1. Introduction
The direct activation of allylic alcohols, without pre-activation of the alcohol moiety, in palladium catalyzed allylic substitution reactions for application in C-C and C-X bond formation, is of growing interest [
1,
2,
3]. Traditionally, allylic alcohols are pre-activated by transforming the alcohol moiety into a better leaving group, such as a halide, tosylate, carboxylate or phosphate. Alternatively they can be activated by stoichiometric Lewis acid adducts, such as BEt
3 and Ti(O-
i-Pr)
4 [
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14]. However, all these approaches lead to formation of substantial (in most cases stoichiometric) amounts of (salt) waste, which can be avoided by the direct catalytic activation of the allylic alcohol [
15,
16,
17,
18,
19]. Ozawa and co-workers reported the first examples in which allylic alcohols were directly used as coupling partners in Pd-catalyzed allylic substitution reactions by the π-allyl palladium complexes of substituted diphosphinidenecyclobutene ligands (DPCB-Y) [
15]. Mechanistic studies demonstrated that C-O bond dissociation is the rate-determining step in these reactions (proposed by Ozawa and Yoshifuji to proceed via a palladium-hydride intermediate) [
20], which was later also confirmed by le Floch and co-workers on the basis of DFT calculations (invoking ammonium promoted protonation of the allyl alcohol) [
21]. The theoretical study further showed that nucleophilic attack on the π-allyl complex first generates a cationic allyl amine, which assists in dissociation of the hydroxyl group in the oxidative addition step. Another prominent example was reported by Oshima and co-workers, showing that a EtOAc/H
2O biphasic system allows the
in situ activation of allylic alcohols with tppts based Pd(0) catalyst by hydration of the alcohol moiety [
22]. Several water molecules assist in delocalization of the developing negative charge in the transition state, and thus lower the activation energy according to the reported DFT calculations. Similarly, the use of methanol as a solvent in allylic alkylation reactions of simple ketones with allylic alcohols was reported to be crucial for the C-O bond cleavage step, which was catalyzed by the [(dppf)Pd(allyl)] in the presence of pyrolidine as co-catalyst [
23]. Solvation of the hydroxyl/hydroxy moiety in the charge-developing transition state was again proposed to lower the activation energy.
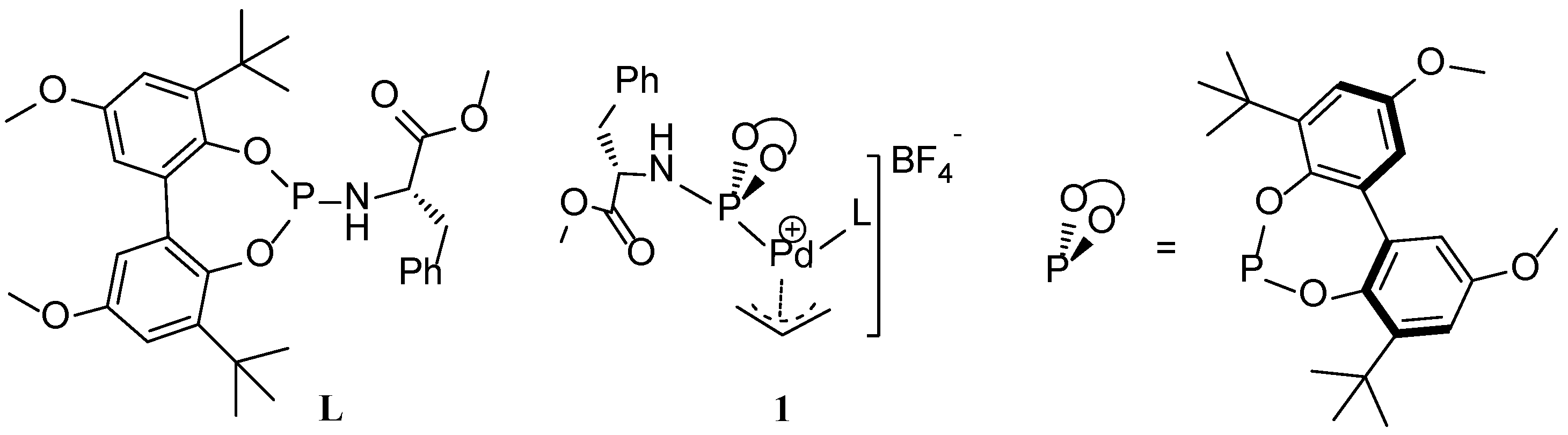
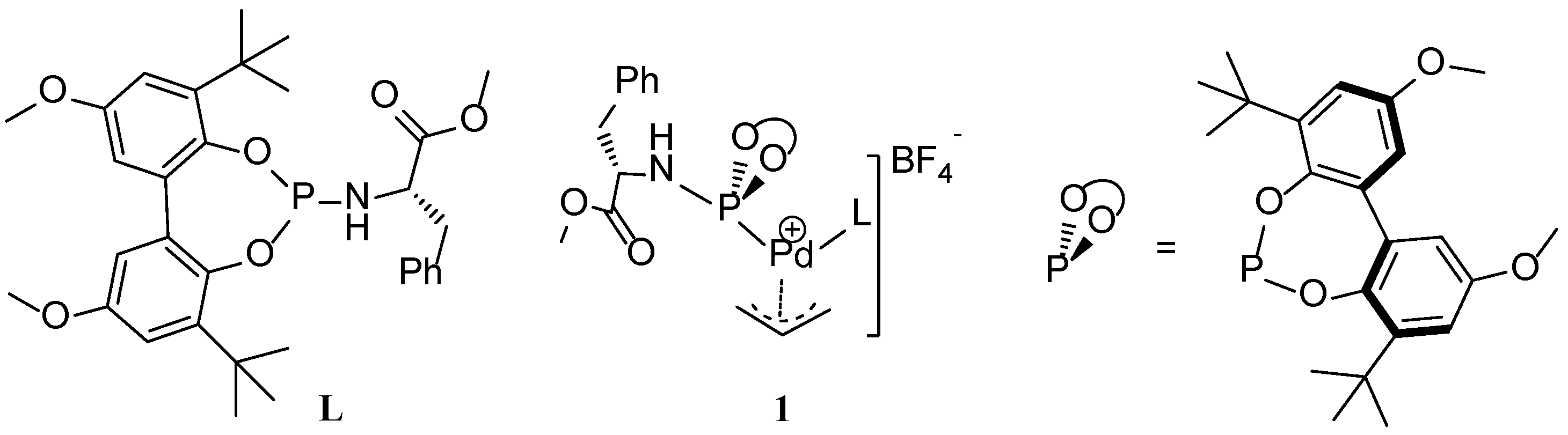
Scheme 1.
The structure of phosphoramidite ligand (L) and schematic representation of Pd-allyl complex, 1.
Scheme 1.
The structure of phosphoramidite ligand (L) and schematic representation of Pd-allyl complex, 1.
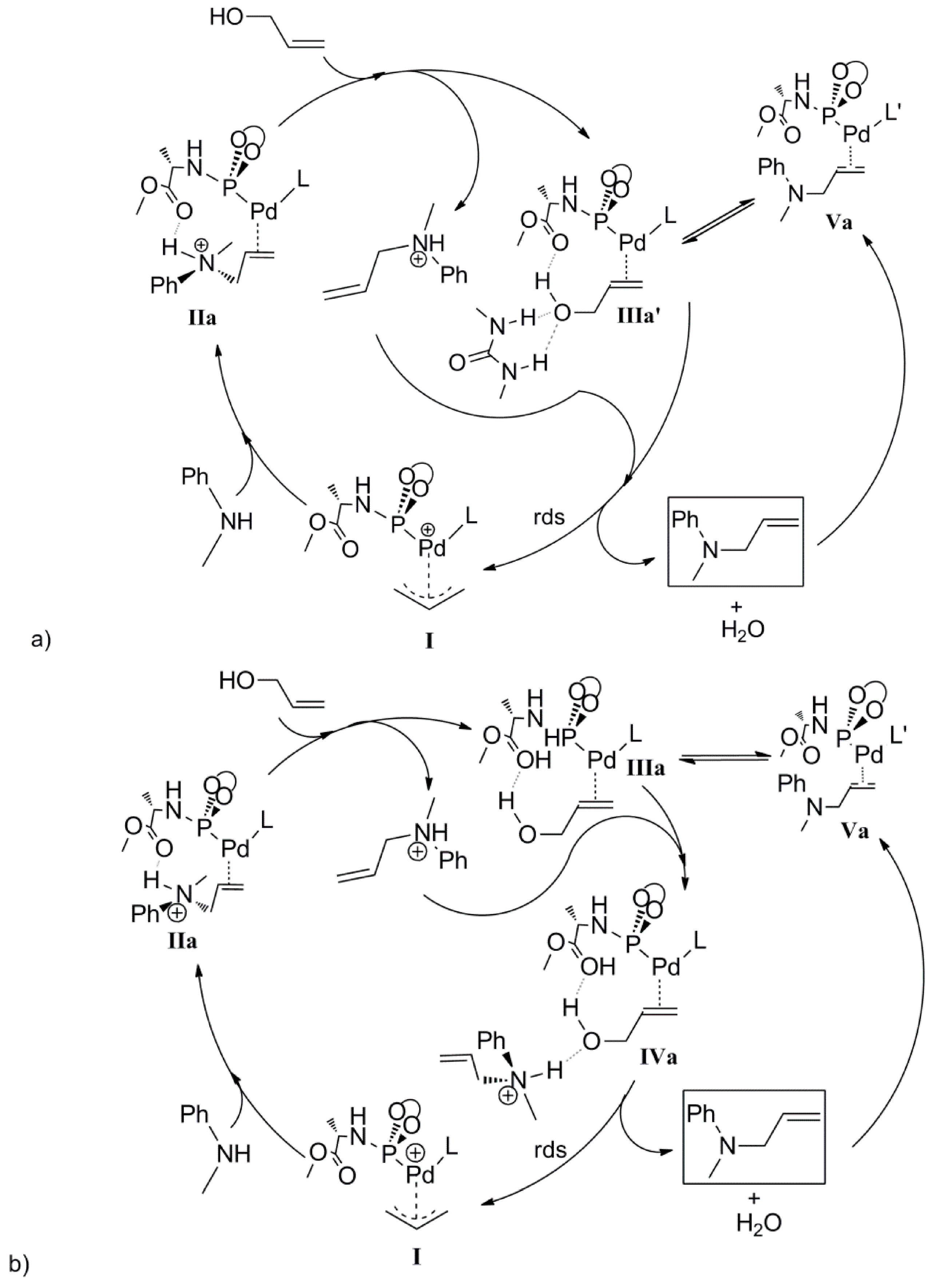
We recently disclosed a new phosphoramidite-based Pd(π-allyl)catalyst (
1) for allylic substitution reactions, using unactivated allylic alcohols to selectively produce linear alkylated and aminated products (
Scheme 1) [
24]. The addition of catalytic amount of 1,3-diethylurea (3 mol%) increased the catalytic activity and improved the reproducibility of these reactions. Detailed kinetic studies revealed zero order kinetics in the nucleophilic amine and first order kinetics in the allylic alcohol and 1,3-diethylurea. On this basis, we proposed the mechanism depicted in
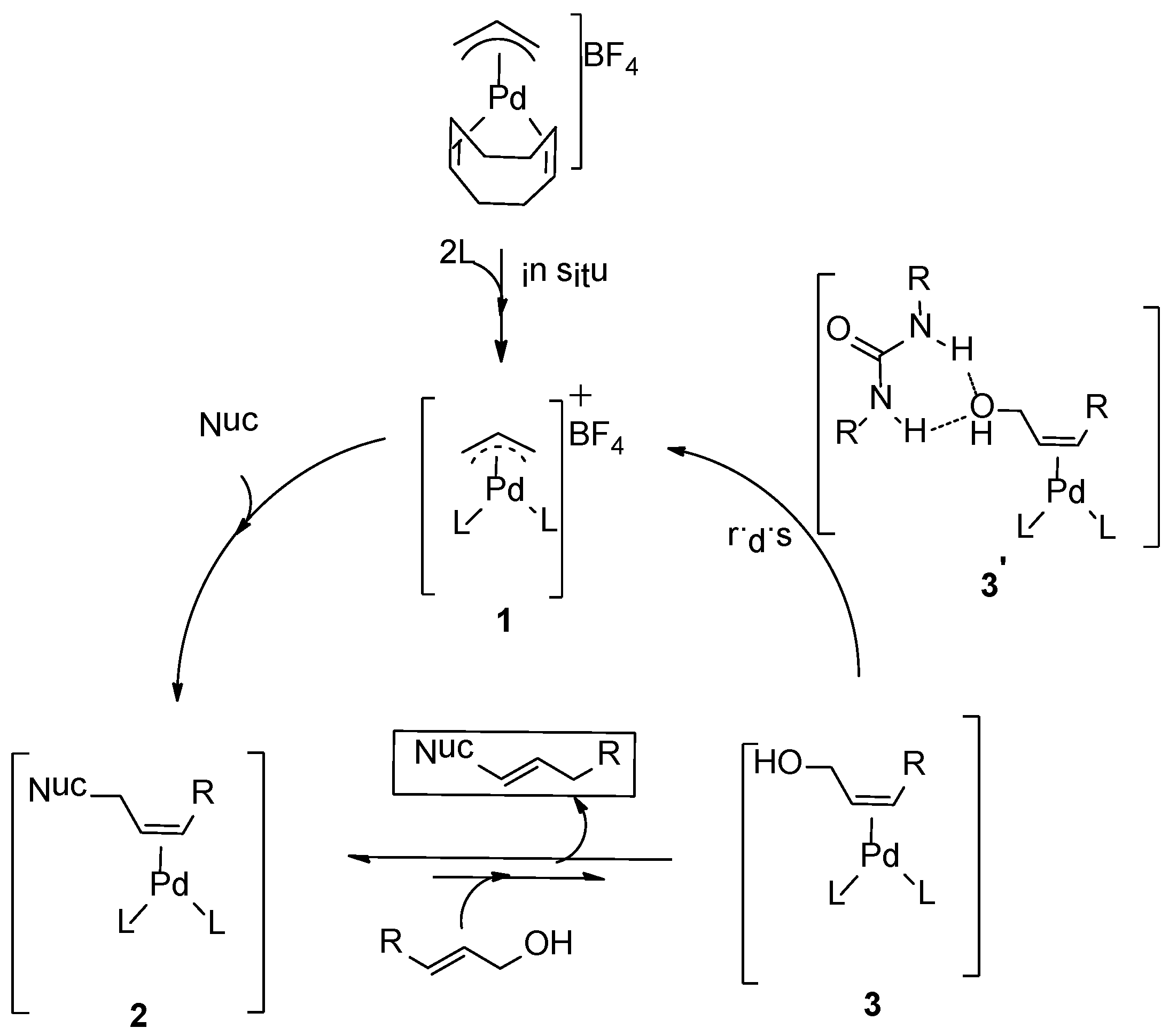
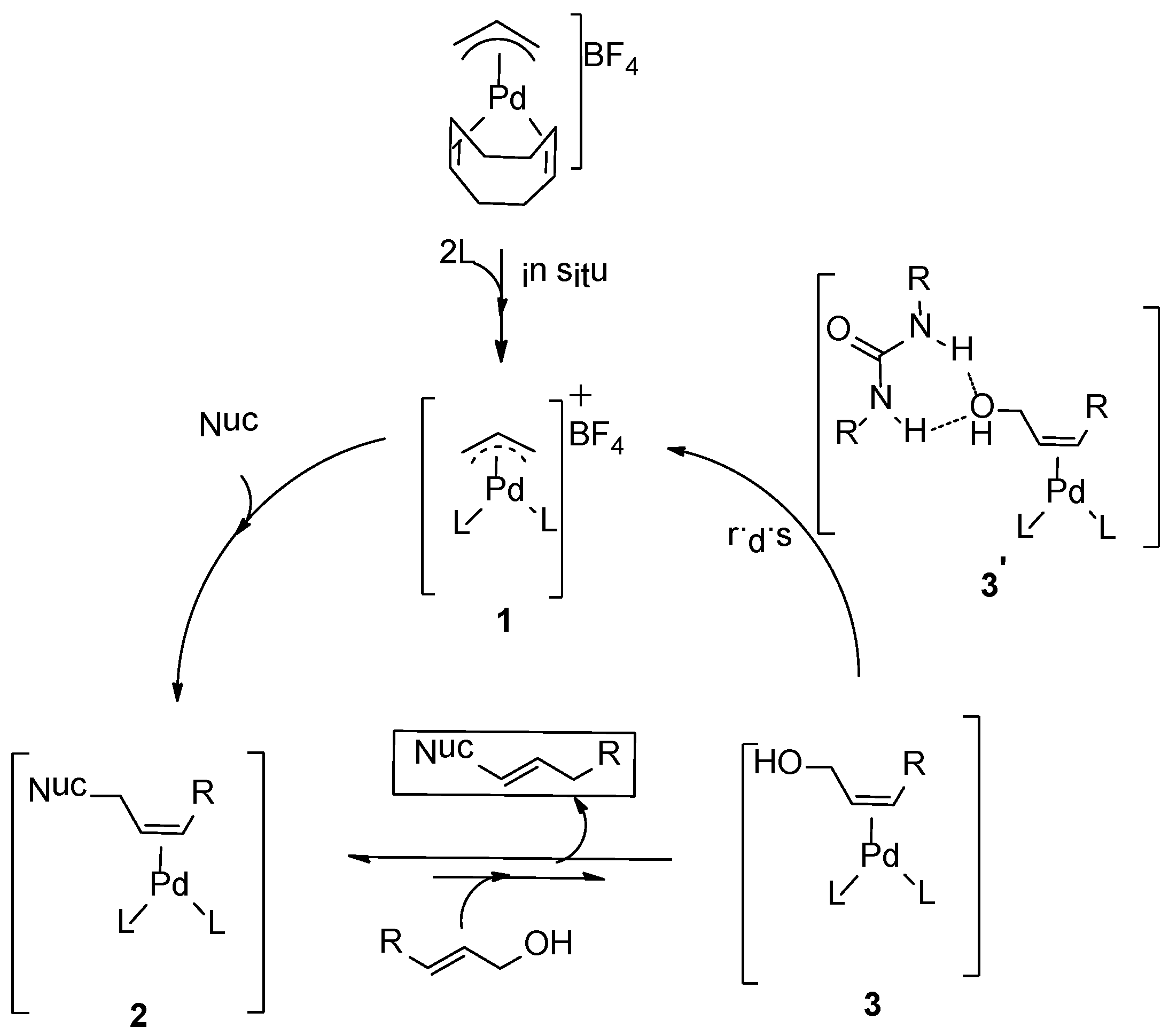
Scheme 2. The catalyst precursor
1 was generated
in situ by mixing [(η
3-allyl)Pd(cod)]BF
4 with ligand
L. Nucleophilic attack of amine is expected to generate intermediate
2, and the coordination of the allyl alcohol to palladium results in formation of intermediate
3 and the allylamine product. Experimentally, we observed product inhibition; the reaction becomes slower with increasing amounts of the product. This is most likely due to a shift in the equilibrium between intermediate
3 and
2 in favor of
2 at higher product concentrations. In line with observations reported by others, the allylic alcohol C-O bond oxidative addition step is likely the rate-determining step. Preliminary DFT calculations suggested that hydrogen-bonding interactions between the hydroxyl group of the alcohol and both the carboxyl group of the ligand and the1,3-diethylurea moiety assist in the C-O bond activation step. Mechanistic details of the direct activation of allylic alcohols by this system, and in particular the role of the 1,3-diethylurea additive in this process, are required to facilitate further development of this type of catalyst systems. Here we report the mechanism of the Pd-catalyzed direct amination of allylic alcohols based on DFT calculations. We also found the mechanism by which the 1,3-diethylurea additive activates the catalyst as it forms H-bonds with the allylalcohol substrate.
Scheme 2.
Proposed mechanism for amination reactions of allylic alcohols [
22].
Scheme 2.
Proposed mechanism for amination reactions of allylic alcohols [
22].
2. Results and Discussion
The mechanism we explored computationally is based on our experimental kinetic studies (
Scheme 2). The rate limiting step derived from the kinetic studies is the C-O bond oxidative addition in intermediate
3, judging from the first order rate dependency on allyl alcohol and 1,3-diethylurea and zero order behavior in the amine (=nucleophile). We took this information as a starting point to build our computational model. For the theoretical calculations, we employed allyl alcohol and
N-methylaniline as the substrate and nucleophile, respectively. The reaction produces the corresponding allyl-amine as the product (
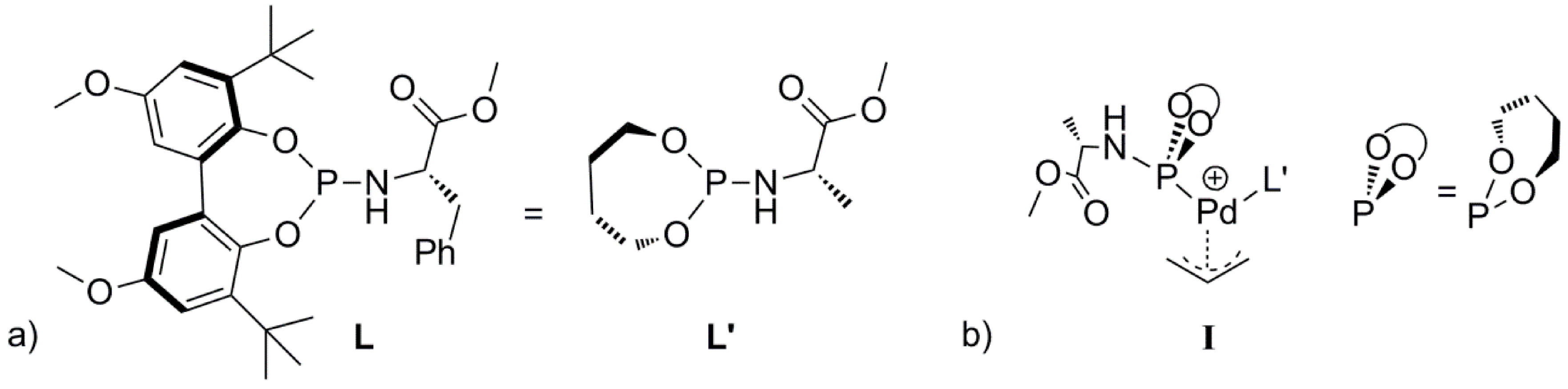
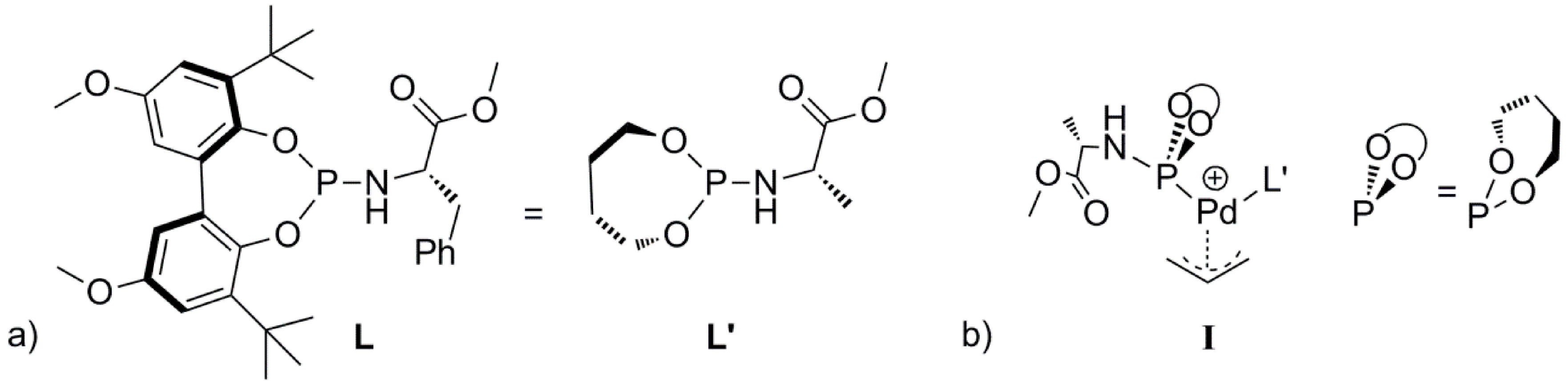
Scheme 2). To avoid complicated conformational searches, the ligand
L was simplified by removing the bulky substituents of the backbone and replacing the chiral benzyl group by a methyl group (
S chirality maintained, see
L’,
Scheme 3a). Notably, the preferred
R chirality on bi-phenyl backbones of the phosphoramidite ligand
L in the palladium homo-complex
1 [
25] complies with the DFT calculations using the simplified ligand
L’ in
I. Further note that the chiral element of the ligand was not used to induce enantioselectivity, neither experimentally nor in the DFT calculations. In the Figures and Schemes, ligand
L’ is drawn in a simplified manner showing only the relevant parts of the ligand for clarity (
Scheme 3b). We performed all calculations in Turbomole with the BP86 functional. We employed the large def2-TZVP basis set and Grimme’s version 3 (DFT-D3, disp3) dispersion corrections.
Scheme 3.
(a) The ligand L and the simplified ligand L’. (b) Schematic representation of the L’ based Pd-allyl complex I.
Scheme 3.
(a) The ligand L and the simplified ligand L’. (b) Schematic representation of the L’ based Pd-allyl complex I.
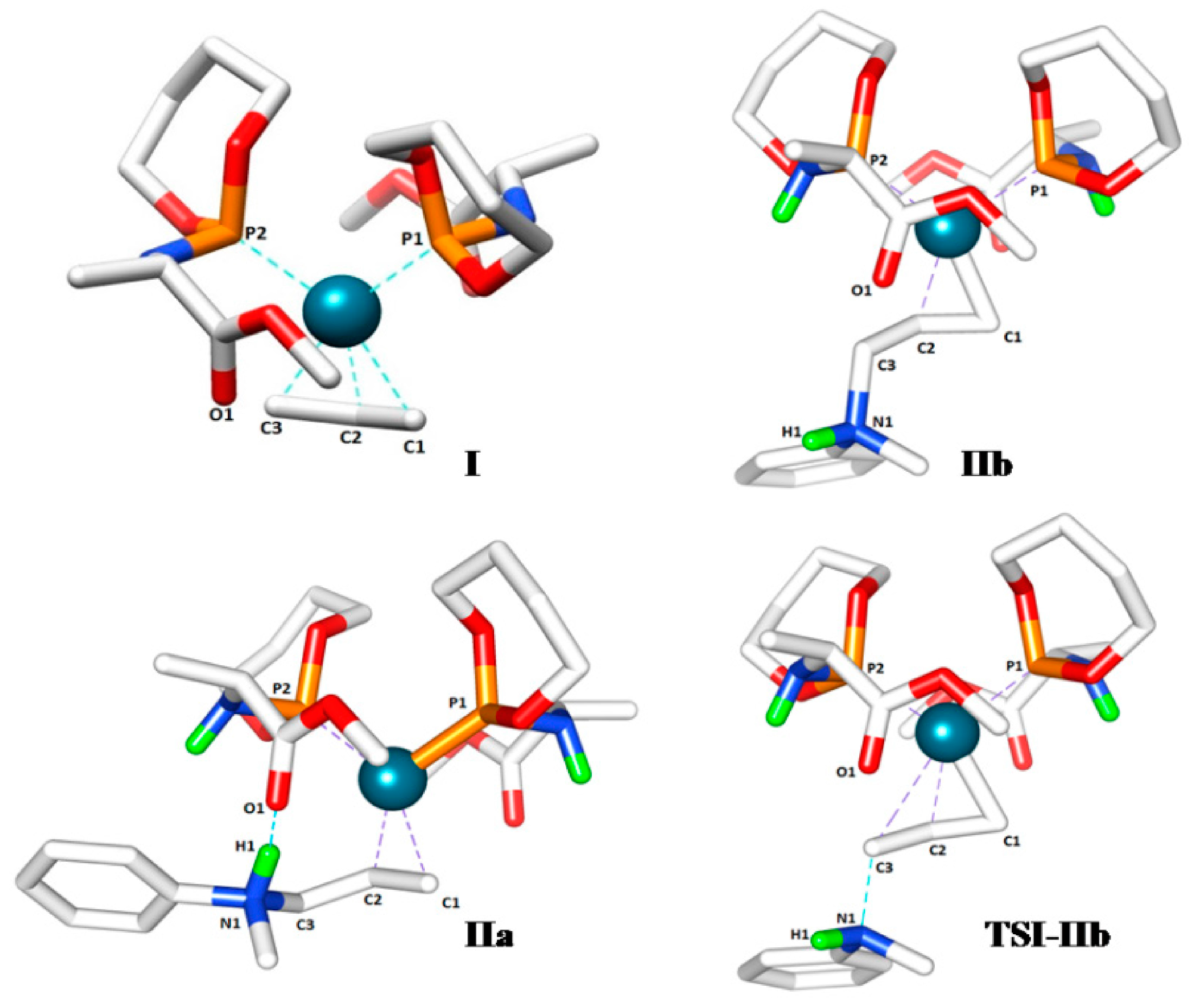
We started by optimizing the geometries of several isomers of the π-allyl complex
I having different conformation of ligand
L’ around the metal center. In subsequent studies we took the structure with the lowest energy as the reference point (
G0I = 0 kcal mol
−1). The coordination geometry of the palladium center in this structure is square planar, with the P-donor of the two ligands L’ and the two donor sites of the bidentate allyl-ligand surrounding the metal. The steric interactions between the two ligands L’ mutually and with the allyl ligand appear to be minimized in
I (
Scheme 4). This geometry of the Pd(
L’) moiety is used for the calculation of succeeding reaction intermediates.
First, we investigated the nucleophilic attack of the amine to the allyl moiety of complex
I. In principle, the nucleophilic attack could follow either an inner-sphere mechanism (first coordination of the nucleophile to the metal, followed by a reductive elimination step) or outer-sphere attack (direct attack of the nucleophile to the allyl moiety, without pre-coordination of the nucleophile to the metal). This strongly depends on the basicity of amines [
26,
27]. Strong nucleophiles (p
Ka > 20) are expected to attack the Pd metal and hence likely follow an inner-sphere mechanism while weaker nucleophiles (p
Ka < 20), such as
N-methylaniline (p
Ka~10), are believed to attacked the allyl-moiety directly and likely follow an outer-sphere mechanism. Thus, in this computational study we focused on the outer-sphere mechanism.
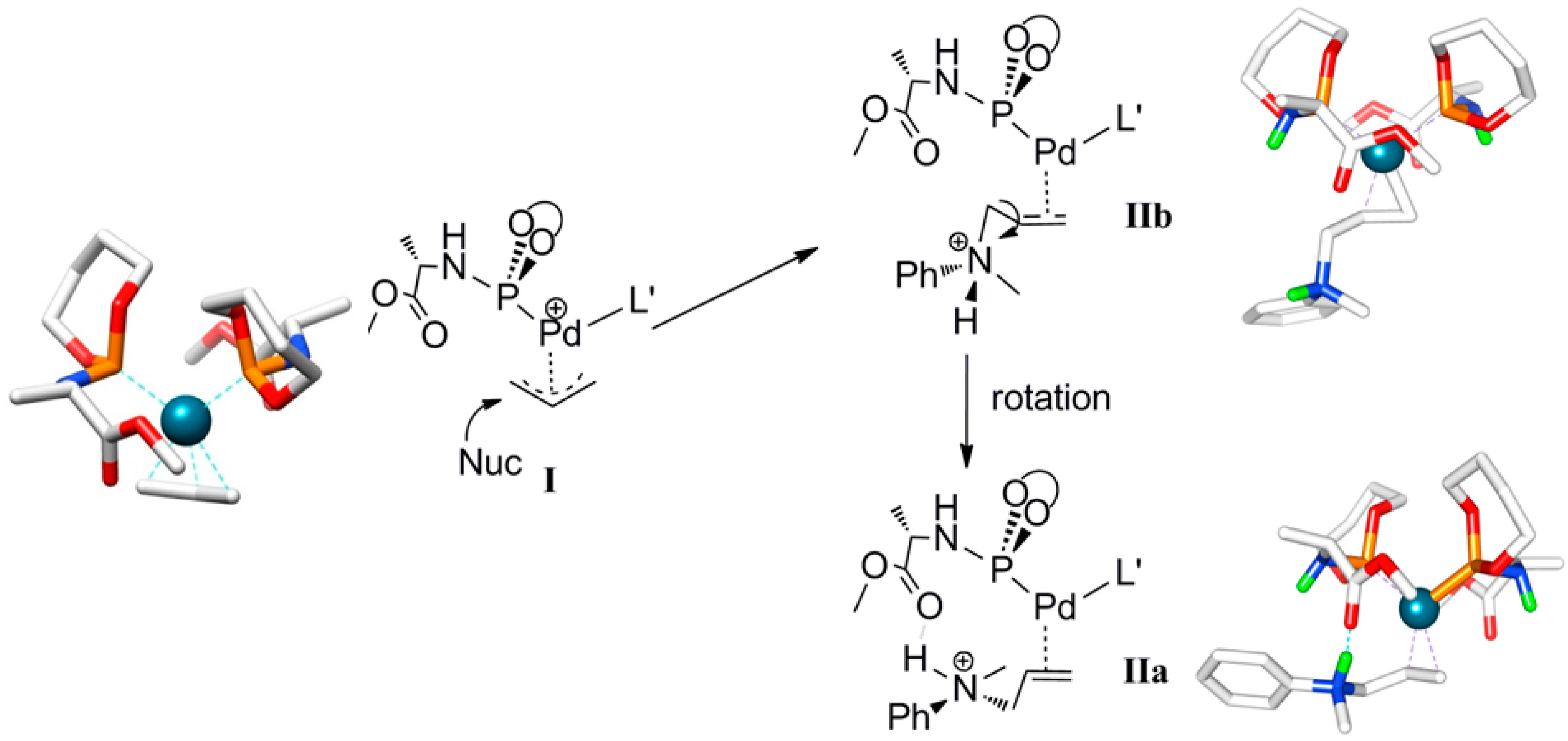
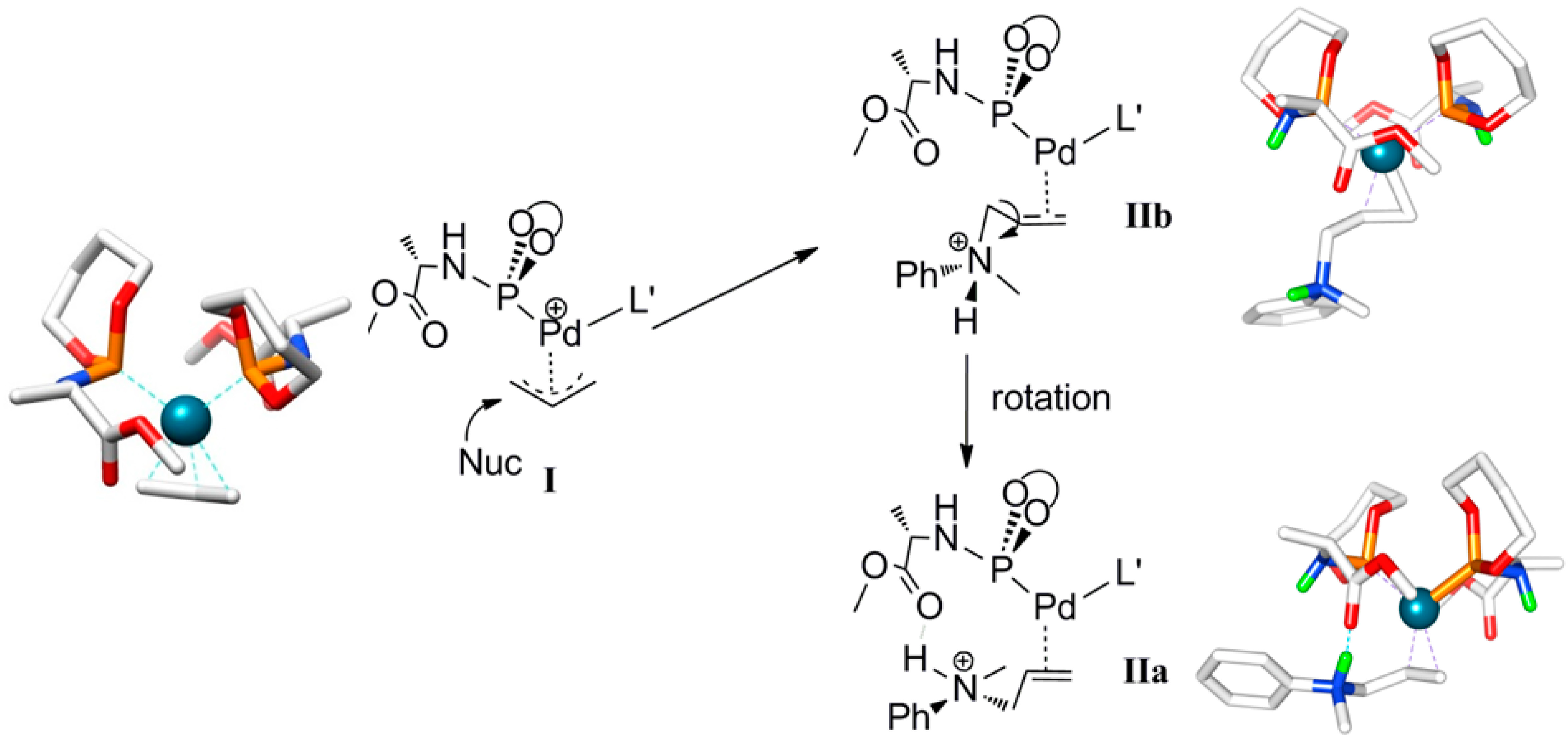
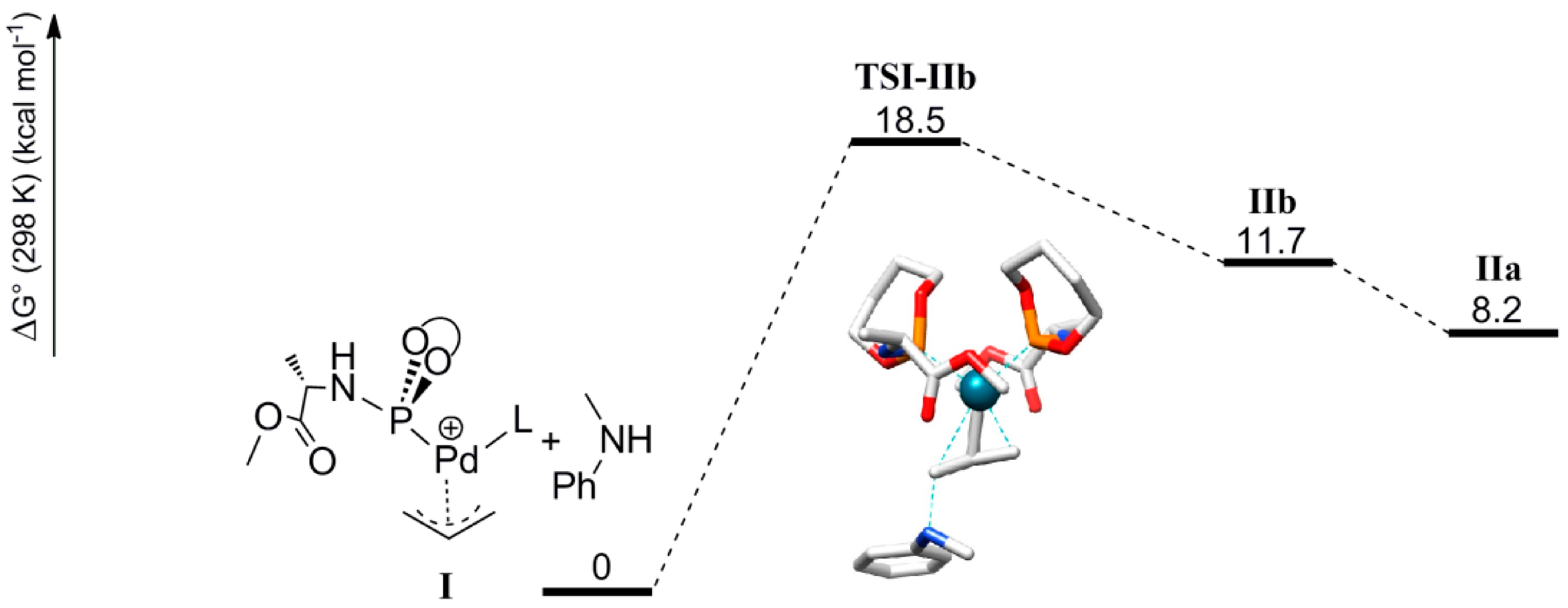
Scheme 4.
Calculated structure and schematic representation of complex I, formation of complex IIbby nucleophilic attack and IIa by rotational isomerization (ΔG0IIa-IIb = −3.5 kcal mol−1).
Scheme 4.
Calculated structure and schematic representation of complex I, formation of complex IIbby nucleophilic attack and IIa by rotational isomerization (ΔG0IIa-IIb = −3.5 kcal mol−1).
Outer-sphere nucleophilic attack at the allyl moiety produces the cationic Pd-allylamine complex
IIb initially. Following this, rapid isomerization of
IIb is expected to form the thermodynamically more stable rotamer
IIa by C-C bond rotation (
Scheme 4). The relative energies of the product
IIa (8.2 kcal mol
−1) is lower than product
IIb (11.7 kcal mol
−1). H-bonding between the N-H moiety of the allyl-amine and the carboxylic group of the ligand
L’ in complex
IIa explains its relative stability (
Scheme 5). Notably, the direct formation of
IIa by the nucleophilic attack at
I is not favorable. Steric hindrance around the palladium-allyl moiety prevents nucleophilic attack in any other direction.
Scheme 5.
Calculated pathway for nucleophilic attack of the amine at complex
I, which forms complex
IIb, which then converts to
IIa via rotation (See
Scheme 4 for the calculated structures and schematic representation of
IIa and
IIb).
Scheme 5.
Calculated pathway for nucleophilic attack of the amine at complex
I, which forms complex
IIb, which then converts to
IIa via rotation (See
Scheme 4 for the calculated structures and schematic representation of
IIa and
IIb).
In the precursor
I, the overall positive charge is delocalized over the Pd
II atom, the P-donors of the ligands
L’ and the allyl moiety, while in
IIa and
IIb the charge is largely concentrated on the quaternary ammonium moieties. Hence, in the absence of stabilization by discrete solvent (or more likely substrate alcohol) molecules, the complexes
IIa and
IIb are artificially destabilized in the applied gas phase DFT calculations. However, when we applied cosmo dielectric solvent corrections, the effects of H-bonding in stabilization of some of the intermediates was partially lost due to overcompensation of dielectric forces [
28], which does not represent the known effects of H-bonding in apolar solvents used experimentally. Thus, despite the abovementioned problems with charge relocation for some steps in this study, we still decided to focus mostly on a description of the mechanistic pathways on the basis of pure gas phase DFT calculations. The cosmo corrected pathways are, however, reported in the supporting information and in
Scheme 8 for comparison, and we will further debate the effects of cosmo corrections in the discussion about the rate limiting step of the catalytic cycle (
vide infra). The overestimated entropies in the gas phase as compared to solution data were corrected by the suggested 6 kcal mol
−1 correction term for each step involving a change in the number of species (see experimental section for details).
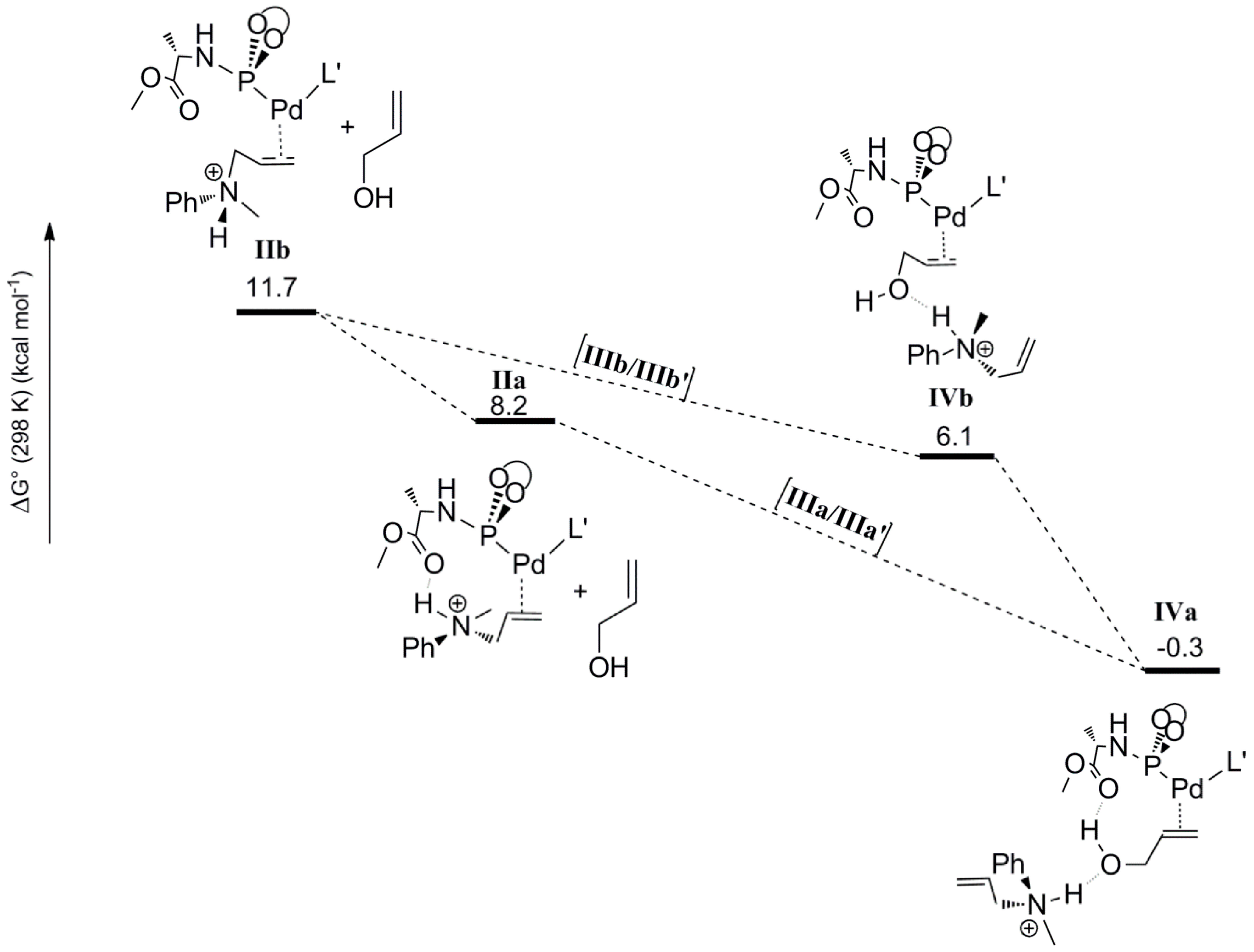
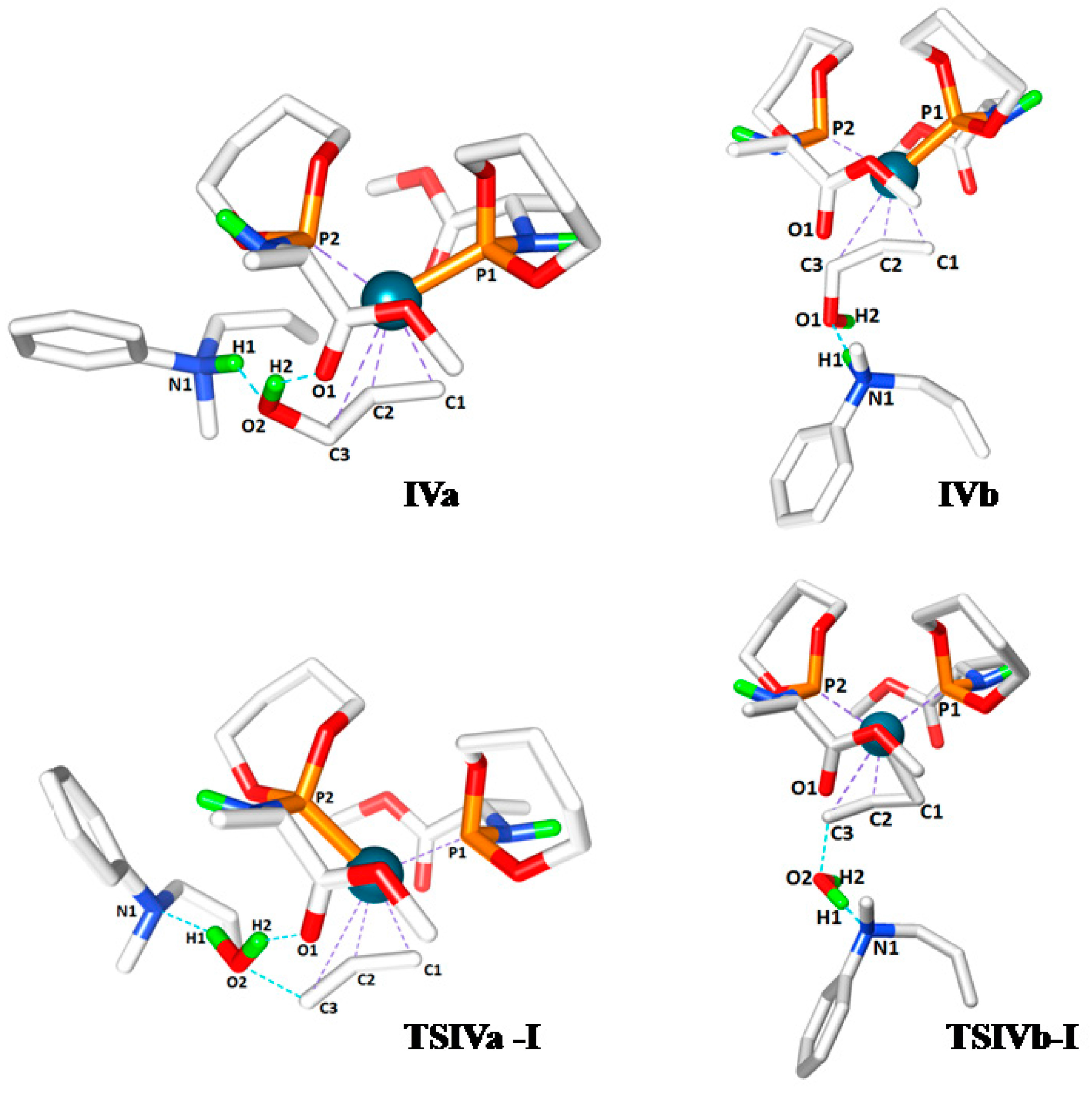
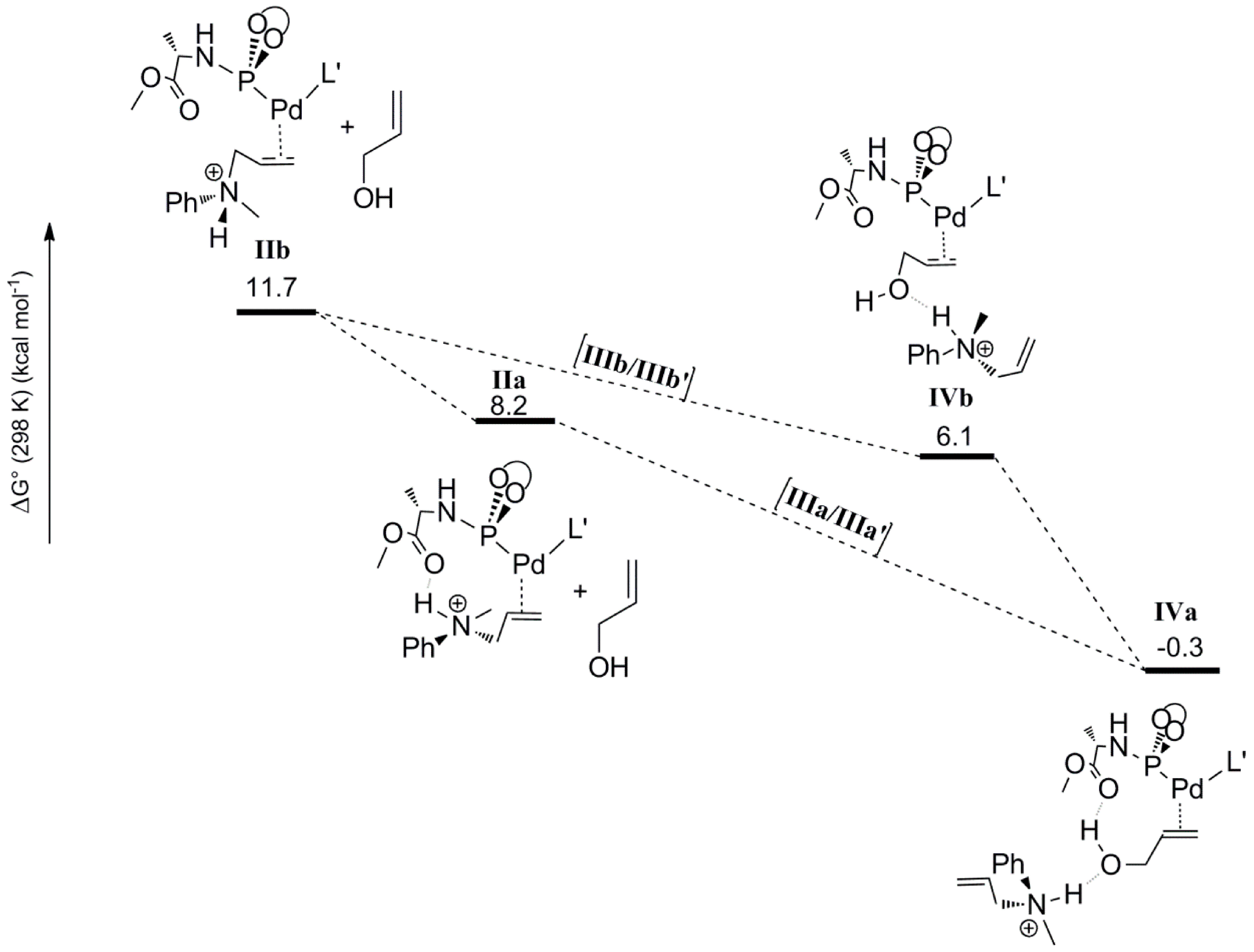
The second step considered in the DFT computed mechanism is the displacement of the protonated allyl-amine coordinated to Pd
0 by the allyl-alcohol substrate (
Scheme 6). Since gas phase DFT calculations become very unreliable when complete charge changes of the computed molecules are involved (e.g., converting a cationic species to a neutral species), we treated this step by keeping the cationic ammonium product H-bonded to the coordinated allyl alcohol. Furthermore, in previous studies, similar treatments (again to avoid charge-associated problems in gas phase calculations) were found to actually have a barrier-lowering effect on the subsequent oxidative addition step due to H‑bond assisted C-O bond breaking [
19]. The coordination of the allyl alcohol substrate to Pd
0 can occur in two manners, leading to two possible configurations
IVa and
IVb based on the position of the hydroxyl group with respect to the carboxyl group of ligand
L’. In
IVa the hydroxyl group of the allylalcohol is H-bonded to the carbonyl group of
L’, whereas in
IVb it is not. In both cases the protonated allylamine is H-bonded to the oxygen atom of the hydroxyl group of the Pd coordinated allyl-alcohol substrate (
Scheme 6). The computed relative free energies of
IVa and
IVb (relative to precursor
I) are –0.3 and 6.1 kcal mol
−1, respectively. Hence species
IVa is the most stable intermediate. Complex
IVa might also form via rotational isomerization of
IVb.
Scheme 6.
Substitution of the coordinated allyl-ammonium product by the allyl alcohol substrate leading to species IVa and IVb.
Scheme 6.
Substitution of the coordinated allyl-ammonium product by the allyl alcohol substrate leading to species IVa and IVb.
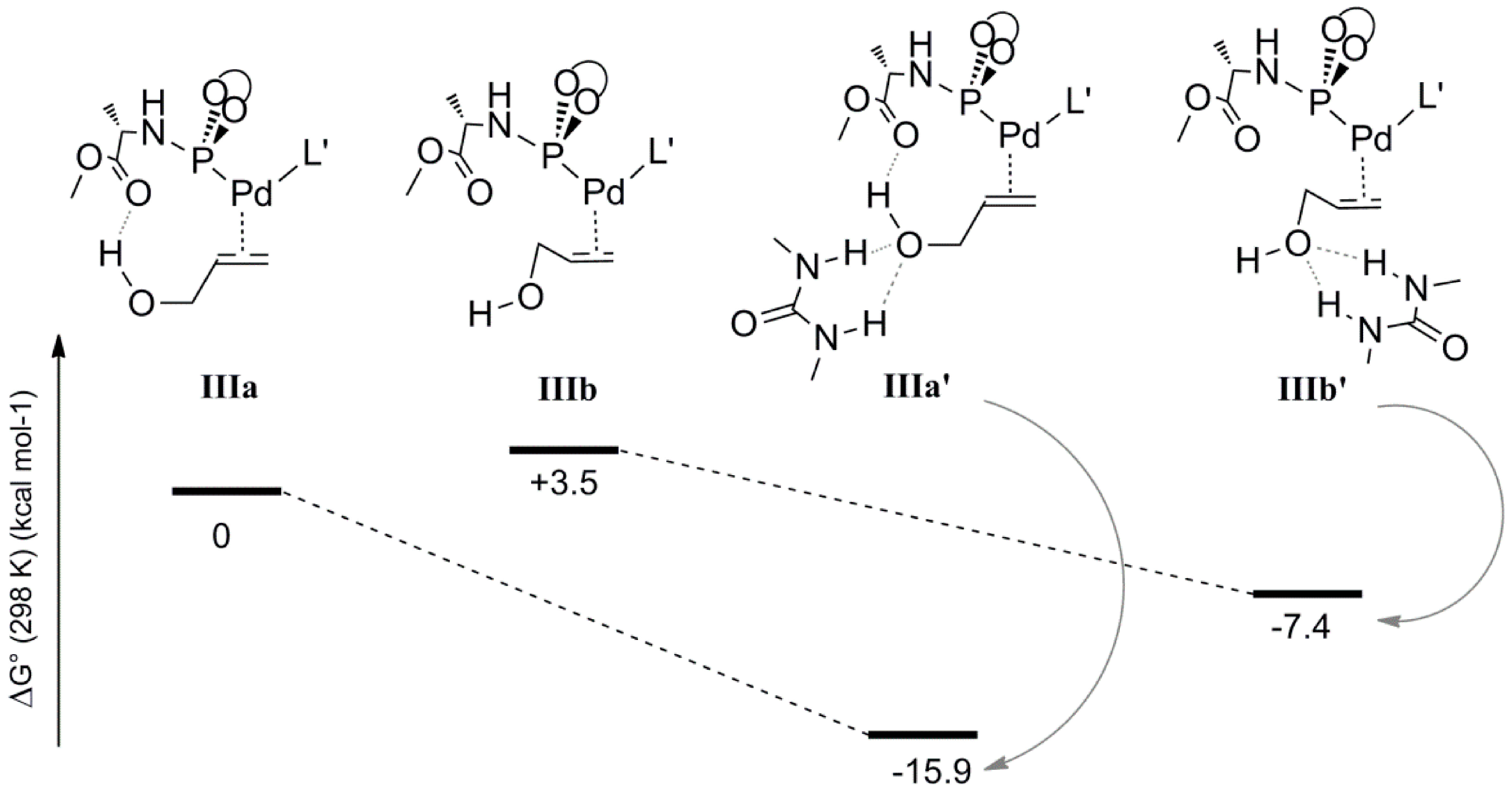
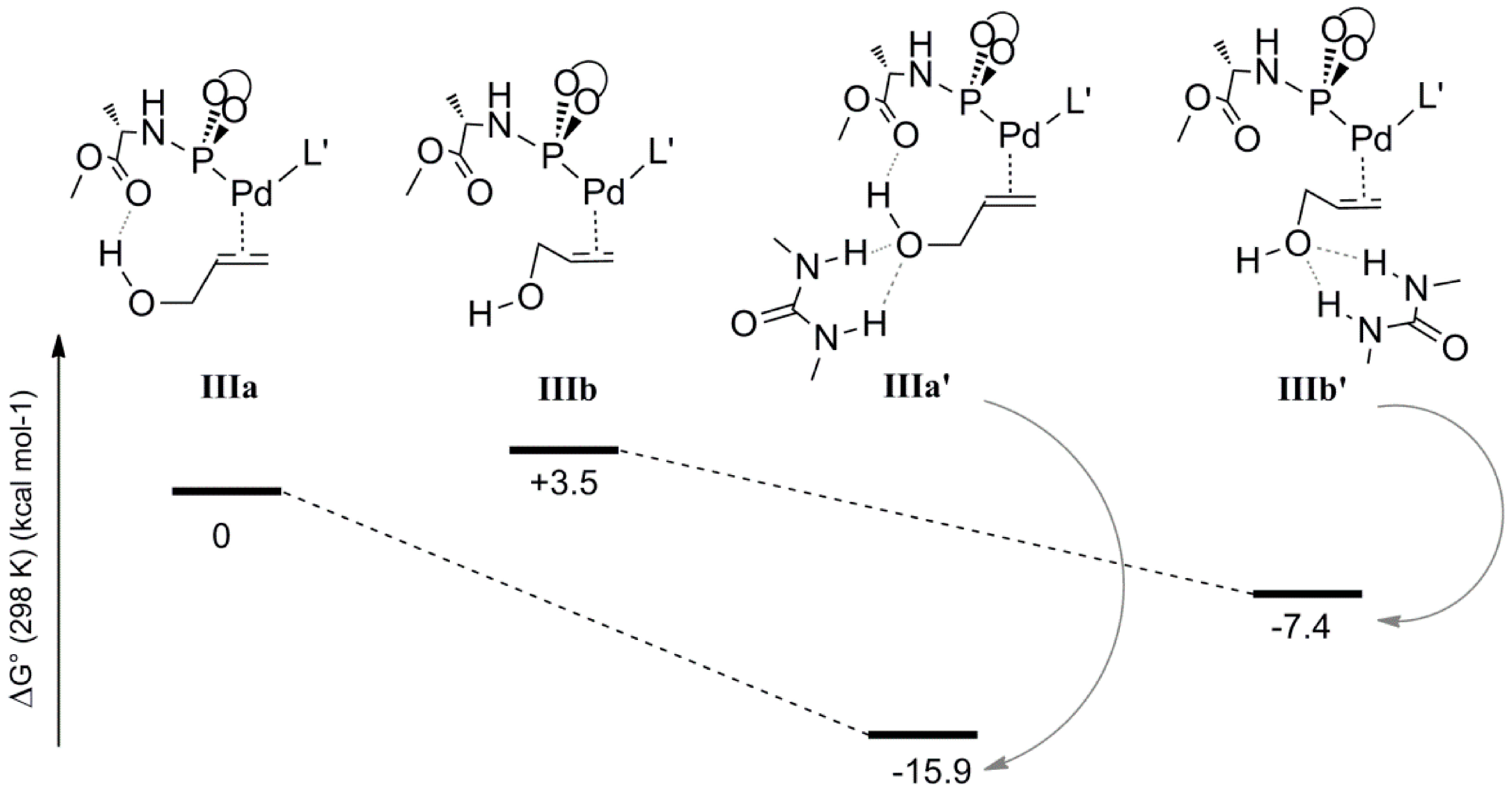
To elucidate the possible role of the 1,3-diethylurea moiety, we also optimized the geometries
IIIa and
IIIb, which are analogues of
IVa and
IVb lacking the H-bonded allyl-ammonium moiety. The energies and geometries of
IIIa and
IIIb were compared to their analogues
IIIa’ and
IIIb’, each containing a 1,3-diethylurea moiety double H-bonded to the hydroxyl of the coordinated allyl-alcohol (
Scheme 7). Complex
IIIa is more stable than
IIIb (relative energies 0 and 3.5 kcal mol
−1, respectively), in line with the fact that
IIIb lacks the internal hydrogen bonding between the hydroxyl moiety of the allylalcohol and the carboxyl group of
L’. The hydrogen bonded 1,3-diethylurea moiety further stabilizes
IIIa’ substantially (Δ
G0 = −15.9 kcal mol
−1) whereas species
IIIb’ shows less stabilization (Δ
G0 = −7.4 kcal mol
−1). Hence, the H-bonding pattern in
IIIa’ based on internal H-bond donation from the hydroxyl-group to ligand
L’ and hydrogen bond acceptation from the 1,3-diethylurea H-bond donor is clearly cooperative, a feature that is commonly observed and is a consequence of polarization. It is very likely that this also plays an important role in the oxidative addition process under the experimental conditions. Unfortunately, it was not possible to compute the effect of 1,3-diethyl urea H-bonding on the barrier of the rate determining oxidative addition process. This is due to the abovementioned requirement to keep the overall charge neutral to prevent any unrealistic charge effects in the gas phase calculations. Hence, to still mimic the effect of a hydrogen bond, we used the associated allyl-ammonium salt product, which can deliver a proton after the transition state to keep an overall 1+ charge for each complex involved in that catalytic cycle.
Scheme 7.
The energy profile of the allyl alcohol coordinated Pd complexes in the presence and absence of urea molecule, IIIa-IIIa’ and IIIb-IIIb’.
Scheme 7.
The energy profile of the allyl alcohol coordinated Pd complexes in the presence and absence of urea molecule, IIIa-IIIa’ and IIIb-IIIb’.
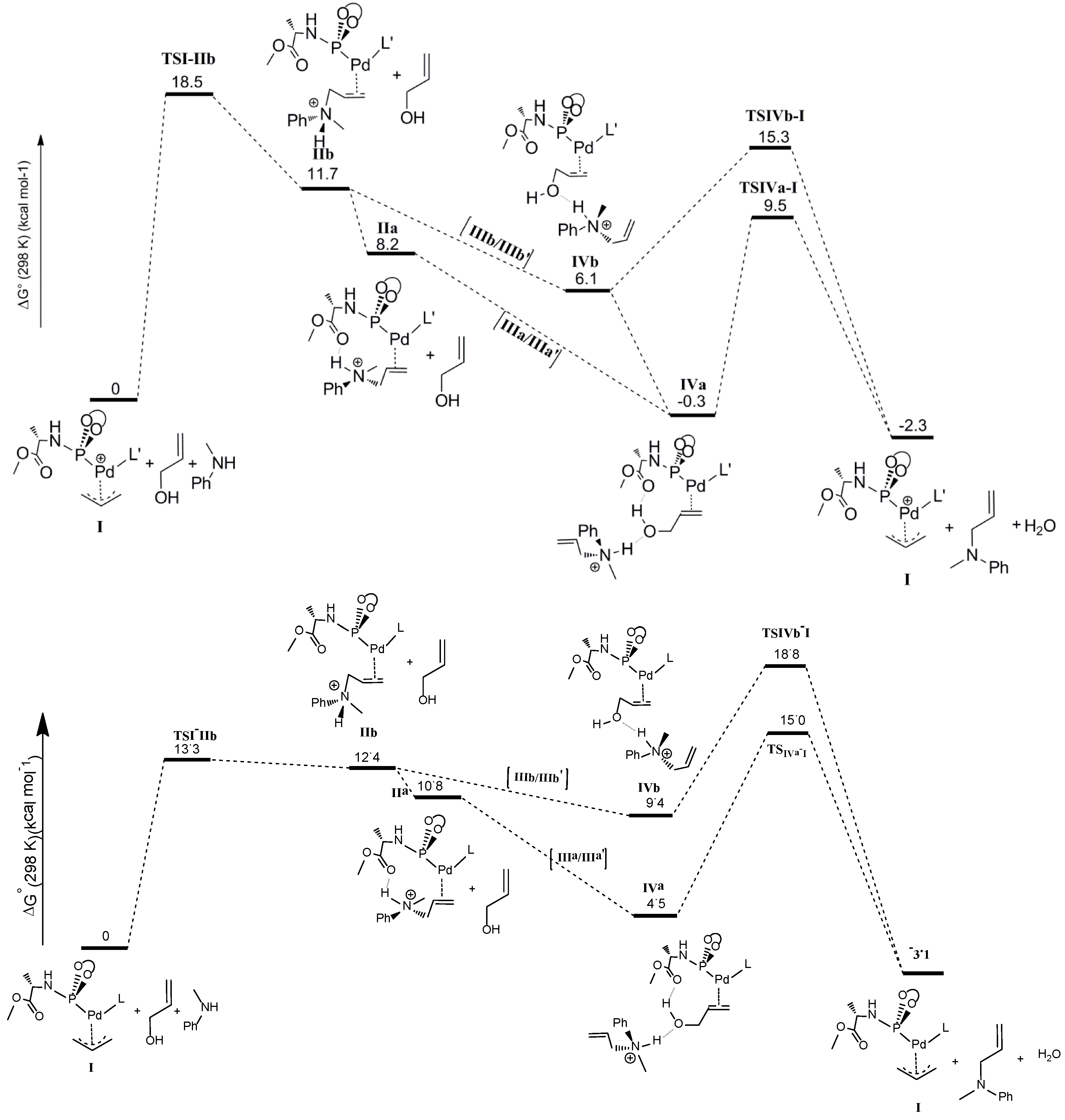
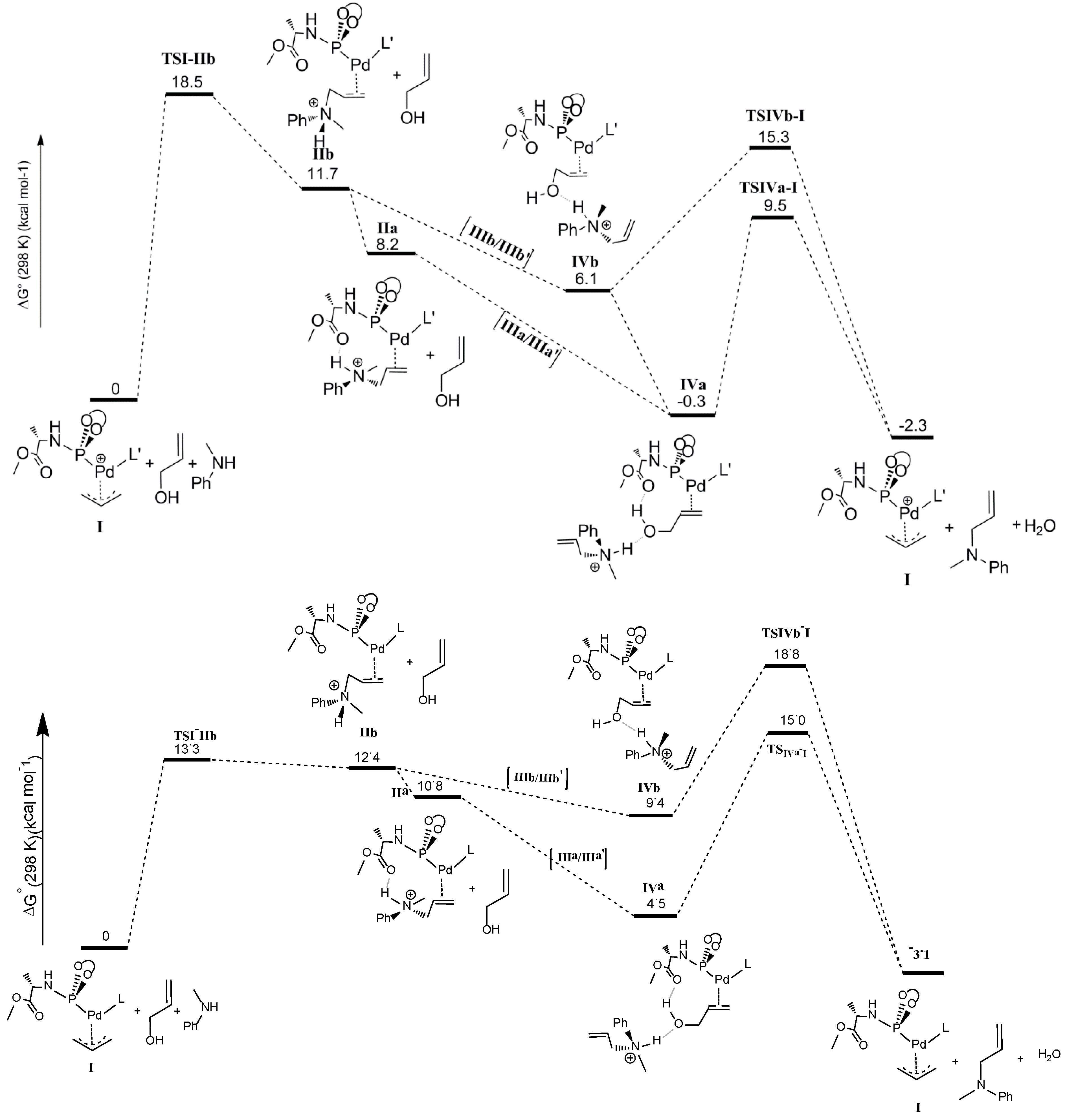
The last step of the computed catalytic cycle involves oxidative addition of the C‑O bond of the allyl alcohol substrate in intermediates
IVa and
IVb to regenerate the palladium-allyl intermediate
I. Since this process involves breaking the C-O bond and liberation of a hydroxide (OH
−) moiety being a poor leaving group, this process is expected to be facilitated by hydrogen bonding. In good agreement, the
TSIVa-I barrier (9.5 kcal mol
−1) for oxidative addition of the C-O bond is lower than
TSIVb‑I (15.3 kcal mol
−1) reflecting the effects of cooperative H-bonding (
Scheme 8). The process leads to elimination of water, because the H-bonded ammonium salt transfers its proton to the hydroxide-leaving group in a concerted manner. Hence, unfavorable uncompensated charge relocations in the gas phase calculations play a smaller role in the transition states
TSIVa-I and
TSIVb-I than in the precursors
IVa and
IVb. Besides, the relative barriers on going from
IVa and
IVb to
TSIVa-I and
TSIVb-I are likely somewhat underestimated, while the absolute barriers of
TSIVa-I and
TSIVb-I versus I suffer less from such uncompensated charge relocation effects.
In the gas phase calculations without cosmo dielectric solvent corrections, the transition state barrier for nucleophilic attack of the amine at the allyl moiety (
TSI-IIb = 18.5 kcal mol
−1) is much higher than the transition state for the oxidative addition step (
TSIVa-I = 9.5 kcal mol
−1), which would indicate that the former should be the rate determining step. However, the experimental kinetic experiments clearly indicate that the oxidative addition is rate determining. Concerning the rate determining step, the experimental data and the computational model without cosmo corrections are therefore not in agreement. This is due to the abovementioned charge concentration effects in this reaction, so that the gas phase DFT numbers do not accurately describe the solution data. Therefore we also investigated the overall pathway including cosmo dielectric solvent corrections. The results taking toluene as the medium are shown in the bottom part of
Scheme 8. Indeed, this leads to a shift in the rate-limiting step from the nucleophilic attack to the oxidative addition step, in good agreement with the experimental data. To describe the effects of H-bonding, however, some of the steps seem to be better described without cosmo, which is the reason we focused on discussing those calculations first.
Scheme 8.
Calculated catalytic pathway of the complete cycle. Top: Free energies without cosmo corrections; Bottom: Free energies with cosmo corrections (ε = 2.38; toluene).
Scheme 8.
Calculated catalytic pathway of the complete cycle. Top: Free energies without cosmo corrections; Bottom: Free energies with cosmo corrections (ε = 2.38; toluene).
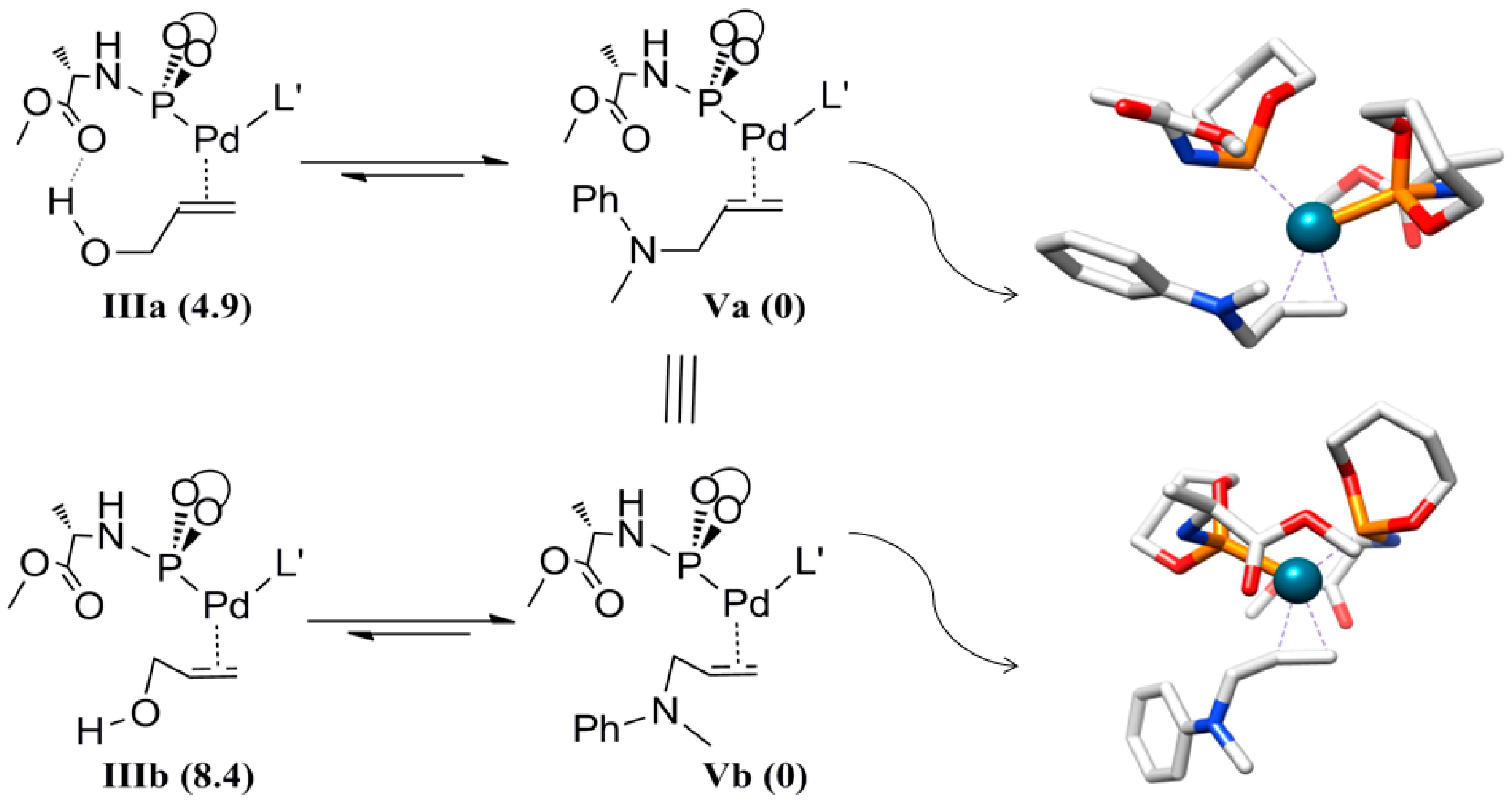
As reported, we have discovered by kinetic analysis that product inhibition takes place during the catalysis. The coordination of the allylamine to Pd is computed to be more favorable than coordination of the allyl alcohol. We have examined this by calculating the product coordinated Pd complexes with two possible configurations and determined the relative energies with respect to allyl alcohol coordinated Pd complexes
IIIa and
IIIb (
Scheme 9). The formation of these complexes is expected to take place along with the formation of isomers
IVa and
IVb (the hydrogen bonded analogues) from
IIa and
IIb, which represent the product-coordinated complexes. Deprotonation of the product in complex
IIa/IIb affords complexes
Va and
Vb.
Scheme 9.
Schematic representation of the product inhibition equilibriums and calculated structures of Va and Vb. The values between parentheses are the relative energies (kcal mol−1). The orientation of the allylamine at Pd does not lead to a significant energy difference (complex Va and Vb have the same relative free energy).
Scheme 9.
Schematic representation of the product inhibition equilibriums and calculated structures of Va and Vb. The values between parentheses are the relative energies (kcal mol−1). The orientation of the allylamine at Pd does not lead to a significant energy difference (complex Va and Vb have the same relative free energy).
Notably, both isomers Va and Vb have the same relative free energy, indicating the poor secondary interaction between the product and the complex, besides the alkene coordination energy. The relative energies of IIIa and IIIb with respect to V were found to be 4.9 and 8.4 kcal mol−1, respectively. Note that the allyl amine adducts Va and Vb are more stable than the allyl alcohol adducts, in agreement with the experimental observations, showing product inhibition. This is likely due to sigma bond inductive effects of nitrogen versus oxygen. The lower energy of IIIa compared to IIIb is due to the hydrogen bond with the carbonyl of the ligand. The same trend is observed for intermediate IVa being lower in energy than the IVb, and the larger difference in these complexes is again a result of cooperative hydrogen bonding.
2.1. Summary of the DFT Computed Pathway
The most interesting finding of the DFT study is the cooperative hydrogen bond between the hydroxyl and carbonyl groups (substrate-ligand) and urea (or the ammonium salt we used as a reference). We found experimentally that phosphoramidite ligands that do not have the ester functionality did not show the urea co-catalyzed activation of allyl alcohols, which is in line with the DFT calculations. The interaction between the coordinated allyl alcohol and ligand
L’ proved to be significant in the whole catalytic cycle, overall lowering the relative free energy of the Pd-intermediates (
Scheme 8). The function of 1,3-diethylurea is determined to further lower the barrier for C-O oxidative addition (
Scheme 10a). In addition, the calculations show a preference for the coordination of the product leading to product inhibition, however, a cooperative urea binding to
IIIa will shift the equilibrium in favor of the substrate complex (
Scheme 7 and
Scheme 9). The computed catalytic cycle complies with the experimental kinetic data if we include cosmo dielectric solvent corrections. Oxidative addition of the C‑O bond of the coordinated allyl alcohol in intermediate
IVa via the rate limiting transition state
TSIVa-I is assisted by H-bonding. This was computed using the ammonium product as the model (to reduce undesired effects of charge changes), but in practice this does explain the rate enhancing effect of the urea (
Scheme 10b). This mechanism deviates from the mechanism proposed by Ozama, Samec and ourself involving allyl alcohol activation with palladium hydrides [
29,
30].
Scheme 10.
(a) Proposed catalytic cycle in agreement with the experimental studies. (b) DFT calculated catalytic cycle using the allyl ammonium product as a model for urea H-bonding (intermediate IVa as a model for IIIa’).
Scheme 10.
(a) Proposed catalytic cycle in agreement with the experimental studies. (b) DFT calculated catalytic cycle using the allyl ammonium product as a model for urea H-bonding (intermediate IVa as a model for IIIa’).
2.2. Bond Length Analysis
We analyzed the bond length changes throughout the calculated catalytic pathway in order to obtain more information about the effect of H-bond interactions on the formation of the isomeric intermediates
a and
b (see
Table 1). First, we examined the nucleophilic attack of the
N-methylaniline to complex
I, which involves intermediates
IIb and its respective transition state structure
TSI-IIb (
Figure 1). The Pd-P1 and Pd-P2 bond lengths show only small changes on going from the precursor
I to the transition state
TSI-IIb and subsequently formed the
IIb, which has virtually same bond distances as
I (
Table 1).
Figure 1.
In all structures the related atoms are labeled such that C1 refers to the allyl carbon trans to P2, C2 is the central allyl carbon and C3 is the terminal allyl carbon trans to P1.
Figure 1.
In all structures the related atoms are labeled such that C1 refers to the allyl carbon trans to P2, C2 is the central allyl carbon and C3 is the terminal allyl carbon trans to P1.
Table 1.
Bond lengths (Å) of the calculated structures of intermediates involved in nucleophilic attack.
Table 1.
Bond lengths (Å) of the calculated structures of intermediates involved in nucleophilic attack.
| Bond | I | IIa | IIb | TSI-IIb |
|---|
| dPd-P1 | 2.261 | 2.274 | 2.261 | 2.256 |
| dPd-P2 | 2.262 | 2.245 | 2.26 | 2.266 |
| dPd-C1 | 2.233 | 2.176 | 2.143 | 2.147 |
| dPd-C2 | 2.205 | 2.149 | 2.162 | 2.176 |
| dPd-C3 | 2.225 | 3.167 | 2.99 | 2.853 |
| dC3-N1 | - | 1.521 | 1.639 | 1.841 |
| dN1-H1 | - | 1.054 | 1.028 | 1.025 |
| dH1-O1 | - | 1.8 | - | - |
The attack of the amine leads to elongation of the Pd-C3 bond (
I Pd-C3 = 2.225 Å,
TSI-IIb Pd-C3 = 2.853 Å) while the Pd-C1 and Pd-C2 bonds shorten, as expected. On the other hand, generated by the rotational isomerization, the coordinated ligands of
IIa require a rearrangement of the ligands around palladium to allow the H-bond interactions between the proton of the amine (H1) and the carboxyl moiety of the ligand (O1). Thus, the bond lengths of Pd-P1 and Pd-P2 show clear changes (compare Pd-P1 = 2.261 Å, Pd-P2 = 2.260 Å in
IIb with Pd-P1 = 2.274 Å, Pd-P2 = 2.245 Å in
IIa) (
Table 1). Besides, the newly formed C3-N1 bond is also affected by the H-bond interactions. The shorter C3‑N1 bond of the lower energy isomer
IIa must be the result of H1-O1 interaction as it directs the positioning of the amine (
IIa C3-N1 = 1.521 Å,
IIb C3-N1 = 1.639 Å) (
Table 1).
Next, we analysed the bond length changes occurring in the oxidative addition step, which involves the simultaneous carbon-hydroxyl bond dissociation (C3-O2), hydroxyl-hydrogen bond formation (H1-O2, water), the release of the product and formation of the complex
I (
Table 2). The intermediate
IVa was found lowest in energy (−0.3 kcal mol
−1), which also needed lower activation energy than
IVb to generate the products (
TSIVa-I = 9.5 and
TSIVa-I = 15.3 kcal mol
−1). Judging from the comparative Pd-C3 distances of
TSIVa-I and
TSIVb-I (3.223 and 2.816 Å, respectively) relative to
I (2.225 Å), the
TSIVa-I represents an early transition state (
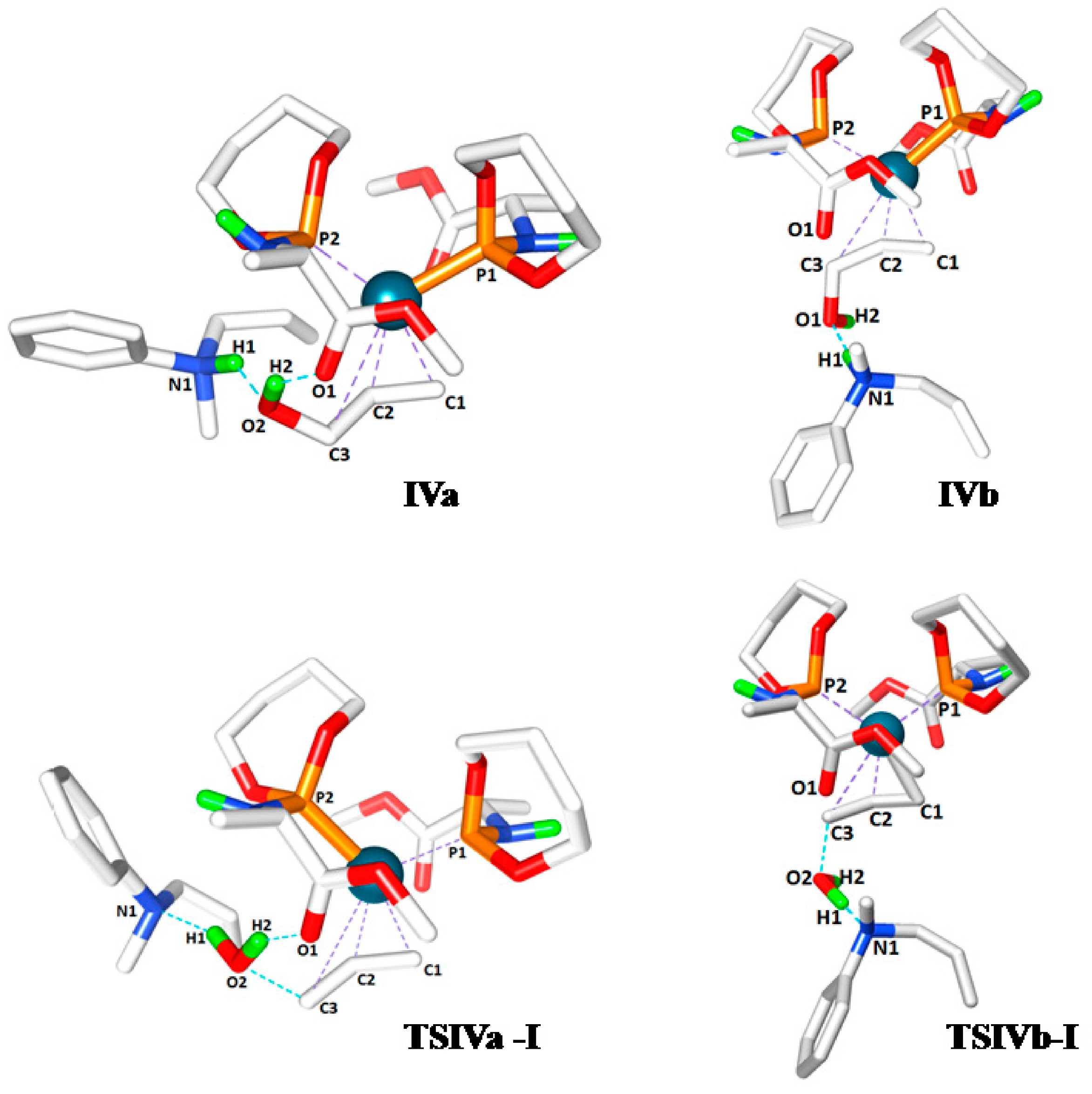
Table 2). Moreover, we determined that the additional H-bond interaction in
TSIVa-I, between the proton acceptor carbonyl (O1) and proton donor hydroxyl group (H2), facilitates the C3-O2, N1-H1 bond dissociation and H1-O2 bond formation processes of the transition state by altering the partial charges of the participating atoms (
Figure 2). The C3-O2 and N1-H1 bonds are longer in
TSIVa-I (1.950 and 1.760 Å) compared to
TSIVb-I (1.844 and 1.360Å) and the H1‑O2 bond distance in
TSIVa-I (1.018 Å) is shorter than in
TSIVb-I (1.171 Å), which might be the effect of complementary H-bonding in
TSIVa-I.
Figure 2.
Transition state structures of IVa, TSIVa-I, IVb and TSIVb-I.
Figure 2.
Transition state structures of IVa, TSIVa-I, IVb and TSIVb-I.
Table 2.
Bond lengths (Å) of the calculated structures of intermediates involved in oxidative addition step.
Table 2.
Bond lengths (Å) of the calculated structures of intermediates involved in oxidative addition step.
| Bond | I | IVa | IVb | TSIVa-I | TSIVb-I |
|---|
| dPd-P1 | 2.261 | 2.272 | 2.259 | 2.244 | 2.26 |
| dPd-P2 | 2.262 | 2.235 | 2.247 | 2.284 | 2.271 |
| dPd-C1 | 2.233 | 2.218 | 2.167 | 2.128 | 2.143 |
| dPd-C2 | 2.205 | 2.261 | 2.158 | 2.334 | 2.199 |
| dPd-C3 | 2.225 | 3.182 | 3.048 | 3.223 | 2.816 |
| dC3-O2 | - | 1.454 | 1.501 | 1.950 | 1.844 |
| dN1-H1 | - | 1.064 | 1.081 | 1.760 | 1.360 |
| dH1-O2 | - | 1.793 | 1.575 | 1.018 | 1.171 |
| dH2-O2 | - | 0.988 | 0.975 | 0.997 | 0.976 |
| dH2-O1 | - | 1.845 | - | 1.720 | - |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}