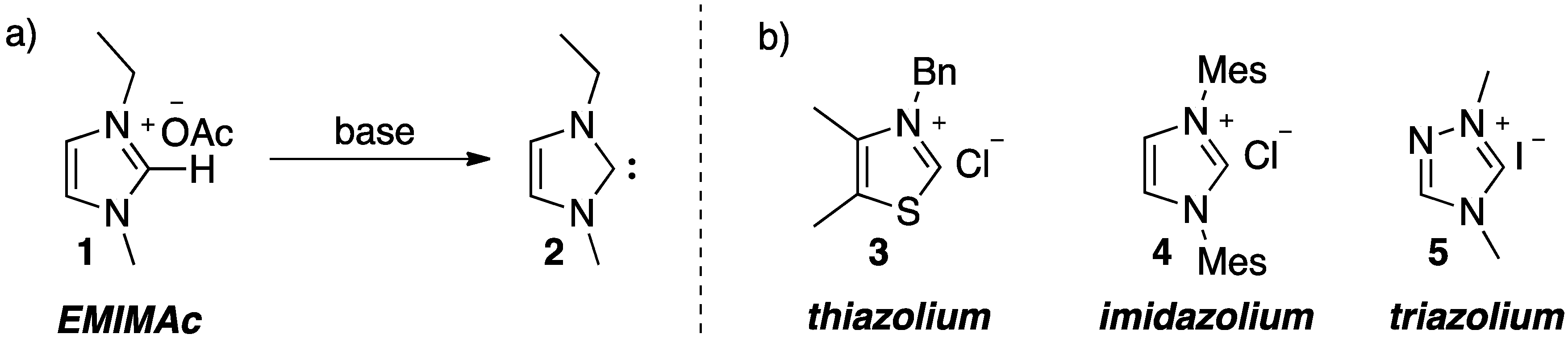

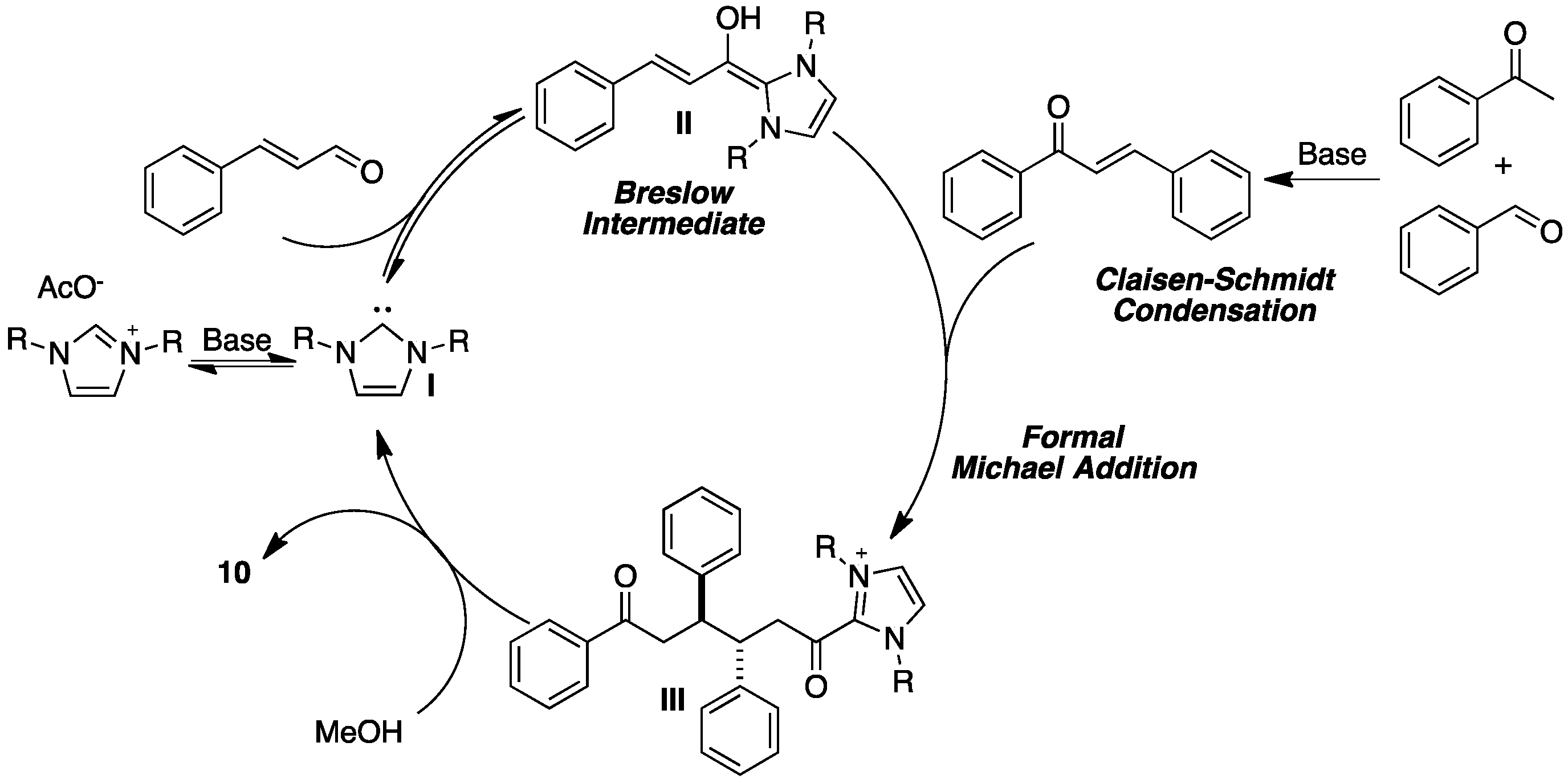
2. Results and Discussion

{kind=link}
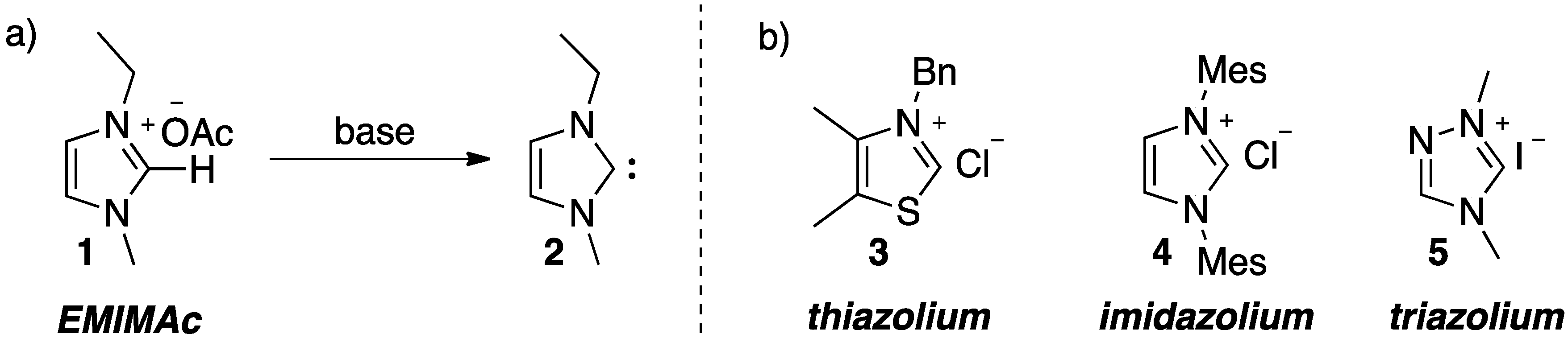{kind=link}
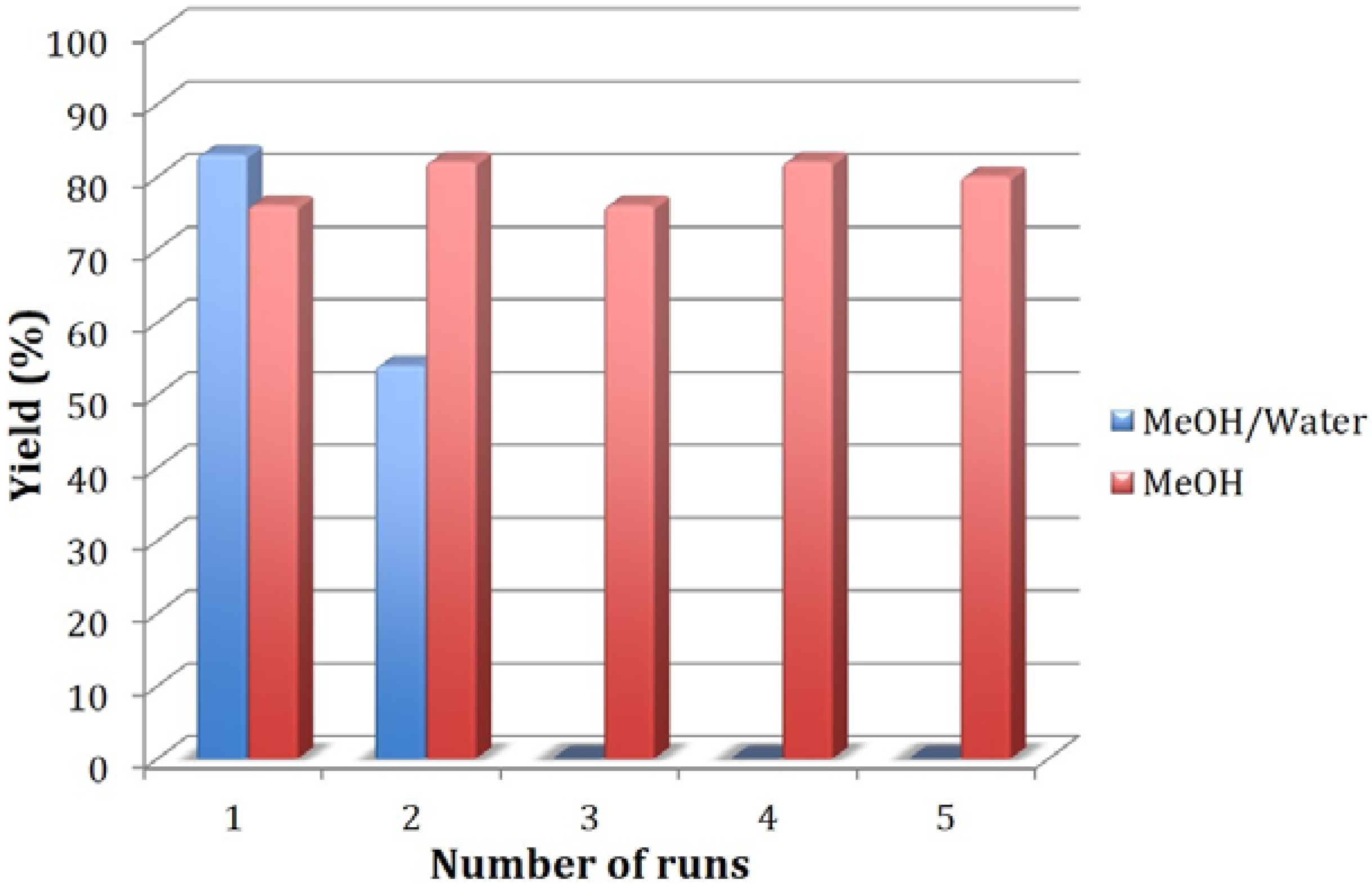{kind=link}
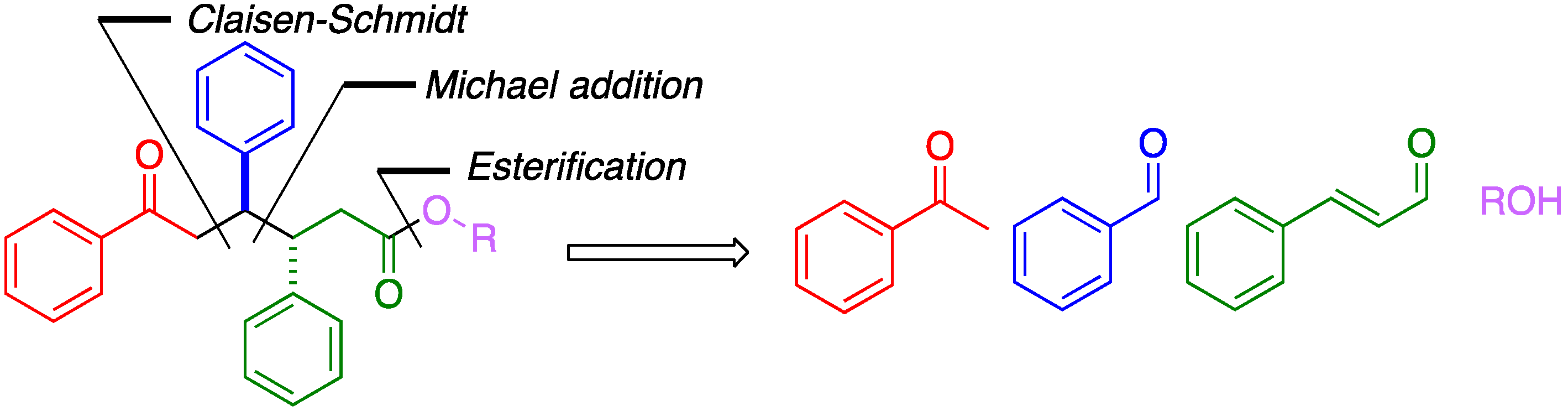{kind=link}
{kind=link}
{kind=link}
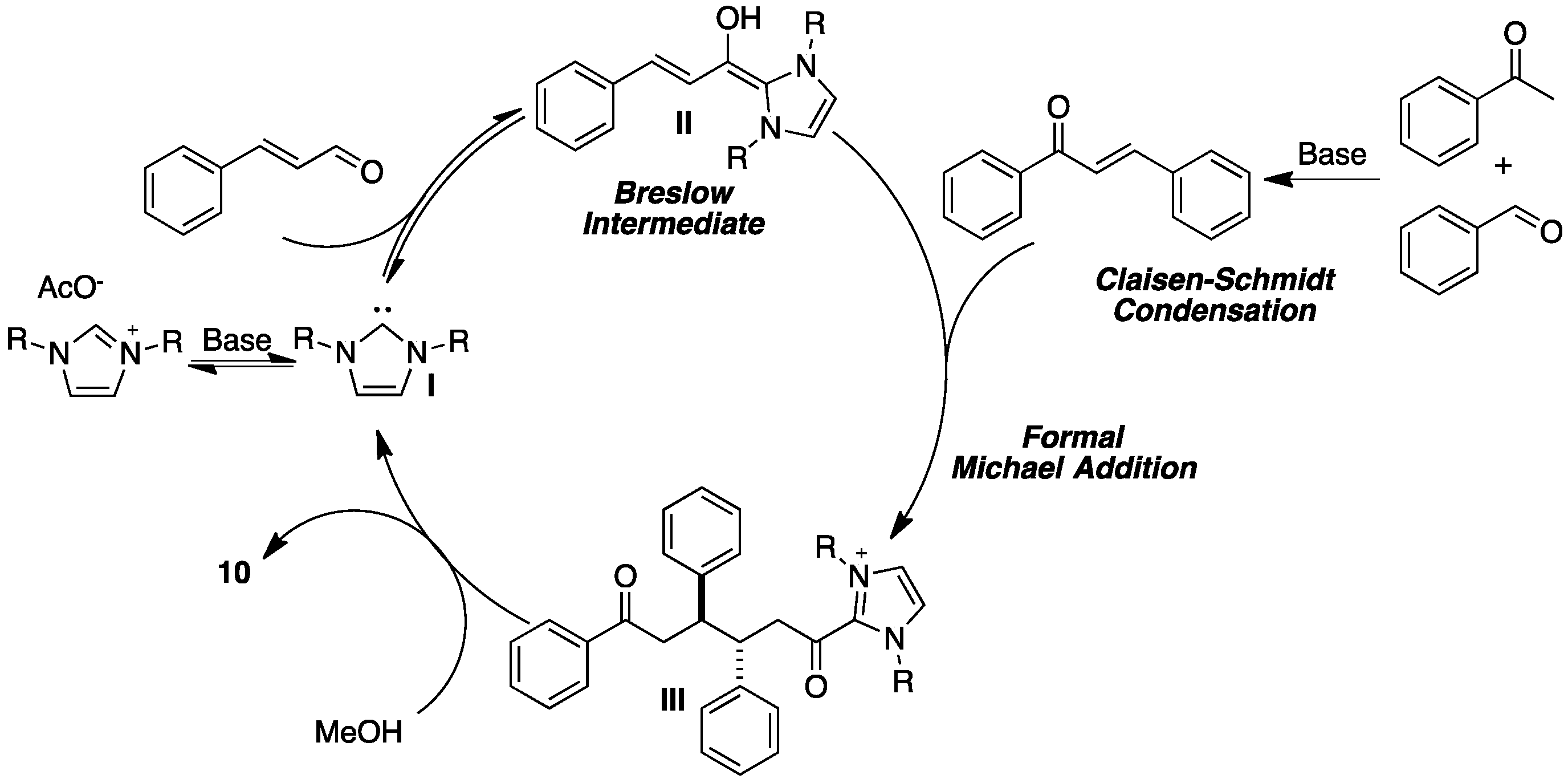{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Solvent | Base | NHC Precatalyst | Yield (%) b |
| 1 | acetonitrile | NaOMe | EMIMAc | 35 |
| 2 | acetonitrile | tBuOK | EMIMAc | 38 |
| 3 | acetonitrile | TBD | EMIMAc | 54 |
| 4 c | acetonitrile | TBD | EMIMAc | 66 |
| 5 c | dichloromethane | TBD | EMIMAc | 65 |
| 6 | acetonitrile | Cs2CO3 | EMIMAc | 45 |
| 7 d | acetonitrile | NaOH | EMIMAc | 43 |
| 8 | acetonitrile | DBU | EMIMAc | 0 |
| 9 c | 1,4-dioxane | TBD | EMIMAc | 20 |
| 10 c | methanol | TBD | EMIMAc | 31 |
| 11 c | acetonitrile | TBD | BMIMCl | 56 |
| 12 c | acetonitrile | TBD | EMIMLactate | 59 |
| 13 c | acetonitrile | TBD | EMIMCl | 52 |
| 14 c | acetonitrile | TBD | 3 | 0 |
| 15 c | acetonitrile | TBD | 4 | 0 |
| 16 c | acetonitrile | TBD | 5 | 0 |
 | ||||
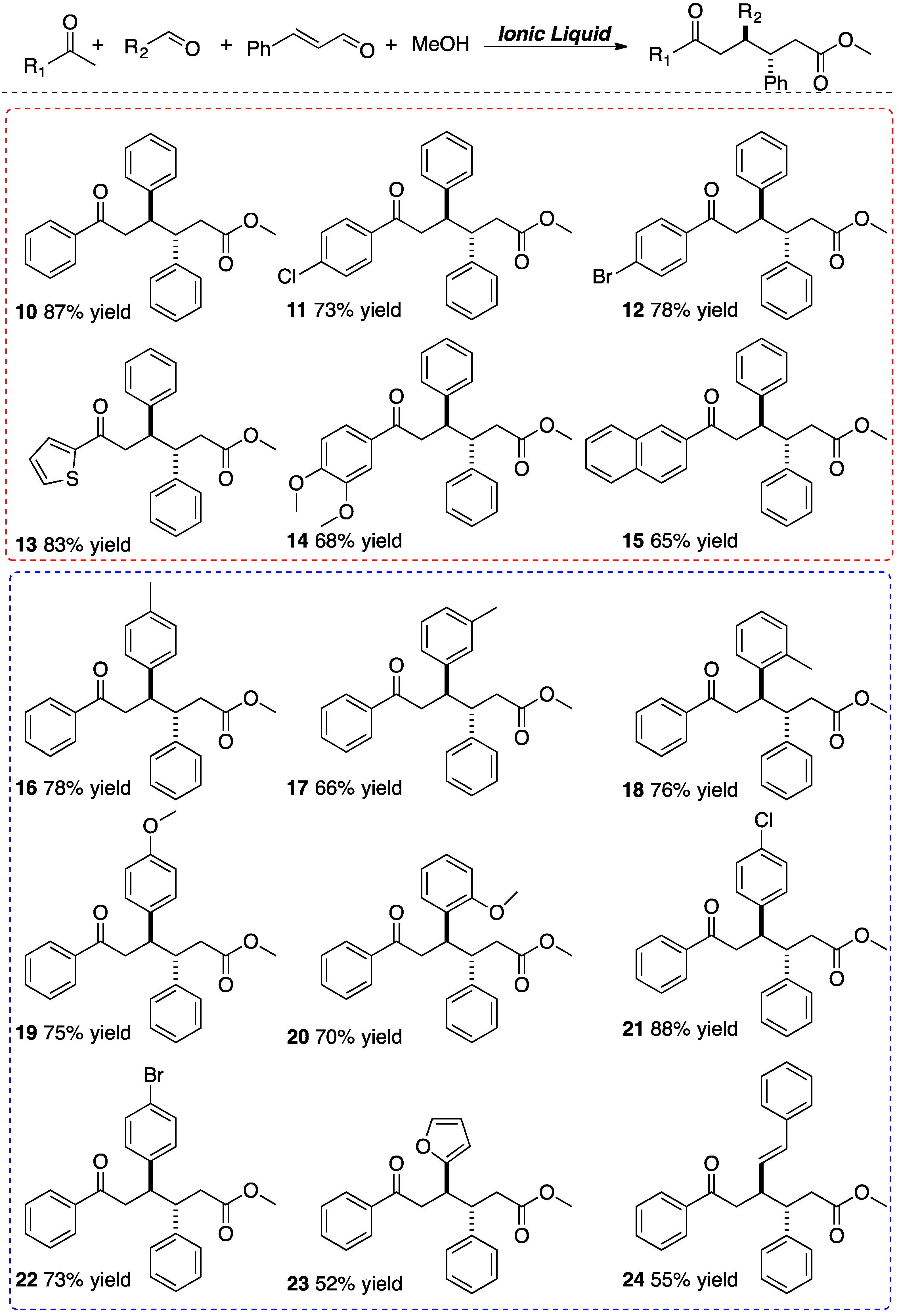

3. Experimental Section
3.1. General Information
3.2. Representative Procedure for the One-Pot Four-Component Synthesis of OTHOs
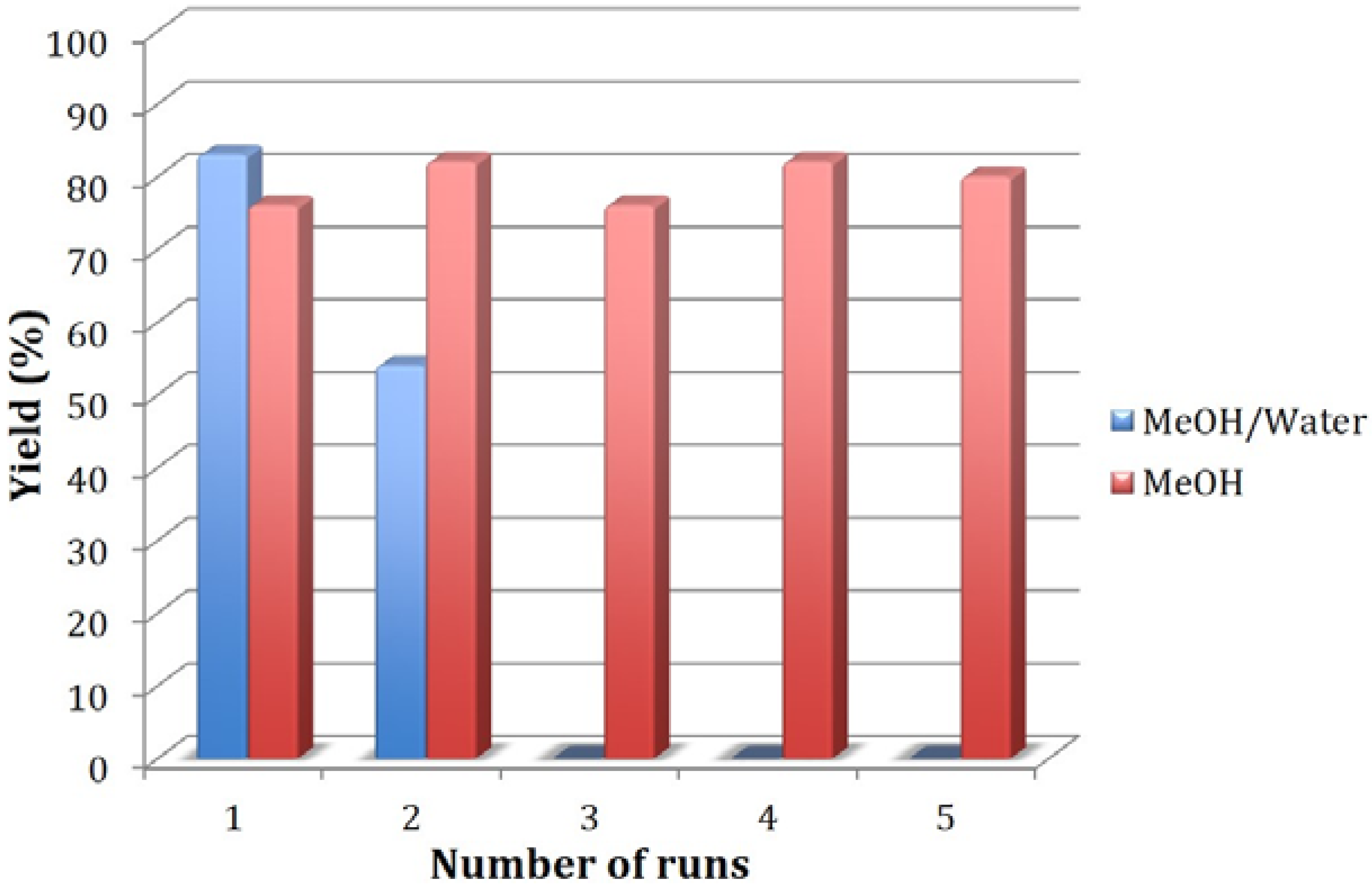
3.3. Procedure for Recycling of EMIMAc
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, Q.; Zhang, S.; Deng, Y. Recent advances in ionic liquid catalysis. Green. Chem. 2011, 13, 2619–2637. [Google Scholar] [CrossRef]
- Closson, A.; Johansson, M.; Backvall, J.-E. Ionic liquid-immobilized catalytic system for biomimetic dihydroxylation of olefins. Chem. Commun. 2004, 7, 1494–1495. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Mi, X.; Zhang, L.; Liu, S.; Xu, H.; Cheng, J.-P. Functionalized chiral ionic liquids as highly efficient asymmetric organocatalysts for michael addition to nitroolefins. Angew. Chem. Int. Ed. 2006, 45, 3093–3097. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Zhang, L.; Cheng, J.-P. Functionalized chiral ionic liquids: A new type of asymmetric organocatalysts and nonclassical chiral ligands. Chem. Asian J. 2009, 4, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Siyutkin, D.E.; Kucherenko, A.S.; Zlotin, S.G. Ionic Liquid Organocatalysts; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp. 617–650. [Google Scholar]
- Zalewska, K.; Branco, L.C. Organocatalysis with Chiral Ionic Liquids. Mini-Rev. Org. Chem. 2014, 11, 141–153. [Google Scholar] [CrossRef]
- Welton, T. Ionic liquids in catalysis. Coord. Chem. Rev. 2004, 248, 2459–2477. [Google Scholar] [CrossRef]
- Welton, T. Room-temperature ionic liquids. Solvents for synthesis and catalysis. Chem. Rev. 1999, 99, 2071–2084. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.A.P.; Frizzo, C.P.; Moreira, D.N.; Zanatta, N.; Bonacorso, H.G. Ionic liquids in heterocyclic synthesis. Chem. Rev. 2008, 108, 2015–2050. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Lancaster, N.L.; Sethi, A.R.; Welton, T. The role of hydrogen bonding in controlling the selectivity of Diels-Alder reactions in room-temperature ionic liquids. Green Chem. 2002, 4, 517–520. [Google Scholar] [CrossRef]
- Gholap, A.R.; Venkatesan, K.; Daniel, T.; Lahoti, R.J.; Srinivasan, K.V. Ultrasound promoted acetylation of alcohols in room temperature ionic liquid under ambient conditions. Green Chem. 2003, 5, 693–696. [Google Scholar] [CrossRef]
- Forsyth, S.A.; MacFarlane, D.R.; Thomson, R.J.; von Itzstein, M. Rapid, clean, and mild O-acetylation of alcohols and carbohydrates in an ionic liquid. Chem. Commun. 2002, 7, 714–715. [Google Scholar] [CrossRef]
- Dubreuil, J.F.; Bazureau, J.P. Rate accelerations of 1,3-dipolar cycloaddition reactions in ionic liquids. Tetrahedron Lett. 2000, 41, 7351–7355. [Google Scholar] [CrossRef]
- Enders, D.; Balensiefer, T. Nucleophilic Carbenes in Asymmetric Organocatalysis. Acc. Chem. Res. 2004, 37, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-heterocyclic carbenes. Chem. Rev. 2007, 107, 5606–5655. [Google Scholar] [CrossRef] [PubMed]
- List, B. Enamine catalysis is a powerful strategy for the catalytic generation and use of carbanion equivalents. Acc. Chem. Res. 2004, 37, 548–557. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Bindu, S.; Sreekumar, V. N-Heterocyclic carbenes: Reagents, not just ligands! Angew. Chem. Int. Ed. 2004, 43, 5130–5135. [Google Scholar] [CrossRef] [PubMed]
- Marion, N.; Díez-González, S.; Nolan, S.P. N-heterocyclic carbenes as organocatalysts. Angew. Chem. Int. Ed. 2007, 46, 2988–3000. [Google Scholar] [CrossRef] [PubMed]
- Alba, A.-N.; Companyo, X.; Viciano, M.; Rios, R. Organocatalytic domino reactions. Curr. Org. Chem. 2009, 13, 1432–1474. [Google Scholar] [CrossRef]
- Biju, A.T.; Kuhl, N.; Glorius, F. Extending NHC-catalysis: Coupling aldehydes with unconventional reaction partners. Acc. Chem. Res. 2011, 44, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Bugaut, X.; Glorius, F. Organocatalytic umpolung: N-heterocyclic carbenes and beyond. Chem. Soc. Rev. 2012, 41, 3511–3522. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.H., Jr.; Forrester, K.J. Thiazolium-ion based organic ionic liquids (OILs).1,2 Novel OILs which promote the benzoin condensation. Tetrahedron Lett. 1999, 40, 1621–1622. [Google Scholar] [CrossRef]
- Xu, L.-W.; Gao, Y.; Yin, J.-J.; Li, L.; Xia, C.-G. Efficient and mild benzoin condensation reaction catalyzed by simple 1-N-alkyl-3-methylimidazolium salts. Tetrahedron Lett. 2005, 46, 5317–5320. [Google Scholar] [CrossRef]
- Jiang, F.S.; Yu, H.; Gao, G.; Xie, R.G. Benzoin condensation in imidazolium based room-temperature ionic liquids. Chin. Chem. Lett. 2005, 16, 321–324. [Google Scholar]
- Estager, J.; Lévêque, J.-M.; Turgis, R.; Draye, M. Solventless and swift benzoin condensation catalyzed by 1-alkyl-3-methylimidazolium ionic liquids under microwave irradiation. J. Mol. Catal. A 2006, 256, 261–264. [Google Scholar] [CrossRef]
- Estager, J.; Lévêque, J.-M.; Turgis, R.; Draye, M. Neat benzoin condensation in recyclable room-temperature ionic liquids under ultrasonic activation. Tetrahedron Lett. 2007, 48, 755–759. [Google Scholar] [CrossRef]
- Orsini, M.; Chiarotto, I.; Elinson, M.N.; Sotgiu, G.; Inesi, A. Benzoin condensation in 1,3-dialkylimidazolium ionic liquids via electrochemical generation of N-heterocyclic carbene. Electrochem. Commun. 2009, 11, 1013–1017. [Google Scholar] [CrossRef]
- Kelemen, Z.; Holloczki, O.; Nagy, J.; Nyulaszi, L. An organocatalytic ionic liquid. Org. Biomol. Chem. 2011, 9, 5362–5364. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.-L.; Zhang, R.-L.; Xie, C.-X.; Yu, S.-T. Synthesis of thermoregulated phase-separable triazolium ionic liquids catalysts and application for Stetter reaction. Tetrahedron 2010, 66, 9145–9150. [Google Scholar] [CrossRef]
- Aupoix, A.; Vo-Thanh, G. Solvent-free synthesis of alkylthiazolium-based ionic liquids and their use as catalysts in the intramolecular Stetter reaction. Synlett 2009, 12, 1915–1920. [Google Scholar]
- Yu, F.-L.; Jiang, J.-J.; Zhao, D.-M.; Xie, C.-X.; Yu, S.-T. Imidazolium chiral ionic liquid derived carbene-catalyzed conjugate umpolung for synthesis of γ-butyrolactones. RSC Adv. 2013, 3, 3996–4000. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, Y.; Chen, E.Y.X. Organocatalytic upgrading of the key biorefining building block by a catalytic ionic liquid and N-heterocyclic carbenes. Green Chem. 2012, 14, 2738–2746. [Google Scholar] [CrossRef]
- Feroci, M.; Chiarotto, I.; Orsini, M.; Pelagalli, R.; Inesi, A. Umpolung reactions in an ionic liquid catalyzed by electrogenerated N-heterocyclic carbenes. Synthesis of saturated esters from activated α, β-unsaturated aldehydes. Chem. Commun. 2012, 48, 5361–5363. [Google Scholar] [CrossRef] [PubMed]
- Forte, G.; Chiarotto, I.; Inesi, A.; Loreto, M.A.; Feroci, M. Electrogenerated N-heterocyclic carbene in ionic liquid: An insight into the mechanism of the oxidative esterification of aromatic aldehydes. Adv. Synth. Catal. 2014. [Google Scholar] [CrossRef]
- Chiarotto, I.; Feroci, M.; Sotgiu, G.; Inesi, A. Electrogenerated N-heterocyclic carbenes in the room temperature parent ionic liquid as an efficient medium for transesterification/acylation reactions. Eur. J. Org. Chem. 2013, 2013, 326–331. [Google Scholar] [CrossRef]
- Constable, D.J.C.; Jimenez-Gonzalez, C.; Henderson, R.K. Perspective on solvent use in the pharmaceutical industry. Org. Process Res. Dev. 2007, 11, 133–137. [Google Scholar] [CrossRef]
- Jiménez-González, C.; Curzons, A.; Constable, D.C.; Cunningham, V. Expanding GSK’s solvent selection guide—Application of life cycle assessment to enhance solvent selections. Clean Technol. Environ. Policy 2004, 7, 42–50. [Google Scholar] [CrossRef]
- Dunn, P.J. The importance of green chemistry in process research and development. Chem. Soc. Rev. 2012, 41, 1452–1461. [Google Scholar] [CrossRef] [PubMed]
- Dömling, A.; Ugi, I. Multicomponent reactions with isocyanides. Angew. Chem. Int. Ed. 2000, 39, 3168–3210. [Google Scholar] [CrossRef]
- Ta , L.; Axelsson , A.; Bijl, J.; Haukka, M.; Sundén, H. Ionic liquids as precatalysts in the highly stereoselective conjugate addition of α,β-unsaturated aldehydes to chalcones. Chem. Eur. J. 2014, 20, 13889–13893. [Google Scholar] [CrossRef] [PubMed]
- Sundén, H.; Ta, L.; Axelsson, A. Highly stereoselective synthesis of 1,6-ketoesters mediated by ionic liquids: A three-component reaction enabling rapid access to a new class of low molecular weight gelators. J. Vis. Exp. 2015, e53213. [Google Scholar] [CrossRef]
- Holloczki, O.; Gerhard, D.; Massone, K.; Szarvas, L.; Nemeth, B.; Veszpremi, T.; Nyulaszi, L. Carbenes in ionic liquids. New J. Chem. 2010, 34, 3004–3009. [Google Scholar] [CrossRef]
- Guo, C.; Fleige, M.; Janssen-Müller, D.; Daniliuc, C.G.; Glorius, F. Switchable selectivity in an NHC-catalysed dearomatizing annulation reaction. Nat. Chem. 2015, 7, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Feroci, M.; Chiarotto, I.; D’Anna, F.; Forte, G.; Noto, R.; Inesi, A. Stability and organocatalytic efficiency of N-heterocyclic carbenes electrogenerated in organic solvents from imidazolium ionic liquids. Electrochim. Acta 2015, 153, 122–129. [Google Scholar] [CrossRef]
- Wendler, F.; Todi, L.-N.; Meister, F. Thermostability of imidazolium ionic liquids as direct solvents for cellulose. Thermochim. Acta 2012, 528, 76–84. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Axelsson, A.; Ta, L.; Sundén, H. Ionic Liquids as Carbene Catalyst Precursors in the One-Pot Four-Component Assembly of Oxo Triphenylhexanoates (OTHOs). Catalysts 2015, 5, 2052-2067. https://doi.org/10.3390/catal5042052
Axelsson A, Ta L, Sundén H. Ionic Liquids as Carbene Catalyst Precursors in the One-Pot Four-Component Assembly of Oxo Triphenylhexanoates (OTHOs). Catalysts. 2015; 5(4):2052-2067. https://doi.org/10.3390/catal5042052
Chicago/Turabian StyleAxelsson, Anton, Linda Ta, and Henrik Sundén. 2015. "Ionic Liquids as Carbene Catalyst Precursors in the One-Pot Four-Component Assembly of Oxo Triphenylhexanoates (OTHOs)" Catalysts 5, no. 4: 2052-2067. https://doi.org/10.3390/catal5042052