
Loss of E2F1 Extends Survival and Accelerates Oral Tumor Growth in HPV-Positive Mice


Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Tissue Specific HPV Oncogene Expression Causes Embryonic Lethality

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parents | Total Mice | Number of Litters | Age of Analysis (Days) | iHPV | Cre | p Value Cre |
|---|---|---|---|---|---|---|
| CMV-Cre × iHPV 1 | 42 | 8 | 12.6 ± 3.7 | 42 (100.0%) | 4 (9.5%) | <0.0001 |
| CMV-Cre × FVB | 100 | 10 | 14.0 ± 1.7 | 0 (0.0%) | 55 (55.0%) | |
| Nestin-Cre × iHPV 2 | 42 | 8 | 12.6 ± 1.7 | 42 (100.0%) | 0 (0.0%) | <0.0001 |
| Nestin-Cre × FVB | 28 | 3 | 14.5 ± 2.2 | 0 (0.0%) | 18 (64.3%) | |
| Albumin-Cre × iHPV 3 | 55 | 6 | 13.3 ± 1.3 | 55 (100.0%) | 26 (47.3%) | N.D. |
| GFAP-Cre × iHPV 3 | 34 | 4 | 9.3 ± 1.3 | 34 (100.0%) | 18 (52.9%) | N.D. |
| K14-Cre × iHPV 4 | 49 | 5 | 11.4 ± 3.7 | 49 (100.0%) | 49 (100.0%) | N.D. |
| PDX1-Cre × iHPV 3 | 34 | 6 | 13.0 ± 2.5 | 34 (100.0%) | 19 (55.9%) | N.D. |
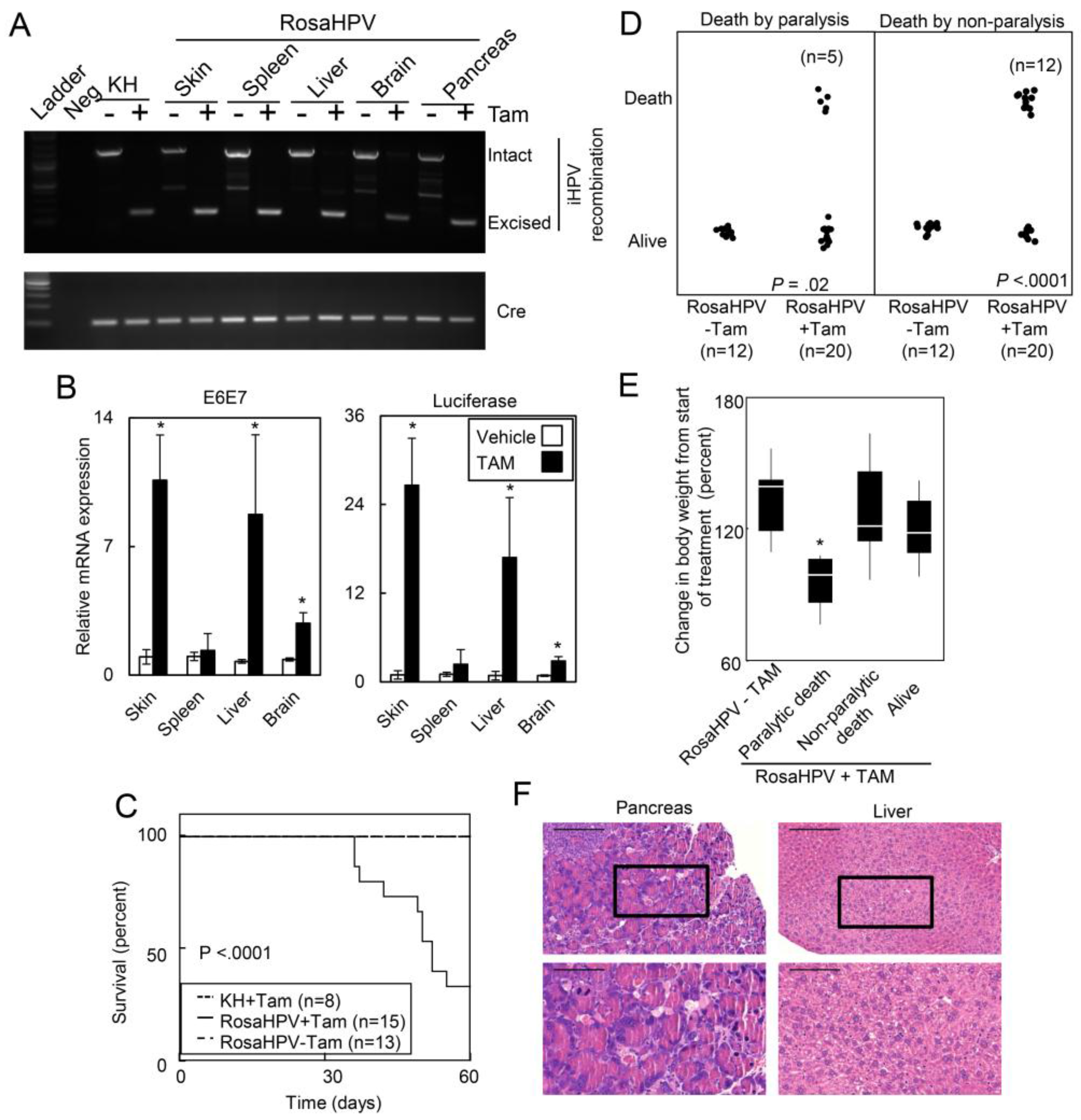
2.1.2. Ubiquitous Expression of HPV Oncogenes Caused Tissue Necrosis and Lethality in Adult Mice
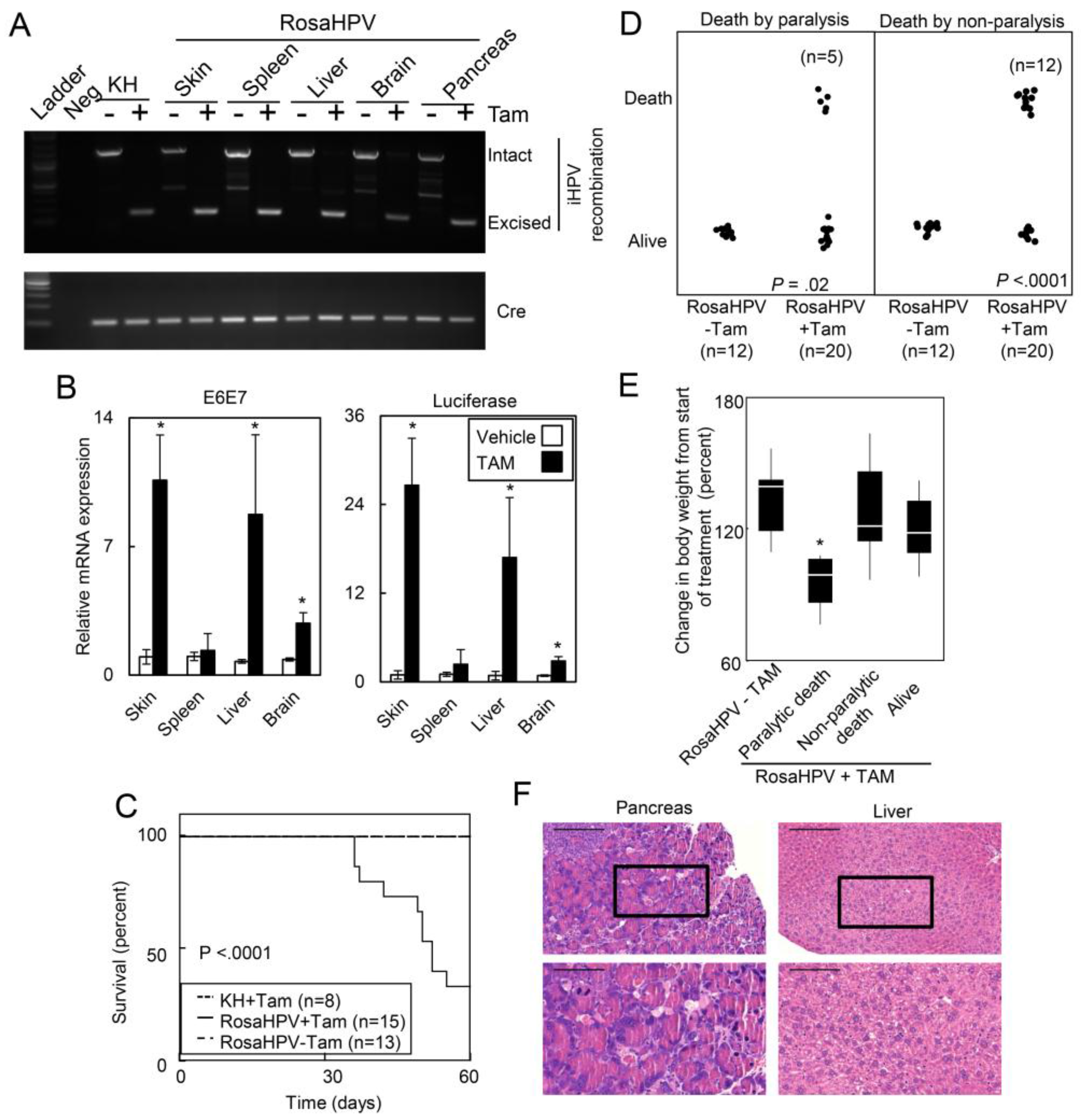
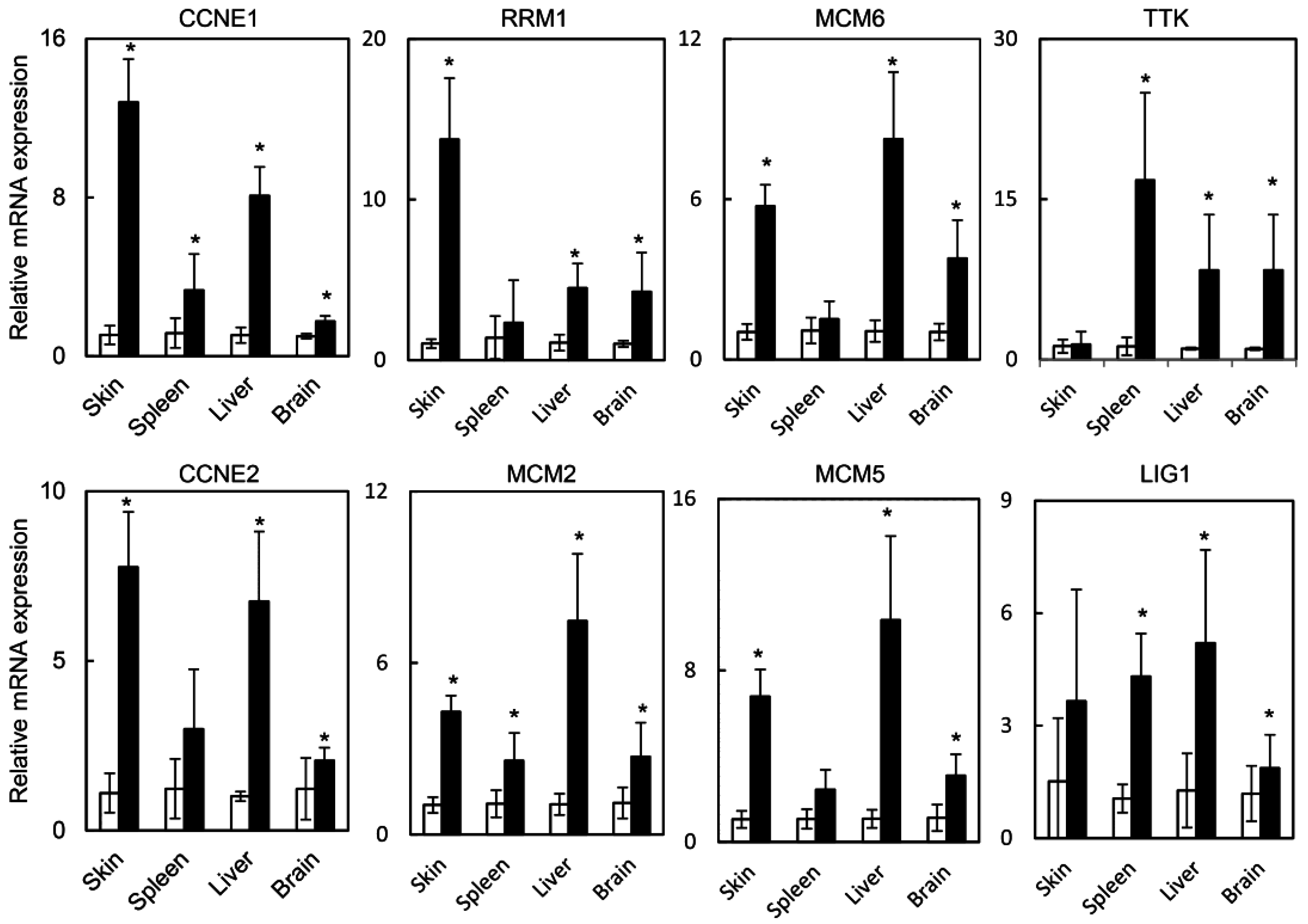
2.1.3. HPV Oncogenes Induce Expression of E2F1 Target Genes in the Skin, Spleen, Liver and Brain
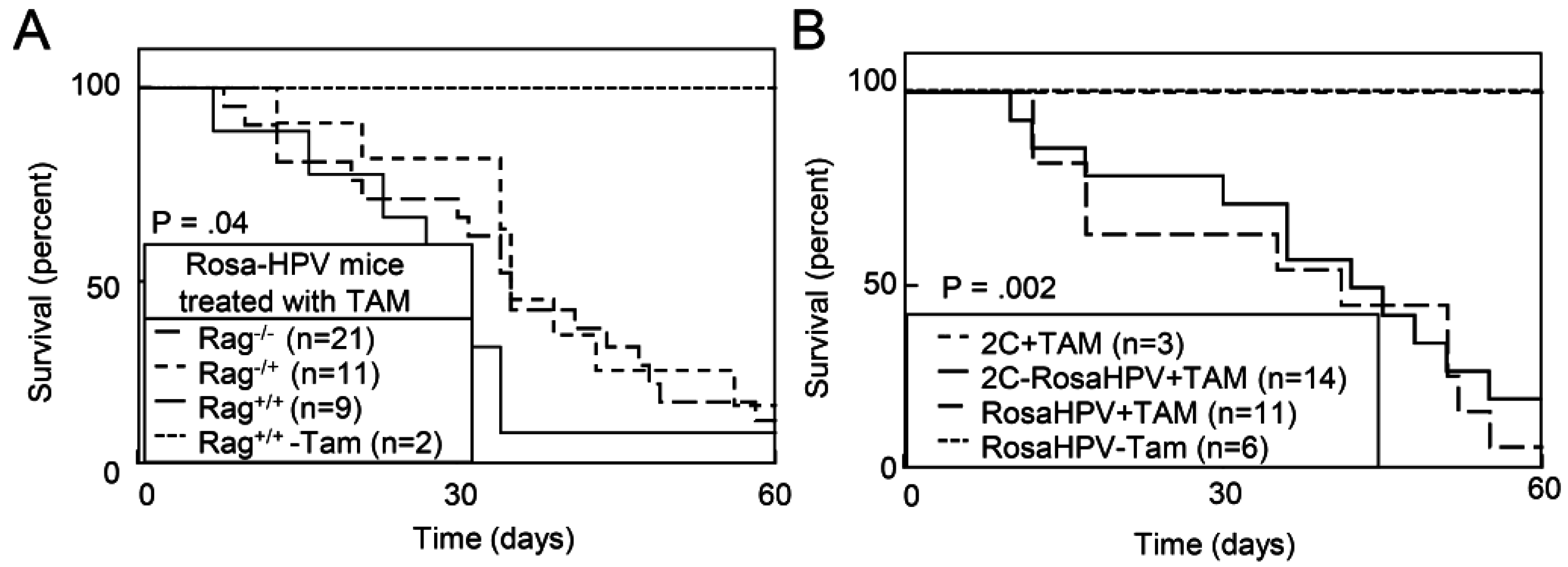
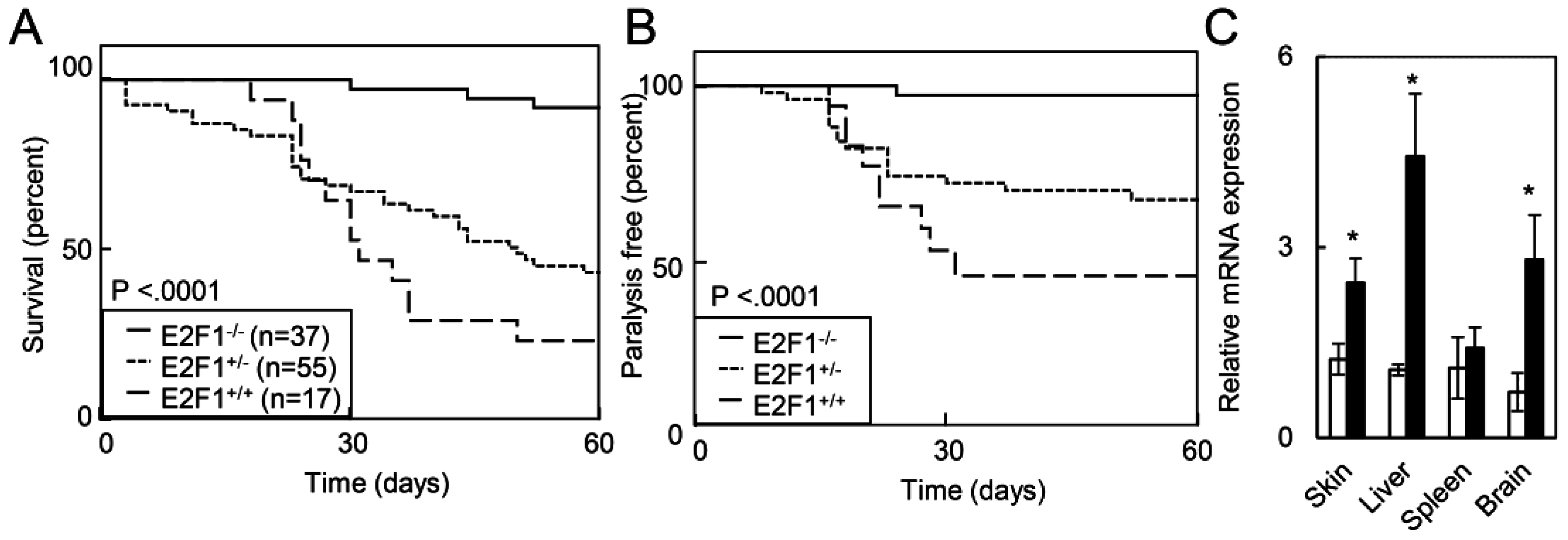
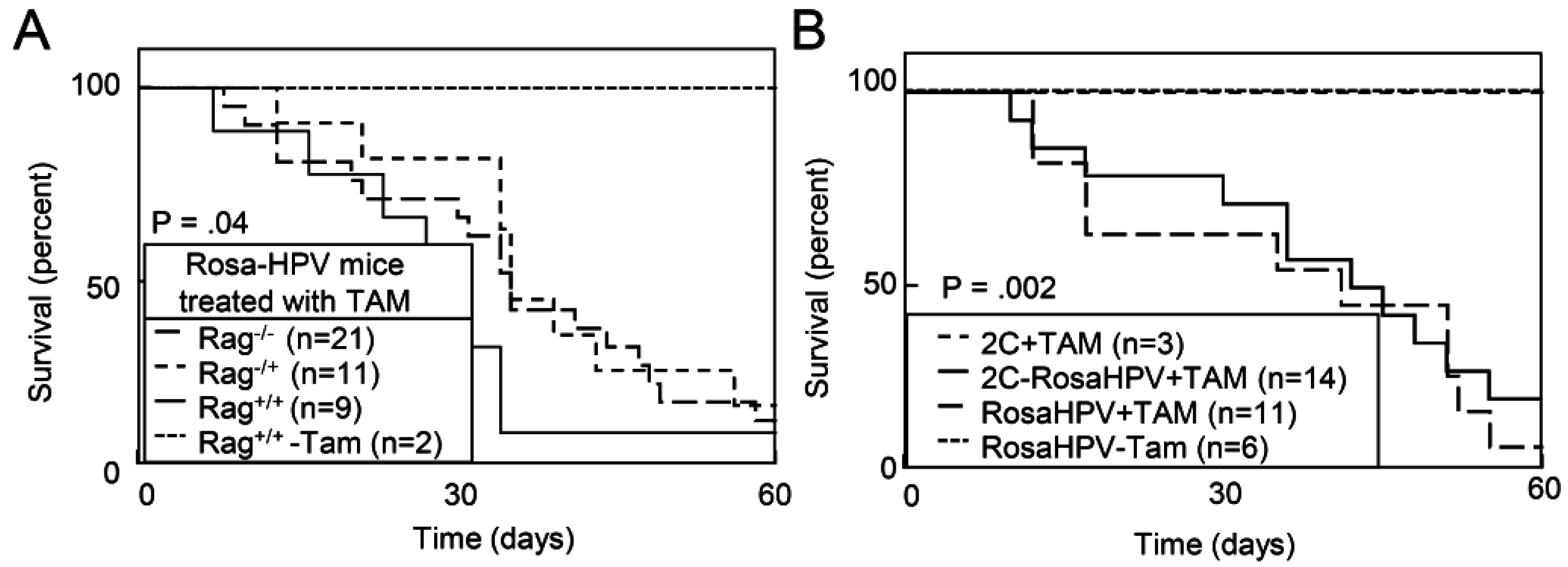
2.1.4. Deletion of E2F1 Rescues HPV Oncogene Lethality in Adult Mice

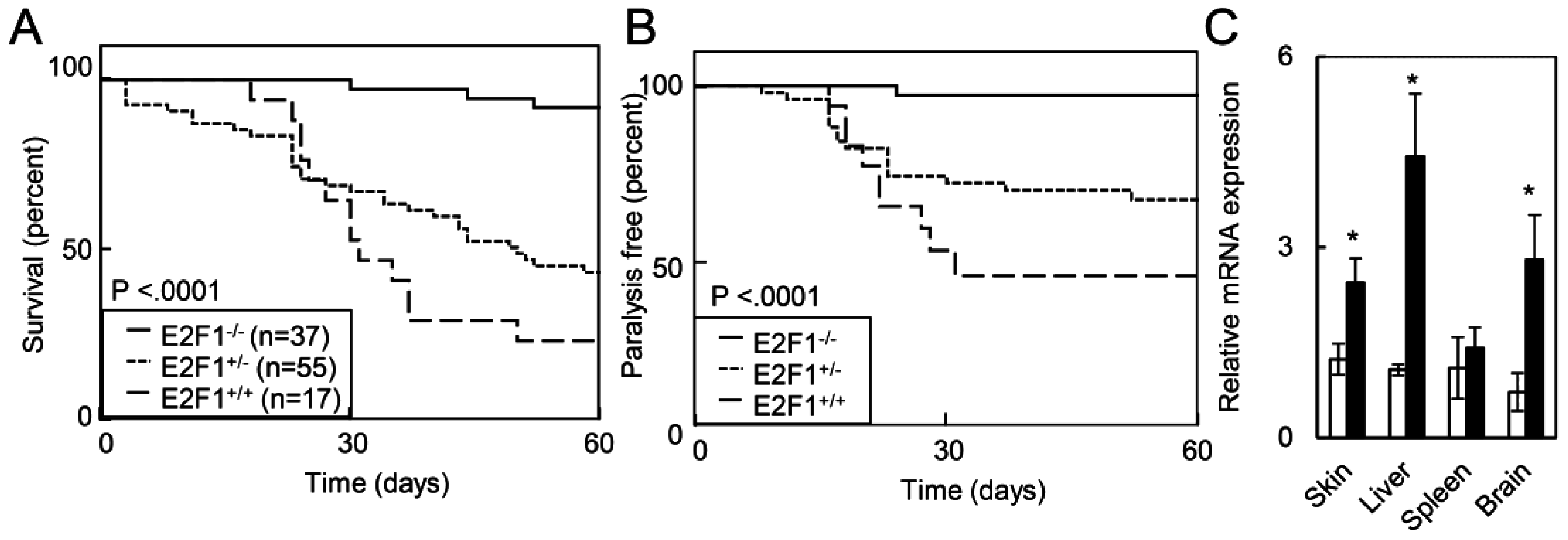
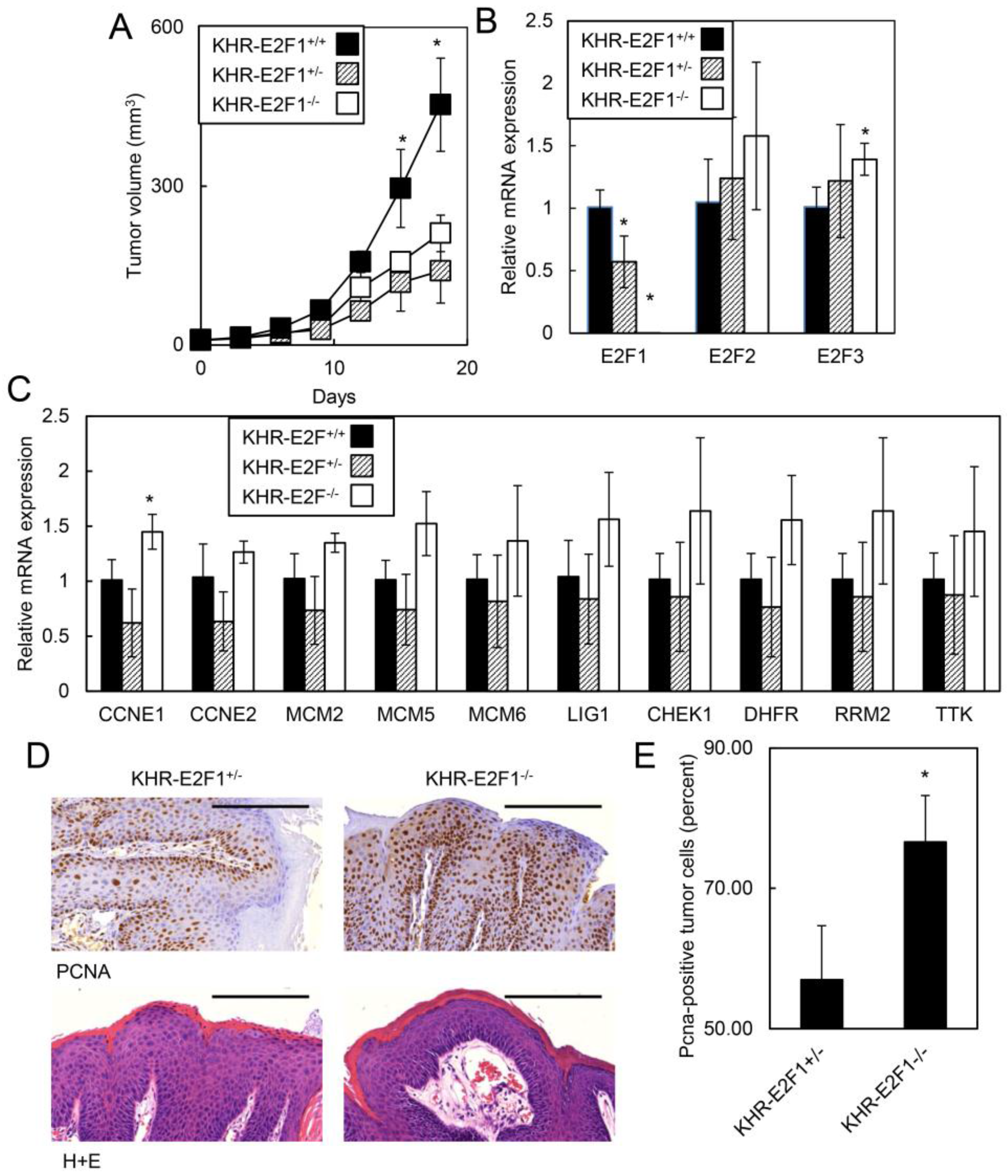
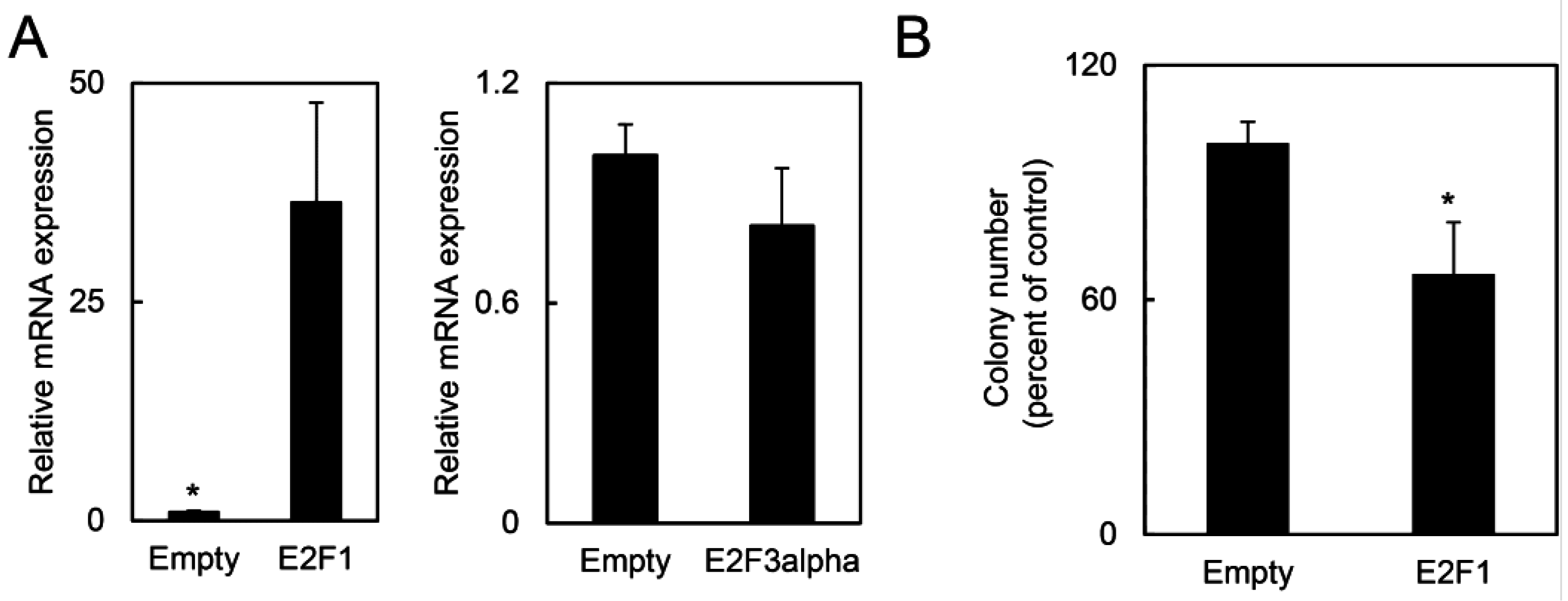
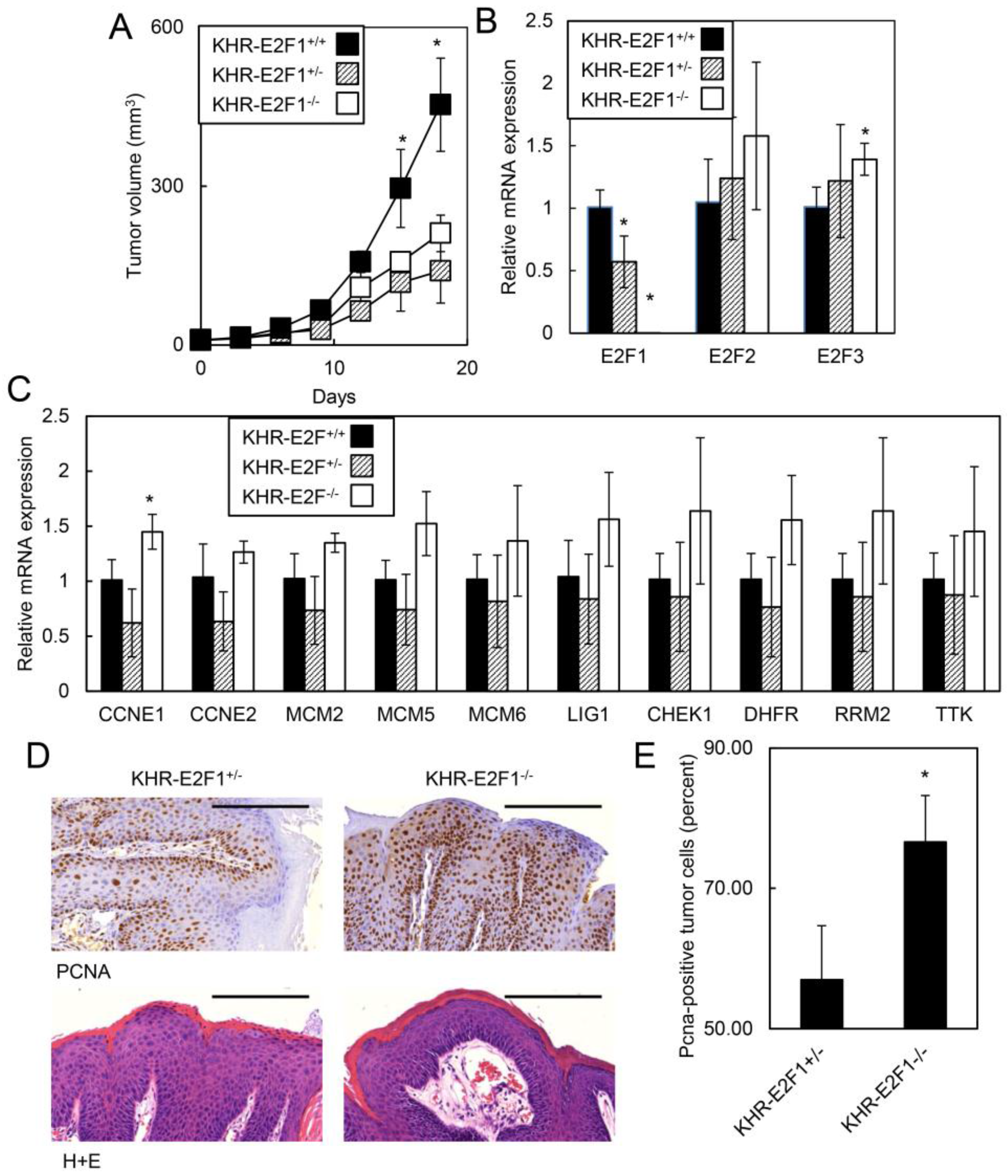
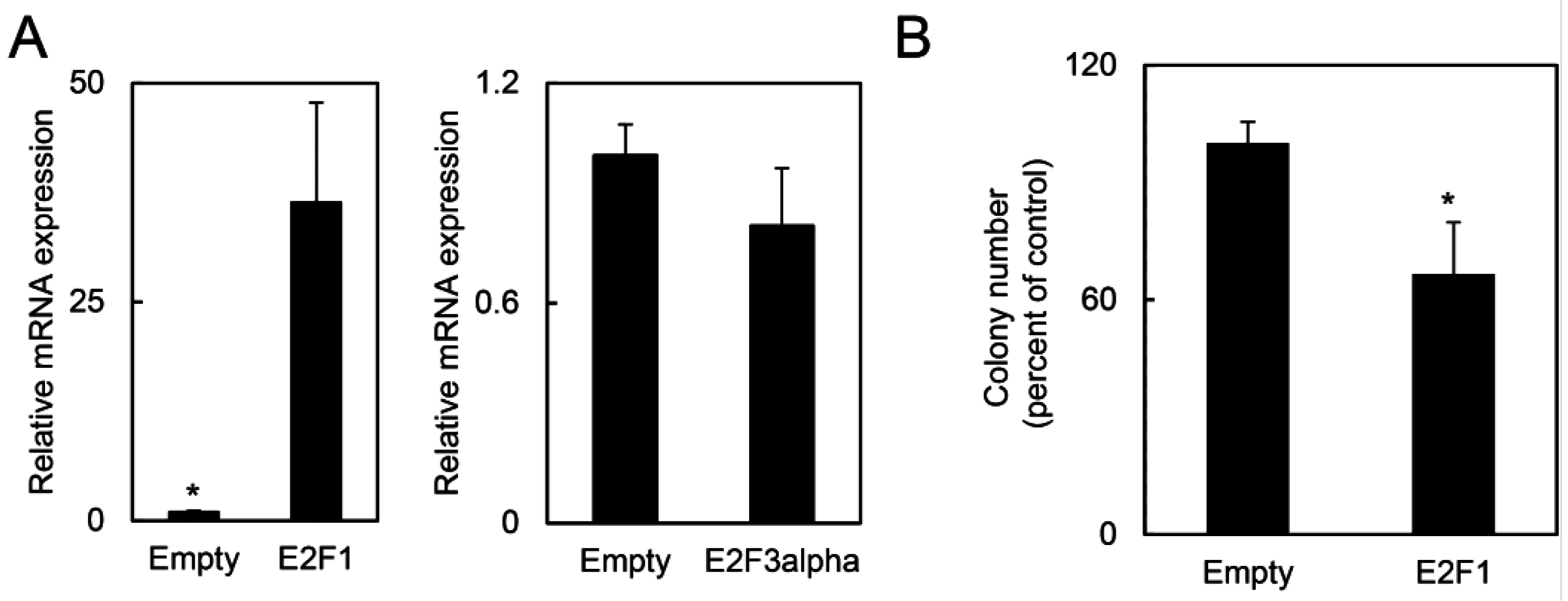
2.1.5. E2F1 Inhibits HPV-Positive Tumor Growth and Clonogenicity
2.2. Discussion
3. Experimental Section
3.1. Animals
3.2. Reagents and Antibodies
3.3. HPV Recombination
3.4. Quantitative RT-PCR
3.5. Colony Formation Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gillison, M.L.; Koch, W.M.; Capone, R.B.; Spafford, M.; Westra, W.H.; Wu, L.; Zahurak, M.L.; Daniel, R.W.; Viglione, M.; Symer, D.E.; et al. Evidence for a causal association between human papillomavirus and a subset of head and neck cancers. J. Natl. Cancer Inst. 2000, 92, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Munoz, N.; Bosch, F.X.; de Sanjose, S.; Herrero, R.; Castellsague, X.; Shah, K.V.; Snijders, P.J.; Meijer, C.J. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 2003, 348, 518–527. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, E.; Freese, U.K.; Gissmann, L.; Mayer, W.; Roggenbuck, B.; Stremlau, A.; zur Hausen, H. Structure and transcription of human papillomavirus sequences in cervical carcinoma cells. Nature 1985, 314, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Bao, R.; Faber, P.W.; Bindokas, V.P.; Bechill, J.; Lingen, M.W.; Spiotto, M.T. Notch1 activation or loss promotes hpv-induced oral tumorigenesis. Cancer Res. 2015, 75, 3958–3969. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Herrero, R.A.M.N. Human papillomavirus and cancer. In Infections & Human Cancer, Cancer Surveys; Newton, R., Beral, V., Weiss, R.A., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1999; Volume 33, pp. 75–98. [Google Scholar]
- Xue, Y.; Bellanger, S.; Zhang, W.; Lim, D.; Low, J.; Lunny, D.; Thierry, F. HPV16 E2 is an immediate early marker of viral infection, preceding E7 expression in precursor structures of cervical carcinoma. Cancer Res. 2010, 70, 5316–5325. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.J.; Hooper, M.L.; Armstrong, J.F.; Clarke, A.R. Effects of heterozygosity for the Rb-1t19neo allele in the mouse. Oncogene 1995, 10, 1615–1620. [Google Scholar] [PubMed]
- Hu, N.; Gutsmann, A.; Herbert, D.C.; Bradley, A.; Lee, W.H.; Lee, E.Y. Heterozygous Rb-1 delta 20/+mice are predisposed to tumors of the pituitary gland with a nearly complete penetrance. Oncogene 1994, 9, 1021–1027. [Google Scholar] [PubMed]
- Jacks, T.; Fazeli, A.; Schmitt, E.M.; Bronson, R.T.; Goodell, M.A.; Weinberg, R.A. Effects of an Rb mutation in the mouse. Nature 1992, 359, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Clarke, A.R.; Maandag, E.R.; van Roon, M.; van der Lugt, N.M.; van der Valk, M.; Hooper, M.L.; Berns, A.; te Riele, H. Requirement for a functional Rb-1 gene in murine development. Nature 1992, 359, 328–330. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.Y.; Chang, C.Y.; Hu, N.; Wang, Y.C.; Lai, C.C.; Herrup, K.; Lee, W.H.; Bradley, A. Mice deficient for Rb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 1992, 359, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, G.; Jacks, T. The retinoblastoma gene family: Cousins with overlapping interests. Trends Genet. 1998, 14, 223–229. [Google Scholar] [CrossRef]
- Giovanni, A.; Keramaris, E.; Morris, E.J.; Hou, S.T.; O'Hare, M.; Dyson, N.; Robertson, G.S.; Slack, R.S.; Park, D.S. E2F1 mediates death of B-amyloid-treated cortical neurons in a manner independent of p53 and dependent on bax and caspase 3. J. Biol. Chem. 2000, 275, 11553–11560. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Talos, F.; Nemajerova, A.; Moll, U.M.; Petrenko, O. E2F1 plays a direct role in rb stabilization and p53-independent tumor suppression. Cell Cycle 2008, 7, 1776–1781. [Google Scholar] [CrossRef] [PubMed]
- Polager, S.; Ginsberg, D. P53 and E2F: Partners in life and death. Nat. Rev. Cancer 2009, 9, 738–748. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Levine, A.J. P53 and E2F-1 cooperate to mediate apoptosis. Proc. Natl. Acad. Sci. USA 1994, 91, 3602–3606. [Google Scholar] [CrossRef] [PubMed]
- Macleod, K.F.; Hu, Y.; Jacks, T. Loss of Rb activates both p53-dependent and independent cell death pathways in the developing mouse nervous system. EMBO J. 1996, 15, 6178–6188. [Google Scholar] [PubMed]
- Yamasaki, L.; Bronson, R.; Williams, B.O.; Dyson, N.J.; Harlow, E.; Jacks, T. Loss of E2F-1 reduces tumorigenesis and extends the lifespan of Rb1+/− mice. Nat. Genet. 1998, 18, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.Y.; Hu, Y.; Macleod, K.F.; Crowley, D.; Yamasaki, L.; Jacks, T. Mutation of E2F-1 suppresses apoptosis and inappropriate S phase entry and extends survival of Rb-deficient mouse embryos. Mol. Cell 1998, 2, 293–304. [Google Scholar] [CrossRef]
- Arbeit, J.M.; Munger, K.; Howley, P.M.; Hanahan, D. Progressive squamous epithelial neoplasia in k14-human papillomavirus type 16 transgenic mice. J. Virol. 1994, 68, 4358–4368. [Google Scholar] [PubMed]
- Auewarakul, P.; Gissmann, L.; Cid-Arregui, A. Targeted expression of the E6 and E7 oncogenes of human papillomavirus type 16 in the epidermis of transgenic mice elicits generalized epidermal hyperplasia involving autocrine factors. Mol. Cell. Biol. 1994, 14, 8250–8258. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, K.S.; Lee, E.J.; Kim, M.O.; Park, J.H.; Cho, K.I.; Imakawa, K.; Hyun, B.H.; Chang, K.T.; Lee, H.T.; et al. Human keratin 14 driven HPV 16 E6/E7 transgenic mice exhibit hyperkeratinosis. Life Sci. 2004, 75, 3035–3042. [Google Scholar] [CrossRef] [PubMed]
- Brooks, L.A.; Sullivan, A.; O’Nions, J.; Bell, A.; Dunne, B.; Tidy, J.A.; Evans, D.J.; Osin, P.; Vousden, K.H.; Gusterson, B.; et al. E7 proteins from oncogenic human papillomavirus types transactivate p73: Role in cervical intraepithelial neoplasia. Br. J. Cancer 2002, 86, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Pytynia, M.; Pelizzari, C.; Spiotto, M. Bioluminescent imaging of hpv-positive oral tumor growth and its response to image-guided radiotherapy. Cancer Res. 2014, 74, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Sha, W.C.; Nelson, C.A.; Newberry, R.D.; Kranz, D.M.; Russell, J.H.; Loh, D.Y. Selective expression of an antigen receptor on cd8-bearing t lymphocytes in transgenic mice. Nature 1988, 335, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Spiotto, M.T.; Rowley, D.A.; Schreiber, H. Bystander elimination of antigen loss variants in established tumors. Nat. Med. 2004, 10, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Eichten, A.; Rud, D.S.; Grace, M.; Piboonniyom, S.O.; Zacny, V.; Munger, K. Molecular pathways executing the “trophic sentinel” response in HPV-16 E7-expressing normal human diploid fibroblasts upon growth factor deprivation. Virology 2004, 319, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Alani, R.M.; Munger, K. The human papillomavirus E7 oncoprotein can uncouple cellular differentiation and proliferation in human keratinocytes by abrogating p21cip1-mediated inhibition of CDK2. Genes Dev. 1997, 11, 2101–2111. [Google Scholar] [CrossRef] [PubMed]
- Field, S.J.; Tsai, F.Y.; Kuo, F.; Zubiaga, A.M.; Kaelin, W.G., Jr.; Livingston, D.M.; Orkin, S.H.; Greenberg, M.E. E2F-1 functions in mice to promote apoptosis and suppress proliferation. Cell 1996, 85, 549–561. [Google Scholar] [CrossRef]
- Russell, J.L.; Weaks, R.L.; Berton, T.R.; Johnson, D.G. E2F1 suppresses skin carcinogenesis via the Arf-p53 pathway. Oncogene 2006, 25, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Paulson, Q.X.; Pusapati, R.V.; Hong, S.; Weaks, R.L.; Conti, C.J.; Johnson, D.G. Transgenic expression of E2F3a causes DNA damage leading to atm-dependent apoptosis. Oncogene 2008, 27, 4954–4961. [Google Scholar] [CrossRef] [PubMed]
- Lazzerini Denchi, E.; Helin, K. E2F1 is crucial for E2F-dependent apoptosis. EMBO Rep. 2005, 6, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, H.I.; Wu, L.; de Bruin, A.; Timmers, C.; Rosol, T.J.; Weinstein, M.; Robinson, M.L.; Leone, G. Specificity of E2F1, E2F2, and E2F3 in mediating phenotypes induced by loss of Rb. Cell Growth Differ. 2002, 13, 215–225. [Google Scholar] [PubMed]
- Zheng, S.; Moehlenbrink, J.; Lu, Y.C.; Zalmas, L.P.; Sagum, C.A.; Carr, S.; McGouran, J.F.; Alexander, L.; Fedorov, O.; Munro, S.; et al. Arginine methylation-dependent reader-writer interplay governs growth control by E2F-1. Mol. Cell 2013, 52, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Sima, N.; Wang, W.; Kong, D.; Deng, D.; Xu, Q.; Zhou, J.; Xu, G.; Meng, L.; Lu, Y.; Wang, S.; et al. RNA interference against HPV16 E7 oncogene leads to viral E6 and E7 suppression in cervical cancer cells and apoptosis via upregulation of Rb and p53. Apoptosis 2008, 13, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Bargou, R.C.; Wagener, C.; Bommert, K.; Arnold, W.; Daniel, P.T.; Mapara, M.Y.; Grinstein, E.; Royer, H.D.; Dorken, B. Blocking the transcription factor E2F/dp by dominant-negative mutants in a normal breast epithelial cell line efficiently inhibits apoptosis and induces tumor growth in scid mice. J. Exp. Med. 1996, 183, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Leone, G.; Sears, R.; Huang, E.; Rempel, R.; Nuckolls, F.; Park, C.H.; Giangrande, P.; Wu, L.; Saavedra, H.I.; Field, S.J.; et al. Myc requires distinct E2F activities to induce S phase and apoptosis. Mol. Cell 2001, 8, 105–113. [Google Scholar] [CrossRef]
- Vasioukhin, V.; Degenstein, L.; Wise, B.; Fuchs, E. The magical touch: Genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc. Natl. Acad. Sci. USA 1999, 96, 8551–8556. [Google Scholar] [CrossRef] [PubMed]
- Badea, T.C.; Wang, Y.; Nathans, J. A noninvasive genetic/pharmacologic strategy for visualizing cell morphology and clonal relationships in the mouse. J. Neurosci. 2003, 23, 2314–2322. [Google Scholar] [PubMed]
- Dassule, H.R.; Lewis, P.; Bei, M.; Maas, R.; McMahon, A.P. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development 2000, 127, 4775–4785. [Google Scholar] [PubMed]
- Postic, C.; Shiota, M.; Niswender, K.D.; Jetton, T.L.; Chen, Y.; Moates, J.M.; Shelton, K.D.; Lindner, J.; Cherrington, A.D.; Magnuson, M.A. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using cre recombinase. J. Biol. Chem. 1999, 274, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, S.R.; Petricoin, E.F.; Maitra, A.; Rajapakse, V.; King, C.; Jacobetz, M.A.; Ross, S.; Conrads, T.P.; Veenstra, T.D.; Hitt, B.A.; et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 2003, 4, 437–450. [Google Scholar] [CrossRef]
- Schwenk, F.; Baron, U.; Rajewsky, K. A cre-transgenic mouse strain for the ubiquitous deletion of loxp-flanked gene segments including deletion in germ cells. Nucleic Acids Res. 1995, 23, 5080–5081. [Google Scholar] [CrossRef] [PubMed]
- Tronche, F.; Kellendonk, C.; Kretz, O.; Gass, P.; Anlag, K.; Orban, P.C.; Bock, R.; Klein, R.; Schutz, G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat. Genet. 1999, 23, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, L.; Theis, M.; Alvarez-Maya, I.; Brenner, M.; Willecke, K.; Messing, A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis 2001, 31, 85–94. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, R.; Bechill, J.; Spiotto, M.T. Loss of E2F1 Extends Survival and Accelerates Oral Tumor Growth in HPV-Positive Mice. Cancers 2015, 7, 2372-2385. https://doi.org/10.3390/cancers7040895
Zhong R, Bechill J, Spiotto MT. Loss of E2F1 Extends Survival and Accelerates Oral Tumor Growth in HPV-Positive Mice. Cancers. 2015; 7(4):2372-2385. https://doi.org/10.3390/cancers7040895
Chicago/Turabian StyleZhong, Rong, John Bechill, and Michael T. Spiotto. 2015. "Loss of E2F1 Extends Survival and Accelerates Oral Tumor Growth in HPV-Positive Mice" Cancers 7, no. 4: 2372-2385. https://doi.org/10.3390/cancers7040895