
Influence of the Synthesis Method for Pt Catalysts Supported on Highly Mesoporous Carbon Xerogel and Vulcan Carbon Black on the Electro-Oxidation of Methanol


Abstract
:1. Introduction
2. Results and Discussion
2.1. Textural and Chemical Characterization
2.1.1. Characterization of the Carbon Supports

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SBET (m2·g−1) | Total pore volume (0.7–180 nm) (cm3·g−1) | Micropore volume (cm3·g−1) | Mesopore volume (cm3·g−1) | Average pore size (nm) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Vmicro (0–0.7 nm) | Vmicro (0.7–2 nm) | Vmicro (0–2 nm) | Vmeso (2–25 nm) | Vmeso (25–300 nm) | VBJH (1.5–300 nm) | ||||
| Vulcan | 224 | 0.47 | 0.01 | 0.03 | 0.04 | 0.24 | 0.17 | 0.41 | 11.0 |
| CXG | 660 | 1.79 | 0.06 | 0.07 | 0.13 | 0.97 | 0.69 | 1.66 | 23.3 |
| Sample | SBET (m2·g−1) | Total pore volume (0.7–180 nm) (cm3·g−1) | Micropore volume (cm3·g−1) | Mesopore volume (cm3·g−1) | Average pore size (nm) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Vmicro (0–0.7 nm) | Vmicro (0.7–2 nm) | Vmicro (0–2 nm) | Vmeso (2–25 nm) | Vmeso (25–300 nm) | VBJH (1.5–300 nm) | ||||
| Pt/Vulcan-SBM | 187 | 0.47 | 0.02 | 0.01 | 0.03 | 0.21 | 0.21 | 0.42 | 14.8 |
| Pt/Vulcan-FAM | 191 | 0.38 | 0.02 | 0.01 | 0.03 | 0.20 | 0.13 | 0.33 | 11.9 |
| Pt/Vulcan-ME | 177 | 0.35 | 0.02 | 0.01 | 0.03 | 0.17 | 0.15 | 0.32 | 10.3 |
| Pt/CXG-SBM | 475 | 1.34 | 0.07 | 0.05 | 0.12 | 0.45 | 0.77 | 1.22 | 24.5 |
| Pt/CXG-FAM | 478 | 1.08 | 0.09 | 0.05 | 0.14 | 0.34 | 0.59 | 0.93 | 23.5 |
| Pt/CXG-ME | 349 | 1.27 | 0.06 | 0.04 | 0.10 | 0.38 | 0.77 | 1.15 | 25.5 |
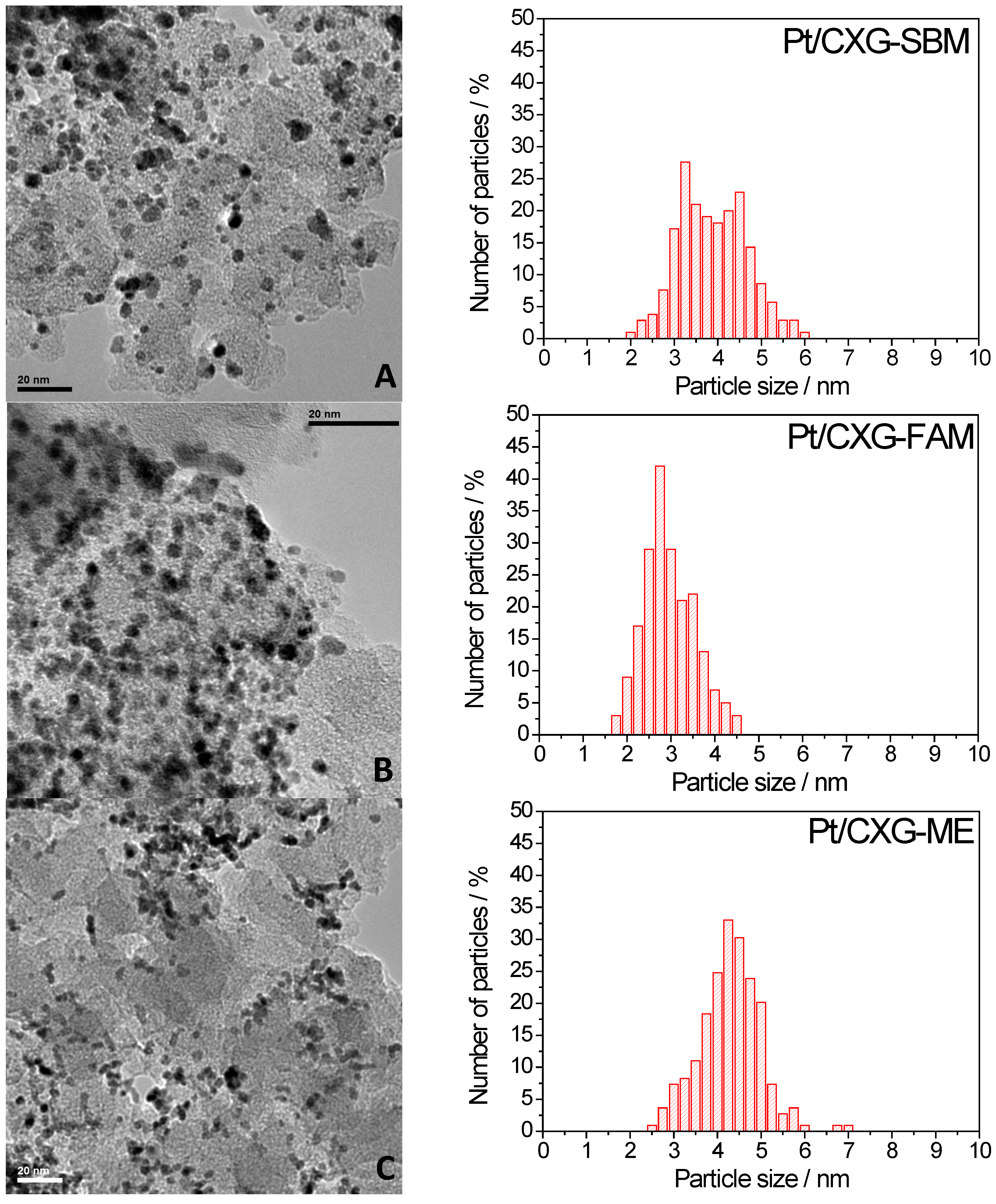
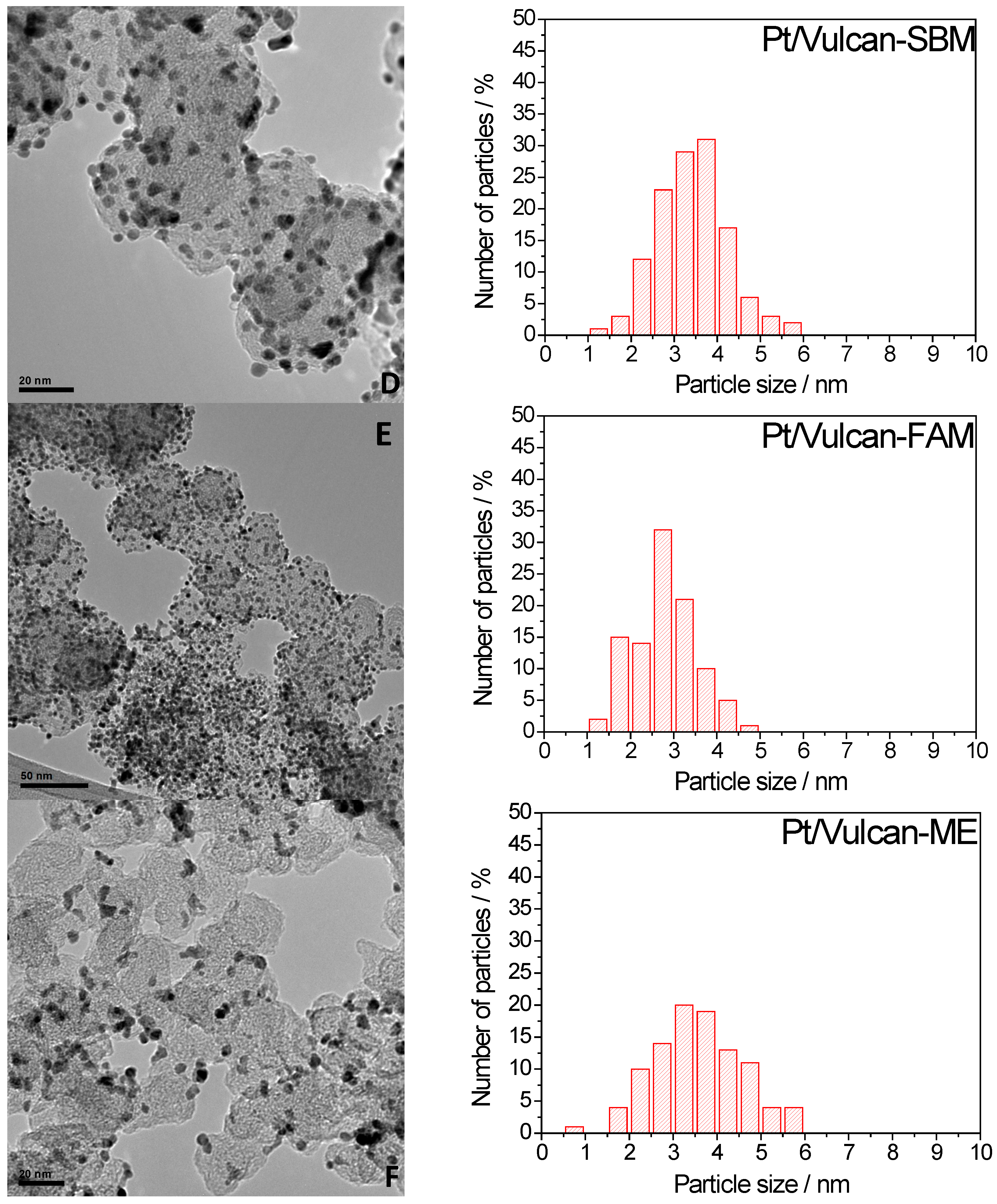
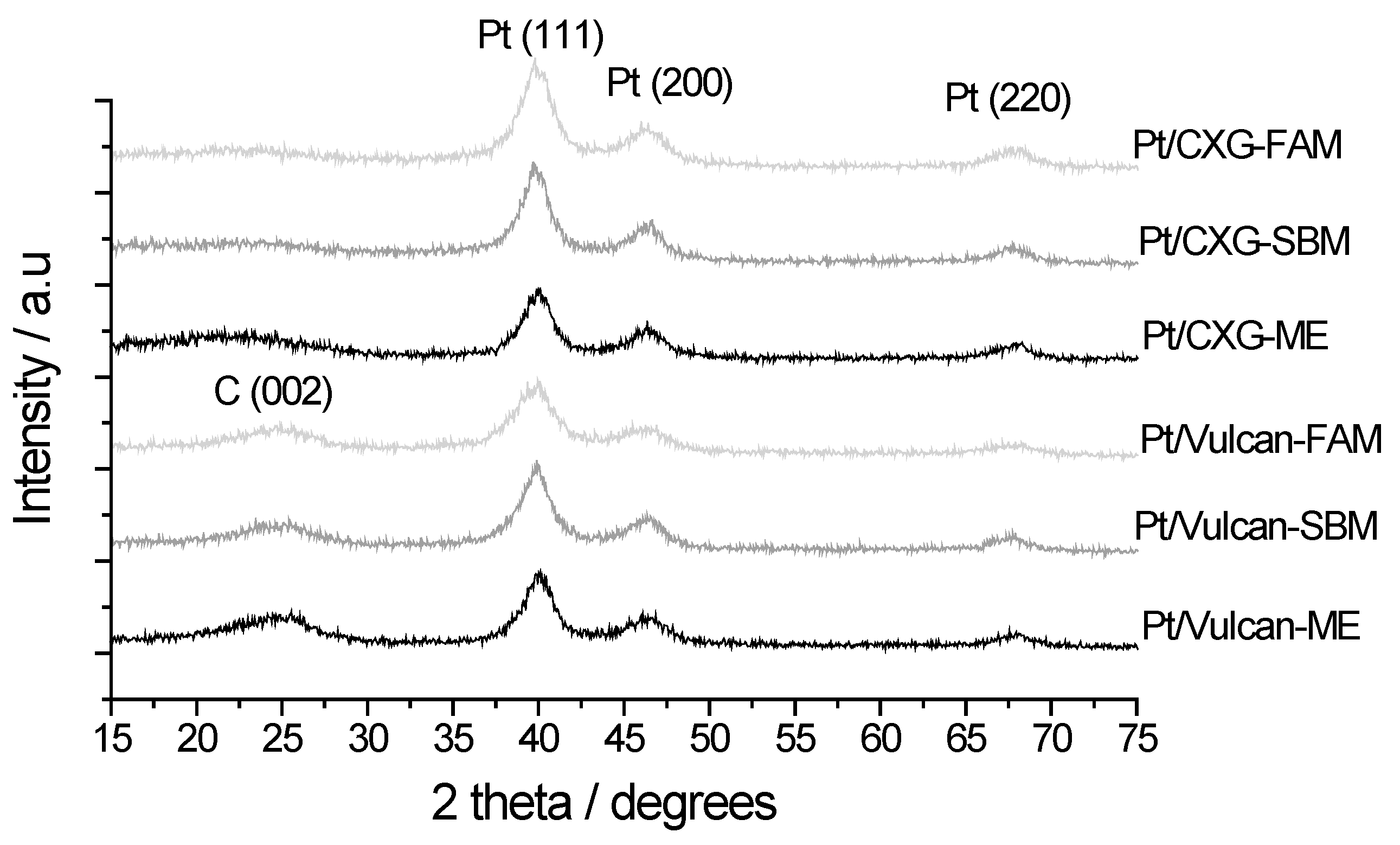
2.1.2. Catalyst Characterization
| Catalysts | Pt crystal size (nm) | % Pt (ICP-AES) |
|---|---|---|
| Pt/CXG-SBM | 4.2 | 20.2 |
| Pt/CXG-FAM | 3.6 | 17.9 |
| Pt/CXG-ME | 3.9 | 17.1 |
| Pt/Vulcan-SBM | 3.4 | 16.8 |
| Pt/Vulcan-FAM | 4.6 | 16.7 |
| Pt/Vulcan-ME | 4.4 | 17.4 |
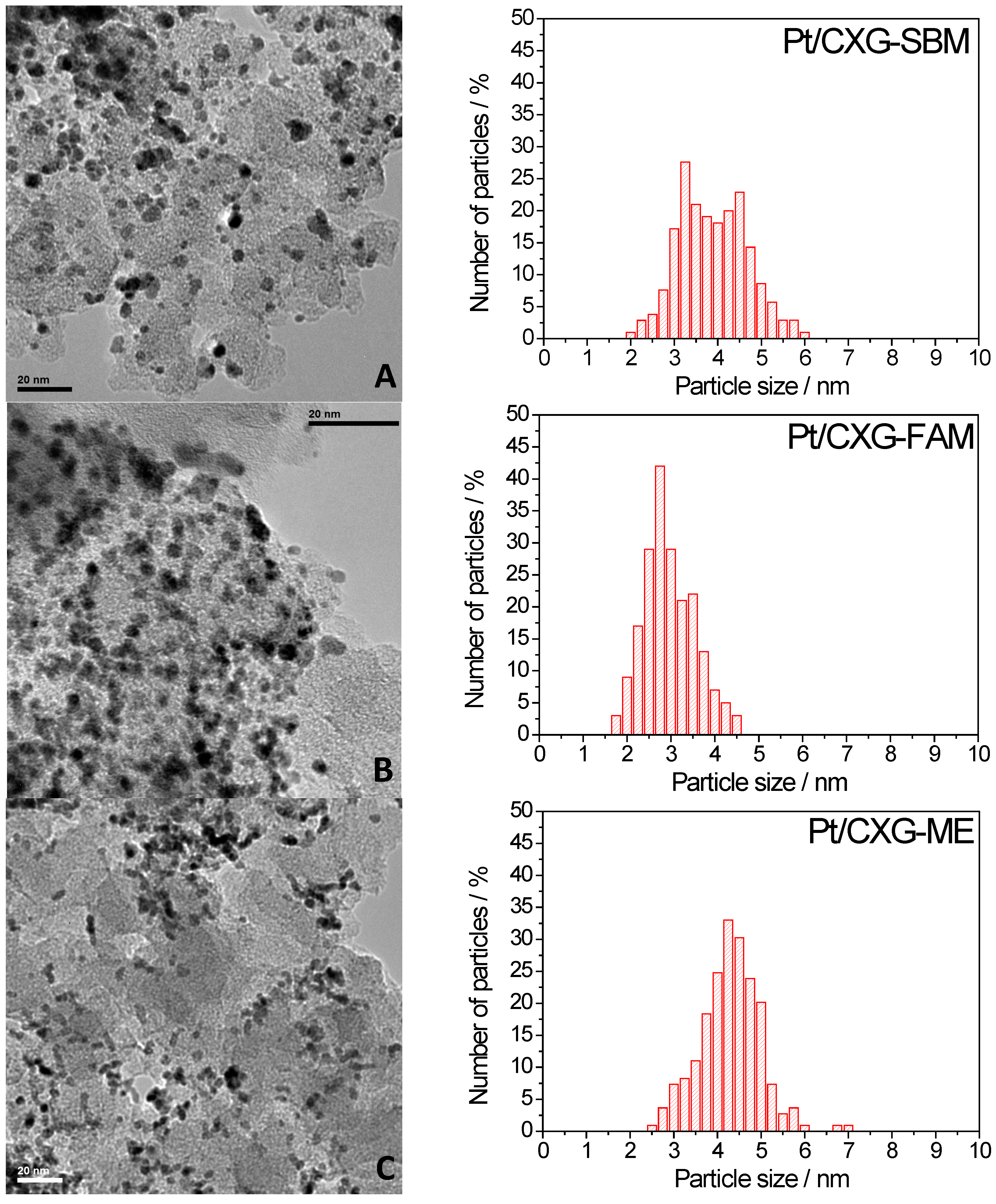
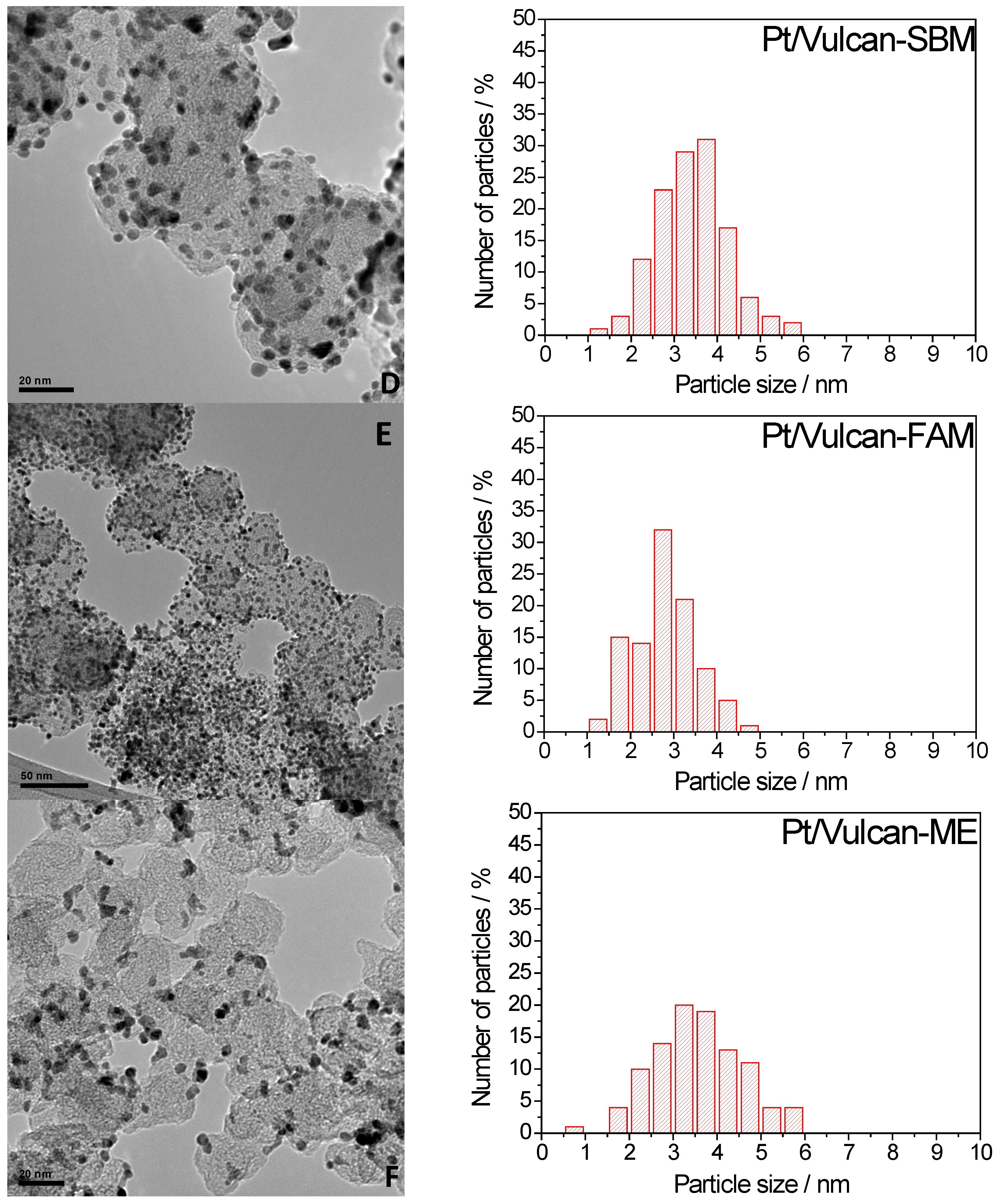
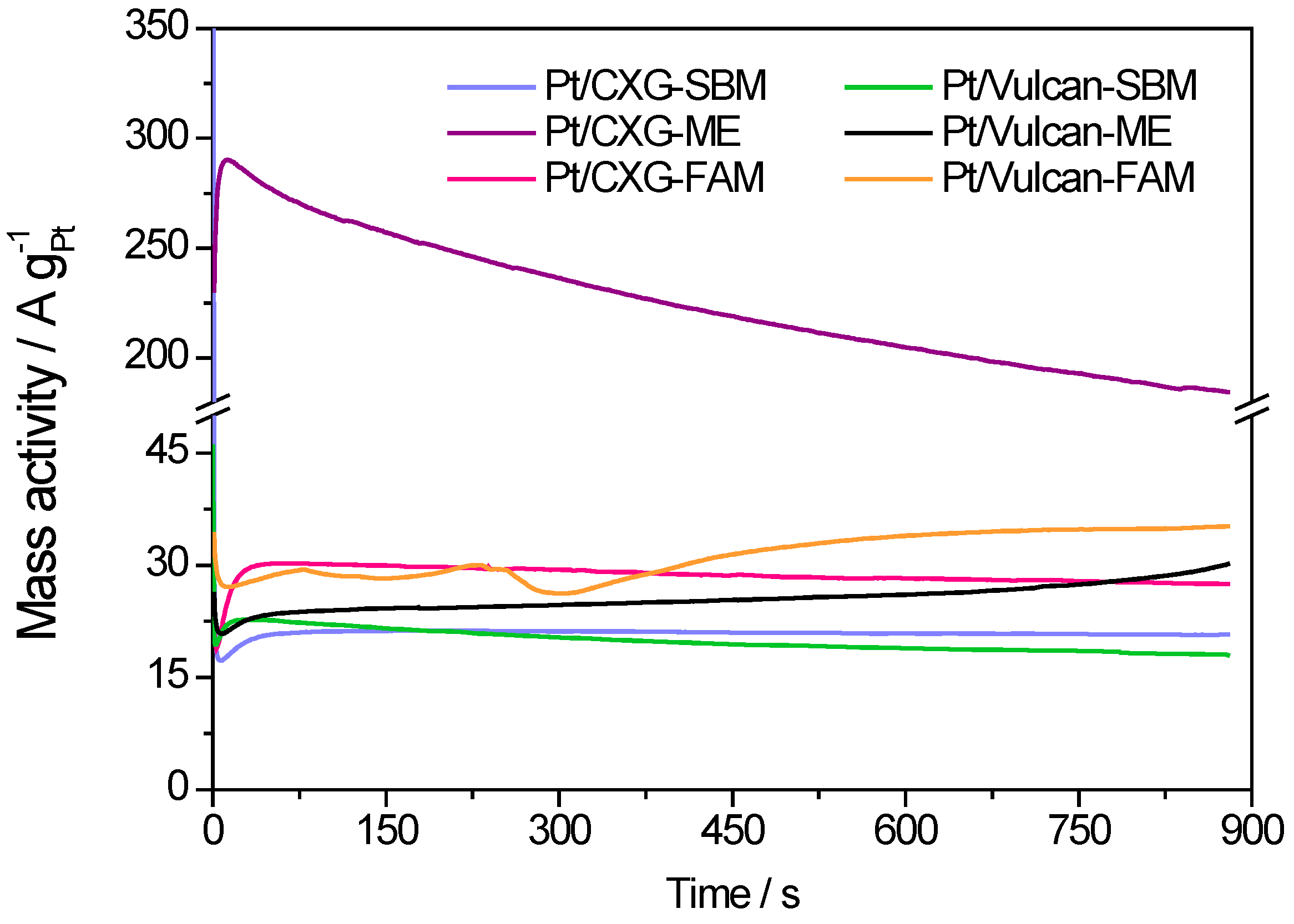
2.2. Electrochemical Characterization

| Catalysts | Pt crystal size (nm) | ECSA (m2·g−1) | Peak mass activity (A·g−1 Pt) | Peak specific activity (mA·cm−2 Pt) |
|---|---|---|---|---|
| Pt/CXG-SBM | 4.2 | 47.9 | 363 | 0.96 |
| Pt/CXG-FAM | 3.6 | 38.6 | 367 | 1.13 |
| Pt/CXG-ME | 3.9 | 36.5 | 470 | 1.29 |
| Pt/Vulcan-SBM | 3.4 | 39.2 | 330 | 0.84 |
| Pt/Vulcan-FAM | 4.6 | 41.4 | 300 | 0.47 |
| Pt/Vulcan-ME | 4.4 | 14.7 | 232 | 1.58 |
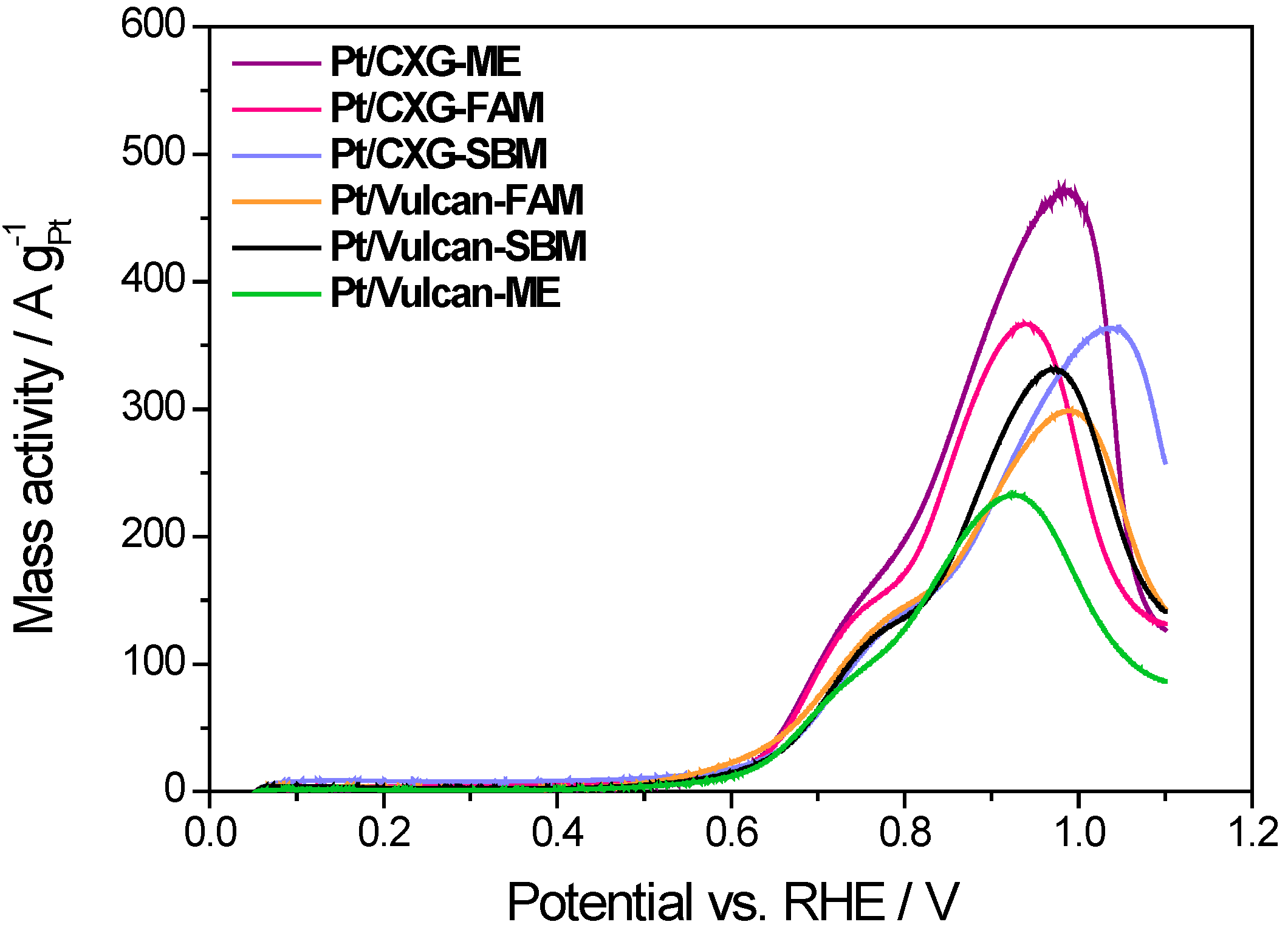
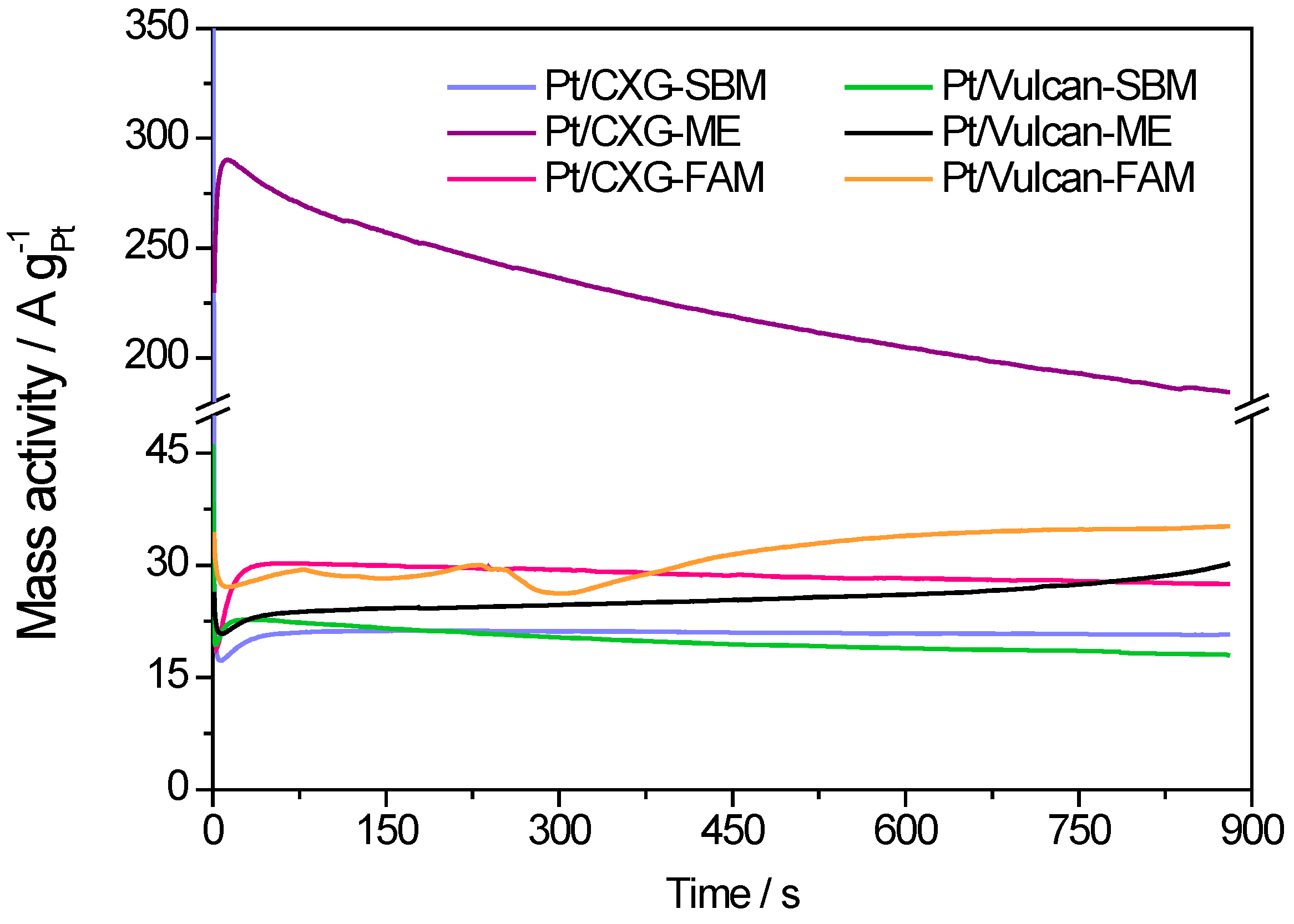
3. Experimental Section
3.1. Synthesis of Carbon Xerogels
3.2. Pt catalysts Synthesis
3.3. Carbon Supports and Catalysts Textural, Structural and Morphological Characterization
3.4. Electrochemical Characterization
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Job, N.; Ribeiro Pereira, M.F.; Lambert, S.; Cabiac, A.; Delahay, G.; Colomer, J.-F.; Marien, J.; Figueiredo, J.L.; Pirard, J.-P. Highly dispersed platinum catalysts prepared by impregnation of texture-tailored carbon xerogels. J. Catal. 2006, 240, 160–171. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Ribeiro Pereira, M.F.; Serp, P.; Kalck, P.; Samant, P.V.; Fernandes, J.B. Development of carbon nanotube and carbon xerogel supported catalysts for the electro-oxidation of methanol in fuel cells. Carbon 2006, 44, 2516–2522. [Google Scholar] [CrossRef]
- Samant, P.V.; Fernandes, J.B.; Rangel, C.M.; Figueiredo, J.L. Carbon xerogel supported Pt and Pt–Ni catalysts for electro-oxidation of methanol in basic medium. Catal. Today 2005, 102–103, 173–176. [Google Scholar] [CrossRef]
- Sebastián, D.; Suelves, I.; Lázaro, M.J.; Moliner, R. Carbon nanofibers as electrocatalyst support for fuel cells: Effect of hydrogen on their properties in CH4 decomposition. J. Power Sources 2009, 192, 51–56. [Google Scholar] [CrossRef]
- Arbizzani, C.; Beninati, S.; Manferrari, E.; Soavi, F.; Mastragostino, M. Cryo- and xerogel carbon supported PtRu for DMFC anodes. J. Power Sources 2007, 172, 578–579. [Google Scholar] [CrossRef]
- Al-Muhtaseb, S.A.; Ritter, J.A. Preparation and Properties of Resorcinol-Formaldehyde Organic and Carbon Gels. Adv. Mater. 2003, 15, 101–114. [Google Scholar] [CrossRef]
- Antolini, E. Carbon supports for low-temperature fuel cell catalysts. Appl. Catal. B 2009, 88, 1–24. [Google Scholar] [CrossRef]
- Pekala, R.W. Organic aerogels from the polycondensation of resorcinol with formaldehyde. J. Mater. Sci. 1989, 24, 3221–3227. [Google Scholar] [CrossRef]
- Alegre, C.; Calvillo, L.; Moliner, R.; González-Expósito, J.A.; Guillén-Villafuerte, O.; Martínez Huerta, M.V.; Pastor, E.; Lázaro, M.J. Pt and PtRu electrocatalysts supported on carbon xerogels for direct methanol fuel cells. J. Power Sources 2011, 196, 4226–4235. [Google Scholar] [CrossRef]
- Yoshizawa, N.; Hatori, H.; Soneda, Y.; Hanzawa, Y.; Kaneko, K.; Dresselhaus, M.S. Structure and electrochemical properties of carbon aerogels polymerized in the presence of Cu2+. J. Non-Cryst. Solids 2003, 330, 99–105. [Google Scholar] [CrossRef]
- Job, N.; Marie, J.; Lambert, S.; Berthon-Fabry, S.; Achard, P. Carbon xerogels as catalyst supports for PEM fuel cell cathode. Energy Convers. Manag. 2008, 49, 2461–2470. [Google Scholar] [CrossRef]
- Job, N.; Lambert, S.; Chatenet, M.; Gommes, C.; Maillard, F.; Berthon-Fabry, S.; Regalbuto, J.R.; Pirard, J.P. Preparation of highly loaded Pt/carbon xerogel catalysts for Proton Exchange Membrane fuel cells by the Strong Electrostatic Adsorption method. Catal. Today 2009, 150, 119–127. [Google Scholar] [CrossRef]
- Maillard, F.; Savinova, E.R.; Simonov, P.A.; Zaikovskii, V.I.; Stimming, U. Infrared Spectroscopic Study of CO Adsorption and Electro-oxidation on Carbon-Supported Pt Nanoparticles: Interparticle versus Intraparticle Heterogeneity. J. Phys. Chem. 2004, 108, 17893–17904. [Google Scholar] [CrossRef]
- Maillard, F.; Schreier, S.; Savinova, E.R.; Weinkauf, S.; Stimming, U. Influence of particle agglomeration on the catalytic activity of carbon-supported Pt nanoparticles in CO monolayer oxidation. Phys. Chem. Chem. Phys. 2005, 7, 385–393. [Google Scholar] [CrossRef]
- Job, N.; Théry, A.; Pirard, R.; Marien, J.; Kocon, L.; Rouzaud, J.N.; Béguin, F.; Pirard, J.P. Carbon aerogels, cryogels and xerogels: Influence of the drying method on the textural properties of porous carbon materials. Carbon 2005, 43, 2481–2494. [Google Scholar] [CrossRef]
- Morales-Torres, S.; Maldonado-Hódar, F.J.; Pérez-Cadenas, A.F.; Carrasco-Marín, F. Structural characterization of carbon xerogels: From film to monolith. Microporous Mesoporous Mater. 2012, 153, 24–29. [Google Scholar] [CrossRef]
- Gorgulho, H.F.; Gonçalves, F.; Pereira, M.F.R.; Figueiredo, J.L. Synthesis and characterization of nitrogen-doped carbon xerogels. Carbon 2009, 47, 2032–2039. [Google Scholar] [CrossRef]
- Sebastián, D.; Alegre, C.; Calvillo, L.; Pérez, M.; Moliner, R.; Lázaro, M.J. Carbon supports for the catalytic dehydrogenation of liquid organic hydrides as hydrogen storage and delivery system. Int. J. Hydrog. Energy 2013, 39, 4109–4115. [Google Scholar] [CrossRef]
- Sebastián, D.; Lázaro, M.J.; Suelves, I.; Moliner, R.; Baglio, V.; Stassi, A.; Aricò, A.S. The influence of carbon nanofiber support properties on the oxygen reduction behavior in proton conducting electrolyte-based direct methanol fuel cells. Int. J. Hydrog. Energy 2012, 37, 6253–6260. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alegre, C.; Gálvez, M.E.; Moliner, R.; Lázaro, M.J. Influence of the Synthesis Method for Pt Catalysts Supported on Highly Mesoporous Carbon Xerogel and Vulcan Carbon Black on the Electro-Oxidation of Methanol. Catalysts 2015, 5, 392-405. https://doi.org/10.3390/catal5010392
Alegre C, Gálvez ME, Moliner R, Lázaro MJ. Influence of the Synthesis Method for Pt Catalysts Supported on Highly Mesoporous Carbon Xerogel and Vulcan Carbon Black on the Electro-Oxidation of Methanol. Catalysts. 2015; 5(1):392-405. https://doi.org/10.3390/catal5010392
Chicago/Turabian StyleAlegre, Cinthia, María Elena Gálvez, Rafael Moliner, and María Jesús Lázaro. 2015. "Influence of the Synthesis Method for Pt Catalysts Supported on Highly Mesoporous Carbon Xerogel and Vulcan Carbon Black on the Electro-Oxidation of Methanol" Catalysts 5, no. 1: 392-405. https://doi.org/10.3390/catal5010392