
Dimethyl-Aluminium Complexes Bearing Naphthyl-Substituted Pyridine-Alkylamides as Pro-Initiators for the Efficient ROP of ε-Caprolactone


Abstract
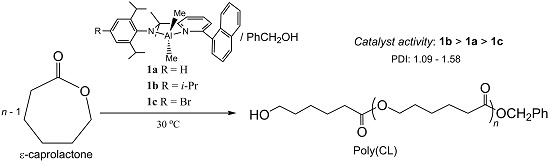
:
1. Introduction

2. Results and Discussion
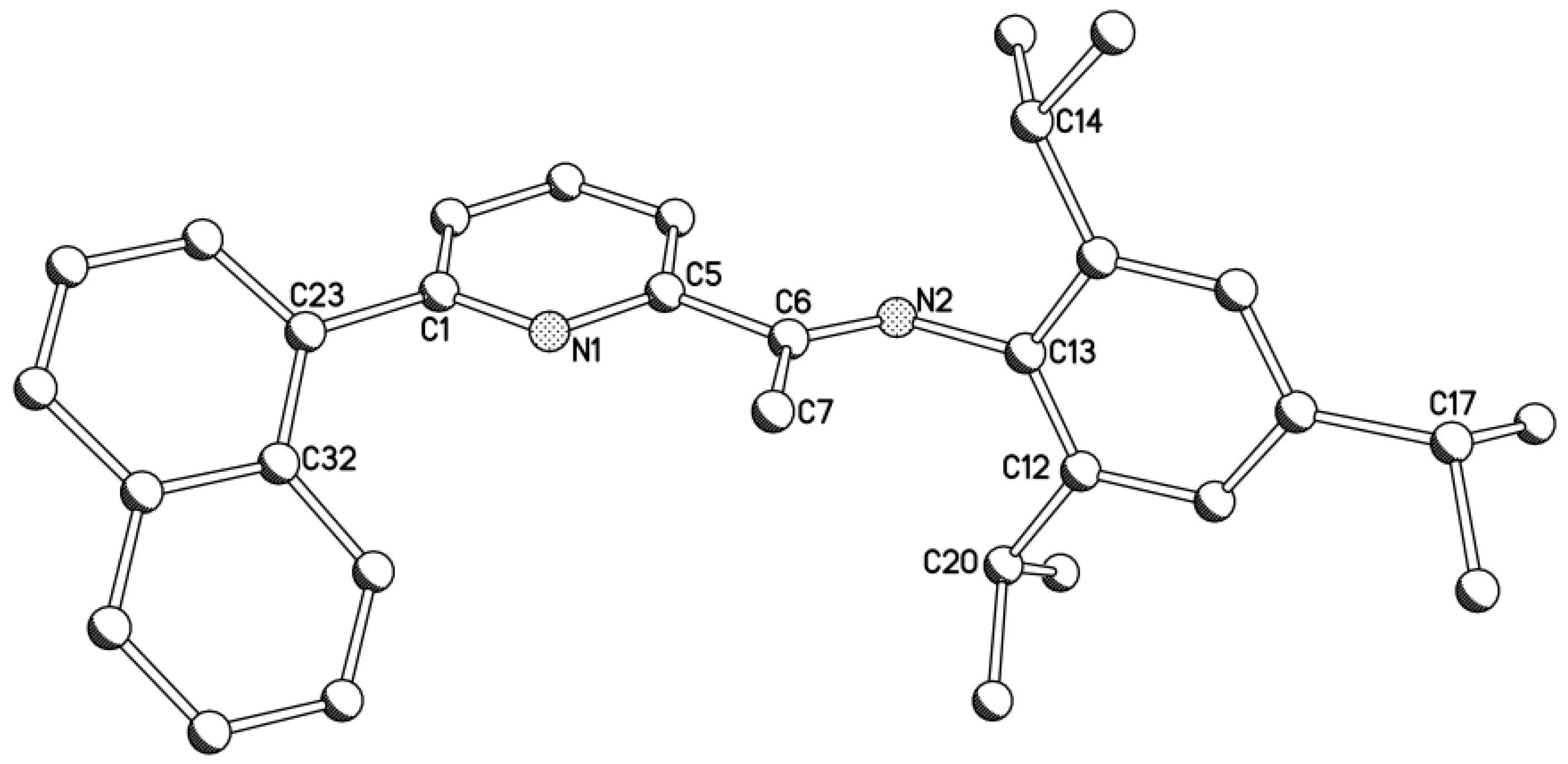
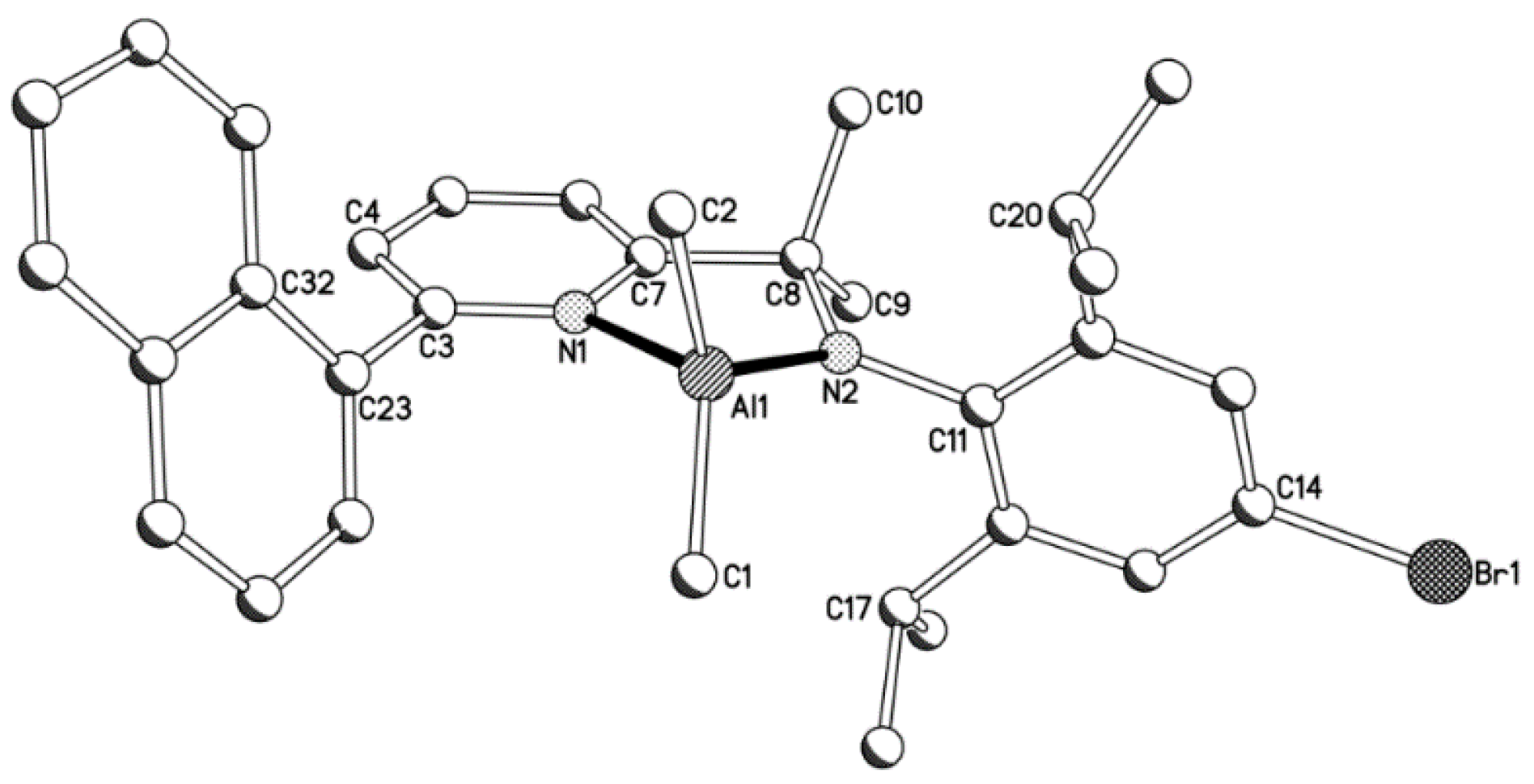
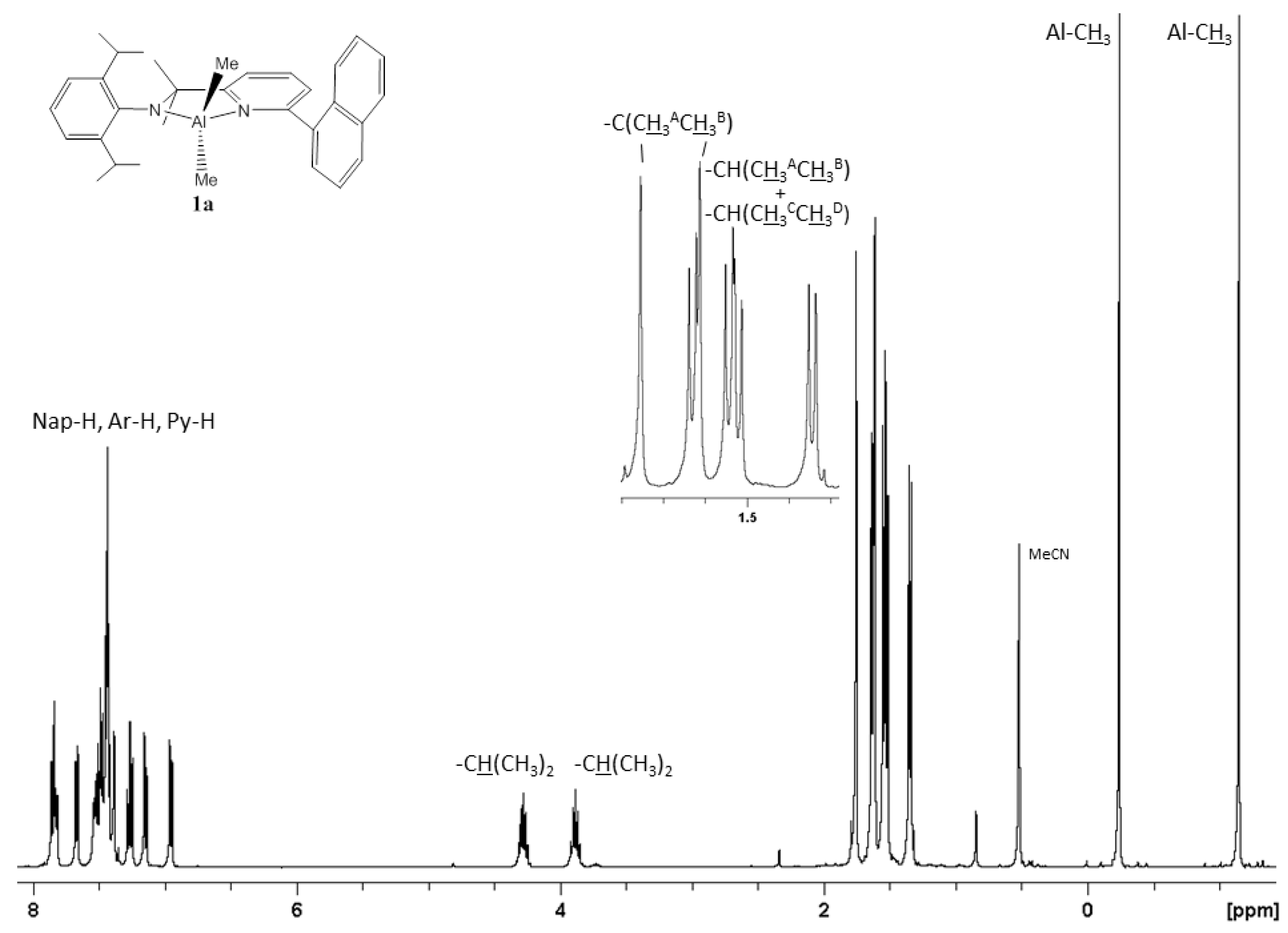
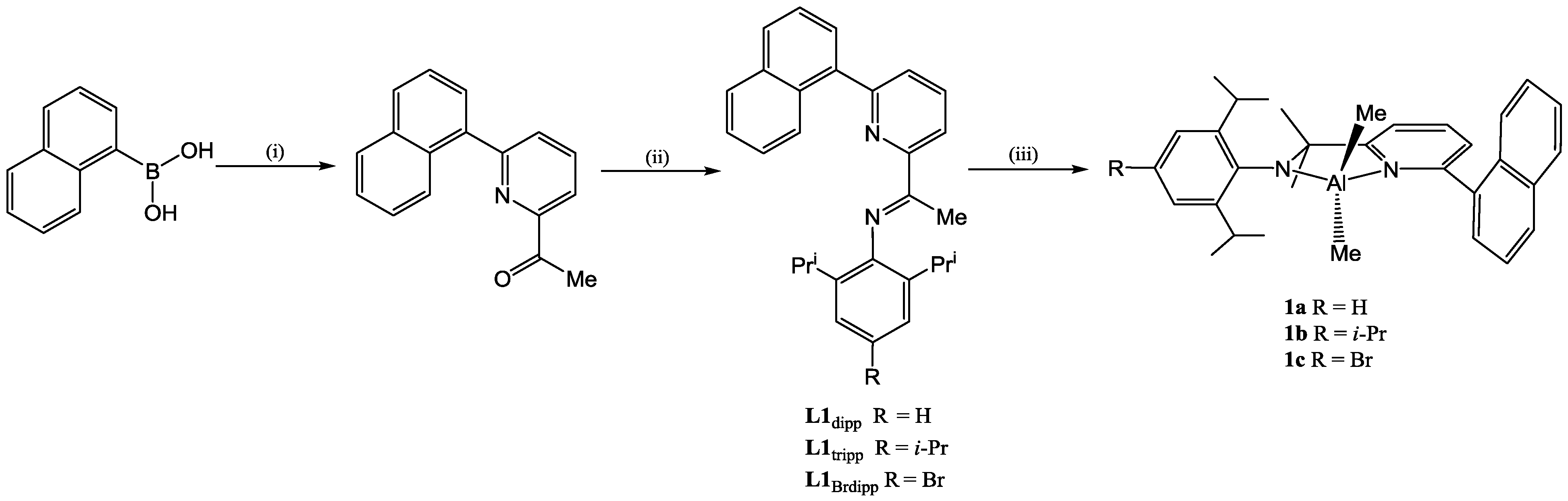
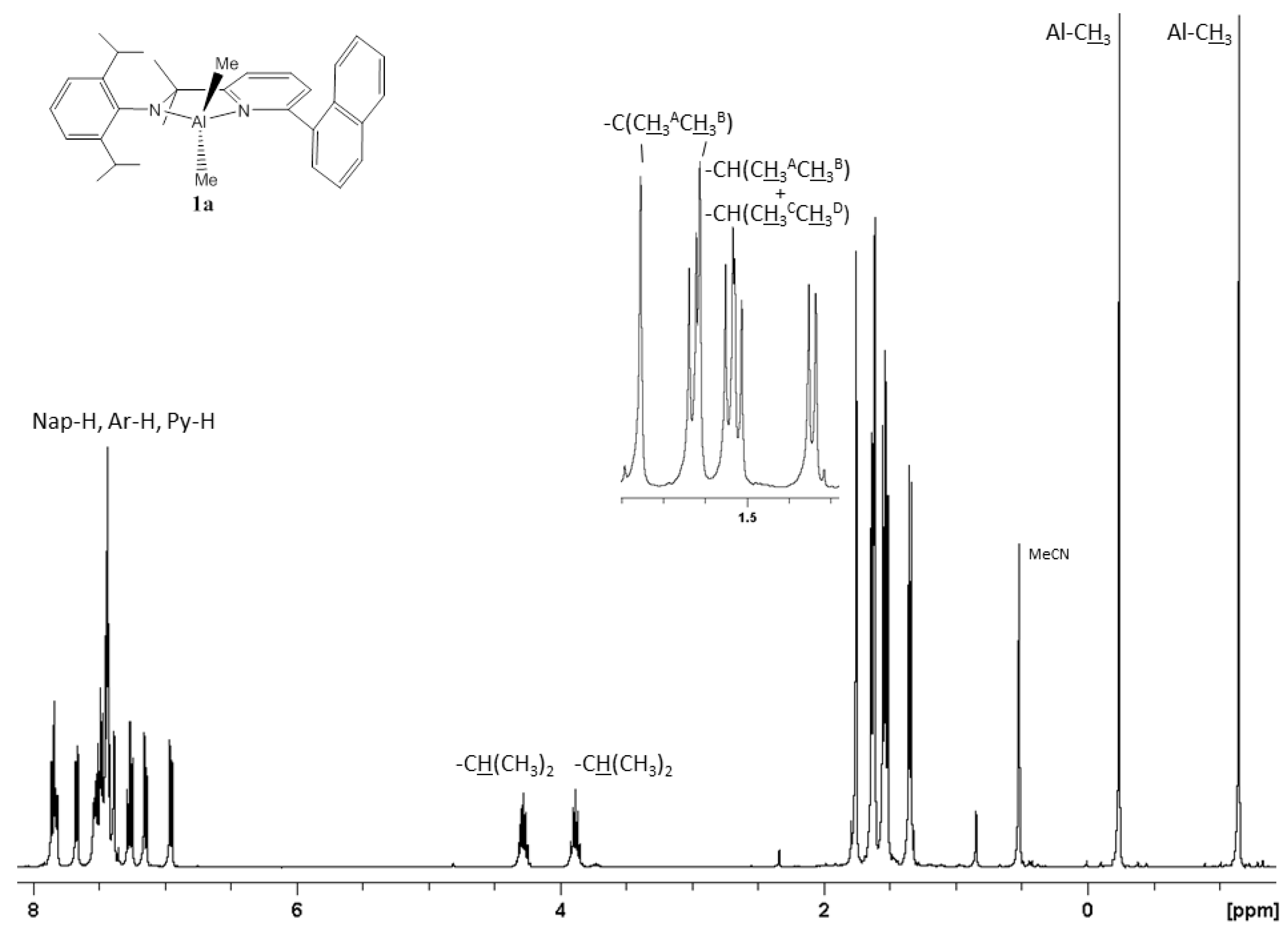
2.1. Synthetic and Structural Aspects

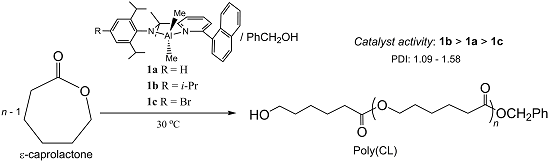{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Bond Distances (Å) | Bond Angles (deg) | ||||||
|---|---|---|---|---|---|---|---|---|
| C(6)–N(2) | C(6)–C(7) | C(1)–C(23) | C(5)–C(6) | N(2)–C(13) | C(6)–N(2)–C(13) | C(7)–C(6)–N(2) | ||
| L1dipp | 1.282(3) | 1.496(4) | 1.503(3) | 1.488(4) | 1.419(3) | 120.6(2) | 125.8(2) | |
| L1tripp | 1.280(5) | 1.488(6) | 1.482(6) | 1.502(5) | 1.430(5) | 121.2(4) | 126.5(4) | |
| Complex | Bond Distances (Å) | |||||||
|---|---|---|---|---|---|---|---|---|
| Al(1)–C(1) | Al(1)–C(2) | Al(1)–N(1) | Al(1)–N(2) | C(8)–N(2) | C(3)–C(23) | C(11)–N(2) | C(14)–R | |
| 1a | 1.950(2) | 1.990(2) | 2.003(2) | 1.838(2) | 1.471(3) | 1.490(3) | 1.429(3) | - |
| 1b | 1.968(6) | 1.961(6) | 1.989(5) | 1.829(4) | 1.482(6) | 1.554(7) | 1.444(6) | 1.518(7) (R = C(33)i-Pr) |
| 1c | 1.936(6) | 1.971(6) | 1.997(5) | 1.824(5) | 1.458(7) | 1.486(8) | 1.440(7) | 1.893(6) (R = Br(1)) |
| Complex | Bond Angles (deg) | |||||||
| C(1)–Al(1)–C(2) | C(1)–Al(1)–N(1) | C(1)–Al(1)–N(2) | C(2)–Al(1)–N(1) | C(2)–Al(1)–N(2) | N(1)–Al(1)–N(2) | C(7)–C(8)–N(2) | ||
| 1a | 113.55(11) | 119.55(10) | 117.79(10) | 98.75(10) | 118.79(10) | 83.40(8) | 106.31(17) | |
| 1b | 111.1(3) | 114.3(2) | 117.5(2) | 106.3(2) | 119.0(3) | 85.4(2) | 107.9(4) | |
| 1c | 113.5(3) | 119.9(3) | 113.4(3) | 101.3(2) | 121.3(2) | 83.9(2) | 107.0(5) | |
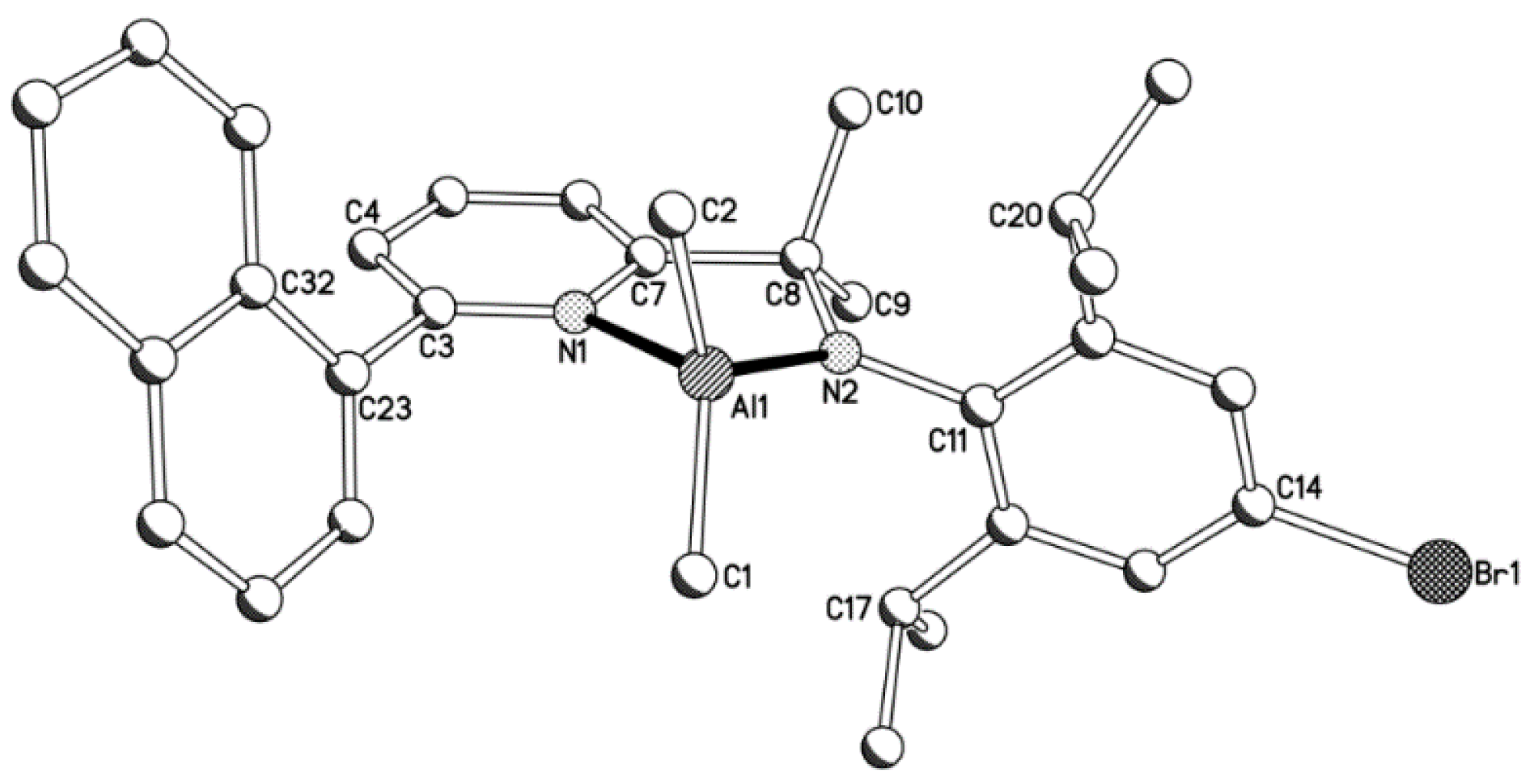
2.2. Polymerization Results
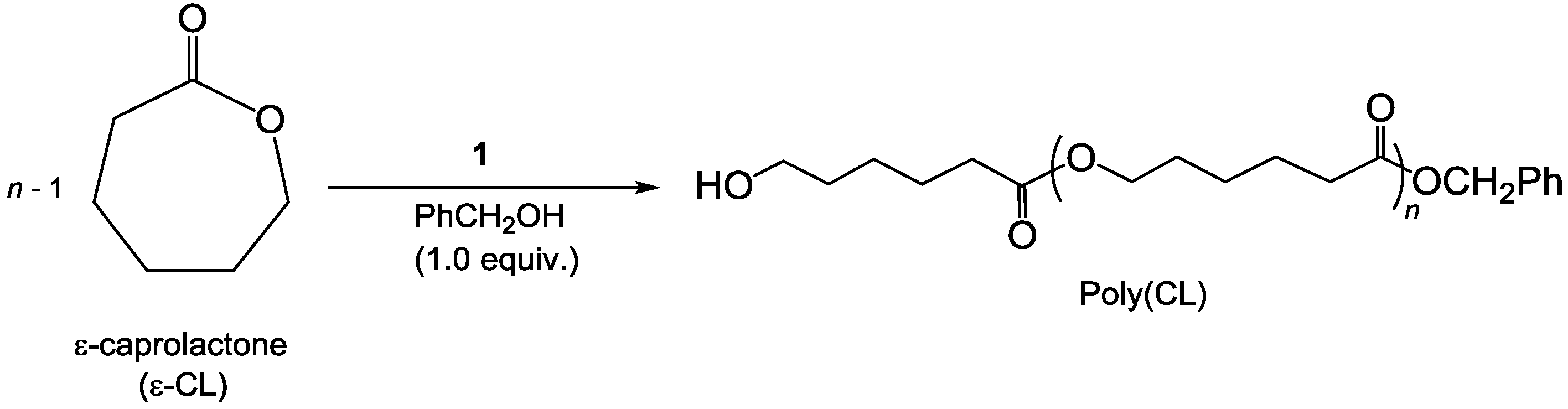
| Entry | Pro-Initiator (R) | Temp./°C | Time/min | Conversion/% b | Mn (SEC) c | Mn (Calcd) d | Đ |
|---|---|---|---|---|---|---|---|
| 1 | 1a (H) | 30 | 30 | 18 | 6750 | 5340 | 1.33 |
| 2 | 1a (H) | 30 | 60 | 36 | 12180 | 10370 | 1.58 |
| 3 | 1a (H) | 30 | 90 | 40 | 11740 | 11510 | 1.57 |
| 4 | 1a (H) | 30 | 120 | 59 | 19010 | 16920 | 1.38 |
| 5 | 1b (i-Pr) | 30 | 30 | 35 | 8700 | 9990 | 1.47 |
| 6 | 1b (i-Pr) | 30 | 60 | 60 | 16540 | 17210 | 1.39 |
| 7 | 1b (i-Pr) | 30 | 90 | 73 | 18850 | 20910 | 1.46 |
| 8 | 1b (i-Pr) | 30 | 120 | 80 | 20790 | 22910 | 1.39 |
| 9 | 1c (Br) | 30 | 30 | 13 | 4250 | 3810 | 1.14 |
| 10 | 1c (Br) | 30 | 60 | 25 | 8270 | 7230 | 1.14 |
| 11 | 1c (Br) | 30 | 90 | 34 | 11060 | 9800 | 1.09 |
| 12 | 1c (Br) | 30 | 120 | 38 | 10810 | 10940 | 1.24 |
| 13 | 1b (i-Pr) | 50 | 30 | 86 | 20270 | 24620 | 1.65 |
| 14 | 1b (i-Pr) | 50 | 60 | 100 | 28880 | 28610 | 1.61 |

3. Experimental Section
3.1. General Procedures
3.2. Synthesis of 2-{CMe=O}-6-(1-C10H7)C5H3N
3.3. Synthesis of 2-{CMe=N(Ar)}-6-(1-C10H7)C5H3N
3.3.1. Ar = 2,6-i-Pr2C6H3 (L1dipp)
3.3.2. Ar = 2,4,6-i-Pr3C6H2 (L1tripp)
3.3.3. Ar = 4-Br-2,6-i-Pr2C6H2 (L1Brdipp)
3.4. Synthesis of Complexes [2-{CMe2N(Ar)}-6-(1-C10H7)C5H3N]AlMe2 (1)
3.4.1. Ar = 2,6-i-Pr2C6H3 (1a)
3.4.2. Ar = 2,4,6-i-Pr3C6H2 (1b)
3.4.3. Ar = 4-Br-2,6-i-Pr2C6H2 (1c)
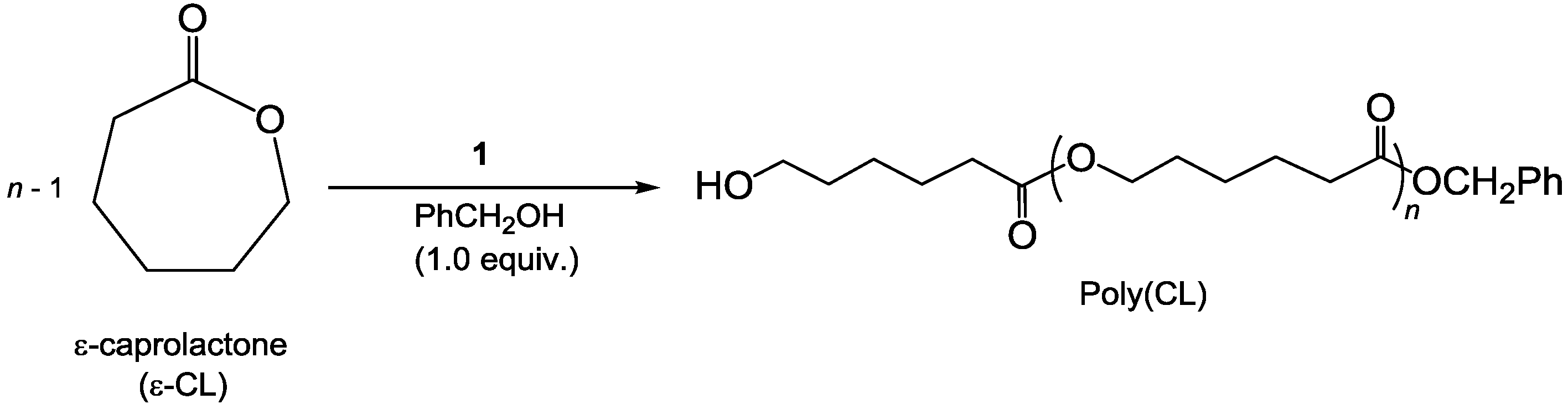
3.5. Procedure for ROP and SEC Details
3.5.1. Catalytic Evaluation
3.5.2. Size Exclusion Chromatography
3.6. Crystallographic Studies
| Complex | L1dipp | L1tripp | 1a | 1b | 1c |
|---|---|---|---|---|---|
| Formula | C29H30N2 | C32H36N2 | C32H39N2Al | C35H45N2Al | C32H40BrN2OAl·1.5OH2 |
| M | 406.55 | 448.63 | 478.63 | 520.71 | 584.56 |
| Crystal size (mm3) | 0.26 × 0.22 × 0.19 | 0.42 × 0.16 × 0.04 | 0.40 × 0.16 × 0.12 | 0.21 × 0.16 × 0.15 | 0.16 × 0.12 × 0.05 |
| Temperature (K) | 150 (2) | 150 (2) | 150 (2) | 150 (2) | 150 (2) |
| Crystal system | monoclinic | triclinic | triclinic | monoclinic | monoclinic |
| Space group | P2(1)/c | P-1 | P-1 | P2 (1)/n | P2 (1)/c |
| a (Å) | 11.490 (8) | 8.991 (4) | 10.662 (4) | 12.133 (5) | 10.967 (4) |
| b (Å) | 13.214 (9) | 11.022 (5) | 10.809 (4) | 17.302 (7) | 15.832 (6) |
| c (Å) | 15.214 (10) | 13.139 (6) | 13.680 (5) | 14.609 (6) | 16.957 (7) |
| α (o) | 90 | 95.509 (8) | 95.348 (7) | 90 | 90 |
| β (o) | 95.243 (13) | 98.517 (10) | 104.935 (7) | 91.243 (9) | 96.335 (9) |
| γ (o) | 90 | 96.640 (9) | 113.807 (6) | 90 | 90 |
| U (Å3) | 2300 (3) | 1270.6 (9) | 1358.5 (9) | 3066 (2) | 2926.3 (19) |
| Z | 4 | 2 | 2 | 4 | 4 |
| Dc (Mg m−3) | 1.174 | 1.173 | 1.170 | 1.128 | 1.327 |
| F(000) | 872 | 484 | 516 | 1128 | 1228 |
| μ (Mo-Kα)(mm−1) | 0.068 | 0.068 | 0.097 | 0.091 | 1.464 |
| Reflections collected | 16270 | 9445 | 10786 | 22006 | 21151 |
| Independent reflections | 4050 | 4460 | 5288 | 5402 | 5145 |
| Rint | 0.2956 | 0.1405 | 0.0603 | 0.2260 | 0.2420 |
| Restraints/parameters | 0/285 | 0/314 | 0/720 | 94/353 | 0/333 |
| Final R indices (I > 2σ(I)) | R1 = 0.0754 wR2 = 0.1433 | R1 = 0.0738 wR2 = 0.1331 | R1 = 0.0578 wR2 = 0.1109 | R1 = 0.0897 wR2 = 0.1838 | R1 = 0.0750 wR2 = 0.1266 |
| All data | R1 = 0.1283 wR2 = 0.1703 | R1 = 0.1990 wR2 = 0.1732 | R1 = 0.0917 wR2 = 0.1235 | R1 = 0.2281 wR2 = 0.2226 | R1 = 0.1690 wR2 = 0.1504 |
| Goodness of fit on F2 (all data) | 0.929 | 0.793 | 0.913 | 0.833 | 0.822 |
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- O’Keefe, B.J.; Hillmyer, M.A.; Tolman, W.B. Polymerization of lactide and related cyclic esters by discrete metal complexes. Dalton Trans. 2001. [Google Scholar] [CrossRef]
- Dechy-Cabaret, O.; Martin-Vaca, B.; Bourissou, D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004, 104, 6147–6176. [Google Scholar] [CrossRef] [PubMed]
- Stridsberg, K.M.; Ryner, M.; Albertsson, A.-C. Controlled Ring-Opening Polymerization: Polymers with designed Macromolecular Architecture. Adv. Polym. Sci. 2002, 157, 41–65. [Google Scholar]
- Wu, J.; Yu, T.-L.; Chen, C.-T.; Lin, C.-C. Recent developments in main group metal complexes catalyzed/initiated polymerization of lactides and related cyclic esters. Coord. Chem. Rev. 2006, 250, 602–626. [Google Scholar] [CrossRef]
- Arbaoui, A.; Redshaw, C. Metal catalysts for ε-caprolactone polymerization. Polym. Chem. 2010, 1, 801–826. [Google Scholar] [CrossRef]
- Aida, T.; Inoue, S. Formation of Poly(lactide) with Controlled Molecular Weight. Polymerization of Lactide by Aluminum Porphyrin. Chem. Lett. 1987, 16, 991–994. [Google Scholar]
- Cameron, P.A.; Jhurry, D.; Gibson, V.C.; White, A.J.P.; Williams, D.J.; Williams, S. Controlled polymerization of lactides at ambient temperature using [5-Cl-salen]AlOMe. Macromol. Rapid Commun. 1999, 20, 616–618. [Google Scholar] [CrossRef]
- Hormnirun, P.; Marshall, E.L.; Gibson, V.C.; White, A.J.P.; Williams, D.J. Remarkable Stereocontrol in the Polymerization of Racemic Lactide Using Aluminum Initiators Supported by Tetradentate Aminophenoxide Ligands. J. Am. Chem. Soc. 2004, 126, 2688–2689. [Google Scholar] [CrossRef] [PubMed]
- Hormnirun, P.; Marshall, E.L.; Gibson, V.C.; Pugh, R.I.; White, A.J.P. A study of ligand substituent effects on the rate and stereoselectivity of lactide polymerization using aluminum salen-type initiators. Proc. Natl. Acad. Sci. USA 2006, 103, 15343–15348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spassky, N.; Wisniewski, M.; Pluta, C.; le Borgne, A. Highly stereoelective polymerization of rac-(d,l)-lactide with a Chiral Schiff’s base/aluminium alkoxide initiator. Macromol. Chem. Phys. 1996, 197, 2627–2637. [Google Scholar] [CrossRef]
- Ovitt, T.M.; Coates, G.W. Stereoselective Ring-Opening Polymerization of meso-Lactide: Synthesis of Syndiotactic Poly (lactic acid). J. Am. Chem. Soc. 1999, 121, 4072–4073. [Google Scholar] [CrossRef]
- Ovitt, T.M.; Coates, G.W. Stereoselective Ring-Opening Polymerization of rac-Lactide with a Single-Site, Racemic Aluminum Alkoxide Catalyst: Synthesis of Stereoblock Poly (lactic acid). J. Polym. Sci. Polym. Chem. 2000, 38, 4686–4692. [Google Scholar] [CrossRef]
- Radano, C.P.; Baker, G.L.; Smith, M.R., III. Stereoselective Polymerization of a Racemic Monomer with a Racemic Catalyst: Direct Preparation of the Polylactic Acid Stereocomplex from Racemic Lactide. J. Am. Chem. Soc. 2000, 122, 1552–1553. [Google Scholar] [CrossRef]
- Zhong, Z.; Dijkstra, P.J.; Feijen, J. [(salen)Al]-Mediated, Controlled and Stereoselective Ring-Opening Polymerization of Lactide in Solution and without Solvent: Synthesis of Highly Isotactic Polylactide Stereocopolymers from Racemic d,l-Lactide. Angew. Chem. Int. 2002, 41, 4510–4513. [Google Scholar] [CrossRef]
- Nomura, N.; Ishii, R.; Akakura, M.; Aoi, K. Stereoselective ring-opening polymerization of racemic lactide using aluminum-achiral ligand complexes: Exploration of a chain-end control mechanism. J. Am. Chem. Soc. 2002, 124, 5938–5939. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Pang, X.; Yu, H.; Zhang, X.; Chen, X.; Cui, D.; Wang, X.; Jing, X. Polymerization of rac-Lactide Using Schiff Base Aluminum Catalysts: Structure, Activity, and Stereoselectivity. Macromolecules 2007, 40, 1904–1913. [Google Scholar] [CrossRef]
- Alcazar-Roman, L.M.; O’Keefe, B.J.; Hillmyer, M.A.; Tolman, W.B. Electronic influence of ligand substituents on the rate of polymerization of ε-caprolactone by single-site aluminium alkoxide catalysts. Dalton Trans. 2003. [Google Scholar] [CrossRef]
- Amgoune, A.; Lavanant, L.; Thomas, C.M.; Chi, Y.; Welter, R.; Dagorne, S.; Carpentier, J.-F. An Aluminum Complex Supported by a Fluorous Diamino-Dialkoxide Ligand for the Highly Productive Ring-Opening Polymerization of ε-Caprolactone. Organometallics 2005, 24, 6279–6282. [Google Scholar] [CrossRef]
- Chen, C.-T.; Huang, C.-A.; Huang, B.-H. Aluminium metal complexes supported by amine bis-phenolate ligands as catalysts for ring-opening polymerization of ε-caprolactone. Dalton Trans. 2003. [Google Scholar] [CrossRef]
- Shen, M.; Zhang, W.; Nomura, K.; Sun, W.-H. Synthesis and characterization of organoaluminum compounds containing quinolin-8-amine derivatives and their catalytic behaviour for ring-opening polymerization of ε-caprolactone. Dalton Trans. 2009. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, J.; Horeglad, P.; Dranka, M.; Justyniak, I. Simple Generation of Cationic Aluminum Alkyls and Alkoxides Based on the Pendant Arm Tridentate Schiff Base. Inorg. Chem. 2004, 43, 5789–5791. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.-A.; Wang, Z.-X. Zinc and Aluminum Complexes Supported by Quinoline-Based N,N,N-Chelate Ligands: Synthesis, Characterization, and Catalysis in the Ring-Opening Polymerization of ε-Caprolactone and rac-Lactide. Organometallics 2011, 30, 4364–4373. [Google Scholar] [CrossRef]
- Peng, K.-F.; Chen, C.-T. Synthesis and catalytic application of aluminium anilido-pyrazolate complexes. Dalton Trans. 2009. [Google Scholar] [CrossRef] [PubMed]
- Bakthavachalam, K.; Reddy, N.D. Synthesis of Aluminum Complexes of Triaza Framework Ligands and Their Catalytic Activity toward Polymerization of ε-Caprolactone. Organometallics 2013, 32, 3174–3184. [Google Scholar] [CrossRef]
- Gao, W.; Zhang, J.; Luo, X.; Liu, X.; Mu, Y. Al and Zn complexes bearing N,N,N-tridentate quinolinyl anilido-imine ligands: Synthesis, characterization and catalysis in l-lactide polymerization. Dalton Trans. 2012, 41, 11454–11463. [Google Scholar]
- Sun, W.-H.; Shen, M.; Zhang, W.; Huang, W.; Liu, S.; Redshaw, C. Methylaluminium 8-quinolinolates: Synthesis, characterization and use in ring-opening polymerization (ROP) of ε-caprolactone. Dalton Trans. 2011, 40, 2645–2653. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.-A.; Wang, Z.-X. Synthesis and characterisation of aluminium (III) and tin (II) complexes bearing quinoline-based N,N,O-tridentate ligands and their catalysis in the ring-opening polymerisation of ε-caprolactone. Dalton Trans. 2011, 40, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.-L.; Chaia, Z.-Y.; Wang, Z.-X. Synthesis of N,N,O-chelate zinc and aluminum complexes and their catalysis in the ring-opening polymerization of ε-caprolactone and rac-lactide. Dalton Trans. 2014, 43, 14470–14480. [Google Scholar] [CrossRef] [PubMed]
- Normand, M.; Dorcet, V.; Kirillov, E.; Capentier, J.-F. {Phenoxy-imine} aluminum vs. -indium Complexes for the Immortal ROP of Lactide: Different Stereocontrol, Different Mechanisms. Organometallics 2013, 32, 1694–1709. [Google Scholar] [CrossRef]
- Gong, S.; Ma, H. β-Diketiminate aluminium complexes: Synthesis, characterization and ring-opening polymerization of cyclic esters. Dalton Trans. 2008. [Google Scholar] [CrossRef] [PubMed]
- Yao, W.; Mu, Y.; Gao, A.; Su, Q.; Liu, Y.; Zhang, Y. Efficient ring-opening polymerization of ɛ-caprolactone using anilido-imine-aluminum complexes in the presence of benzyl alcohol. Polymer 2008, 49, 2486–2491. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, Y.; Cao, J.; Wang, L.; Redshaw, C.; Sun, W.-H. Synthesis and Characterization of Dialkylaluminum Amidates and Their Ring-Opening Polymerization of ε-Caprolactone. Organometallics 2011, 30, 6253–6261. [Google Scholar] [CrossRef]
- Shen, M.; Huang, W.; Zhang, W.; Hao, X.; Sun, W.-H.; Redshaw, C. Synthesis and characterisation of alkylaluminium benzimidazolates and their use in the ring-opening polymerisation of ε-caprolactone. Dalton Trans. 2010, 39, 9912–9922. [Google Scholar] [CrossRef] [PubMed]
- Kampova, H.; Riemlova, E.; Klikarova, J.; Pejchal, V.; Merna, J.; Vlasak, P.S.; Svec, P.; Ruzickova, Z.; Ruzicka, A. Aluminium complexes containing N,N’-chelating amino-amide hybrid ligands applicable for preparation of biodegradable polymers. J. Organomet. Chem. 2015, 778, 35–41. [Google Scholar] [CrossRef]
- Iwasa, N.; Katao, S.; Liu, J.; Fujiki, M.; Furukawa, Y.; Nomura, K. Notable Effect of Fluoro Substituents in the Imino Group in Ring-Opening Polymerization of ε-Caprolactone by Al Complexes Containing Phenoxyimine Ligands. Organometallics 2009, 28, 2179–2187. [Google Scholar] [CrossRef]
- Liu, J.; Iwasa, N.; Nomura, K. Synthesis of Al complexes containing phenoxy-imine and their use as the precursors for efficient living of ε-caprolactone. Dalton Trans. 2008. [Google Scholar] [CrossRef]
- Tseng, H.-T.; Chiang, M.Y.; Lu, W.-Y.; Chen, Y.-J.; Lian, C.-J.; Chen, Y.-H.; Tsai, H.-Y.; Lai, Y.-C.; Chen, H.-Y. A closer look at ε-caprolactone polymerization catalyzed by alkyl aluminum complexes: The effect of induction period on overall catalytic activity. Dalton Trans. 2015, 44, 11763–11773. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.-C.; Huang, C.-H.; Huang, J.-H.; Lee, H.-Y.; Chen, J.-T. Four- and Five-Coordinate Aluminum Ketiminate Complexes: Synthesis, Characterization, and Ring-Opening Polymerization. Inorg. Chem. 2002, 41, 6450–6455. [Google Scholar] [CrossRef] [PubMed]
- Nomura, N.; Aoyama, T.; Ishii, R.; Kondo, T. Salicylaldimine-Aluminum Complexes for the Facile and Efficient Ring-Opening Polymerization of ε-Caprolactone. Macromolecules 2005, 38, 5363–5366. [Google Scholar] [CrossRef]
- Bouyayi, M.; Roisnel, T.; Carpentier, J.-F. Aluminum Complexes of Bidentate Fluorinated Alkoxy-Imino Ligands: Syntheses, Structures, and Use in Ring-Opening Polymerization of Cyclic Esters. Organometallics 2012, 31, 1458–1466. [Google Scholar] [CrossRef]
- Chapurina, Y.; Roisnel, T.; Carpentier, J.-F.; Kirillov, E. Zinc, aluminum and group 3 metal complexes of sterically demanding naphthoxy-pyridine ligands: Synthesis, structure, and use in ROP of racemic lactide and β-butyrolactone. Inorg. Chim. Acta 2015, 431, 161–175. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhao, K.-Q.; Elsegood, M.R.J.; Prior, T.J.; Sun, X.; Mo, S.; Redshaw, C. Organoaluminium complexes of ortho-, meta-, para-anisidines: Synthesis, structural studies and ROP of ε-caprolactone (and rac-lactide). Catal. Sci. Technol. 2014, 4, 3025–3031. [Google Scholar] [CrossRef]
- Myers, T.W.; Berben, L.A. A sterically demanding iminopyridine ligand affords redox-active complexes of aluminum(III) and gallium(III). Inorg. Chem. 2012, 51, 1480–1488. [Google Scholar] [CrossRef] [PubMed]
- Budzelaar, P.H.M. Radical Chemistry of Iminepyridine Ligands. Eur. J. Inorg. Chem. 2012, 2012, 530–534. [Google Scholar] [CrossRef]
- Nienkemper, K.; Kehr, G.; Kehr, S.; Froehlich, R.; Erker, G. (Amidomethyl) pyridine zirconium and hafnium complexes: Synthesis and structural characterization. J. Organomet. Chem. 2008, 693, 1572–1589. [Google Scholar] [CrossRef]
- Gibson, V.C.; Redshaw, C.; White, A.J.P.; Williams, D.J. Synthesis and structural characterisation of aluminium imino-amide and pyridyl-amide complexes: Bulky monoanionic N,N chelate ligands via methyl group transfer. J. Organomet. Chem. 1998, 550, 453–456. [Google Scholar] [CrossRef]
- Wei, Y.; Wang, S.; Zhou, S.; Feng, Z.; Guo, L.; Zhu, X.; Mu, X.; Yao, F. Aluminum Alkyl Complexes Supported by Bidentate N,N Ligands: Synthesis, Structure, and Catalytic Activity for Guanylation of Amines. Organometallics 2015, 34, 1882–1889. [Google Scholar] [CrossRef]
- Bruce, M.; Gibson, V.C.; Redshaw, C.; Solan, G.A.; White, A.J.P.; Williams, D.J. Cationic alkyl aluminium ethylene polymerization catalysts based on monoanionic N,N,N-pyridyliminoamide ligands. Chem. Commun. 1998. [Google Scholar] [CrossRef]
- Arbaoui, A.; Redshaw, C.; Hughes, D.L. Multinuclear alkylaluminium macrocyclic Schiff base complexes: Influence of procatalyst structure on the ring opening polymerisation of ε-caprolactone. Chem. Commun. 2008. [Google Scholar] [CrossRef] [PubMed]
- Arbaoui, A.; Redshaw, C.; Hughes, D.L. One-pot conversion of tetraiminodiphenols to diiminodiaminodiphenols via methyl transfer at aluminium. Supramol. Chem. 2009, 21, 35–43. [Google Scholar] [CrossRef]
- Knijnenburg, Q.; Smits, J.M.M.; Budzelaar, P.H.M. Reaction of the Diimine Pyridine Ligand with Aluminum Alkyls: An Unexpectedly Complex Reaction. Organometallics 2006, 25, 1036–1046. [Google Scholar] [CrossRef]
- Davies, C.J.; Gregory, A.; Griffith, P.; Perkins, T.; Singh, K.; Solan, G.A. Use of Suzuki cross-coupling as a route to 2-phenoxy-6-iminopyridines and chiral 2-phenoxy-6-(methanamino) pyridines. Tetrahedron 2008, 64, 9857–9864. [Google Scholar] [CrossRef]
- Cross, W.B.; Hope, E.G.; Lin, Y.-H.; Macgregor, S.A.; Singh, K.; Solan, G.A.; Yahya, N. N,N-Chelate-control on the regioselectivity in acetate-assisted C–H activation. Chem. Commun. 2013, 49, 1918–1920. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, C.; Lenoble, G.; Oberhauser, W.; Parisel, S.; Zanobini, F. Synthesis, Characterization, and Reactivity of Neutral and Cationic Pd–C,N,N Pincer Complexes. Eur. J. Inorg. Chem. 2005, 2005, 4794–4800. [Google Scholar] [CrossRef]
- Annunziata, L.; Pragliola, S.; Pappalardo, D.; Tedesco, C.; Pellecchia, C. New (Anilidomethyl)pyridine Titanium(IV) and Zirconium(IV) Catalyst Precursors for the Highly Chemo- and Stereoselective cis-1,4-Polymerization of 1,3-Butadiene. Macromolecules 2011, 44, 1934–1941. [Google Scholar] [CrossRef]
- Annunziata, L.; Pappalardo, D.; Tedesco, C.; Pellecchia, C. Bis[(amidomethyl)pyridine] Zirconium(IV) Complexes: Synthesis, Characterization, and Activity as Olefin Polymerization Catalysts. Organometallics 2009, 28, 688–697. [Google Scholar] [CrossRef]
- Frazier, K.A.; Froese, R.D.; He, Y.; Klosin, J.; Theriault, C.N.; Vosejpka, P.C.; Zhou, Z.; Abboud, K.A. Pyridylamido Hafnium and Zirconium Complexes: Synthesis, Dynamic Behavior, and Ethylene/1-Octene and Propylene Polymerization Reactions. Organometallics 2011, 30, 3318–3329. [Google Scholar] [CrossRef]
- Zuccaccia, C.; Macchioni, A.; Busico, V.; Cipullo, R.; Talarico, G.; Alfano, F.; Boone, H.W.; Frazier, K.A.; Hustad, P.D.; Stevens, J.C.; Vosejpka, P.C.; Abboud, K.A. Intra- and Intermolecular NMR Studies on the Activation of Arylcyclometallated Hafnium Pyridyl-Amido Olefin Polymerization Precatalysts. J. Am. Chem. Soc. 2008, 130, 10354–10368. [Google Scholar] [CrossRef] [PubMed]
- Luconi, L.; Rossin, A.; Tuci, G.; Tritto, I.; Boggioni, L.; Klosin, J.J.; Theriault, C.N.; Giambastiani, G. Facing Unexpected Reactivity Paths with ZrIV-Pyridylamido Polymerization Catalysts. Chem.-Eur. J. 2012, 18, 671–687. [Google Scholar] [CrossRef] [PubMed]
- Domski, G.J.; Edson, J.B.; Keresztes, I.; Lobkovsky, E.B.; Coates, G.W. Synthesis of a new olefin polymerization catalyst supported by an sp3-C donor via insertion of a ligand-appended alkene into the Hf–C bond of a neutral pyridylamidohafnium trimethyl complex. Chem. Commun. 2008. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Kijima, T.; Izumi, T. The synthesis and application of 2-acetyl-6-(1-naphthyl)-pyridine oxime as a new ligand for palladium precatalyst in Suzuki coupling reaction. J. Heterocycl. Chem. 2009, 46, 116–118. [Google Scholar] [CrossRef]
- Coates, G.W.; Domski, G.J. Pyridlyamidohafnium catalyst precursors, active species from this and uses thereof to polymerize alkenes. WO 2008112133 A2, 7 March 2008. [Google Scholar]
- Patel, P.; Chang, S. N-Substituted hydroxylamines as synthetically versatile amino sources in the iridium-catalyzed mild C–H amidation reaction. Org. Lett. 2014, 16, 3328–3331. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, F.; Kochi, T.; Mutsutani, H.; Kibbayashi, N.; Urano, S.; Sato, M.; Nishiyamo, S.; Tanabe, T. Palladium-Catalyzed Aromatic C–H Halogenation with Hydrogen Halides by Means of Electrochemical Oxidation. J. Am. Chem. Soc. 2009, 131, 11310–11311. [Google Scholar] [CrossRef] [PubMed]
- Saver, M.; Schappacher, M.; Soum, A. Controlled Ring-Opening Polymerization of Lactones and Lactides Initiated by Lanthanum Isopropoxide, 1. General Aspects and Kinetics. Macromol. Chem. Phys. 2002, 203, 889–899. [Google Scholar] [CrossRef]
- Duda, A.; Kowalski, A.; Penczek, S. Polymerization of l,l-Lactide Initiated by Aluminum Isopropoxide Trimer or Tetramer. Macromolecules 1998, 31, 2114–2122. [Google Scholar]
- Alkarekshi, W.; Armitage, A.P.; Boyron, O.; Davies, C.J.; Govere, M.; Gregory, A.; Singh, K.; Solan, G.A. Phenolate Substituent Effects on Ring-Opening Polymerization of ε-Caprolactone by Aluminum Complexes Bearing 2- (Phenyl-2-olate)-6-(1-amidoalkyl)pyridine Pincers. Organometallics 2013, 32, 249–259. [Google Scholar] [CrossRef]
- Nomura, N.; Ishii, R.; Yamamoto, Y.; Kondo, T. Stereoselective Ring-Opening Polymerization of a Racemic Lactide by Using Achiral Salen- and Homosalen-Aluminum Complexes. Chem. Eur. J. 2007, 13, 4433–4451. [Google Scholar] [CrossRef] [PubMed]
- Armarego, W.L.F.; Perrin, D.D. Purification of Laboratory Chemicals, 4th ed.; Butterworth Heinemann: Oxford, UK, 1996. [Google Scholar]
- Grubert, L.; Jacobi, D.; Buck, K.; Abraham, W.; Mügge, C.; Krause, E. Pseudorotaxanes and Rotaxanes Incorporating Cycloheptatrienyl Stations—Synthesis and Co-Conformation. Eur. J. Org. Chem. 2001, 2001, 3921–3932. [Google Scholar] [CrossRef]
- Oskam, J.H.; Fox, H.H.; Yap, K.B.; McConville, D.H.; Odell, R.; Lichtenstein, B.J.; Schrock, R.R. Ligand variation in alkylidene complexes of the type Mo(CHR)(NR′)(OR″)2. J. Organomet. Chem. 1993, 459, 185–198. [Google Scholar] [CrossRef]
- So, C.M.; Lau, C.P.; Chan, A.S.C.; Kwong, F.Y. Suzuki-Miyaura Coupling of Aryl Tosylates Catalyzed by an Array of Indolyl Phosphine-Palladium Catalysts. J. Org. Chem. 2008, 73, 7731–7734. [Google Scholar] [CrossRef] [PubMed]
- Parks, J.E.; Wagner, B.E.; Holm, R.H. Syntheses employing pyridyllithium reagents: New routes to 2,6-disubstituted pyridines and 6,6′-disubstituted 2,2′-bipyridyls. J. Organomet. Chem. 1973, 56, 53–66. [Google Scholar] [CrossRef]
- Tellier, F.; Sauvêtre, R.; Normant, J.-F. Synthese stereospecifique de dienes fluores. J. Organomet. Chem. 1985, 292, 19–28. [Google Scholar] [CrossRef]
- SHELXTL, version 6.10; Bruker AXS, Inc.: Madison, WI, USA, 2000.
- Spek, A.L. PLATON, An Integrated Tool for the Analysis of the Results of a Single Crystal Structure Determination. Acta Cryst. Sect. A 1990, A46, C34. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armitage, A.P.; Boyron, O.; Champouret, Y.D.M.; Patel, M.; Singh, K.; Solan, G.A. Dimethyl-Aluminium Complexes Bearing Naphthyl-Substituted Pyridine-Alkylamides as Pro-Initiators for the Efficient ROP of ε-Caprolactone. Catalysts 2015, 5, 1425-1444. https://doi.org/10.3390/catal5031425
Armitage AP, Boyron O, Champouret YDM, Patel M, Singh K, Solan GA. Dimethyl-Aluminium Complexes Bearing Naphthyl-Substituted Pyridine-Alkylamides as Pro-Initiators for the Efficient ROP of ε-Caprolactone. Catalysts. 2015; 5(3):1425-1444. https://doi.org/10.3390/catal5031425
Chicago/Turabian StyleArmitage, Andrew P., Olivier Boyron, Yohan D. M. Champouret, Mehzabin Patel, Kuldip Singh, and Gregory A. Solan. 2015. "Dimethyl-Aluminium Complexes Bearing Naphthyl-Substituted Pyridine-Alkylamides as Pro-Initiators for the Efficient ROP of ε-Caprolactone" Catalysts 5, no. 3: 1425-1444. https://doi.org/10.3390/catal5031425