Carbon-Supported PtRuMo Electrocatalysts for Direct Alcohol Fuel Cells



Abstract
:
1. Introduction

2. Results and Discussion
2.1. Preparation and Characterization of Carbon Supported PtRuMo Catalysts
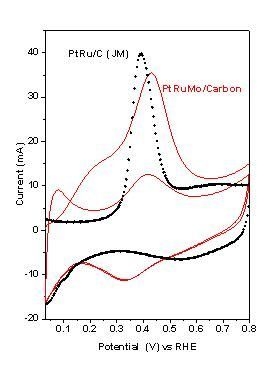
2.1.1. Effect of Mo Precursor

{kind=link}
{kind=link}
{kind=link}
{kind=link}
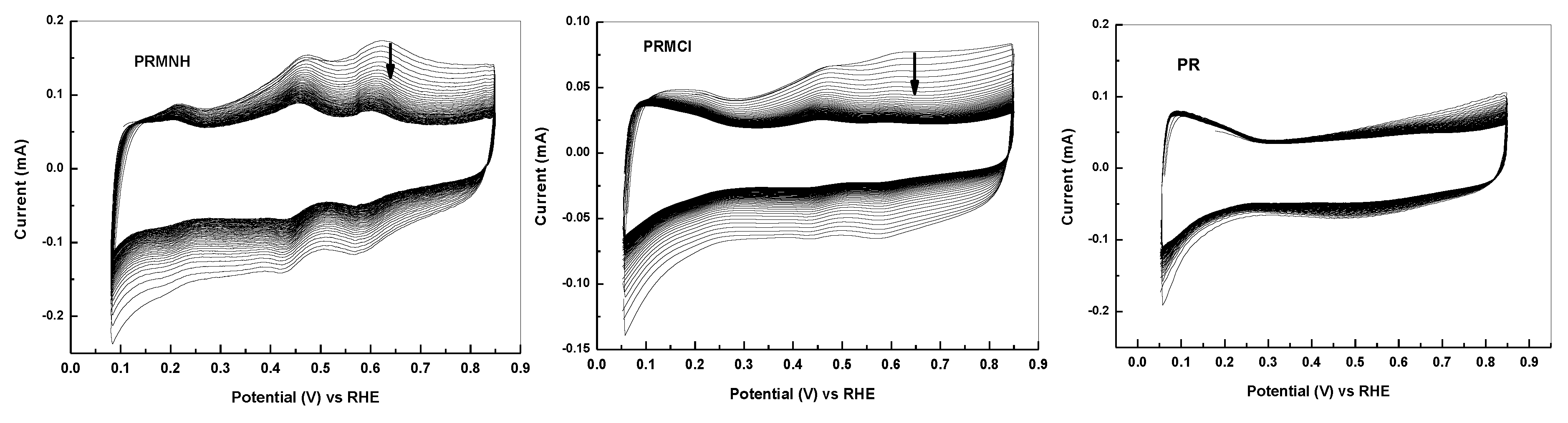{kind=link}
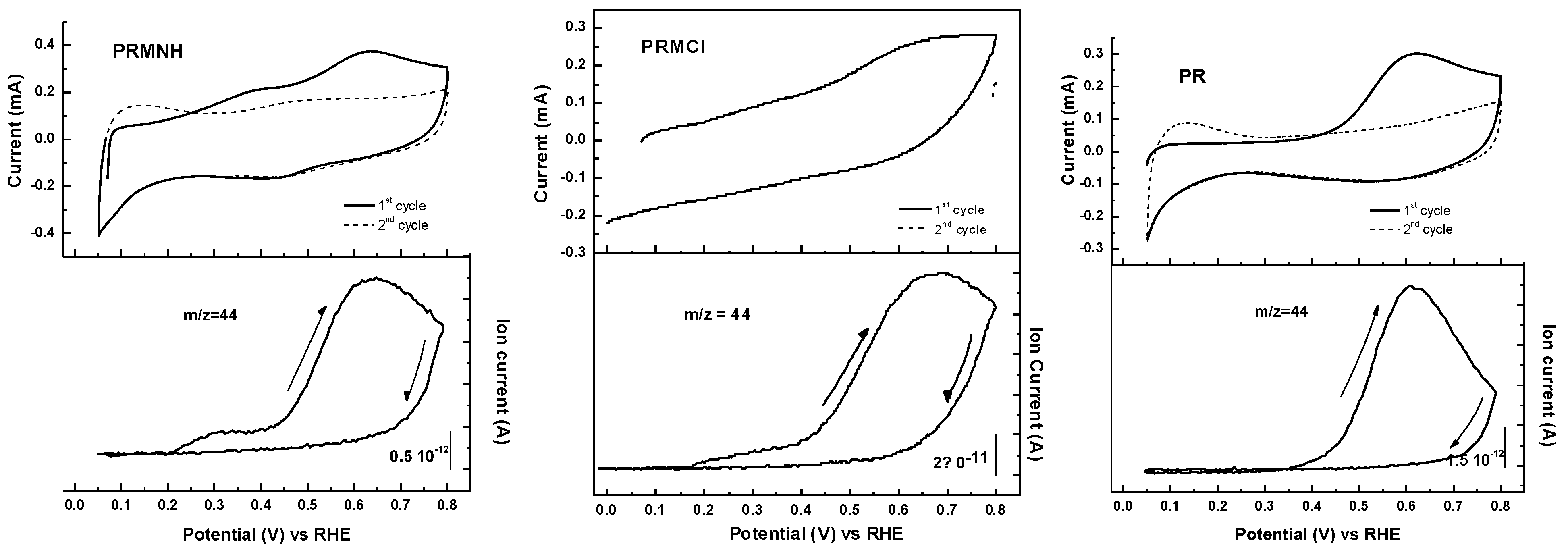{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Average particle size (nm) a | Atomic ratio Pt:Ru:Mo | ||
|---|---|---|---|---|
| Bulk composition b | Surface composition c | Bulk composition after stability test b | ||
| PRMCl | 3.8 ± 0.5 | 1:1:0.4 | 1:1.3:0.4 | 1:0.24:0.05 |
| PRMNH | 3.9 ± 0.5 | 1:1:1.2 | 1:1.5:1.3 | 1:0.35:0.3 |
| PR | 3.2 ± 0.5 | 1:0.9 | 1:1 | 1:0.6 |

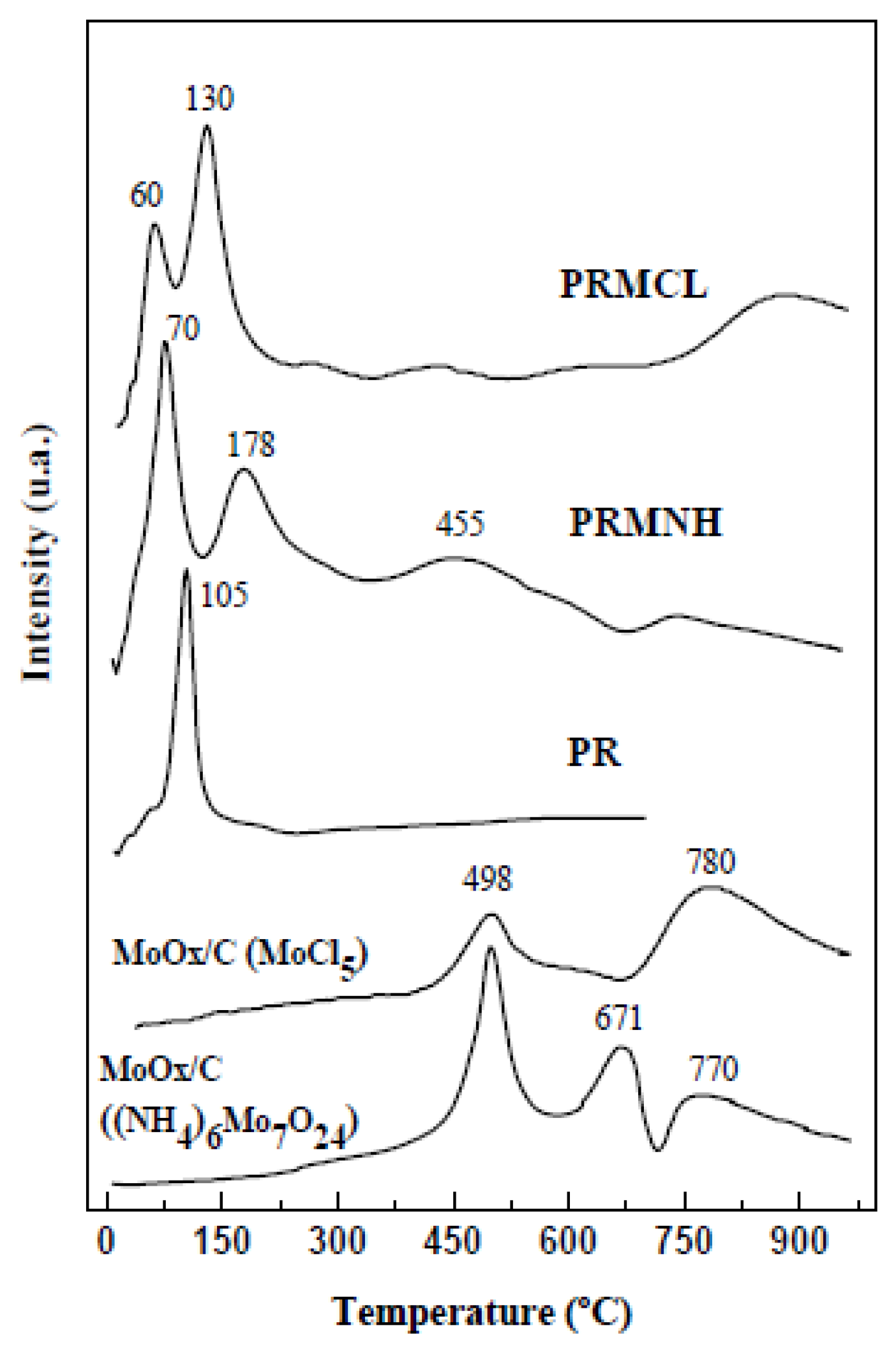
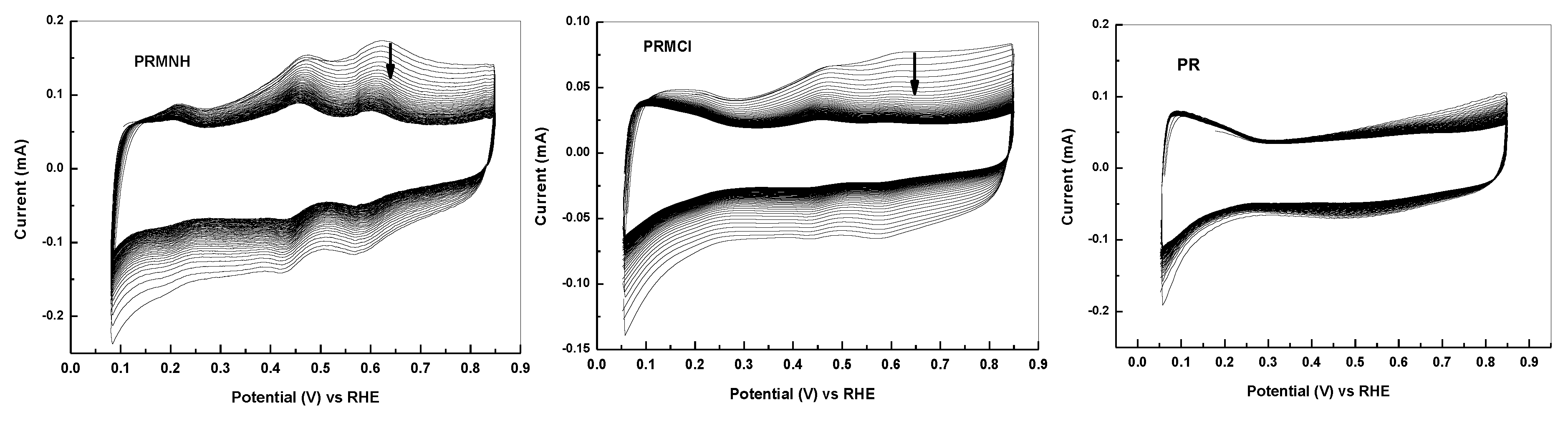
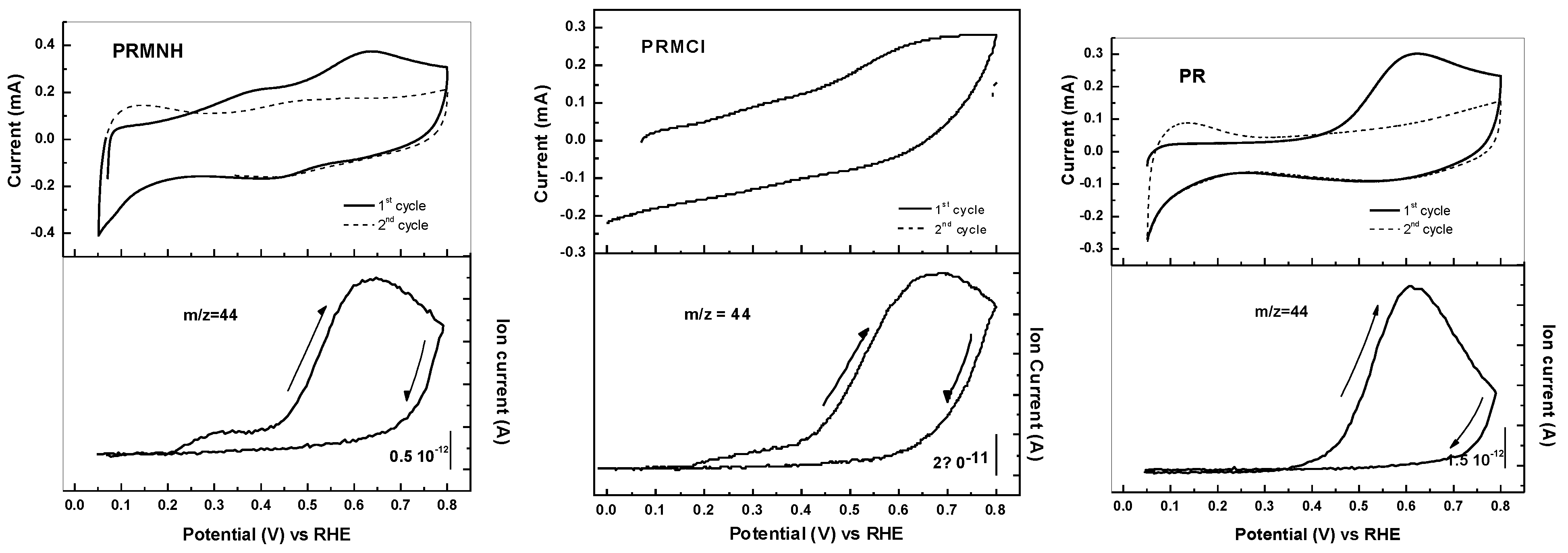
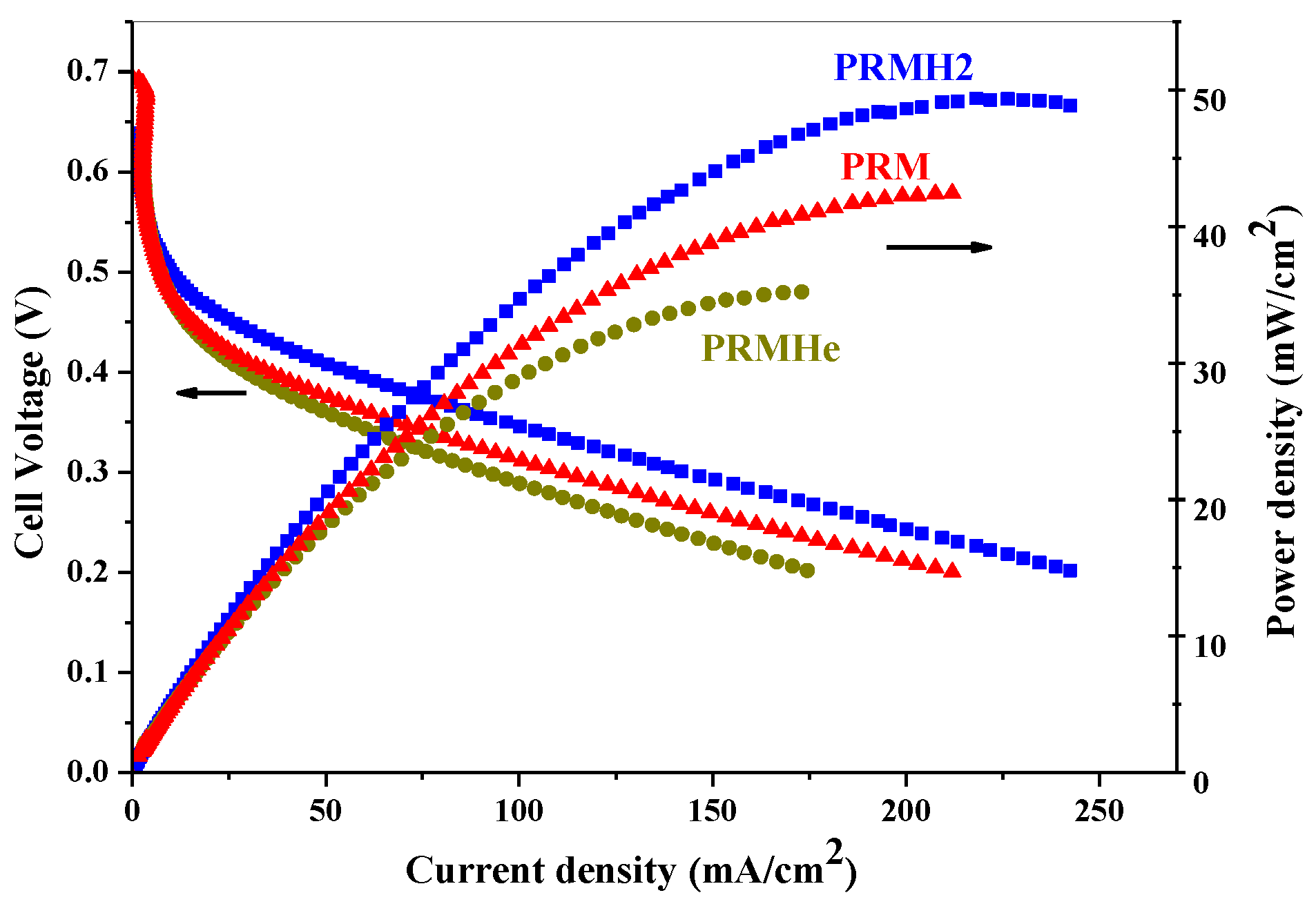
2.1.2. Effect of Thermal Treatment
| Catalysts | Pt:Ru:Mo
atomic ratio | Average particle size (nm) (TEM) | Lattice parameter (afcc) (Å) |
|---|---|---|---|
| PRM | 1:0.8:0.3 | 2.5 | 3.920 |
| PRMHe | 1:0.8:0.3 | 3.2 | 3.909 |
| PRMH2 | 1:0.8:0.3 | 3.7 | 3.894 |


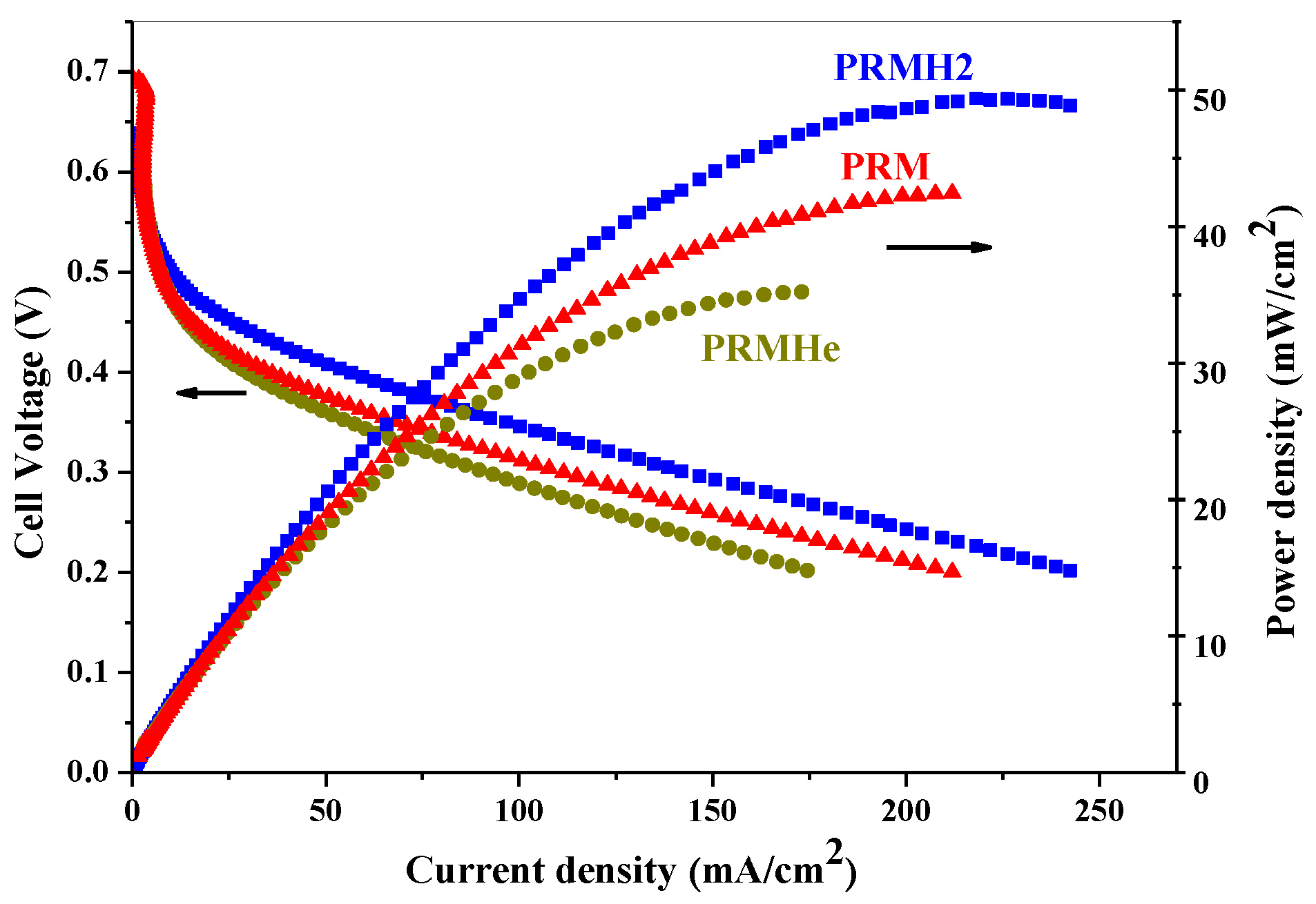
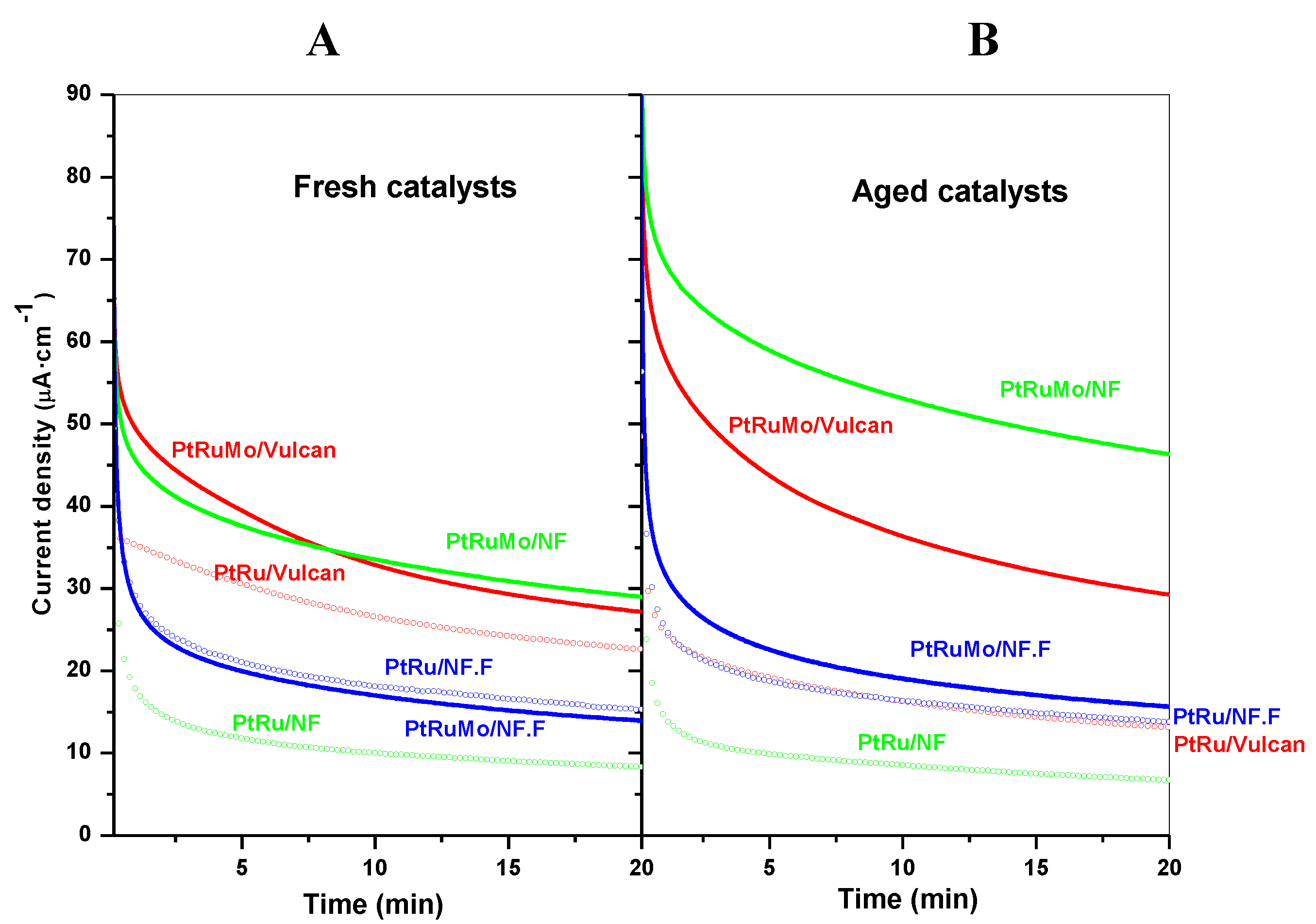
2.2. Effect of Carbon Support
| Catalysts | Bulk compositionPt:Ru (TXRF) | Average particle size (nm) (XRD) | Catalysts | Bulk composition Pt:Ru:Mo (TXRF) | Average particle size (nm) (XRD) |
|---|---|---|---|---|---|
| PtRu/Vulcan | 1:1 | 3.1 | PtRuMo/Vulcan | 1:0.9:0.7 | 2.6 |
| PtRu/NF | 1:0.9 | 2.5 | PtRuMo/NF | 1:0.9:1.2 | n.d. |
| PtRu/NF.F | 1:0.9 | 3.4 | PtRuMo/NF.F | 1:0.8:0.5 | 3.1 |
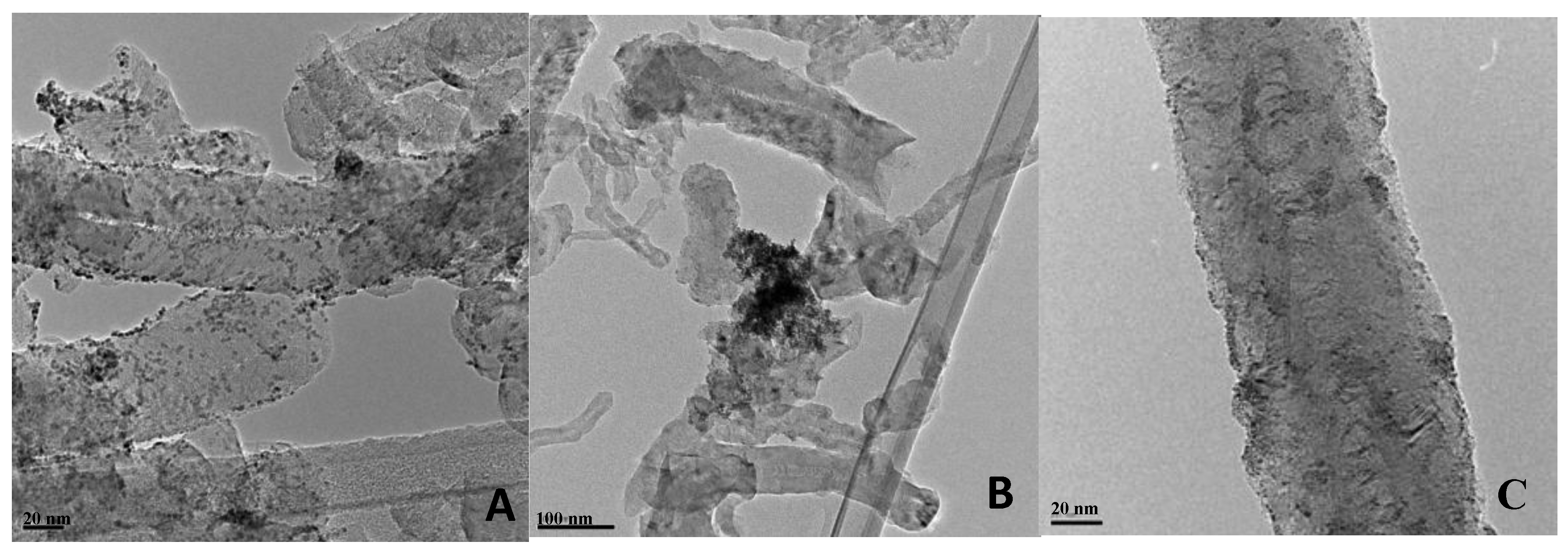
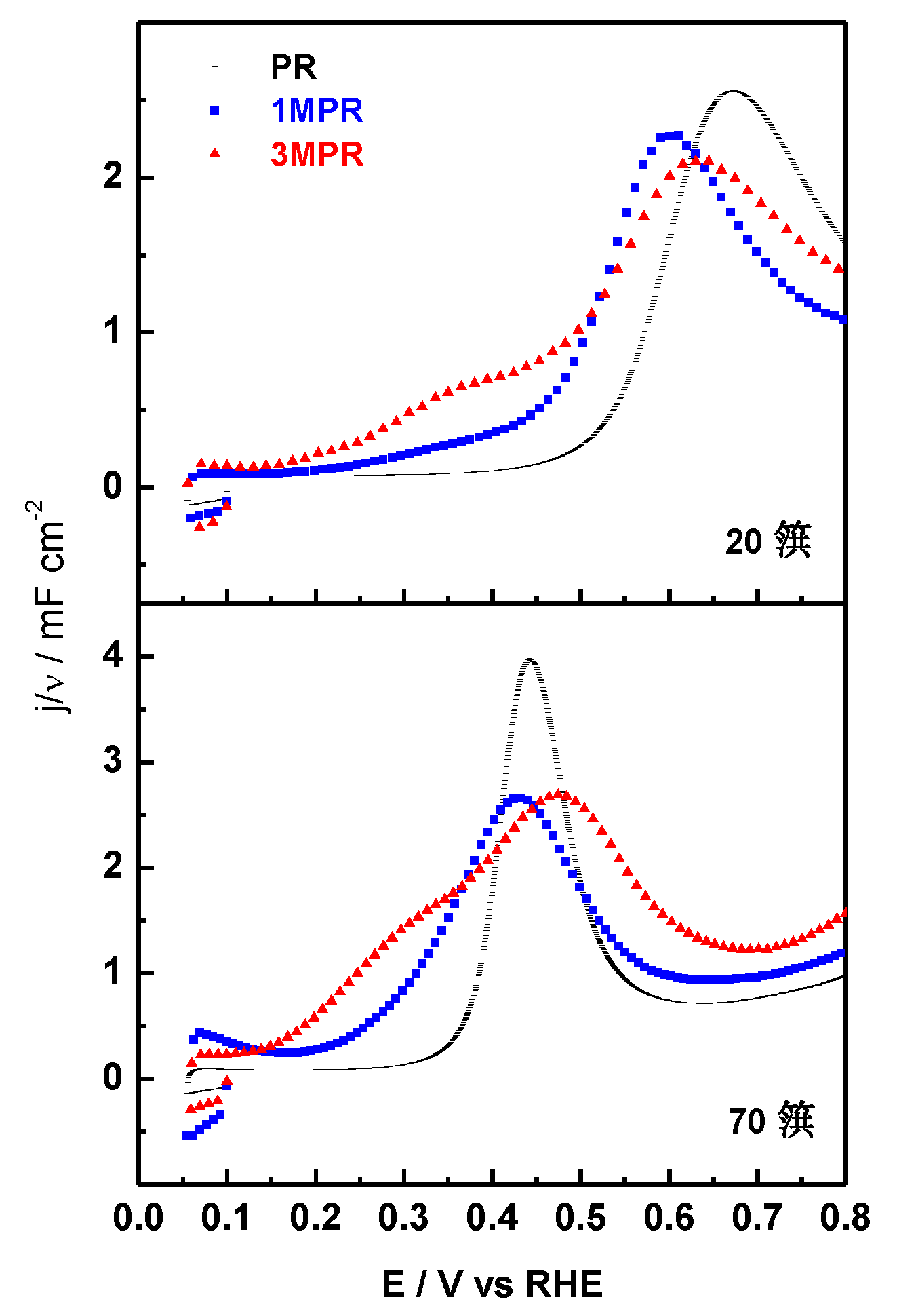
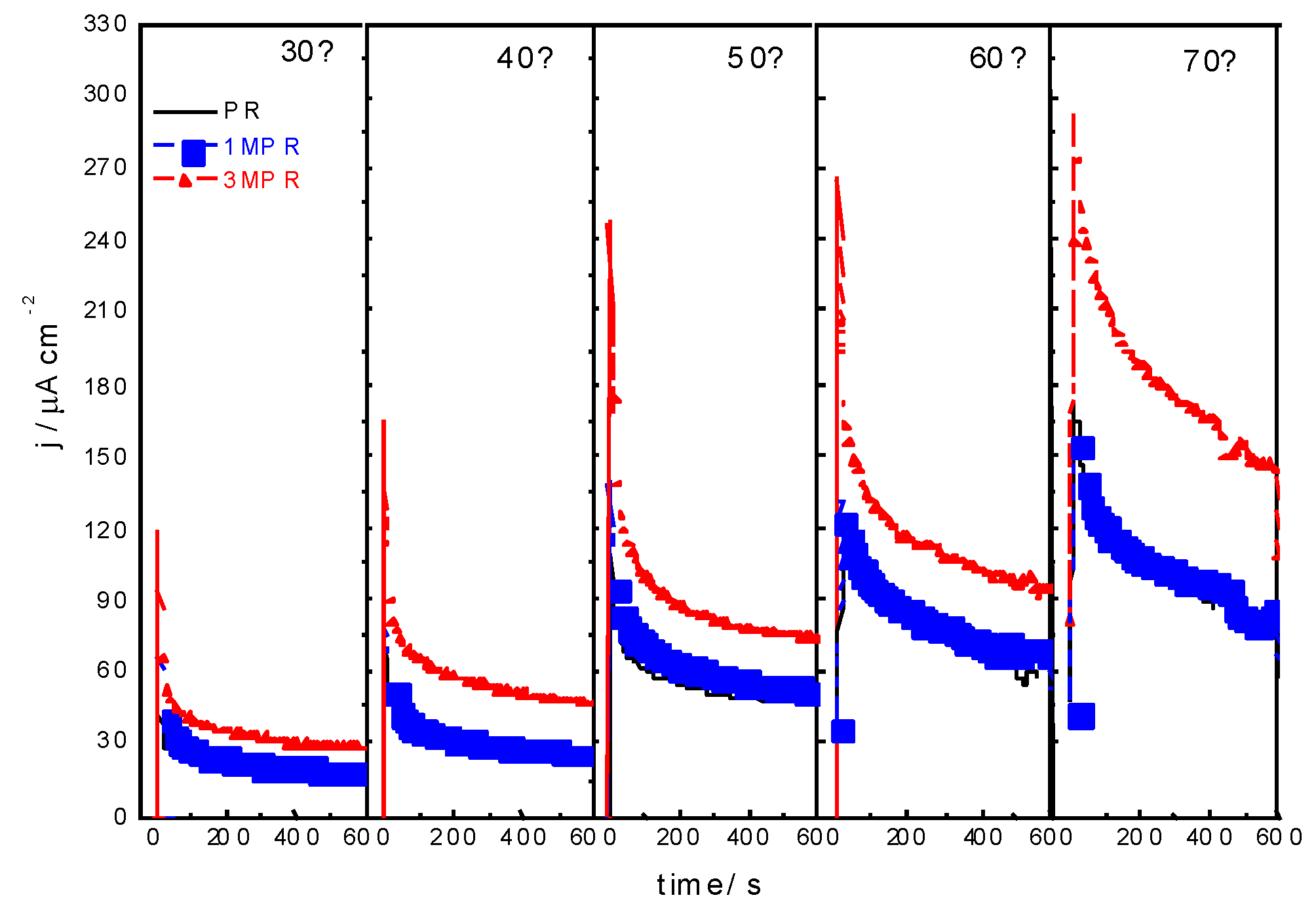
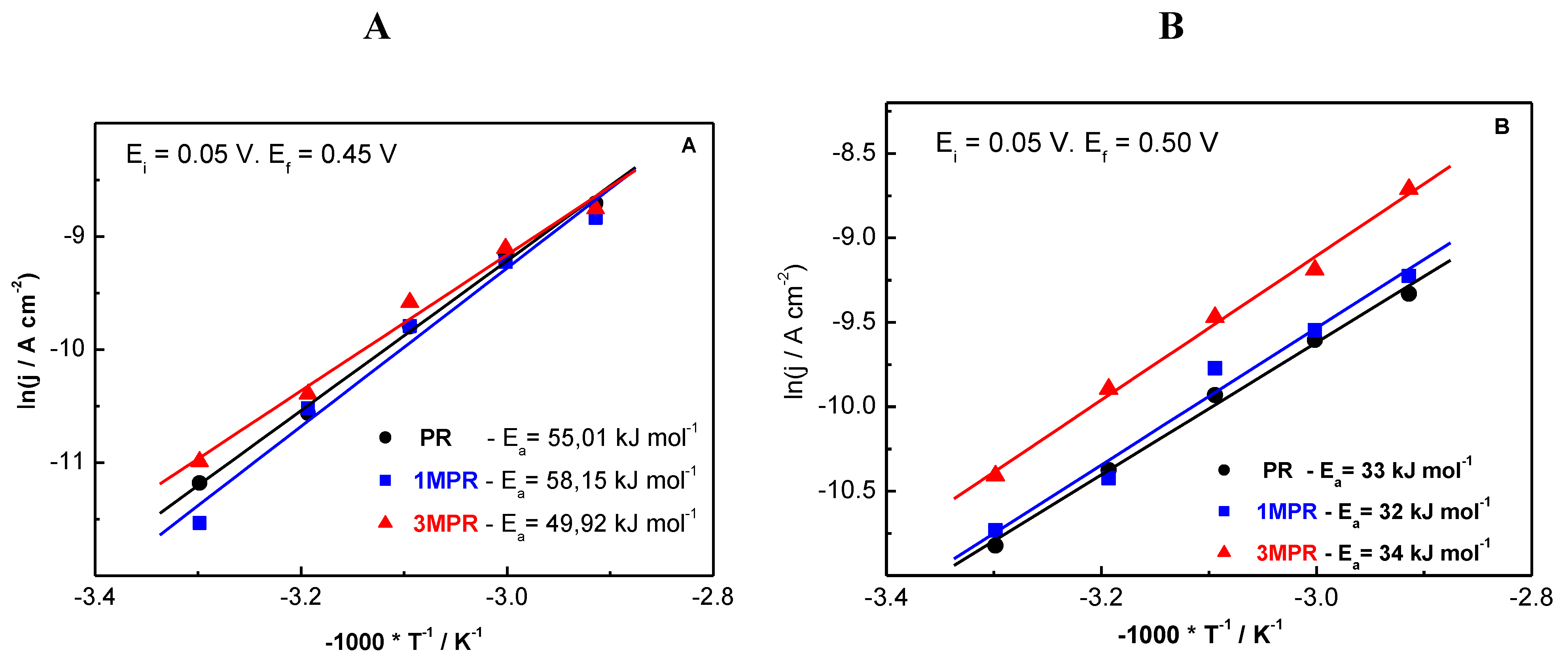
2.3. Effect of Composition
| Catalysts | Bulk compositionPt:Ru (TXRF) | Average particle size (nm) (TEM) | Lattice parameter (fcc) (Å) | Metal loading (wt.%) a Pt:Ru:Mo | Surface metal loading (wt.%) (XPS)
Pt:Ru:Mo |
|---|---|---|---|---|---|
| PR | 1:0.7 | 2.6 | 3.899 | 21:8 | 17:11 |
| 1MPR | 1:0.8:0.07 | 2.7 | 3.898 | 19:9:0.6 | 18:9:0.4 |
| 3MPR | 1:0.8:0.3 | 2.6 | 3.894 | 18:7:3 | 12:8:3 |
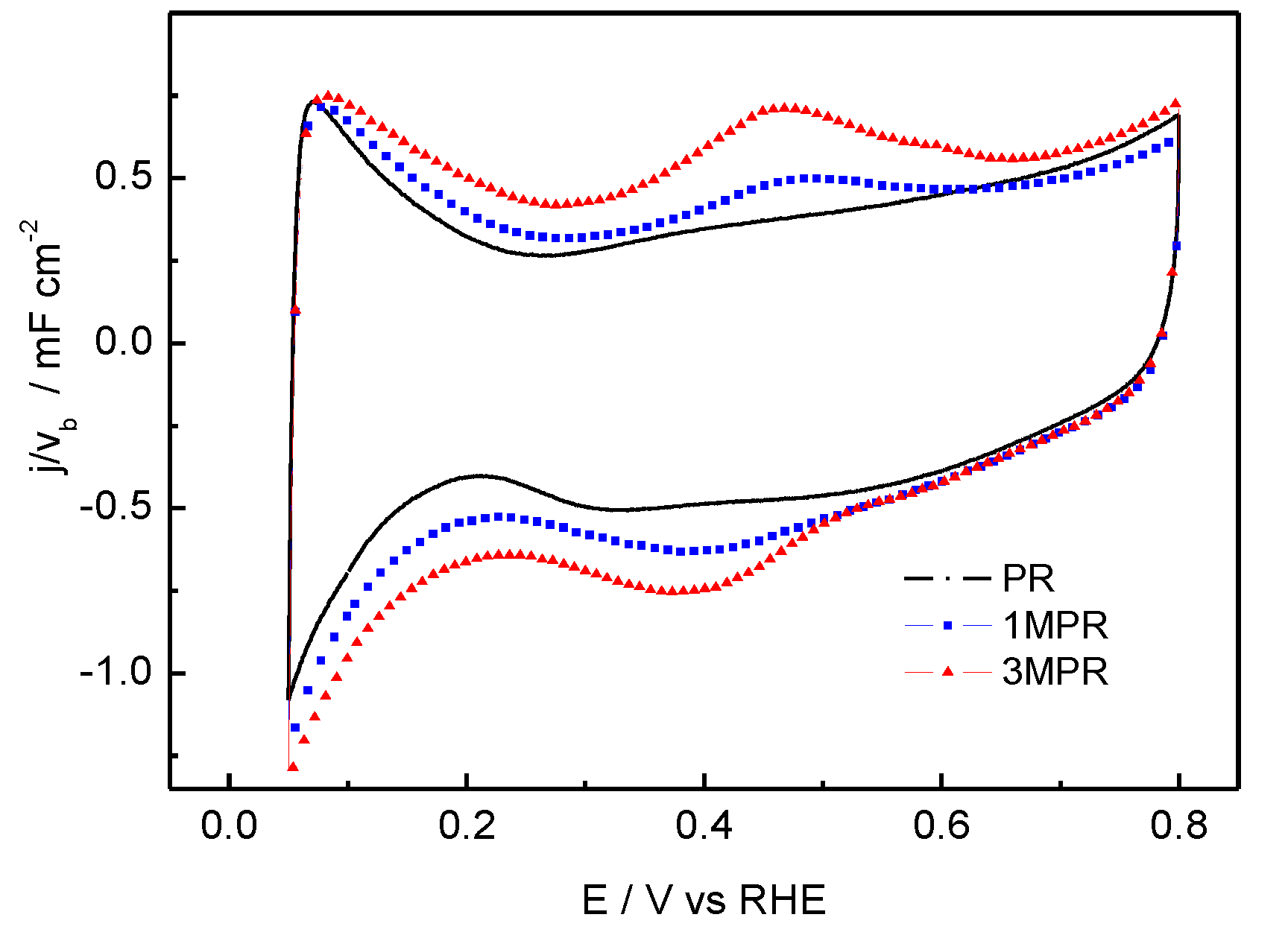

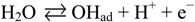

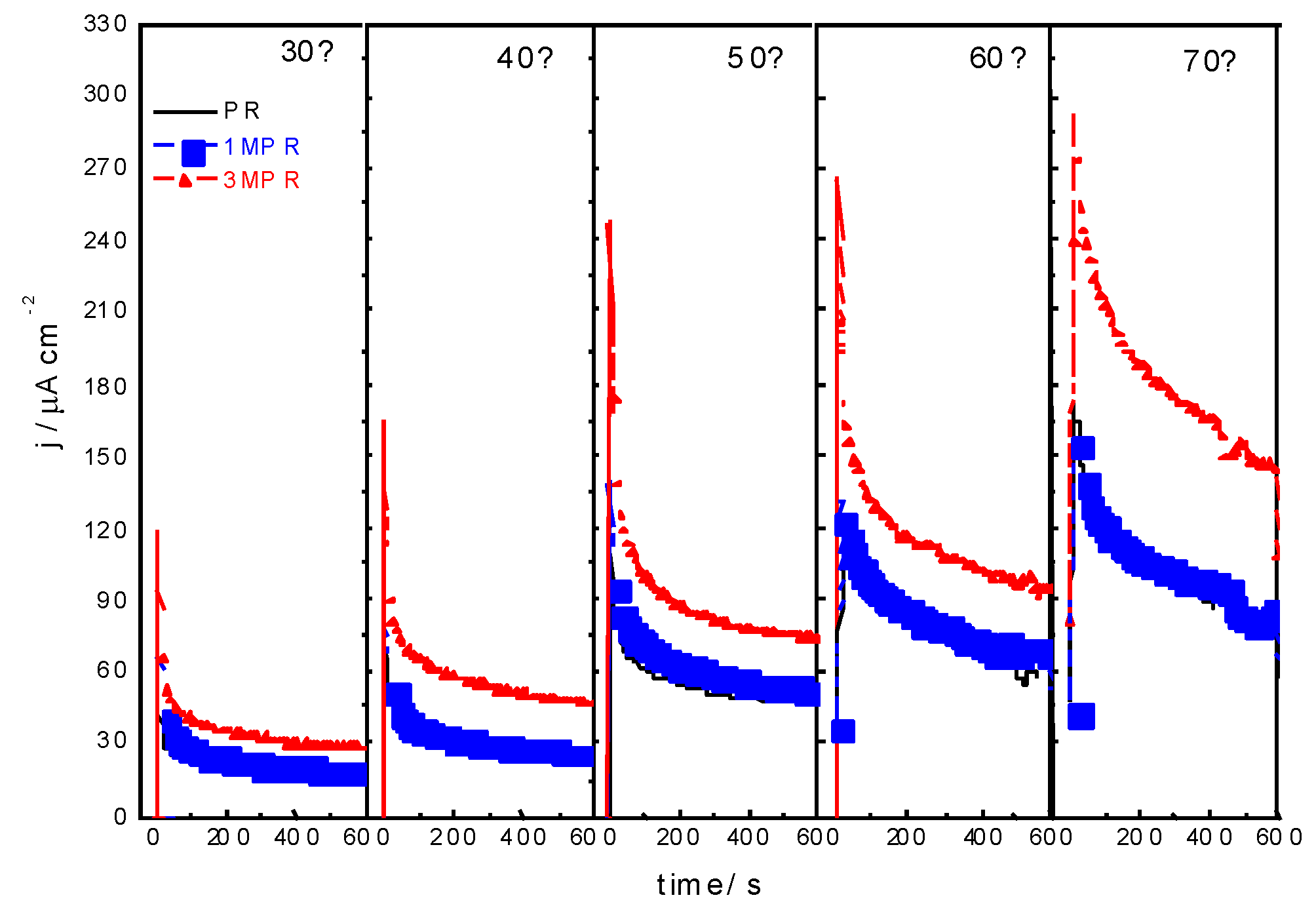
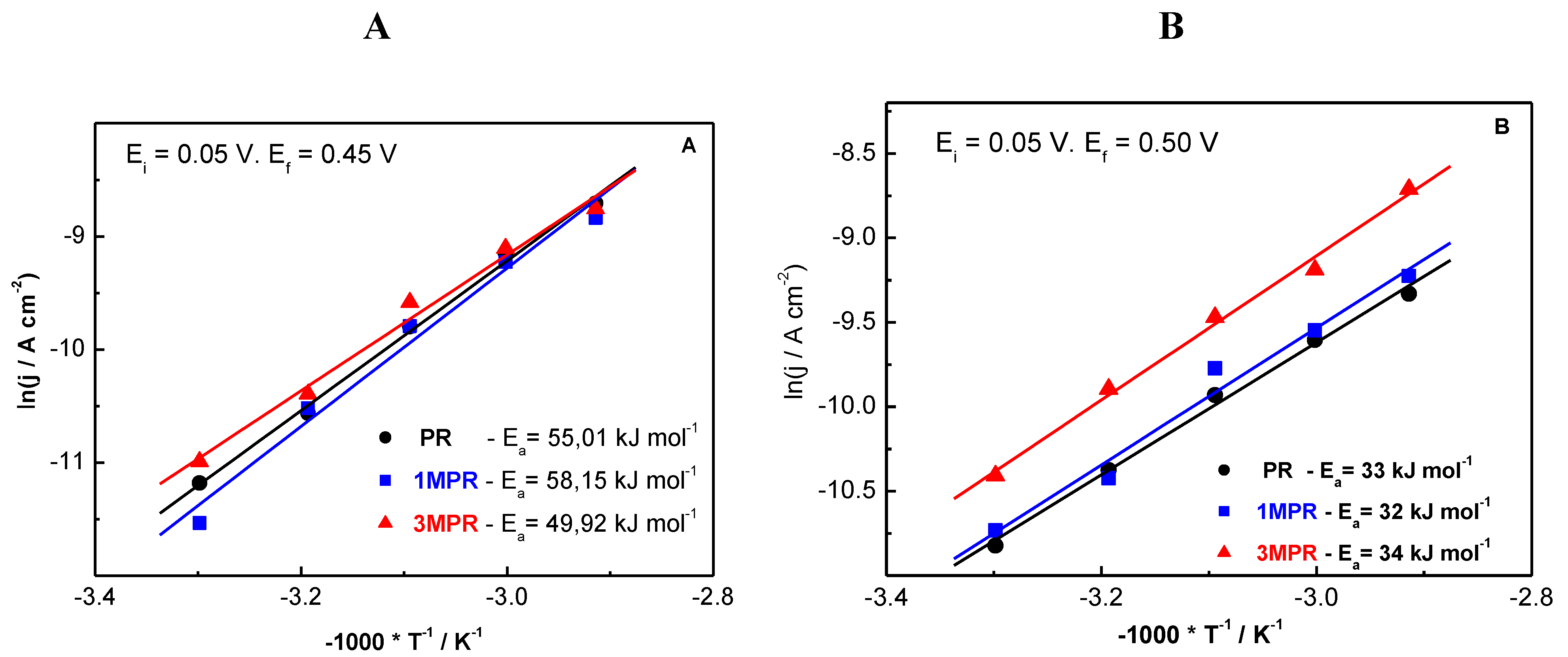

3. Experimental Section
3.1. Catalyst Preparation
- To study the effect of the Mo precursor [44], two ternary catalysts were prepared using MoCl5 (Aldrich) and (NH4)6Mo7O24 (Aldrich) as precursors. Catalysts, with a nominal atomic ratio of Pt/Ru/Mo of 1/1/1, total metal loading of 30 wt.% and supported on Vulcan XC 72R, were labeled PRMCl and PRMNH, using MoCl5 and (NH4)6Mo7O24, respectively. For comparison purpose, a binary catalyst PtRu (1:1)/C, labeled PR, was obtained following the same colloidal methodology.
- To study the influence of thermal treatment [45], 30 wt.% PtRuMo (1:1:0.6)/Vulcan XC 72R catalyst (labeled PRM) was synthesized using (NH4)6Mo7O24 as Mo precursor. This catalyst was subjected to two thermal treatments: an aliquot part of the ternary catalyst was subjected to heating at 300 °C during 1 h in 50 cm3 min−1 of 10% H2/Ar (PRMH2); another aliquot of the ternary catalyst was heated at 300 °C for 1 h in He gas flow, 50 cm3 min−1 (PRMHe). Commercially available electrocatalyst containing 30 wt% PtRu (1:1)/carbon (Johnson-Matthey), labeled PR/C (JM), and 20 wt.% Pt/carbon (ETEK), labeled Pt/C (ETEK) were used for comparison.
- To study the effect of support [43], three supports were compared: Vulcan XC 72R, carbon nanofiber (NF) and carbon nanofiber functionalized with HNO3 (NF.F). Six catalysts were synthesized, with three ternary PtRuMo (1:1:1) catalysts: PRM/Vulcan, PRM/NF and PRM/NF.F, and the respective binary PtRu (1:1) counterparts: PR/Vulcan, PR/NF and PR/NF.F.
- To study the effect of composition [47], two ternary 30 wt.% PtRuMo catalysts supported on Vulcan XC 72R were prepared using (NH4)6Mo7O24 as Mo precursor and treated in H2 at 300 °C for 1 h. Two different PtRuMo atomic ratios, 1:1:0.1 and 1:1:0.5, were investigated, and catalysts obtained were labeled as 1MPR and 3MPR, respectively.
3.2. Physicochemical Characterization
3.3. Electrochemical Characterization
3.3.1. Three Electrode Electrochemical Cell
3.3.2. Electrochemical DMFC Cell Experiments
3.3.3. In Situ Spectroelectrochemical Techniques
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Neergat, M.; Leveratto, D.; Stimming, U. Catalysts for direct methanol fuel cells. Fuel Cells 2002, 2, 25–30. [Google Scholar] [CrossRef]
- Demirci, U.B. Direct liquid-feed fuel cells: Thermodynamic and environmental concerns. J. Power Sources 2007, 169, 239–246. [Google Scholar] [CrossRef]
- Zhao, X.; Yin, M.; Ma, L.; Liang, L.; Liu, C.; Liao, J.; Lu, T.; Xing, W. Recent advances in catalysts for direct methanol fuel cells. Energy Environ. Sci. 2011, 4, 2736–2753. [Google Scholar] [CrossRef]
- Antolini, E. Catalysts for direct ethanol fuel cells. J. Power Sources 2007, 170, 1–12. [Google Scholar] [CrossRef]
- Petrii, O. PtRu electrocatalysts for fuel cells: A representative review. J. Solid State Electrochem. 2008, 12, 609–642. [Google Scholar] [CrossRef]
- De la Fuente, J.L.G.; Martínez-Huerta, M.V.; Rojas, S.; Hernández-Fernández, P.; Terreros, P.; Fierro, J.L.G.; Peña, M.A. Tailoring and structure of PtRu nanoparticles supported on functionalized carbon for DMFC applications: New evidence of the hydrous ruthenium oxide phase. Appl. Catal. B 2009, 88, 505–514. [Google Scholar] [CrossRef]
- Alegre, C.; Calvillo, L.; Moliner, R.; González-Expósito, J.A.; Guill-Villafuerte, O.; Martínez-Huerta, M.V.; Pastor, E.; Lázaro, M.J. Pt and PtRu electrocatalysts supported on carbon xerogels for direct methanol fuel cells. J. Power Sources 2011, 196, 4226–4235. [Google Scholar] [CrossRef]
- García, G.; Florez-Montaño, J.; Hernandez-Creus, A.; Pastor, E.; Planes, G.A. Methanol electrooxidation at mesoporous Pt and Pt-Ru electrodes: A comparative study with carbon supported materials. J. Power Sources 2011, 196, 2979–2986. [Google Scholar] [CrossRef]
- Maiyalagan, T.; Alaje, T.O.; Scott, K. Highly Stable Pt-Ru Nanoparticles Supported on Three-Dimensional Cubic Ordered Mesoporous Carbon (Pt-Ru/CMK-8) as Promising Electrocatalysts for Methanol Oxidation. J. Phys. Chem. C 2011, 116, 2630–2638. [Google Scholar] [CrossRef]
- De la Fuente, J.L.G.; Martínez-Huerta, M.V.; Rojas, S.; Terreros, P.; Fierro, J.L.G.; Peña, M.A. Enhanced methanol electrooxidation activity of PtRu nanoparticles supported on H2O2-functionalized carbon black. Carbon 2005, 43, 3002–3005. [Google Scholar] [CrossRef]
- Antolini, E.; Gonzalez, E.R. A simple model to assess the contribution of alloyed and non-alloyed platinum and tin to the ethanol oxidation reaction on Pt-Sn/C catalysts: Application to direct ethanol fuel cell performance. Electrochim. Acta 2010, 55, 6485–6490. [Google Scholar] [CrossRef]
- Sieben, J.M.; Duarte, M.M.E. Nanostructured Pt and PtSn catalysts supported on oxidized carbon nanotubes for ethanol and ethylene glycol electro-oxidation. Int. J. Hydrogen Energy 2011, 36, 3313–3321. [Google Scholar] [CrossRef]
- Jiang, L.; Colmenares, L.; Jusys, Z.; Sun, G.Q.; Behm, R.J. Ethanol electrooxidation on novel carbon supported Pt/SnOx/C catalysts with varied Pt:Sn ratio. Electrochim. Acta 2007, 53, 377–389. [Google Scholar] [CrossRef]
- Bambagioni, V.; Bianchini, C.; Marchionni, A.; Filippi, J.; Vizza, F.; Teddy, J.; Serp, P.; Zhiani, M. Pd and Pt-Ru anode electrocatalysts supported on multi-walled carbon nanotubes and their use in passive and active direct alcohol fuel cells with an anion-exchange membrane (alcohol = methanol, ethanol, glycerol). J. Power Sources 2009, 190, 241–251. [Google Scholar] [CrossRef]
- Chatterjee, M.; Chatterjee, A.; Ghosh, S.; Basumallick, I. Electro-oxidation of ethanol and ethylene glycol on carbon-supported nano-Pt and -PtRu catalyst in acid solution. Electrochim. Acta 2009, 54, 7299–7304. [Google Scholar] [CrossRef]
- Wang, M.Y.; Chen, J.H.; Fan, Z.; Tang, H.; Deng, G.H.; He, D.L.; Kuang, Y.F. Ethanol electro-oxidation with Pt and Pt-Ru catalysts supported on carbon nanotubes. Carbon 2004, 42, 3251–3272. [Google Scholar] [CrossRef]
- Koper, M.T.M.; Lukkien, J.J.; Jansen, A.P.J.; van Santen, R.A. Lattice gas model for CO electrooxidation on Pt-Ru bimetallic surfaces. J. Phys. Chem. B 1999, 103, 5522–5529. [Google Scholar]
- Watanabe, M.; Motoo, S. Electrocatalysis by ad-atoms: Part II. Enhancement of the oxidation of methanol on platinum by ruthenium ad-atoms. J. Electroanal. Chem. 1975, 60, 267–273. [Google Scholar] [CrossRef]
- Iwasita, T. Electrocatalysis of methanol oxidation. Electrochim. Acta 2002, 47, 3663–3674. [Google Scholar] [CrossRef]
- Rabis, A.; Rodriguez, P.; Schmidt, T.J. Electrocatalysis for Polymer Electrolyte Fuel Cells: Recent Achievements and Future Challenges. ACS Catal. 2012, 2, 864–890. [Google Scholar] [CrossRef]
- Antolini, E. Carbon supports for low temperature fuel cell catalysts. Appl. Catal. B 2009, 88, 1–24. [Google Scholar] [CrossRef]
- Fei, Y.; Cao, X.; Yu, L.; Chen, S.; Lin, W. Synthesis and Catalytic Performance of PtRuMo Nanoparticles Supported on Graphene-Carbon Nanotubes Nanocomposites for Methanol Electro-Oxidation. Int. J. Electrochem. Sci. 2012, 7, 1251–1265. [Google Scholar]
- Kakati, N.; Maiti, J.; Oh, J.Y.; Yoon, Y.S. Study of methanol oxidation of hydrothermally synthesized PtRuMo on multi wall carbon nanotubes. Appl. Surf. Sci. 2011, 257, 8433–8437. [Google Scholar] [CrossRef]
- Chen, S.; Ye, F.; Lin, W. Effect of operating conditions on the performance of a direct methanol fuel cell with PtRuMo/CNTs as anode catalyst. Int. J. Hydrogen Energy 2010, 35, 8225–8233. [Google Scholar] [CrossRef]
- Lee, K.R.; Jeon, M.K.; Woo, S.I. Composition optimization of PtRuM/C (M = Fe and Mo) catalysts for methanol electro-oxidation via combinatorial method. Appl. Catal. B 2009, 91, 428–433. [Google Scholar] [CrossRef]
- Wang, Z.B.; Zuo, P.J.; Yin, G.P. Investigations of compositions and performance of PtRuMo/C ternary catalysts for methanol electrooxidation. Fuel Cells 2009, 2, 106–113. [Google Scholar] [CrossRef]
- Morante-Catacora, T.Y.; Ishikawa, Y.; Cabrera, C.R. Sequential electrodeposition of Mo at Pt and PtRu methanol oxidation catalyst particles on HOPG surfaces. J. Electroanal. Chem. 2008, 621, 103–112. [Google Scholar] [CrossRef]
- Pasupathi, S.; Tricoli, V. Effect of third metal on the electrocatalytic activity of PtRu/Vulcan for methanol electro-oxidation. J. Solid State Electrochem. 2008, 12, 1093–1100. [Google Scholar] [CrossRef]
- Bauer, A.; Gyenge, E.L.; Oloman, C.W. Direct methanol fuel cell with extended reaction zone anode: PtRu and PtRuMo supported on graphite felt. J. Power Sources 2007, 167, 281–287. [Google Scholar] [CrossRef]
- Wang, Z.B.; Yin, G.P.; Lin, Y.G. Synthesis and characterization of PtRuMo/C nanoparticle electrocatalyst for direct ethanol fuel cell. J. Power Sources 2007, 170, 242–250. [Google Scholar] [CrossRef]
- Benker, N.; Roth, C.; Mazurek, M.; Fuess, H. Synthesis and characterization of ternary Pt/Ru/Mo catalysts for the anode of the PEM fuel cell. J. New Mater. Electrochem. Syst. 2006, 9, 121–126. [Google Scholar]
- Hou, Z.; Yi, B.; Yu, H.; Lin, Z.; Zhang, H. CO tolerance electrocatalyst of PtRu-HxMeO3/C (Me = W, Mo) made by composite support method. J. Power Sources 2003, 123, 116–125. [Google Scholar] [CrossRef]
- Oliveira Neto, A.; Franco, E.G.; Arico, E.; Linardi, M.; Gonzalez, E.R. Electro-oxidation of methanol and ethanol on Pt-Ru/C and Pt-Ru-Mo/C electrocatalysts prepared by Bönnemann's method. J. Eur. Ceramic Soc. 2003, 23, 2987–2992. [Google Scholar] [CrossRef]
- Pinheiro, A.L.N.; Oliveira Neto, A.; de Souza, E.C.; Perez, J.; Paganin, V.A.; Ticianelli, E.; Gonzalez, E.R. Electrocatalysis on noble metal and noble metal alloys dispersed on high surface area carbon. J. New Mater. Electrochem. Syst. 2003, 6, 1–8. [Google Scholar]
- Franco, E.G.; Neto, A.O.; Linardi, M.; Arico, E. Synthesis of electrocatalysts by the Bonnemann method for the oxidation of methanol and the mixture H2/CO in a proton exchange membrane fuel cell. J. Braz. Chem. Soc. 2002, 13, 516–521. [Google Scholar] [CrossRef]
- Papageorgopoulos, D.C.; Keijzer, M.; de Bruijn, F.A. The inclusion of Mo, Nb and Ta in Pt and PtRu carbon supported 3electrocatalysts in the quest for improved CO tolerant PEMFC anodes. Electrochim. Acta 2002, 48, 197–204. [Google Scholar] [CrossRef]
- Götz, M.; Wendt, H. Bynary and ternary anode catalyst formulations including the elements W, Sn and Mo for PEMFCs operated on methanol or reformate gas. Electrochim. Acta 1998, 43, 3637–3644. [Google Scholar] [CrossRef]
- Anderson, A.B.; Grantscharova, E.; Seong, S. Systematic Theoretical Study of Alloys of Platinum for Enhanced Methanol Fuel Cell Performance. J. Electrochem. Soc. 1996, 143, 2075–2082. [Google Scholar] [CrossRef]
- Ji, Z.; Jalbout, A.F.; Li, J.Q. Adsorption and diffusion of OH on Mo modified Pt(111) surface: First-principles theory. Solid State Commun. 2007, 142, 148–153. [Google Scholar] [CrossRef]
- Horkans, J.; Shafer, M.W. Effect of orientation, composition and electronic factors in the reduction of O2 on single crystal electrodes of the conducting oxides of molybdenum and tungsten. J. Electrochem. Soc. 1977, 124, 1196–1202. [Google Scholar] [CrossRef]
- Martínez-Huerta, M.V.; Rodríguez, J.L.; Tsiouvaras, N.; Peña, M.A.; Fierro, J.L.G.; Pastor, E. Novel synthesis method of CO-tolerant PtRu-MoOx nanoparticles: structural characteristics and performance for methanol electrooxidation. Chem. Mater. 2008, 20, 4249–4259. [Google Scholar] [CrossRef]
- Watanabe, M.; Uchida, M.; Motoo, S. Preparation of highly dispersed Pt + Ru clusters and the activity for the electrooxidation of methanol. J. Electroanal. Chem. 1987, 229, 395–406. [Google Scholar] [CrossRef]
- Tsiouvaras, N.; Martínez-Huerta, M.V.; Moliner, R.; Lázaro, M.J.; Rodríguez, J.L.; Pastor, E.; Peña, M.A.; Fierro, J.L.G. CO tolerant PtRu-MoOx nanoparticles supported on carbon nanofibers for direct methanol fuel cells. J. Power Sources 2009, 186, 299–304. [Google Scholar] [CrossRef]
- Tsiouvaras, N.; Peña, M.A.; Fierro, J.L.G.; Pastor, E.; Martínez-Huerta, M.V. The effect of the Mo precursor on the nanostructure and activity of PtRuMo electrocatalysts for Proton Exchange Membrane Fuel Cells. Catal. Today 2010, 158, 12–21. [Google Scholar] [CrossRef]
- Tsiouvaras, N.; Martínez-Huerta, M.V.; Paschos, O.; Stimming, U.; Fierro, J.L.G.; Peña, M.A. PtRuMo/C catalysts for direct methanol fuel cells: Effect of the pretreatment on the structural characteristics and methanol electrooxidation. Int. J. Hydrogen Energy 2010, 35, 11478–11488. [Google Scholar] [CrossRef]
- Martínez-Huerta, M.V.; Tsiouvaras, N.; Peña, M.A.; Fierro, J.L.G.; Rodriguez, J.L.; Pastor, E. Electrochemical activation of nanostructured carbon-supported PtRuMo electrocatalyst for methanol oxidation. Electrochim. Acta 2010, 55, 7634–7642. [Google Scholar]
- García, G.; Tsiouvaras, N.; Pastor, E.; Peña, M.A.; Fierro, J.L.G.; Martínez-Huerta, M.V. Ethanol oxidation on PtRuMo/C catalysts: In situ FTIR spectroscopy and DEMS studies. Int. J. Hydrogen Energy 2012, 37, 7131–7140. [Google Scholar] [CrossRef]
- Alcaide, F.; Álvarez, G.; Tsiouvaras, N.; Peña, M.A.; Fierro, J.L.; Martínez-Huerta, M.V. Electrooxidation of H2/CO on carbon-supported PtRu-MoOx nanoparticles for polymer electrolyte fuel cells. Int. J. Hydrogen Energy 2011, 36, 14590–14598. [Google Scholar]
- Grgur, B.N.; Zhuang, G.; Markovic, N.M.; Ross, J.P.N. Electrooxidation of H2/CO Mixtures on a Well-Characterized Pt75Mo25 Alloy Surface. J. Phys. Chem. B 1997, 101, 3910–3913. [Google Scholar]
- Grgur, B.N.; Markovic, N.M.; Ross, P.N., Jr. Electrooxidation of H2, CO and H2/CO mixtures on a well-characterized Pt70Mo30 bulk alloy electrode. J. Phys. Chem. B 1998, 102, 2494–2501. [Google Scholar] [CrossRef]
- Holleman, A.F.; Wiberg, E. Inorganic Chemistry; Wiberg, N., Ed.; Academic Press: San Diego, CA, USA, 2001; pp. 1382–1402. [Google Scholar]
- Briggs, D.; Seah, M.P. Practical Surface Analysis by Auger and X-ray Photoelectron Spectroscopy; Wiley: New York, NY, USA, 1990. [Google Scholar]
- Hobbs, B.S.; Tseung, A.C.C. High performance, platinum activated tungsten oxide fuel cell electrodes. Nature 1969, 222, 556–558. [Google Scholar] [CrossRef]
- Bond, G.C.; Tripathi, J.B.P. Studies of Hydrogen Spillover. J. Chem. Soc. Faraday Trans. 1 1976, 72, 933–941. [Google Scholar] [CrossRef]
- Grgur, B.N.; Markovic, N.M.; Ross, P.N. The electrooxidation of H2 and H2/CO mixtures on carbon supported PtxMoy alloy catalysts. J. Electrochem. Soc. 1999, 146, 1613–1619. [Google Scholar] [CrossRef]
- Mukerjee, S.; Urian, R.C. Bifunctionality in Pt alloy nanocluster electrocatalysts for enhanced methanol oxidation and CO tolerance in PEM fuel cells: Electrochemical and in situ synchrotron spectroscopy. Electrochim. Acta 2002, 47, 3219–3231. [Google Scholar] [CrossRef]
- Lebedeva, N.P.; Janssen, G.J.M. On the preparation and stability of bimetallic PtMo/C anodes for protno-exchange membrane fuel cells. Electrochim. Acta 2005, 51, 29–40. [Google Scholar] [CrossRef]
- Serp, P.; Corrias, M.; Kalck, P. Carbon nanotubes and nanofibers in catalysis. Appl. Catalysis A 2003, 253, 337–358. [Google Scholar] [CrossRef]
- García, G.; Koper, M.T.M. Carbon Monoxide Oxidation on Pt Single Crystal Electrodes: Understanding the Catalysis for Low Temperature Fuel Cells. Chemphyschem 2011, 12, 2064–2072. [Google Scholar] [CrossRef]
- Gilman, S. The Mechanism of Electrochemical Oxidation of Carbon Monoxide and Methanol on Platinum. II. The Reactant-Pair Mechanism for Electrochemical Oxidation of Carbon Monoxide and Methanol 1. J. Phys. Chem. 1964, 68, 70–80. [Google Scholar] [CrossRef]
- Arico, A.S.; Bruce, P.; Scrosati, B.; Tarascon, J.-M.; Schalkwijk, W.V. Nanostructured materials for advanced energy conversion and storage devices. Nat. Mater. 2005, 4, 366–377. [Google Scholar] [CrossRef]
- Hamnett, A. Interfacial Electrochemistry; Theory, Experiment and Applications; Wieckowski, A., Ed.; Marcell Dekker: New York, NY, USA, 1999; p. 843. [Google Scholar]
- Sun, S.G.; Clavilier, J. Electrochemical study on the poisoning intermediate formed from methanol dissociation at low index and stepped platinum surfaces. J. Electroanal. Chem. 1987, 236, 95–112. [Google Scholar] [CrossRef]
- Cuesta, A. At Least Three Contiguous Atoms Are Necessary for CO Formation during Methanol Electrooxidation on Platinum. J. Am. Chem. Soc. 2006, 128, 13332–13333. [Google Scholar] [CrossRef]
- Camara, G.A.; Iwasita, T. Parallel pathways of ethanol oxidation: The effect of ethanol concentration. J. Electroanal. Chem. 2005, 578, 315–321. [Google Scholar] [CrossRef]
- Lamy, C.; Lima, A.; LeRhun, V.; Delime, F.; Coutanceau, C.; Leger, J.M. Recent advances in the development of direct alcohol fuel cells (DAFC). J. Power Sources 2002, 105, 283–296. [Google Scholar] [CrossRef]
- Colmenares, L.; Wang, H.; Jusys, Z.; Jiang, L.; Yan, S.; Sun, G.Q.; Behm, R.J. Ethanol oxidation on novel, carbon supported Pt alloy catalysts—Model studies under defined diffusion conditions. Electrochim. Acta 2006, 52, 221–233. [Google Scholar] [CrossRef]
- Neurock, M. Advances in Electrocatalysis, Materials, Diagnostics and Durability. In First-Principles Modeling for the Electro-Oxidation of Small Molecules—Handbook of Fuel Cells—Fundamentals, Technology and Applications; Vielstich, W., Lamm, A., Gasteiger, H.A., Eds.; Wiley: West Sussex, UK, 2009; Volume 5. [Google Scholar]
- Zhu, J.; Cheng, F.; Tao, Z.; Chen, J. Electrocatalytic Methanol Oxidation of Pt0.5Ru0.5-xSnx/C (x = 0–0.5). J. Phys. Chem. C 2008, 112, 6337–6345. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, Z.; Song, S.; Li, W.; Sun, G.; Tsiakaras, P.; Xin, Q. Pt based anode catalysts for direct ethanol fuel cells. Appl. Catal. B 2003, 46, 273–285. [Google Scholar] [CrossRef]
- Ioroi, T.; Fujiwara, N.; Siroma, Z.; Yasuda, K.; Miyazaki, Y. Platinum and molybdenum oxide deposited carbon electrocatalyst for oxidation of hydrogen containing carbon monoxide. Electrochem. Commun. 2002, 4, 442–446. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Martínez-Huerta, M.V.; Tsiouvaras, N.; García, G.; Peña, M.A.; Pastor, E.; Rodriguez, J.L.; Fierro, J.L.G. Carbon-Supported PtRuMo Electrocatalysts for Direct Alcohol Fuel Cells. Catalysts 2013, 3, 811-838. https://doi.org/10.3390/catal3040811
Martínez-Huerta MV, Tsiouvaras N, García G, Peña MA, Pastor E, Rodriguez JL, Fierro JLG. Carbon-Supported PtRuMo Electrocatalysts for Direct Alcohol Fuel Cells. Catalysts. 2013; 3(4):811-838. https://doi.org/10.3390/catal3040811
Chicago/Turabian StyleMartínez-Huerta, María V., Nikolaos Tsiouvaras, Gonzalo García, Miguel A. Peña, Elena Pastor, José L. Rodriguez, and José L.G. Fierro. 2013. "Carbon-Supported PtRuMo Electrocatalysts for Direct Alcohol Fuel Cells" Catalysts 3, no. 4: 811-838. https://doi.org/10.3390/catal3040811