2.2. Epoxidation of Propylene
Propylene oxide (PO) is an important chemical intermediate for the production of a variety of chemicals and polymers. Commercially, PO is produced via the chlorohydrin process and several hydroperoxide processes. However, the chlorohydrin process results in large amounts of chlorinated compounds, and the hydroperoxide processes typically generate stoichiometric quantities of co-products. The discovery by Haruta and co-workers [
5] that nanoscale gold particles on titania supports provide a highly selective (∼99%) route to vapor-phase PO production, with the use of a mixture of propylene, oxygen, and hydrogen under ambient pressure, has been a significant breakthrough in heterogeneous catalysis. A direct, single-step epoxidation of propylene to PO has both practical and fundamental implications (
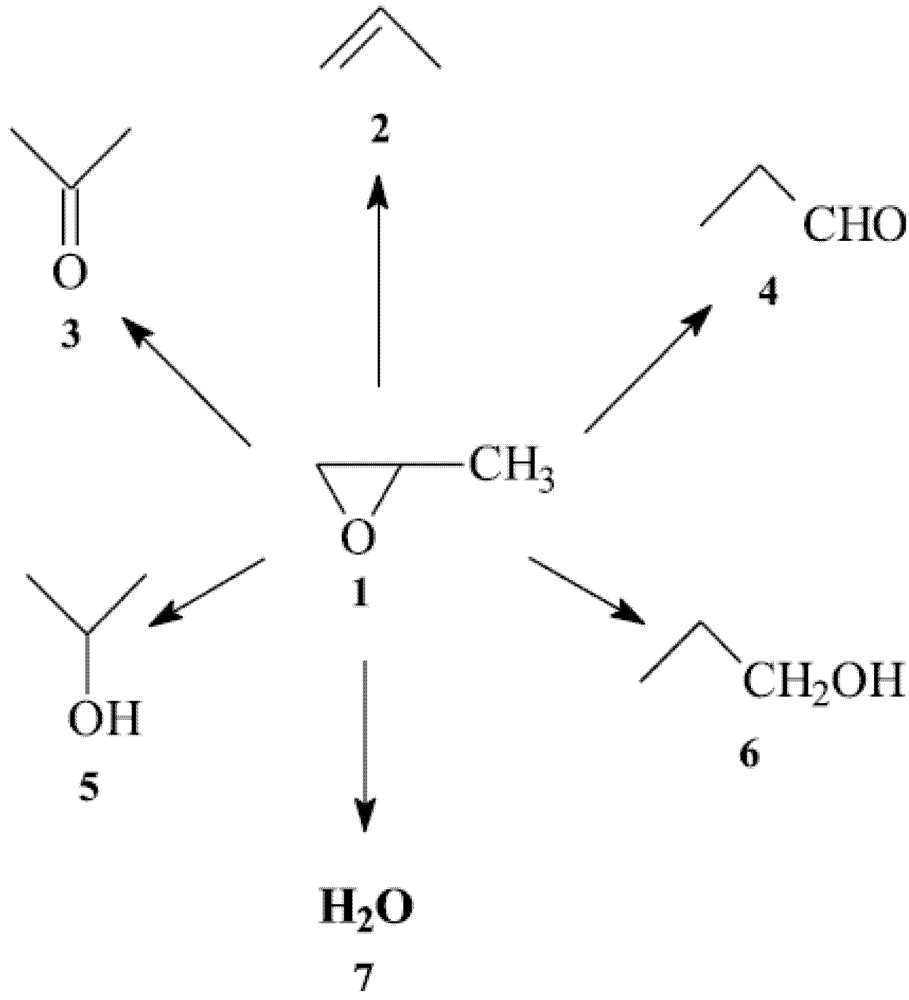
Scheme 2).
Scheme 2.
Epoxidation of propene.
Scheme 2.
Epoxidation of propene.
Since then, among sporadic works of various researchers, five groups displayed intense activity.
Haruta’s group remained a major player in this area [
6,
7,
8,
9,
10].
In the epoxidation reaction they have used Au/Ti-MCM-41 and Au/TiO
2-SiO
2 catalysts with various Ti/Si molar ratios (0.25/100–6.0/100) [
6]. Two different structure-directing organic templates (dodecyltrimethylammonium hydroxide and cetyltrimethylammonium hydroxide) were used in the synthesis of the Ti-MCM-41 support. TiO
2-SiO
2 mixed oxides were prepared by a sol-gel process. Gold nanoparticles were loaded by deposition-precipitation (DP) using various precipitating agents like LiOH, NaOH, KOH, RbOH, and CsOH. Au/Ti-MCM-41 with a Ti/Si = 3/100 molar ratio provided with the optimum performance in terms of conversion and PO selectivity: 3.2% and 93.5%, respectively at 373 K. At higher than 393 K PO selectivity decreased with an increase in the Ti/Si ratio. At 423 and 473 K, effective catalysts in terms of the above factors, had to have Ti/Si ratios of 2/100 and 1.5/100, respectively. A critical balance among Ti-content, Au loading and the reaction temperature was important to achieve higher propylene conversion, PO selectivity above 90% and lower H
2 (and O
2) consumption. Gold supported on TiO
2-SiO
2 mixed oxides showed very poor performance, however, PO selectivity appreciably increased with the increase in the calcination temperature of the support. IR and HRTEM analyses revealed that with the increase in the calcination temperature more isolated TiO
4 units with tetrahedral coordination was formed on the surfaces of Ti-SiO
2 supports. These isolated TiO
4 sites incorporated in the silica surface layers seemed to be an important structural factor for the selective deposition of Au particles and the selective PO formation [
7].
After similar treatments a better performance was observed with Au/Ti-MCM-48 (initial conversion = 5.6%, PO selectivity = 92%), due to its three-dimensional pore system at 423 K [
8]. GC-MS investigation of the extracted species by organic solvent from the used catalyst revealed that acidic as well as oligomeric species accumulated on the catalyst surfaces. These species were assumed to cause catalyst deactivation. Silylation of the catalyst Au/Ti-MCM-48 slowed down deactivation and helped to improve PO selectivity and decreased H
2 consumption as well.
Au/Ti-doped nonporous silica catalyst dried under vacuum at room temperature and calcined at 573 K in air, exhibited catalytic activity to form PO even at a temperature as low as 323 K [
9]. The calcined catalysts were more stable and more active at higher temperatures than the dried catalysts. Simple filtration after aging the suspension resulted in catalysts more active and selective but less stable than the complete washing of the solid precursors. Pretreatment in argon increased the catalytic activity and H
2 efficiency appreciably over the Au catalysts deposited on Ti-doped nonporous silica as opposed to Au/Ti-MCM catalysts. Similar deactivation rate of Au catalysts supported on a variety of titania-silica supports with different porosities suggested that quick deactivation of Au catalysts within hours was mainly affected by the surface properties rather than the pore structure and diffusion limitation of the supports.
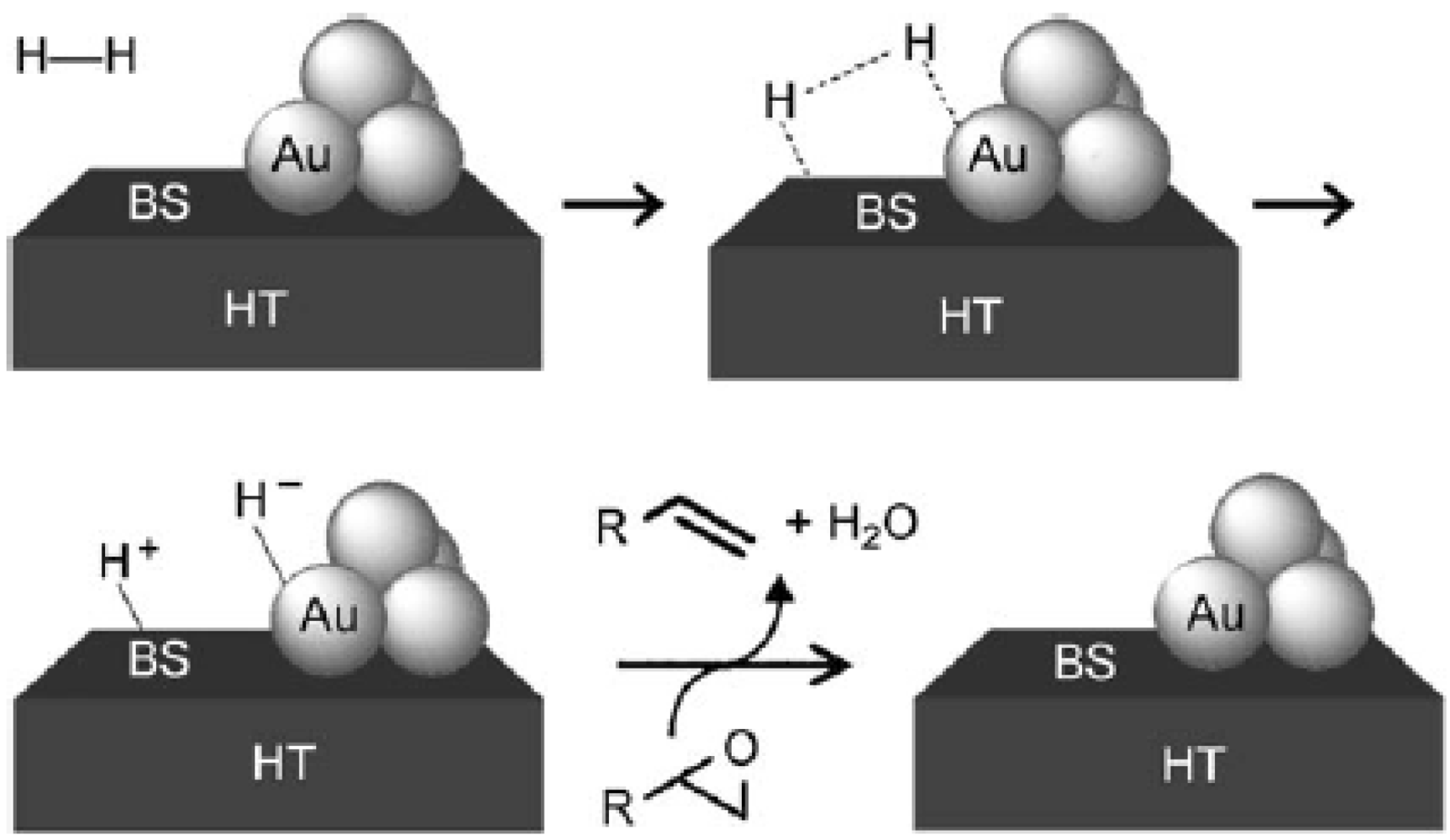
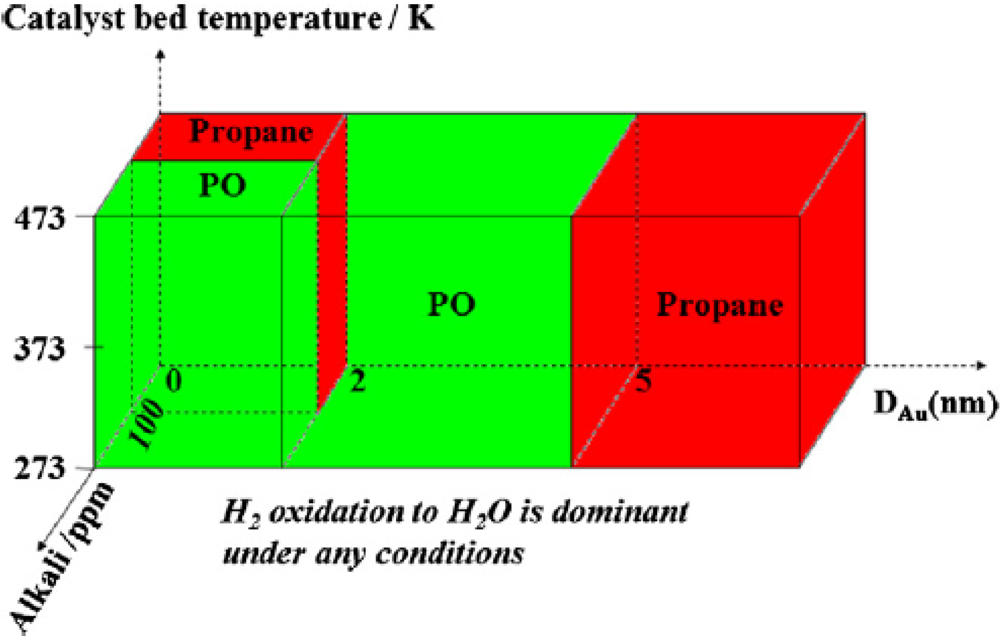
It has been observed that in a H
2-O
2 mixture, the reaction of propene could be switched between hydrogenation and epoxidation over Au/Ti-based oxides [
10] (
Figure 1). Reaction pathways were strongly dependent on the size of Au particles and on the presence of alkalis. Hydrogenation prevailed over Au clusters smaller than 2.0 nm (in the absence of alkalis) and over Au nanoparticles larger than 5.0 nm. Alkali contamination could switch hydrogenation to epoxidation over Au clusters and promoted epoxidation over Au nanoparticles with diameters of 2.0–5.0 nm. It was also confirmed that the hydrogenation of propene was enhanced by the addition of a small amount of O
2.
Figure 1.
The role of factors in deciding between the hydrogenation and epoxidation of propene. Reproduced with permission from Reference [
10]. Copyright (2011) Elsevier.
Figure 1.
The role of factors in deciding between the hydrogenation and epoxidation of propene. Reproduced with permission from Reference [
10]. Copyright (2011) Elsevier.
Oyama
et al. studied the oxidation of propylene to PO with H
2-O
2 mixtures over gold supported on mesoporous titanium silicates, Ti-TUD [
11,
12,
13] and TS-1 [
14]. The gold catalyst supported on Ti-TUD gave stable activity at low conversions of propylene (<6%) and high selectivity to PO (>95%) [
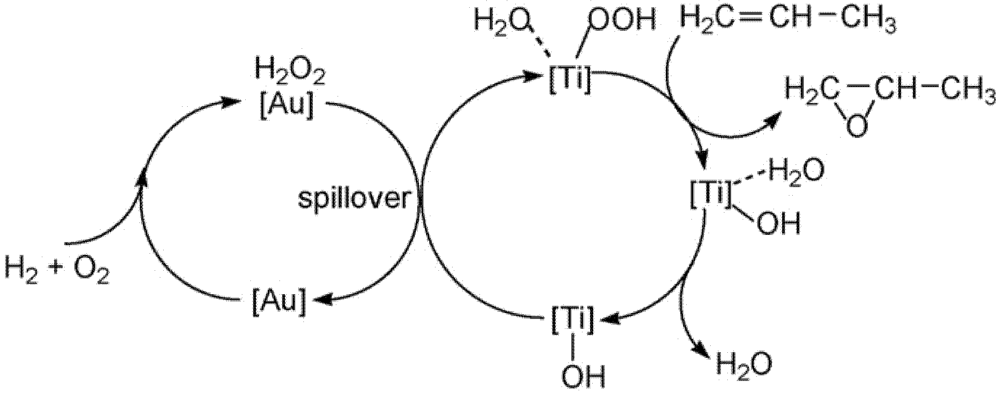
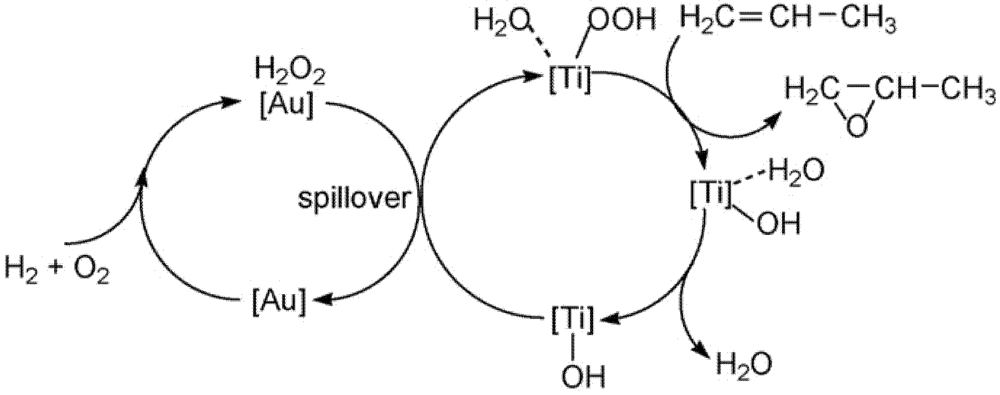
11]. On the basis of kinetic data it was stated that the catalyst operated by the commonly accepted mechanism of hydrogen peroxide production on gold sites supported by
in situ measured UV spectra, and epoxidation on titanium centers (
Scheme 3). Carbon dioxide was formed primarily from further oxidation of PO rather than the oxidation of propylene, while water was produced from the reaction of hydrogen and oxygen. FTIR of the spent catalyst showed the formation of carbon species (probably a bidentate propoxy) due to propylene oxide decomposition, which is a probable cause for catalyst deactivation. Surface formate and acetate species were also observed as oxidation products of the bidentate species in the spent catalyst.
7% Au loading could be achieved on promoting the Ti-TUD support with Ba
2+ at pH 7. The Au particle size was 2.0 nm by transmission electron microscopy, whereas at pH 9 much lower Au loading of 0.11 wt.% was produced with particle size of about 0.9 nm estimated by X-ray absorption fine structure measurements. At 423 K and 0.1 MPa total pressure, the catalyst prepared at pH 7 gave a steady-state propylene conversion of 2.1%, a propylene oxide selectivity of 79%, and a H
2 efficiency of 3.8%, whereas that prepared at pH 9 gave a conversion of 1.4%, a propylene oxide selectivity of 99%, and a H
2 efficiency of 17%. It is concluded that very small Au particles (about 1 nm) were the most active for epoxidation, whereas larger Au particles (about 2 nm) were less active because they promote direct H
2 oxidation to H
2O. X-ray absorption near-edge spectroscopy results indicated that under reaction conditions, the small particles had partially oxidized gold but the larger particles had metallic gold, suggesting that the smaller particles had high coverage of oxygen or oxygen-derived species [
12].
Scheme 3.
A schematic mechanism for propene oxidation over Au/Ti silicalites. Reproduced with permission from Reference [
11]. Copyright (2007) Elsevier.
Scheme 3.
A schematic mechanism for propene oxidation over Au/Ti silicalites. Reproduced with permission from Reference [
11]. Copyright (2007) Elsevier.
A series of Au/titanium silicalite-1 (TS-1) catalysts with different Si/Ti ratios and promoted with alkali and alkaline earth cations were prepared by the DP technique and tested for direct propylene epoxidation. It was found that the gold loading and catalytic activity were highly dependent on the pH of the DP synthesis solution and the final composition of the catalyst. Addition of the cations of group 1 metals such as K or Cs had little effect on the gold content, but increased the activity, while those of group 2 metals such as Mg, Ca, Sr, and Ba increased both the gold content and the catalytic activity. The highest improvement was provided by a Mg
2+-promoted catalyst giving 50% enhancement of activity at 443 K and 0.1 MPa with a H
2/O
2/C
3H
6/Ar = 1/1/1/7 feed mixture. Ammonia temperature-programmed desorption (NH
3-TPD) measurements indicated little change in adsorption amount with promotion indicating that the yield increase was not due to the elimination of acidic sites on the catalyst. Instead, the improved catalytic performance was ascribed to increased Au capture efficiency and dispersion by the catalyst [
13].
Cyanide treatment of Au/TS-1 provided with unexpected results. Catalysts treated with dilute solutions of sodium cyanide resulted in preferential removal of small gold particles, while catalysts treated with concentrated solutions resulted in dissolution of gold and its re-precipitation as Au(I) cyanide. X-ray absorption spectroscopy demonstrated that catalysts that produce PO in the presence of hydrogen and oxygen mixtures had supported Au(III) oxide nanoparticles of 3 nm size after synthesis, which were reduced to gold metal under reaction conditions. Samples treated with concentrated solutions of sodium cyanide resulted in supported Au(I) cyanide particles of large size (9–11 nm). These particles did not produce PO, but, surprisingly, showed high selectivity toward propylene hydrogenation. Increasing Au(I) cyanide particle size resulted in a decrease in hydrogenation activity [
14].
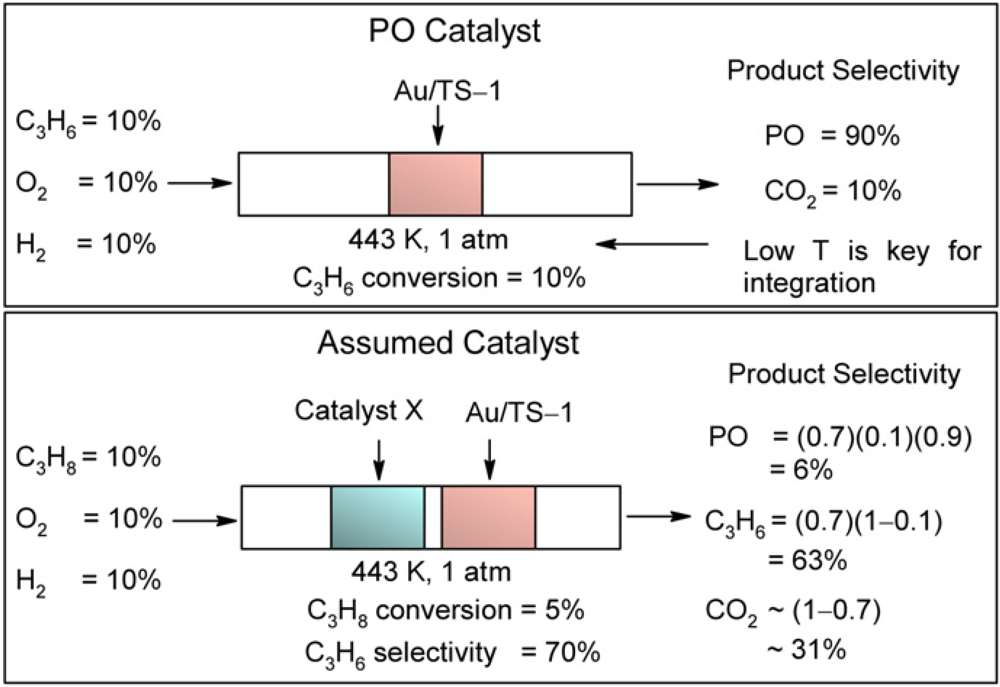
Propane epoxidation has also been attempted (
Figure 2).
Figure 2.
Hypothetical propane to propylene oxide (PO) oxidation process. Reproduced with permission from Reference [
15]. Copyright (2008) Elsevier.
Figure 2.
Hypothetical propane to propylene oxide (PO) oxidation process. Reproduced with permission from Reference [
15]. Copyright (2008) Elsevier.
It was carried out by sequential propane dehydrogenation, propylene epoxidation steps using a two-catalyst bed and H
2 and O
2 as the oxidant mixture. The propane dehydrogenation step used an Au/TiO
2 catalyst that was active at low temperature (443 K), while for the propylene epoxidation step an Au/TS-1 catalyst was applied.
In situ Au L3-edge X-ray absorption near-edge (NEXAFS) structure and UV-visible measurements on Au/TiO
2 under propane dehydrogenation conditions showed activation of oxygen on gold nanoparticles and evidence for the formation of adsorbed oxygen intermediate species responsible for the production of propylene. Propane epoxidation with H
2 and O
2 at 443 K and 0.1 MPa with the dual Au/TiO
2 and Au/TS-1 catalysts resulted in an overall propane conversion of 2%, propylene selectivity of 57%, and PO selectivity of 8%. The catalysts showed little deactivation and maintained their conversion and selectivity levels for the 12 h duration of the measurements [
15].
Delgass and co-workers have also been deeply involved in studying propylene epoxidation over gold-based catalysts [
16,
17,
18,
19,
20].
The epoxidation reaction was investigated over gold particles prepared by the DP method on various modified titanium silicalite-1 (TS-1) supports over a reaction time of 24–36 h in a flow reactor at temperatures of 413, 443, and 473 K. Gold deposition at pH 9–10 allowed for a consistent amount of 1–3 wt.% of the gold available in solution to be deposited, while still maintaining gold particle diameters in the 2–5 nm range, as observed by TEM [
16]. These Au/TS-1 catalysts achieved propylene conversions of 2.5–6.5% and PO selectivities of 60–85% at 443 K. A key result of the work is that PO rates were not highly influenced by the TS-1 particle size and were thus not proportional to the specific external surface area of the support. The conclusion that activity may have resided in the channels of the TS-1 was supported by the finding that the observable gold particles decorating the TS-1 particles only accounted for
ca. 30% of the total gold content of the catalyst. Increasing the gold loading up to 0.74 wt.% did not increase PO rates proportionally, suggesting that the active Au-Ti PO forming centers were limited. In contrast to the prevailing interpretation of this catalyst that a critical Au particle diameter of 2–5 nm is essential for PO activity, the results were consistent with a molecular cluster model where extremely small gold clusters are located near Ti sites inside the TS-1 pores or on the external surface. These clusters were thought to be active for propylene epoxidation. Treatment of the calcined TS-1 support with 1 M NH
4NO
3 at 80 °C, followed by vacuum drying, produced a modified TS-1 support material having a four-fold increase in Au capture efficiency and produced catalysts with 5–10% conversion of propylene with 75–85% selectivity for PO at 200 °C in a flow reactor [
17].
The superior activity and stability of Au/TS-1 catalysts allowed the first comprehensive kinetic analysis of the propylene epoxidation system in the absence of significant deactivation. A unique design of experiments combining the best features of factorial experiments with one-at-a-time experimentation over the nonflammable range was used to collect kinetic information, from which a power rate law was extracted. Explaining the resultant fractional reactant orders (O
2 = 0.31 ± 0.04, H
2 = 0.60 ± 0.03, and C
3H
6 = 0.18 ± 0.04) required a sequence of elementary kinetic steps having a minimum of two active sites participating in the rate-determining step [
18,
19]. A reaction sequence was proposed that accounted for the experimentally determined reaction orders and was consistent with DFT calculations [
20] (
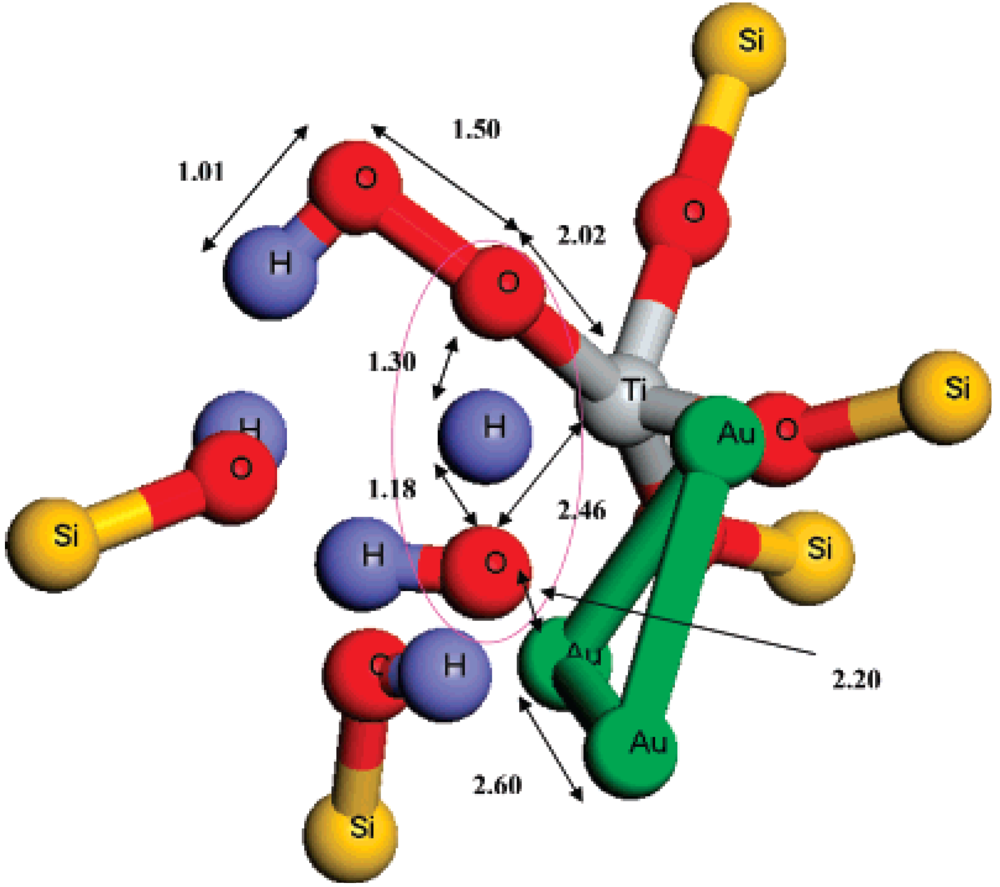
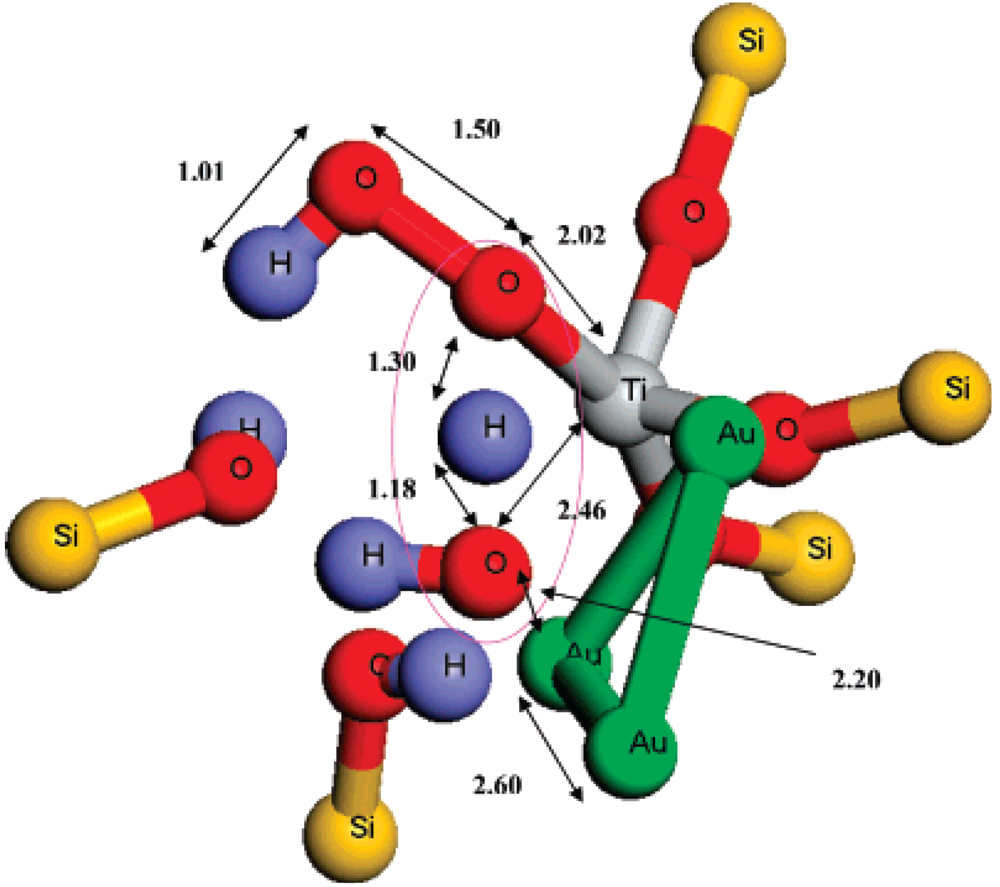
Figure 3). This mechanism suggests that titanium and gold sites must generate and use the epoxidation oxidant simultaneously rather than sequentially, as previously suggested in the literature (
Figure 3).
Figure 3.
The transition state geometry for attack of H
2O
2 on Au
3/T6-Ti-defect site to form Ti-OOH species inside the TS-1 pores. All of the MM atoms and some atoms in the QM region were removed for clarity. The atomic distances in Å are indicated. Reproduced with permission from Reference [
20]. Copyright (2007) American Chemical Society.
Figure 3.
The transition state geometry for attack of H
2O
2 on Au
3/T6-Ti-defect site to form Ti-OOH species inside the TS-1 pores. All of the MM atoms and some atoms in the QM region were removed for clarity. The atomic distances in Å are indicated. Reproduced with permission from Reference [
20]. Copyright (2007) American Chemical Society.
The Weckhuysen group synthesized highly dispersed gold nanoparticles within the channels of a mesoporous Ti-SBA-15 support, followed by thorough catalyst characterization and testing in the selective epoxidation of propene to PO [
21]. For this purpose, two series of Ti-SBA-15 materials differing in their Ti content were prepared by either grafting or direct synthesis. It was found that the Au/Ti-SBA-15 materials obtained by Ti grafting had higher catalytic activity than the samples in which Ti-SBA-15 was obtained by direct synthesis. These differences in catalytic behavior were attributed to differences in the amount and dispersion of Ti within the mesoporous silica support as well as to differences in the size of Au nanoparticles. The adsorption of propene on supported gold nanoparticles has been experimentally identified as a reaction step in the hydro-epoxidation of propene [
22]. This new finding was made possible by applying a detailed analysis of
in situ measured XANES spectra. For this purpose, Au/SiO
2 catalysts were investigated since this support was more inert and propene was not converted. Propene adsorption was investigated by using the oxidation of hydrogen as probe reaction. It was shown that co-feeding of propene dramatically decreased the hydrogen oxidation rate. Since it has been reported in the literature that the oxidation of hydrogen occurs exclusively over gold nanoparticles, this inhibition by propene can be attributed to adsorption of propene on the gold nanoparticles. Analysis of the
in situ XANES spectra confirmed the adsorption of propene on the gold nanoparticles and the mode of adsorption was determined to be π-bonding. Comparative experiments with ethene and propane confirmed this π-bonded adsorption, since ethene similarly inhibited the oxidation of hydrogen, while propane had only a minor effect.
Titanium-containing hexagonal mesoporous silicas (Ti-HMS) with wormhole structure and Si/Ti molar ratios ranging from 10 to 40 have also been studied [
23]. Catalytic results showed that the Au/Ti-HMS catalyst exhibited superior performance in terms of propylene conversion, PO selectivity, and H
2 efficiency in comparison with the Au catalysts supported on the conventional Ti-containing mesoporous materials. Beside the Si/Ti molar ratio, the chain length of alkylamine for the Ti-HMS preparation was crucial for the enhancement of catalytic performance. Specifically, 9.0% of propylene conversion, 97.3% of propylene oxide selectivity, and 30.4% of H
2 efficiency could be obtained at 373 K in the initial 30 min of time-on-stream on the Au/Ti-HMS catalyst. The Ti-HMS having a Si/Ti molar ratio at 20 was prepared by using tetradecylamine as the templating agent. Regeneration of the spent catalyst by calcination in air gave almost no change in PO selectivity but about 25% loss in propylene conversion. The enhanced catalytic performance of Au/Ti-HMS catalyst may be essentially attributed to the homogeneous dispersion and uniformity of titanium species in combination with accessible pore structure.
The works of all groups indicate that in the preparation of gold catalysts that are highly selective in PO formation Ti-containing supports must be used, the particle size of Au is decisive, to achieve its optimum the deposition-precipitation technique is the best and the alkali content of the catalyst also influences catalytic activity. The calcination temperature is important as well and the reaction temperature should not be too high.
2.3. Epoxidation of Styrene
The epoxidation of styrene proceeds in the liquid phase over supported gold catalysts using peroxides, mostly
tert-butyl hydroperoxide, as co-reactant (
Scheme 4).
Scheme 4.
Epoxidation of styrene.
Scheme 4.
Epoxidation of styrene.
In Choudry’s group many different oxides (MgO, CaO, SrO, BaO, Al
2O
3, Ga
2O
3, In
2O
3, Tl
2O
3, TiO
2, Cr
2O
3, MnO
2, Fe
2O
3, CoO
x, NiO, CuO, ZnO, Y
2O
3, ZrO
2, La
2O
3, Ce
2O
3, Nd
2O
3, Sm
2O
3, Eu
2O
3, Tb
2O
3, Er
2O
3, Yb
2O
3 and U
3O
8) as support have been tested [
24,
25,
26,
27,
28]. The supported catalysts were prepared by depositing gold on the support by the DP and the homogeneous DP methods. The catalysts were calcined at various temperatures (400 °C–900 °C) and the catalytic activity of the supported nanogold catalysts in the epoxidation of styrene to styrene oxide by
tert-butyl hydroperoxide were correlated with the Au loading and/or Au particle size of the catalysts. The reaction was strongly influenced by a number of parameters, such as the metal oxide support, the method of gold deposition, the gold loading as well as the catalyst calcination temperature. For characterizing the catalytic performance (both the activities and the selectivities) some activity orders (the selectivity values changed in the same sequence) could be set up, like Au/MgO > Au/Tl
2O
3 > Au/Yb
2O
3 > Au/Tb
2O
3 > Au/CaO (or TiO
2); Au/Tl
2O
3 > Au/In
2O
3 > Au/Ga
2O
3 > Au/Al
2O
3. The nanogold particles-support interactions seem to play an important role in controlling the deposition of gold (amount of gold deposited and size and morphology of gold particles), formation of different surface gold species (Au
0, Au
+ and Au
3+), consequently, they control the catalytic performance (both the activity and selectivity) of the supported nanogold catalysts [
25,
26,
27,
28].
Three kinds of mesoporous alumina (denoted as
meso-Al
2O
3-
x (
x = a, b, c)) supports with different surface basicities were synthesized by various structure-directing agents and different assembly pathways [
29]. It has been found that the mesoporous alumina had more abundant surface basic sites than γ-Al
2O
3. Au nanoparticles were deposited on these supports via the homogeneous DP method using urea as the precipitating agent. The dispersion and average size of the gold particles were found to be dependent on the number of surface basic sites on the supports. Transmission electron microscopy (TEM) observations showed a homogeneous distribution of gold particles lower than 4 nm on
meso-Al
2O
3-b and
meso-Al
2O
3-c having more basic sites on the surface. XPS spectra revealed that only metallic gold was present on the supports irrespective to the surface properties. The catalysts were employed as highly active/selective and reusable catalysts for the epoxidation of styrene with anhydrous
tert-butyl hydroperoxide. Au nanoparticles as well as the surface basic sites of the support were thought to be responsible for the epoxidation.
Somewhat unusual supports were also used in preparing the gold-based catalysts like (
S)-(−)-2-pyrrolidinone-5-carboxylic acid (Py)-modified SBA-15 [
30], S-containing organic-inorganic hybrid mesoporous silicas [
31] or carbon nanotubes [
32].
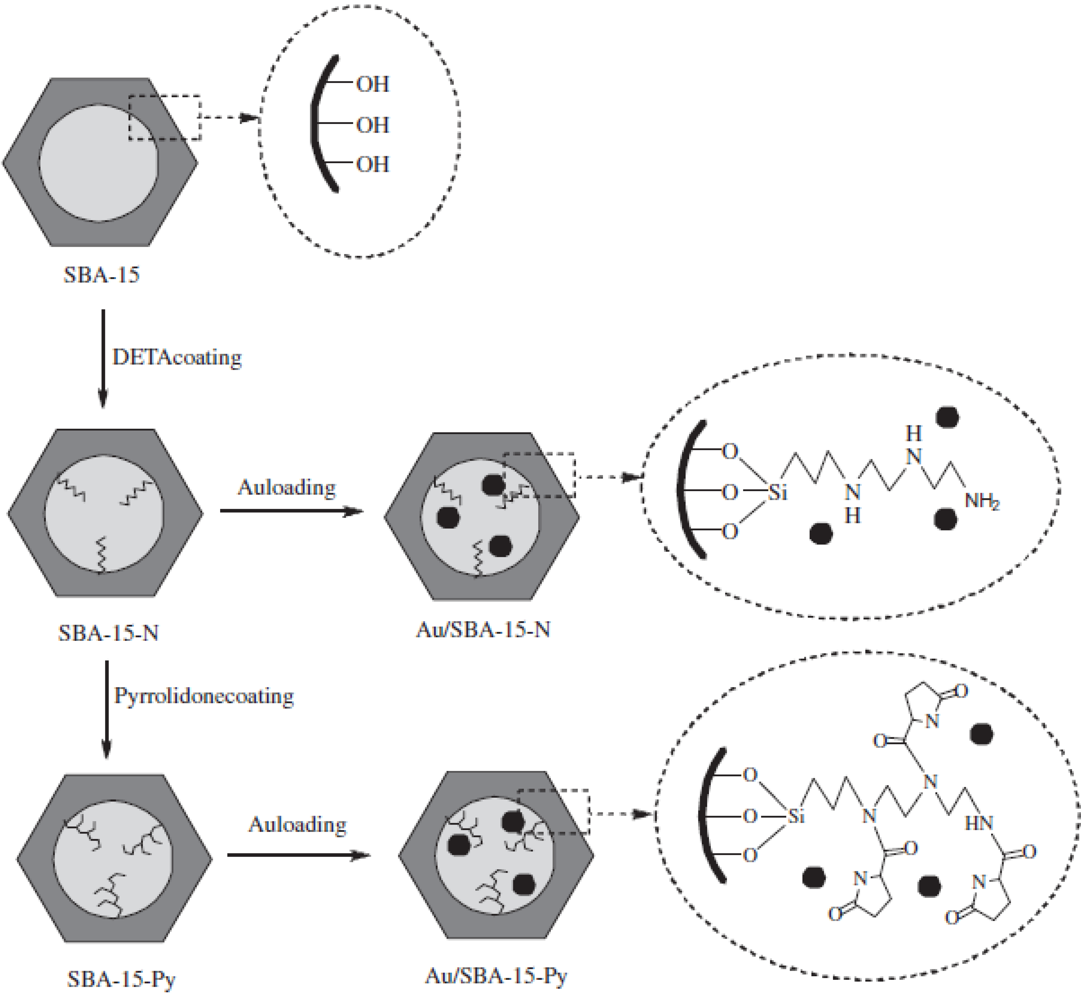
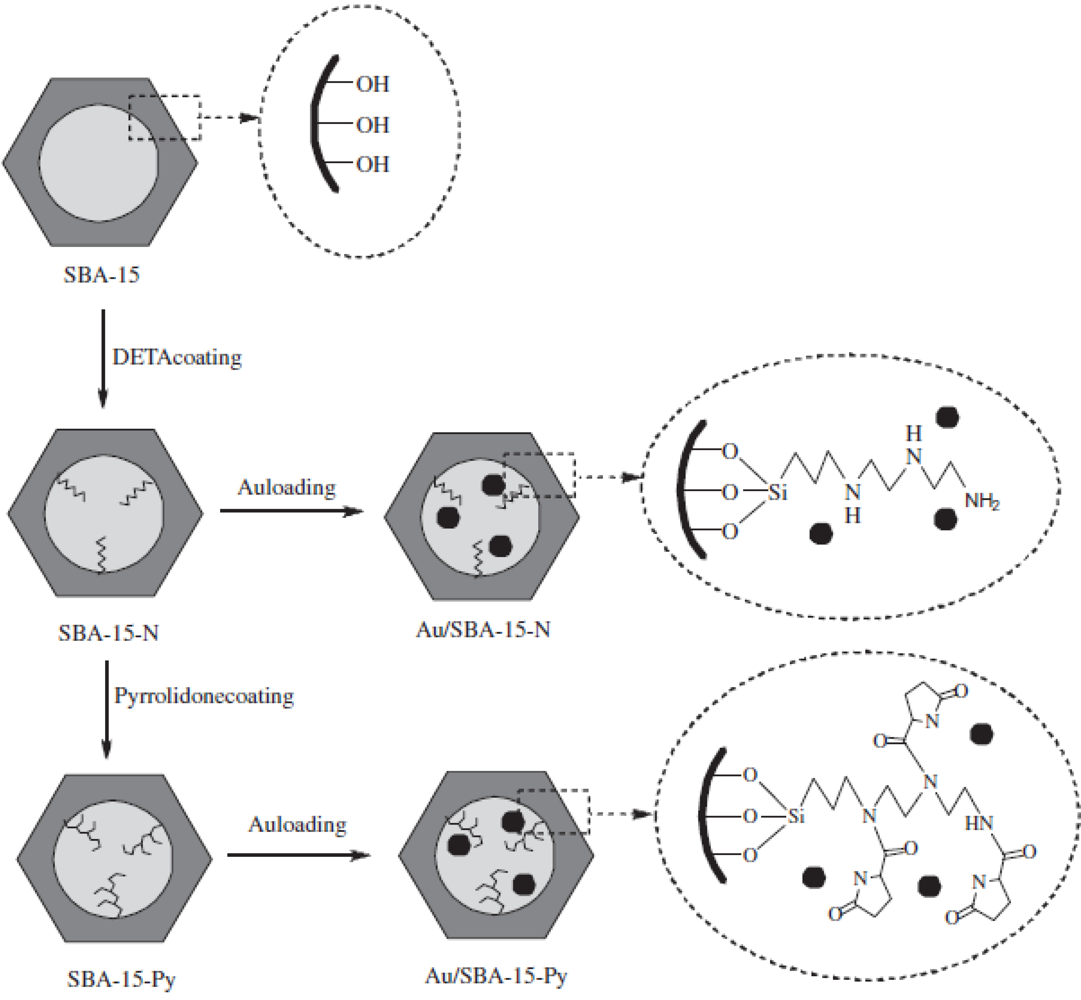
Highly dispersed Au nanoparticles could be deposited in the mesopores of (
S)-(−)-2-pyrrolidinone-5-carboxylic acid (Py)-modified SBA-15 (Au/SBA-15-Py) [
30] (
Figure 4).
13C NMR and IR spectroscopies indicated that Py species were successfully grafted on the surface of mesopores in SBA-15; XRD patterns and N2 adsorption isotherms showed that the mesostructures were well preserved; TEM images clearly confirmed the uniform Au nanoparticles in the mesopores; and XPS suggested an interaction between Au nanoparticles and Py species. Interestingly, and importantly, Au/SBA-15-Py catalysts always exhibited superior catalytic properties in the oxidation of cyclohexene and styrene by molecular oxygen at atmospheric pressure, compared with the pyrrolidone-free SBA-15 supported Au catalyst (Au/SBA-15-N). This phenomenon was reasonably related to the interaction between Au nanoparticles and Py species, which was consistent with results of density functional theory (DFT) calculations.
Figure 4.
Procedure for the preparation of Au/SBA-15-N and Au/SBA-15-Py catalysts (DETA:
N-[3-(trimethoxysilyl)-propyl]diethylenetriamine). Reproduced with permission from Reference [
30]. Copyright (2011) Elsevier.
Figure 4.
Procedure for the preparation of Au/SBA-15-N and Au/SBA-15-Py catalysts (DETA:
N-[3-(trimethoxysilyl)-propyl]diethylenetriamine). Reproduced with permission from Reference [
30]. Copyright (2011) Elsevier.
For the mesoporous organosilica it was found that the location and structure of organic moieties in the mesostructures played a crucial role in the dispersion level of gold nanoparticles, and thus, had a significant effect on the catalytic performance. Under optimum conditions, the gold nanoparticles supported on a periodic mesoporous organosilica with bridging disulfide-ionic liquid moieties (
Scheme 5) showed excellent catalytic performance and reusability in the epoxidation of styrene in the presence of H
2O
2–acetonitrile [
31].
Scheme 5.
The bridging organosilica precursor.
Scheme 5.
The bridging organosilica precursor.
The catalytic activity of carbon nanotube supported gold catalysts prepared by the DP method using urea for precipitation was examined for the oxidation of styrene using
tert-butyl hydroperoxide as oxidant. The system showed good epoxide selectivity. The other factors, such as solvent, reaction time, concentrations of oxidant and catalyst, have also been investigated and reaction conditions were optimized. It is a novel highly active/selective and reusable heterogeneous catalyst for styrene epoxidation [
32].
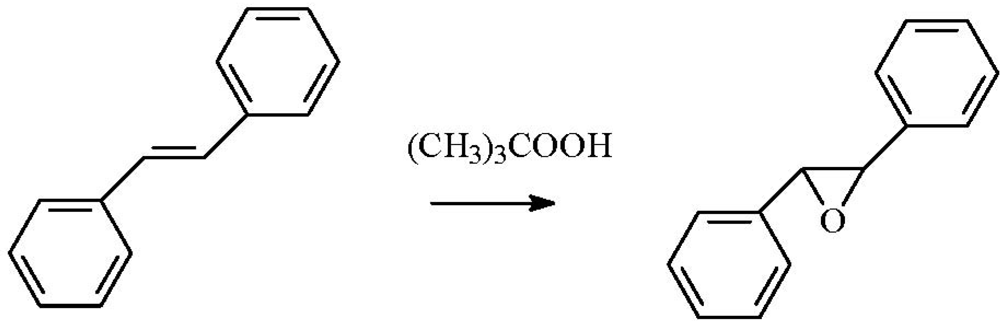
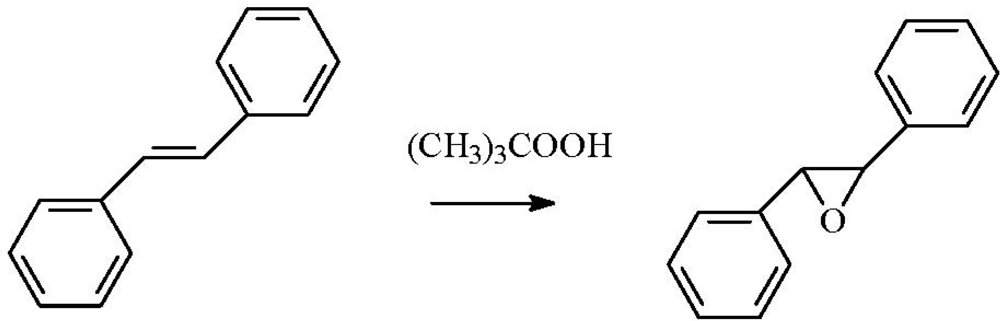
2.4. Epoxidation of Stilbene
The stilbene epoxidation reaction can be efficiently catalyzed by supported gold nanoparticles (
Scheme 6).
Scheme 6.
Epoxidation of trans-stilbene.
Scheme 6.
Epoxidation of trans-stilbene.
A free-radical mechanism has been evidenced in the liquid-phase stereoselective epoxidation of
trans-stilbene using methylcyclohexane as solvent, limited amount of
tert-butyl hydroperoxide, and supported gold catalysts [
33,
34].
Trans-stilbene oxide was the major reaction product observed, with selectivities up to 88% over the Au/TiO
2 reference catalyst. However, the selectivity decreased significantly on using Au/C instead of oxide-supported gold catalysts or H
2O
2 instead of
tert-butyl hydroperoxide. It seemed that
tert-butyl hydroperoxide was the radical source, while methylcyclohexane was propagating the active radical. XPS measurements showed the presence of Au
0 (90%) and Au
+ (10%) on the Au/C catalyst and Au
δ− (90%) and Au
+ (10%) on the Au/TiO
2 catalyst. Both gold and, to a minor extent, titania seemed to be involved in the reaction cycle.
Citrate-functionalized titania nanocrystallites were synthesized from a heteroleptic titanium alkoxide precursor in a low-temperature hydrolytic process and used as gold catalyst support for CO oxidation and aerobic stilbene epoxidation [
35].
The support had little influence on the intrinsic activity of gold but more on the apparent reaction rates which are a combination of catalytic activity and diffusion limitations. These were here minimized by using gadolinium-doped titania nanocrystallites as support for gold nanoparticles [
36]. This material was obtained by the mild hydrolysis of a novel Gd
4TiO(OiPr)
14 bimetallic oxo-alkoxide. It led to enhanced wettability of the <3 nm gold particles in the
tert-butyl hydroperoxide initiated epoxidation of stilbene in methylcyclohexane. The rate-determining step of this reaction was identified as the gold-catalyzed homolytic decomposition of
tert-butyl hydroperoxide generating radicals and initiating the methylcyclohexane-mediated epoxidation of stilbene, yielding a methylcyclohexan-1-ol/
trans-stilbene oxide mixture.
The preparation and characterization and catalytic testing in the aerobic oxidation of cyclohexene and
trans-stilbene in the liquid phase of highly dispersed gold nanoparticles in ordered mesoporous carbons have also been reported [
37]. The carbon supports were prepared using gold-containing functionalized SBA-15 silica templates. Two series of Au/SBA-15 templates were made. In the first series the size of the gold particles was determined by the pore size in the ammonium-functionalized silicas, while the mercaptopropyltrimethoxysilane grafting agent allowed the possibility of controlling the particle size inside the mesopores. Both series provided with highly ordered mesoporous carbons with gold particles incorporated in the carbon nanorods. Their catalytic activities and selectivities in epoxidation were appreciable and practically did not depend on the support.



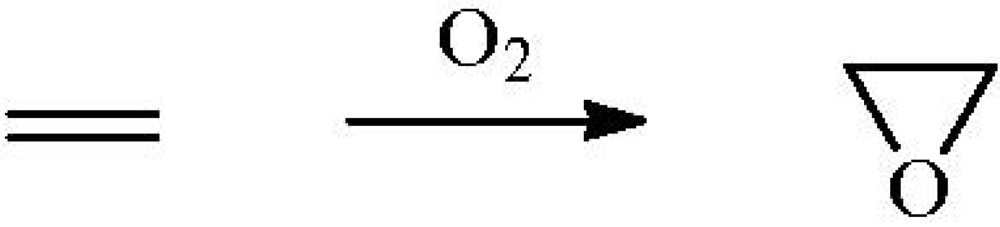


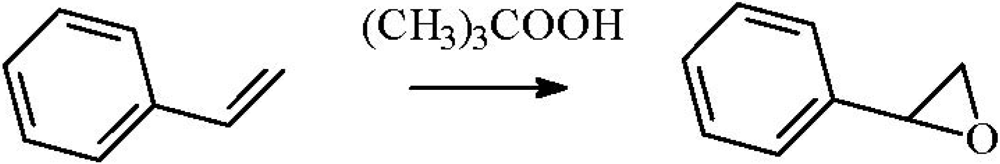




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
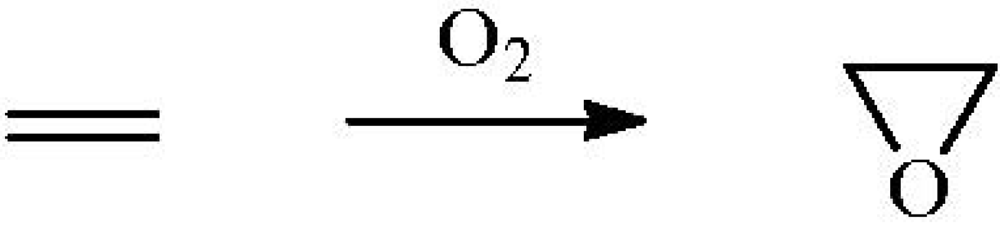{kind=link}
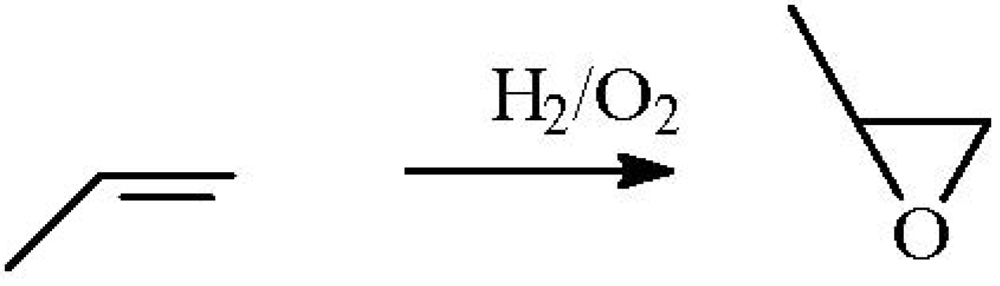{kind=link}
{kind=link}
{kind=link}
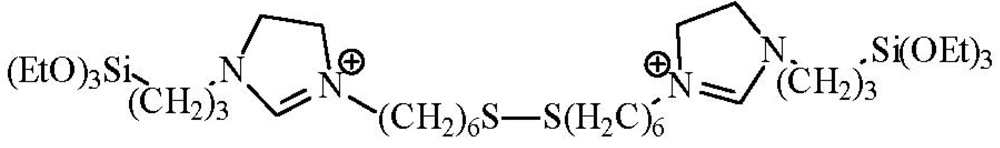{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}