
Hedgehog Signaling in the Maintenance of Cancer Stem Cells

Abstract
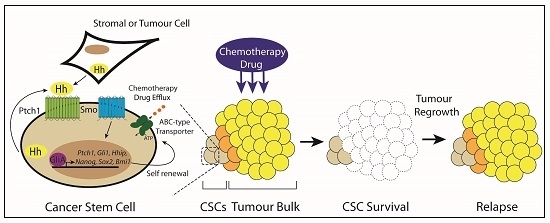
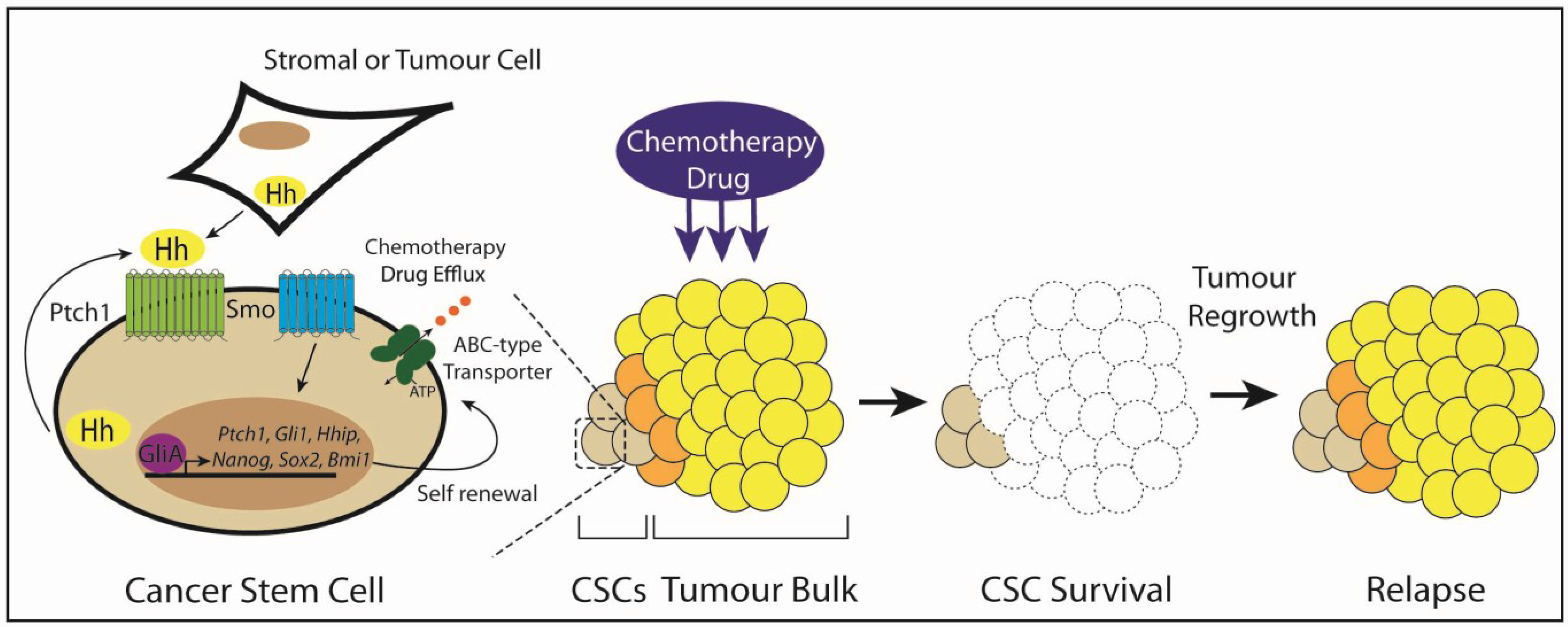
:
1. Introduction
2. The Hedgehog Signaling Network
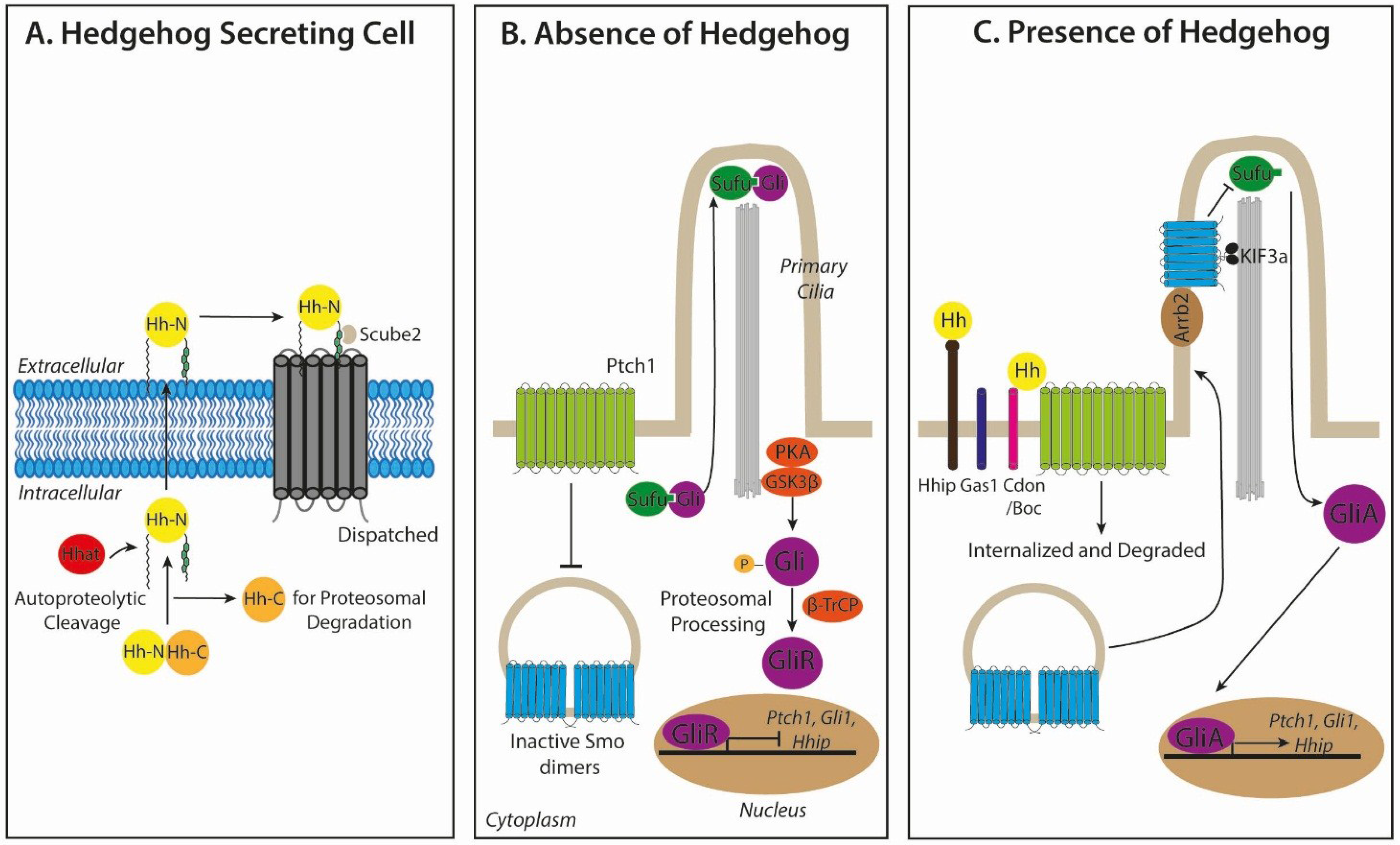
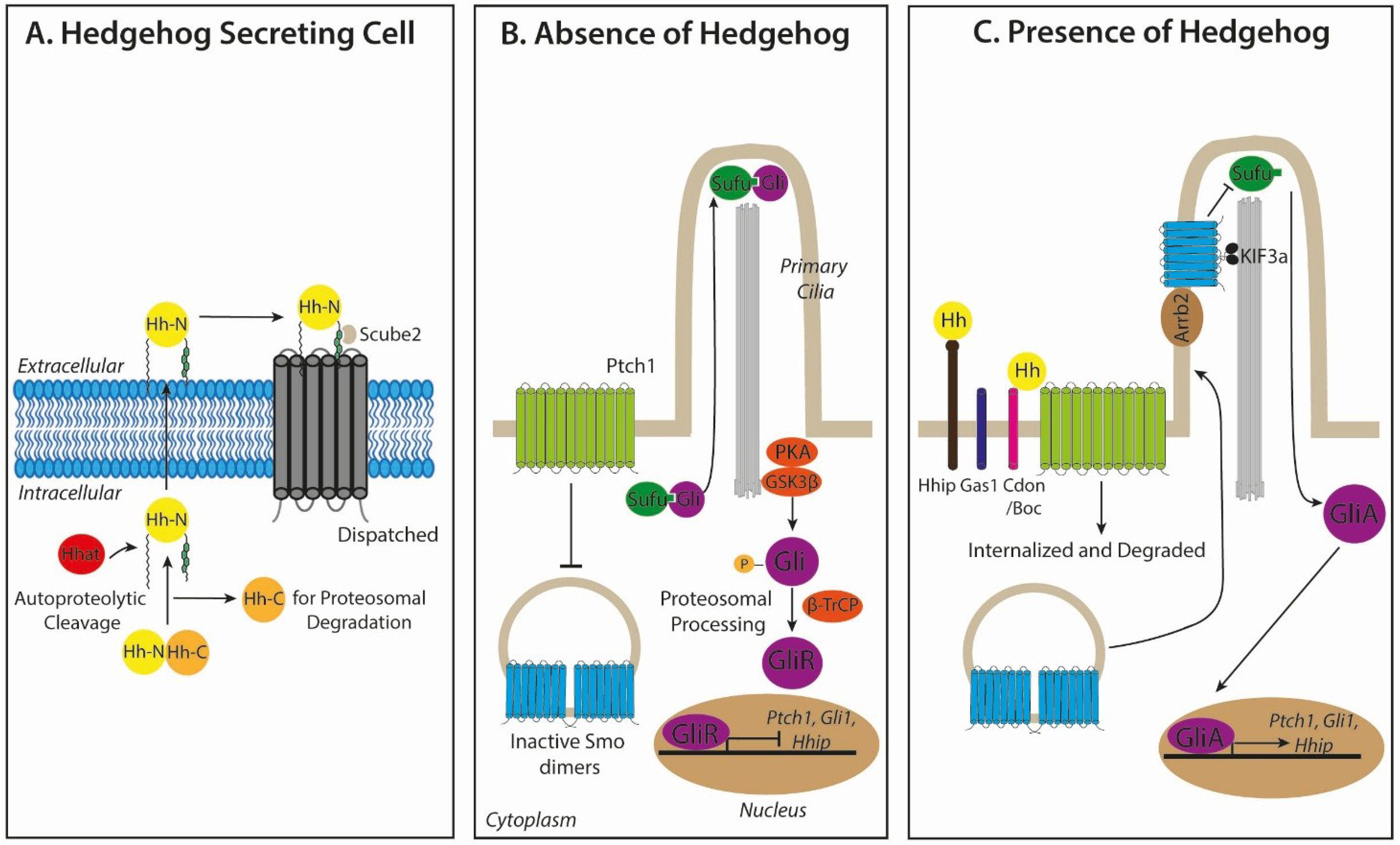
2.1. Hedgehog Biogenesis and Secretion
2.2. Hedgehog Signal Transduction
3. Roles for Hedgehog Signaling in Cancer
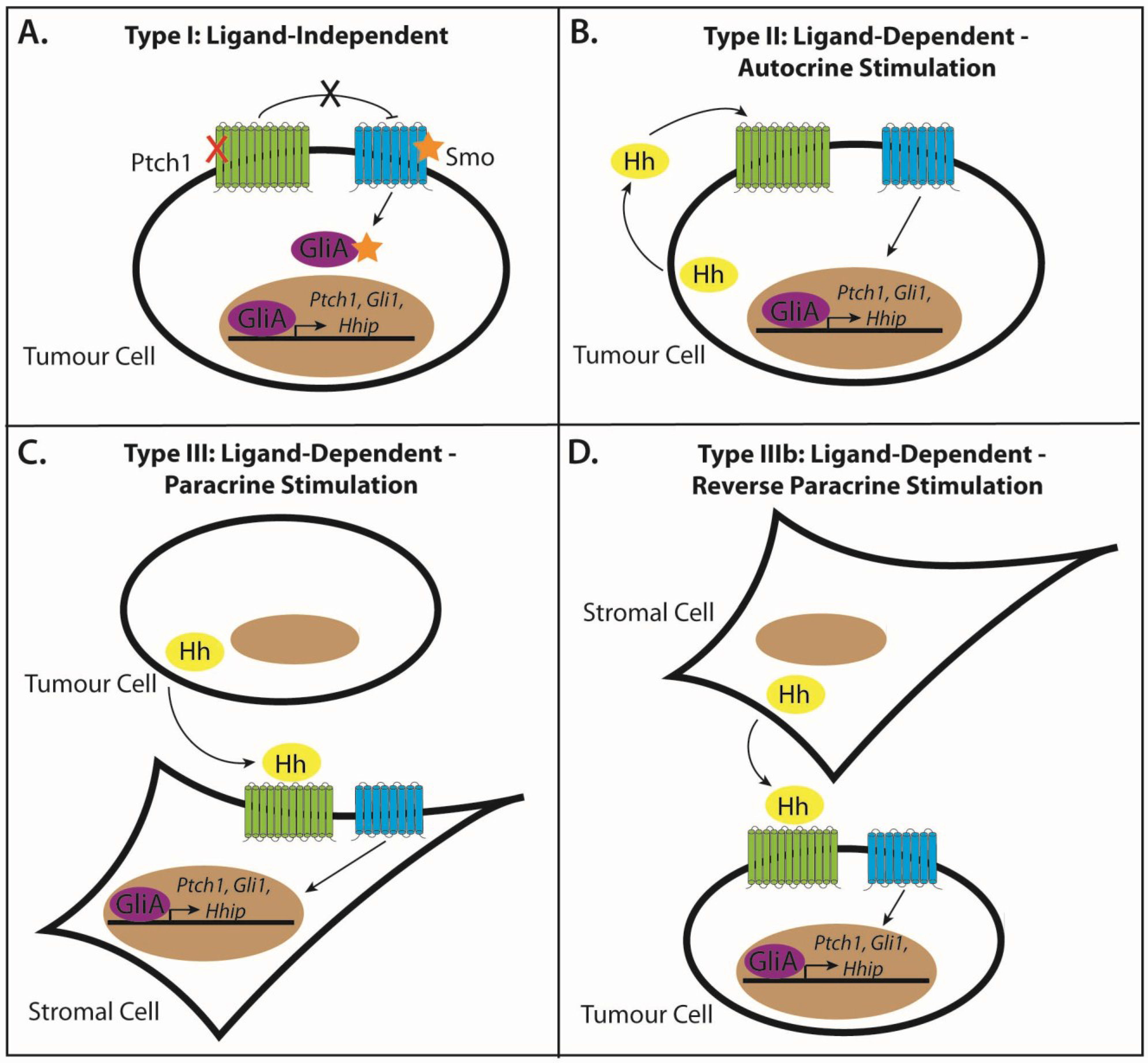
3.1. Modes of Signaling in Hh-Pathway-Dependent Cancers
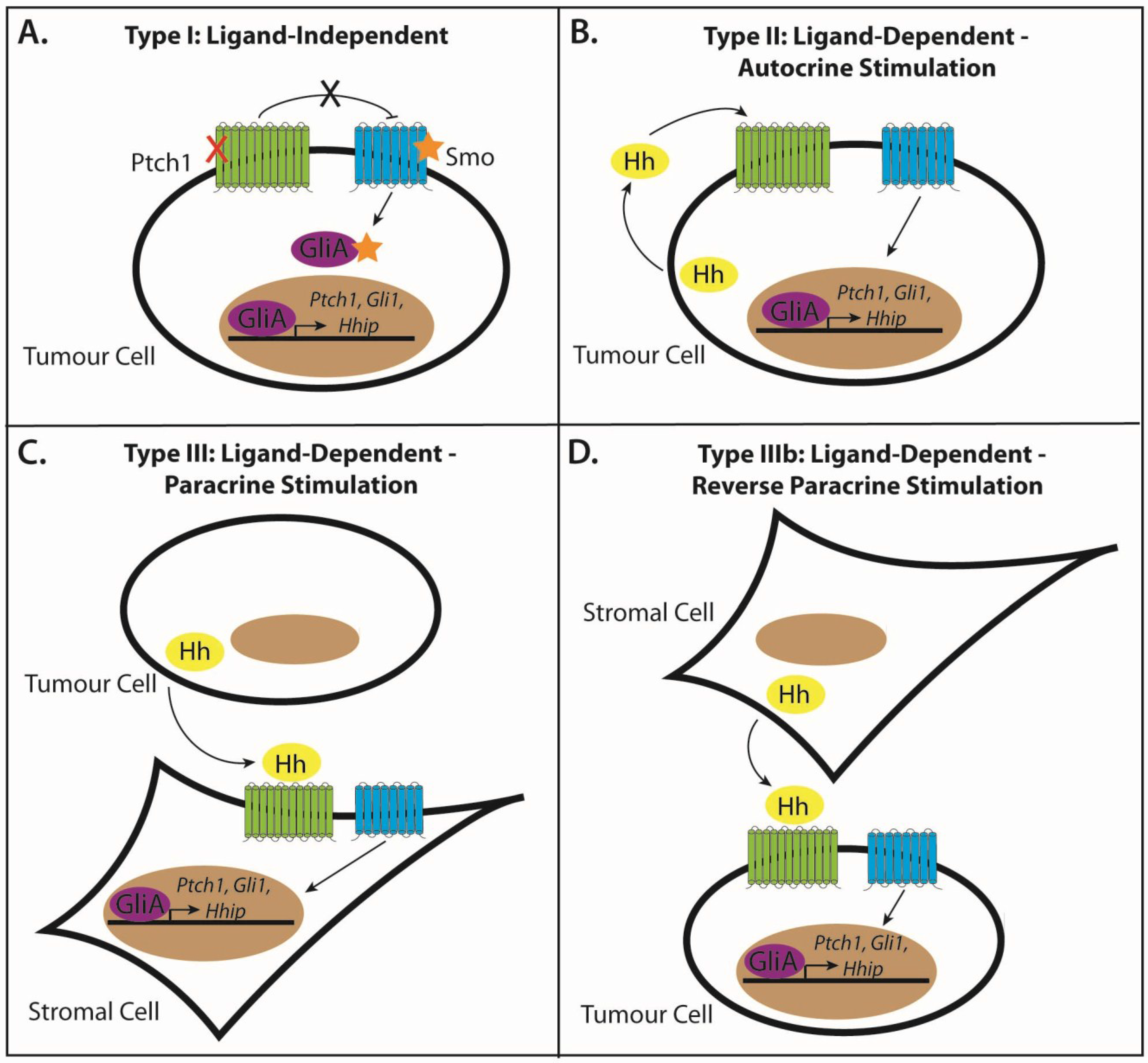
3.2. Type I: Ligand-Independent, Tumour Cell-Intrinsic Signaling
3.3. Type II: Ligand-Dependent, Autocrine Signaling
3.4. Type III: Ligand-Dependent, Paracrine Signaling
3.5. Type IIIb: Ligand-Dependent, Reverse Paracrine Signaling
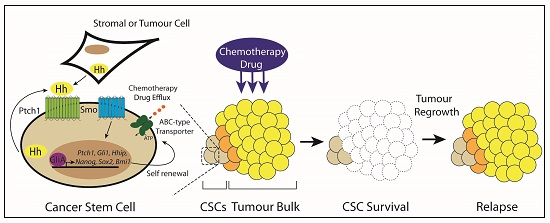
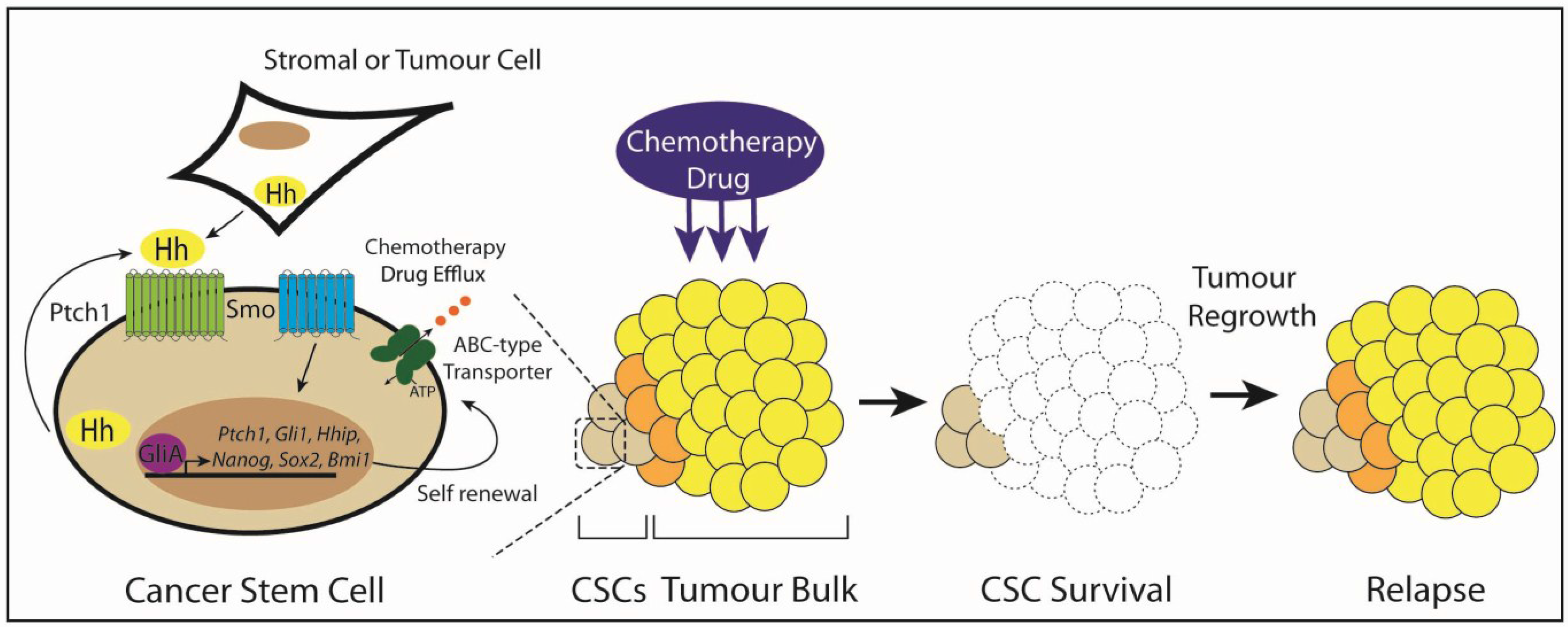
3.6. Type IV: Cancer Stem Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumour Type | CSC Marker | Stemness Genes Expressed | Mode of Hedgehog Inhibition | Experimental Evidence | Combination Therapy | Refs. |
|---|---|---|---|---|---|---|
| Chronic Myeloid Leukaemia (CML) | CD34+, Lin−, Sca+, cKit+ | - | Cyclopamine, Bcr-Abl infected Smo−/− embryonic liver cells, Smo KO in CML mouse model, PF-04449913 | 14-fold reduction in CML LSCs, 60% of mice survived after 7 weeks | Cyclopamine and nilotinib, PF-04449913 and dasatinib | [104,105,106,107,108] |
| Acute Myeloid Leukaemia (AML) | - | - | IPI-926, PF-04449913, Cyclopamine, Endogenous Hhip, 5E1 | Inhibits self-renewal and promotes myelomonocytic differentiation | Sorafenib and IPI-926, cyclopamine or Hhip or 5E1 and cytarabine | [109,110,111,112] |
| Acute Lymphoblastic Leukaemia(ALL) | - | - | Cyclopamine, IPI-926, KAAD-cyclopamine, SANT-1 | Reduces long-term self-renewal in B-ALL, promotes apoptosis in T-ALL | - | [113,114] |
| Multiple Myeloma | CD138neg, CD19+ | - | Cyclopamine, 5E1 | Reduces CD138neg self-renewal by inducing plasma cell differentiation | - | [98] |
| Glioma | CD133+, ALDH1+, ABCG2+ | NANOG, OCT4, SOX2, NESTIN, BMI1 | Cyclopamine | Abolishes tumour engraftment | Cyclopamine, temozolomide and/or 10 Gys of radiation | [101,115,116,117] |
| Breast Cancer | CD44+, CD24−/low, Lin−, ALDH1+ | p63, OCT4, NESTIN, NANOG, BMI1 | Cyclopamine | Reduces mammosphere self-renewal and secondary formation | - | [118,119,120] |
| Small Cell Lung Cancer | - | BMP4, NESTIN, ASH-1 | Cyclopamine, LDE-225, shSMO, 5E1 | Prevents tumour relapse in LX22 xenografts | LDE-225, carboplatin and etoposide or, GDC-0449 and cisplatin | [82,121,122] |
| Non-Small Cell Lung Cancer | - | SOX2, OCT4, NANOG, ALDHA1 | siSHH GDC-0449 | Decreases colony formation and growth in soft agar | GDC-0440, erlotinib and cisplatin | [103,122,123] |
| Gastric Cancer | CD44+, CD24+ | SOX2, NANOG | Cyclopamine, Vismodegib, 5E1, shSMO | Reduces CD44+ tumourspheres and number and diameter of colonies | Vismodegib, 5-flurouracil and/or cisplatin or cyclopamine, oxaliplatin and mitomycin | [124,125] |
| Colon Cancer | CD133+ | NANOG, OCT4 | shSMO Cyclopamine | Reduction of the CD133+ CSC population | - | [83,102] |
| Pancreatic Cancer | CD44+, CD24+, ESA+ | NANOG, OCT4 | GDC-0449, Cyclopamine derivative - CyT | Reduces tumoursphere viability and chemoresistance | CyT and 2 Gys of radiation | [86,126,127,128,129,130,131,132] |
| Prostate Cancer | - | NANOG, OCT4 | Cyclopamine, shGLI1,2, GANT61 | Suppresses tumoursphere and colony formation | Cyclopamine and paclitaxel | [133,134,135,136] |
| Metastatic Melanoma | ALDH+ | SOX2, NANOG, OCT4, KLF4 | shSMO, shGLI1 | Reduces ALDH+ melanospheres fraction, clonogenicity and xenograft growth | - | [137,138,139] |
4. Evidence for the Role of Hedgehog Signaling in Cancer Stem Cell Maintenance
4.1. Leukemic Stem Cells
4.2. Chronic Myeloid Leukaemia
4.3. Acute Myeloid Leukaemia
4.4. Acute Lymphoblastic Leukaemia
4.5. Multiple Myeloma
4.6. Glioma
4.7. Breast Cancer
4.8. Gastrointestinal Cancers
4.9. Pancreatic Cancer
4.10. Prostate Cancer
4.11. Lung Cancer
4.12. Melanoma
5. Targeting Hedgehog Signaling in Cancer Stem Cells
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar]
- Karamboulas, C.; Ailles, L. Developmental signaling pathways in cancer stem cells of solid tumors. Biochim. Biophys. Acta—Gen. Subj. 2013, 1830, 2481–2495. [Google Scholar]
- O’Brien, C.A.; Kreso, A.; Jamieson, C.H.M. Cancer stem cells and self-renewal. Clin. Cancer Res. 2010, 16, 3113–3120. [Google Scholar] [PubMed]
- Chen, K.; Huang, Y.-H.; Chen, J.-L. Understanding and targeting cancer stem cells: Therapeutic implications and challenges. Acta Pharmacol. Sin. 2013, 34, 732–740. [Google Scholar] [PubMed]
- Medema, J.P. Cancer stem cells: The challenges ahead. Nat. Cell Biol. 2013, 15, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Warren, R.Q.; Ivy, S.P. Breast cancer growth and metastasis: interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 2011, 13, 211. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.P.; Ingham, P.W.; Tabin, C.J. Developmental roles and clinical significance of Hedgehog signaling. Curr. Top. Dev. Biol. 2003, 53, 1–114. [Google Scholar] [PubMed]
- Van Den Brink, G.R.; Peppelenbosch, M.P. Expression of hedgehog pathway components in the adult colon. Gastroenterology 2006, 130, 619. [Google Scholar] [CrossRef] [PubMed]
- Wicking, C.; McGlinn, E. The role of hedgehog signalling in tumorigenesis. Cancer Lett. 2001, 173, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.-C.; Levine, C.M.; Zahid, S.; Wilson, E.L.; Joyner, A.L. Sonic hedgehog signals to multiple prostate stromal stem cells that replenish distinct stromal subtypes during regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 20611–20616. [Google Scholar] [CrossRef] [PubMed]
- Ihrie, R.A.; Shah, J.K.; Harwell, C.C.; Levine, J.H.; Guinto, C.D.; Lezameta, M.; Kriegstein, A.R.; Alvarez-Buylla, A. Persistent sonic hedgehog signaling in adult brain determines neural stem cell positional identity. Neuron 2011, 71, 250–262. [Google Scholar] [CrossRef] [PubMed]
- Perler, F.B. Protein splicing of inteins and hedgehog autoproteolysis: Structure, function, and evolution. Cell 1998, 92, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Tukachinsky, H.; Huang, C.H.; Jao, C.; Chu, Y.R.; Tang, H.Y.; Mueller, B.; Schulman, S.; Rapoport, T.A.; Salic, A. Processing and turnover of the Hedgehog protein in the endoplasmic reticulum. J. Cell Biol. 2011, 192, 825–838. [Google Scholar] [CrossRef] [PubMed]
- Chamoun, Z.; Mann, R.K.; Nellen, D.; von Kessler, D.P.; Bellotto, M.; Beachy, P.A.; Basler, K. Skinny hedgehog, an acyltransferase required for palmitoylation and activity of the hedgehog signal. Science 2001, 293, 2080–2084. [Google Scholar] [CrossRef] [PubMed]
- Pepinsky, R.B.; Zeng, C.; Went, D.; Rayhorn, P.; Baker, D.P.; Williams, K.P.; Bixler, S.A.; Ambrose, C.M.; Garber, E.A.; Miatkowski, K.; et al. Identification of a palmitic acid-modified form of human Sonic hedgehog. J. Biol. Chem. 1998, 273, 14037–14045. [Google Scholar] [CrossRef] [PubMed]
- Taylor, F.R.; Wen, D.; Garber, E.A.; Carmillo, A.N.; Baker, D.P.; Arduini, R.M.; Williams, K.P.; Weinreb, P.H.; Rayhorn, P.; Hronowski, X.; et al. Enhanced potency of human Sonic hedgehog by hydrophobic modification. Biochemistry 2001, 40, 4359–4371. [Google Scholar] [CrossRef] [PubMed]
- Callejo, A.; Torroja, C.; Quijada, L.; Guerrero, I. Hedgehog lipid modifications are required for Hedgehog stabilization in the extracellular matrix. Development 2006, 133, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Tukachinsky, H.; Kuzmickas Ryan, P.; Jao, C.Y.; Liu, J.; Salic, A. Dispatched and scube mediate the efficient secretion of the cholesterol-modified hedgehog ligand. Cell Rep. 2012, 2, 308–320. [Google Scholar] [CrossRef] [PubMed]
- Amanai, K.; Jiang, J. Distinct roles of central missing and dispatched in sending the Hedgehog signal. Development 2001, 128, 5119–5127. [Google Scholar] [PubMed]
- Chen, M.-H.; Li, Y.-J.; Kawakami, T.; Xu, S.-M.; Chuang, P.-T. Palmitoylation is required for the production of a soluble multimeric Hedgehog protein complex and long-range signaling in vertebrates. Genes Dev. 2004, 18, 641–659. [Google Scholar] [CrossRef] [PubMed]
- Goetz, J.A.; Singh, S.; Suber, L.M.; Kull, F.J.; Robbins, D.J. A highly conserved amino-terminal region of sonic hedgehog is required for the formation of its freely diffusible multimeric form. J. Biol. Chem. 2006, 281, 4087–4093. [Google Scholar] [CrossRef] [PubMed]
- Gallet, A.; Rodriguez, R.; Ruel, L.; Therond, P.P. Cholesterol modification of hedgehog is required for trafficking and movement, revealing an asymmetric cellular response to hedgehog. Dev. Cell 2003, 4, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Bishop, B.; Aricescu, A.R.; Harlos, K.; O’Callaghan, C.A.; Jones, E.Y.; Siebold, C. Structural insights into hedgehog ligand sequestration by the human hedgehog-interacting protein HHIP. Nat. Struct. Mol. Biol. 2009, 16, 698–703. [Google Scholar] [CrossRef] [PubMed]
- Bosanac, I.; Maun, H.R.; Scales, S.J.; Wen, X.; Lingel, A.; Bazan, J.F.; de Sauvage, F.J.; Hymowitz, S.G.; Lazarus, R.A. The structure of SHH in complex with HHIP reveals a recognition role for the Shh pseudo active site in signaling. Nat. Struct. Mol. Biol. 2009, 16, 691–697. [Google Scholar] [CrossRef] [PubMed]
- Capurro, M.I.; Shi, W.; Filmus, J. LRP1 mediates Hedgehog-induced endocytosis of the GPC3-Hedgehog complex. J. Cell Sci. 2012, 125, 3380–3389. [Google Scholar] [CrossRef] [PubMed]
- Capurro, M.I.; Xu, P.; Shi, W.; Li, F.; Jia, A.; Filmus, J. Glypican-3 inhibits hedgehog signaling during development by competing with patched for hedgehog binding. Dev. Cell 2008, 14, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.-C.; Mulloy, B.; Magee, A.I.; Couchman, J.R. Two distinct sites in sonic hedgehog combine for heparan sulfate interactions and cell signaling functions. J. Biol. Chem. 2011, 286, 44391–44402. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Struhl, G. Dual roles for patched in sequestering and transducing Hedgehog. Cell 1996, 87, 553–563. [Google Scholar] [CrossRef]
- Briscoe, J.; Chen, Y.; Jessell, T.M.; Struhl, G. A hedgehog-insensitive form of Patched provides evidence for direct long-range morphogen activity of Sonic hedgehog in the neural tube. Mol. Cell 2001, 7, 1279–1291. [Google Scholar] [CrossRef]
- Allen, B.; Song, J.; Izzi, L.; Althaus, I.; Kang, J.S.; Charron, F.; Krauss, R.; McMahon, A. Overlapping roles and collective requirement for the coreceptors GAS1, CDO, and BOC in SHH pathway function. Dev. Cell 2011, 20, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Izzi, L.; Lévesque, M.; Morin, S.; Laniel, D.; Wilkes, B.C.; Mille, F.; Krauss, R.S.; McMahon, A.P.; Allen, B.L.; Charron, F. Boc and Gas1 each form distinct Shh receptor complexes with Ptch1 and are required for Shh-mediated cell proliferation. Dev. Cell 2011, 20, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Huangfu, D.; Liu, A.; Rakeman, A.S.; Murcia, N.S.; Niswander, L.; Anderson, K.V. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature 2003, 426, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, R.; Milenkovic, L.; Corcoran, R.B.; Scott, M.P. Hedgehog signal transduction by Smoothened: Pharmacologic evidence for a 2-step activation process. Proc. Natl. Acad. Sci. USA 2009, 106, 3196–3201. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Tong, C.; Jiang, J. Hedgehog regulates smoothened activity by inducing a conformational switch. Nature 2007, 450, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kato, M.; Beachy, P.A. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc. Natl. Acad. Sci. USA 2009, 106, 21666–21671. [Google Scholar] [CrossRef] [PubMed]
- Barnfield, P.C.; Zhang, X.; Thanabalasingham, V.; Yoshida, M.; Hui, C.-C. Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple mechanisms. Differentiation 2005, 73, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Goetz, S.C.; Anderson, K.V. The primary cilium: A signalling centre during vertebrate development. Nat. Rev. Genet. 2010, 11, 331–344. [Google Scholar] [CrossRef] [PubMed]
- Niewiadomski, P.; Kong, J.H.; Ahrends, R.; Ma, Y.; Humke, E.W.; Khan, S.; Teruel, M.N.; Novitch, B.G.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate Hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Zhang, L.; Zhang, Q.; Tong, C.; Wang, B.; Hou, F.; Amanai, K.; Jiang, J. Phosphorylation by double-time/CKIε and CKIα targets cubitus interruptus for Slimb/β-TRCP-mediated proteolytic processing. Dev. Cell 2005, 9, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Shi, Q.; Chen, Y.; Yue, T.; Li, S.; Wang, B.; Jiang, J. Multiple Ser/Thr-rich degrons mediate the degradation of Ci/Gli by the Cul3-HIB/SPOP E3 ubiquitin ligase. Proc. Natl. Acad. Sci. USA 2009, 106, 21191–21196. [Google Scholar] [CrossRef] [PubMed]
- Ruiz I Altaba, A.; Palma, V.; Dahmane, N. Hedgehog-Gli signalling and the growth of the brain. Nat. Rev. Neurosci. 2002, 3, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sasai, N.; Ma, G.; Yue, T.; Jia, J.; Briscoe, J.; Jiang, J. Sonic hedgehog dependent phosphorylation by CK1α and GRK2 is required for ciliary accumulation and activation of smoothened. PLoS Biol. 2011, 9, e1001083. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Ren, X.R.; Nelson, C.D.; Barak, L.S.; Chen, J.K.; Beachy, P.A.; de Sauvage, F.; Lefkowitz, R.J. Activity-dependent internalization of smoothened mediated by β-Arrestin 2 and GRK2. Science 2004, 306, 2257–2260. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Wang, B.; Niswander, L.A. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development 2005, 132, 3103–3111. [Google Scholar] [CrossRef] [PubMed]
- Vokes, S.A.; Ji, H.; Wong, W.H.; McMahon, A.P. A genome-scale analysis of the cis-regulatory circuitry underlying sonic hedgehog-mediated patterning of the mammalian limb. Genes Dev. 2008, 22, 2651–2663. [Google Scholar] [CrossRef] [PubMed]
- Okolowsky, N.; Furth, P.A.; Hamel, P.A. Oestrogen receptor-alpha regulates non-canonical Hedgehog-signalling in the mammary gland. Dev. Biol. 2014, 391, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gradilone, S.A.; Smoot, R.L.; Mertens, J.C.; Bronk, S.F.; Sirica, A.E.; Gores, G.J. Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J. Hepatol. 2014, 60, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D. Hedgehog signalling: Emerging evidence for non-canonical pathways. Cell. Signal. 2009, 21, 1023–1034. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Li, Q.; Moraes, R.C.; Lewis, M.T.; Hamel, P.A. Activation of Erk by sonic hedgehog independent of canonical hedgehog signalling. Int. J. Biochem. Cell Biol. 2010, 42, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Mille, F.; Thibert, C.; Fombonne, J.; Rama, N.; Guix, C.; Hayashi, H.; Corset, V.; Reed, J.C.; Mehlen, P. The Patched dependence receptor triggers apoptosis through a DRAL-caspase-9 complex. Nat. Cell Biol. 2009, 11, 739–746. [Google Scholar] [CrossRef] [PubMed]
- Thibert, C.; Teillet, M.A.; Lapointe, F.; Mazelin, L.; Le Douarin, N.M.; Mehlen, P. Inhibition of neuroepithelial patched-induced apoptosis by Sonic hedgehog. Science 2003, 301, 843–846. [Google Scholar] [CrossRef] [PubMed]
- Barnes, E.A.; Kong, M.; Ollendorff, V.; Donoghue, D.J. Patched1 interacts with cyclin B1 to regulate cell cycle progression. EMBO J. 2001, 20, 2214–2223. [Google Scholar] [CrossRef] [PubMed]
- Adolphe, C.; Hetherington, R.; Ellis, T.; Wainwright, B. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 2006, 66, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.; Winyard, P.J.D.; Woolf, A.S. Immunohistochemical analysis of Sonic hedgehog signalling in normal human urinary tract development. J. Anat. 2007, 211, 620–629. [Google Scholar] [CrossRef] [PubMed]
- Polizio, A.H.; Chinchilla, P.; Chen, X.; Kim, S.; Manning, D.R.; Riobo, N.A. Heterotrimeric Gi proteins link hedgehog signaling to activation of Rho small GTPases to promote fibroblast migration. J. Biol. Chem. 2011, 286, 19589–19596. [Google Scholar] [CrossRef] [PubMed]
- Chinchilla, P.; Xiao, L.; Kazanietz, M.G.; Riobo, N.A. Hedgehog proteins activate pro-angiogenic responses in endothelial cells through non-canonical signaling pathways. Cell Cycle 2010, 9, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Bijlsma, M.F.; Borensztajn, K.S.; Roelink, H.; Peppelenbosch, M.P.; Spek, C.A. Sonic hedgehog induces transcription-independent cytoskeletal rearrangement and migration regulated by arachidonate metabolites. Cell. Signal. 2007, 19, 2596–2604. [Google Scholar] [CrossRef] [PubMed]
- Yam, P.T.; Langlois, S.D.; Morin, S.; Charron, F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron 2009, 62, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Belgacem, Y.H.; Borodinsky, L.N. Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. Proc. Natl. Acad. Sci. 2011, 108, 4482–4487. [Google Scholar] [CrossRef] [PubMed]
- Marini, K.D.; Payne, B.J.; Watkins, D.N.; Martelotto, L.G. Mechanisms of Hedgehog signalling in cancer. Growth Factors 2011, 29, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Brennan, D.; Chen, X.; Cheng, L.; Mahoney, M.; Riobo, N.A. Noncanonical hedgehog signaling. Vitam. Horm. 2012, 88, 55–72. [Google Scholar] [PubMed]
- Teglund, S.; Toftgård, R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim. Biophys. Acta (BBA)—Rev. Cancer 2010, 1805, 181–208. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wicking, C.; Zaphiropoulos, P.G.; Gailani, M.R.; Shanley, S.; Chidambaram, A.; Vorechovsky, I.; Holmberg, E.; Unden, A.B.; Gillies, S.; et al. Mutations of the human homolog of drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996, 85, 841–851. [Google Scholar] [CrossRef]
- Johnson, R.L.; Rothman, A.L.; Xie, J.; Goodrich, L.V.; Bare, J.W.; Bonifas, J.M.; Quinn, A.G.; Myers, R.M.; Cox, D.R.; Epstein, E.H., Jr.; et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996, 272, 1668–1671. [Google Scholar] [CrossRef] [PubMed]
- Pietsch, T.; Waha, A.; Koch, A.; Kraus, J.; Albrecht, S.; Tonn, J.; Sörensen, N.; Berthold, F.; Henk, B.; Schmandt, N.; et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res. 1997, 57, 2085–2088. [Google Scholar] [PubMed]
- Taylor, M.D.; Liu, L.; Raffel, C.; Hui, C.C.; Mainprize, T.G.; Zhang, X.; Agatep, R.; Chiappa, S.; Gao, L.; Lowrance, A.; et al. Mutations in SUFU predispose to medulloblastoma. Nat. Genet. 2002, 31, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, L.; Ghiorzo, P.; Nasti, S.; Battistuzzi, L.; Cusano, R.; Marzocchi, C.; Garré, M.L.; Clementi, M.; Bianchi Scarrá, G. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am. J. Med. Genet. Part A 2009, 149, 1539–1543. [Google Scholar] [CrossRef] [PubMed]
- Tostar, U.; Malm, C.J.; Meis-Kindblom, J.M.; Kindblom, L.G.; Toftgård, R.; Undén, A.B. Deregulation of the hedgehog signalling pathway: A possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol. 2006, 208, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [PubMed]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar] [PubMed]
- Kinzler, K.W.; Bigner, S.H.; Bigner, D.D.; Trent, J.M.; Law, M.L.; O’Brien, S.J.; Wong, A.J.; Vogelstein, B. Identification of an amplified, highly expressed gene in a human glioma. Science 1987, 236, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Wetmore, C.; Eberhart, D.E.; Curran, T. Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res. 2001, 61, 513–516. [Google Scholar] [PubMed]
- Barakat, M.T.; Humke, E.W.; Scott, M.P. Learning from Jekyll to control Hyde: Hedgehog signaling in development and cancer. Trends Mol. Med. 2010, 16, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Johnson, R.L.; Zhang, X.; Bare, J.W.; Waldman, F.M.; Cogen, P.H.; Menon, A.G.; Warren, R.S.; Chen, L.C.; Scott, M.P.; et al. Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer Res. 1997, 57, 2369–2372. [Google Scholar] [PubMed]
- Goodrich, L.V.; Milenković, L.; Higgins, K.M.; Scott, M.P. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997, 277, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Hahn, H.; Wojnowski, L.; Zimmer, A.M.; Hall, J.; Miller, G.; Zimmer, A. Rhabdomyosarcomas and radiation hypersensitivity in a mouse model of Gorlin syndrome. Nat. Med. 1998, 4, 619–622. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Guicherit, O.M.; Zaharian, B.I.; Xu, Y.; Chai, L.; Wichterle, H.; Kon, C.; Gatchalian, C.; Porter, J.A.; Rubin, L.L.; et al. Identification of a small molecule inhibitor of the hedgehog signaling pathway: Effects on basal cell carcinoma-like lesions. Proc. Natl. Acad. Sci. USA 2003, 100, 4616–4621. [Google Scholar] [CrossRef] [PubMed]
- Kawahira, H.; Scheel, D.W.; Smith, S.B.; German, M.S.; Hebrok, M. Hedgehog signaling regulates expansion of pancreatic epithelial cells. Dev. Biol. 2005, 280, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Talpale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Duquet, A.; Malerba, M.; Zbinden, M.; Mas, C.; Gervaz, P.; Ruiz I Altaba, A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol. Med. 2009, 1, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Maun, H.R.; Wen, X.; Lingel, A.; de Sauvage, F.J.; Lazarus, R.A.; Scales, S.J.; Hymowitz, S.G. Hedgehog pathway antagonist 5E1 binds hedgehog at the pseudo-active site. J. Biol. Chem. 2010, 285, 26570–26580. [Google Scholar] [CrossRef] [PubMed]
- El Khatib, M.; Kalnytska, A.; Palagani, V.; Kossatz, U.; Manns, M.P.; Malek, N.P.; Wilkens, L.; Plentz, R.R. Inhibition of hedgehog signaling attenuates carcinogenesis in vitro and increases necrosis of cholangiocellular carcinoma. Hepatology 2013, 57, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Wicking, C.; Smyth, I.; Bale, A. The hedgehog signalling pathway in tumorigenesis and development. Oncogene 1999, 18, 7844–7851. [Google Scholar] [CrossRef] [PubMed]
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed]
- Theunissen, J.-W.; de Sauvage, F.J. Paracrine Hedgehog Signaling in Cancer. Cancer Res. 2009, 69, 6007–6010. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Gould, S.E.; Scales, S.J.; Tang, T.; Tian, H.; Ahn, C.P.; Marshall, D.; Fu, L.; Januario, T.; Kallop, D.; et al. A paracrine requirement for hedgehog signalling in cancer. Nature 2008, 455, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Scales, S.J.; de Sauvage, F.J. Mechanisms of Hedgehog pathway activation in cancer and implications for therapy. Trends Pharmacol. Sci. 2009, 30, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Callahan, C.A.; Dupree, K.J.; Darbonne, W.C.; Ahn, C.P.; Scales, S.J.; de Sauvage, F.J. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proc. Natl. Acad. Sci. USA 2009, 106, 4254–4259. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Tang, T.; Eastham-Anderson, J.; Dunlap, D.; Alicke, B.; Nannini, M.; Gould, S.; Yauch, R.; Modrusan, Z.; DuPree, K.J.; et al. Canonical hedgehog signaling augments tumor angiogenesis by induction of VEGF-A in stromal perivascular cells. Proc. Natl. Acad. Sci. USA 2011, 108, 9589–9594. [Google Scholar] [CrossRef] [PubMed]
- Moran, C.M.; Myers, C.T.; Lewis, C.M.; Krieg, P.A. Hedgehog regulates angiogenesis of intersegmental vessels through the VEGF signaling pathway. Dev. Dyn. 2012, 241, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Lindemann, R.K. Stroma-initiated Hedgehog signaling takes center stage in B-cell lymphoma. Cancer Res. 2008, 68, 961–964. [Google Scholar] [CrossRef] [PubMed]
- Dierks, C.; Grbic, J.; Zirlik, K.; Beigi, R.; Englund, N.P.; Guo, G.R.; Veelken, H.; Engelhardt, M.; Mertelsmann, R.; Kelleher, J.F.; et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nat. Med. 2007, 13, 944–951. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef] [PubMed]
- Becher, O.J.; Hambardzumyan, D.; Fomchenko, E.I.; Momota, H.; Mainwaring, L.; Bleau, A.M.; Katz, A.M.; Edgar, M.; Kenney, A.M.; Cordon-Cardo, C.; et al. Gli activity correlates with tumor grade in platelet-derived growth factor-induced gliomas. Cancer Res. 2008, 68, 2241–2249. [Google Scholar] [CrossRef] [PubMed]
- Po, A.; Ferretti, E.; Miele, E.; De Smaele, E.; Paganelli, A.; Canettieri, G.; Coni, S.; di Marcotullio, L.; Biffoni, M.; Massimi, L.; et al. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J. 2010, 29, 2646–2658. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Batsaikhan, B.E.; Yoshikawa, K.; Kurita, N.; Iwata, T.; Takasu, C.; Kashihara, H.; Shimada, M. Cyclopamine decreased the expression of sonic hedgehog and its downstream genes in colon cancer stem cells. Anticancer Res. 2014, 34, 6339–6344. [Google Scholar] [PubMed]
- Justilien, V.; Walsh, M.P.; Ali, S.; Thompson, E.; Murray, N.; Fields, A. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate hedgehog signaling in lung squamous cell carcinoma. Cancer Cell 2014, 25, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Chen, A.; Jamieson, C.H.; Fereshteh, M.; Abrahamsson, A.; Blum, J.; Kwon, H.Y.; Kim, J.; Chute, J.P.; Rizzieri, D.; et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 2009, 458, 776–779. [Google Scholar] [PubMed]
- Dierks, C.; Beigi, R.; Guo, G.-R.; Zirlik, K.; Stegert, M.R.; Manley, P.; Trussell, C.; Schmitt-Graeff, A.; Landwerlin, K.; Veelken, H.; et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on hedgehog pathway activation. Cancer Cell 2008, 14, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Babashah, S.; Sadeghizadeh, M.; Hajifathali, A.; Tavirani, M.R.; Zomorod, M.S.; Ghadiani, M.; Soleimani, M. Targeting of the signal transducer Smo links microRNA-326 to the oncogenic Hedgehog pathway in CD34+ CML stem/progenitor cells. Int. J. Cancer 2013, 133, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, K.C.S.; Ruela-de-Sousa, R.R.; Fuhler, G.M.; Aberson, H.L.; Ferreira, C.V.; Peppelenbosch, M.P.; Spek, C.A. Hedgehog signaling maintains chemoresistance in myeloid leukemic cells. Oncogene 2010, 29, 6314–6322. [Google Scholar] [PubMed]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.-J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.; Gondek, L.; Li, L.; Wang, Q.; Ma, H.; Chang, E.; Huso, D.L.; Foerster, S.; Marchionni, L.; McGovern, K.; et al. Integration of Hedgehog and mutant FLT3 signaling in myeloid leukemia. Sci. Transl. Med. 2015, 7, 291ra296. [Google Scholar]
- Minami, Y.; Fukushima, N.; Kakiuchi, S.; Minami, H.; Naoe, T. Treatment with hedgehog inhibitor PF-913 attenuates leukemia-initiation potential in acute myeloid leukemic cells. Cancer Res. 2014, 74, 1884. [Google Scholar] [CrossRef]
- Takahashi, T.; Kawakami, K.; Mishima, S.; Akimoto, M.; Takenaga, K.; Suzumiya, J.; Honma, Y. Cyclopamine induces eosinophilic differentiation and upregulates CD44 expression in myeloid leukemia cells. Leuk. Res. 2011, 35, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Kobune, M.; Takimoto, R.; Murase, K.; Iyama, S.; Sato, T.; Kikuchi, S.; Kawano, Y.; Miyanishi, K.; Sato, Y.; Niitsu, Y.; et al. Drug resistance is dramatically restored by hedgehog inhibitors in CD34+ leukemic cells. Cancer Sci. 2009, 100, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.L.; Wang, Q.H.; Brown, P.; Peacock, C.; Merchant, A.A.; Brennan, S.; Jones, E.; McGovern, K.; Watkins, D.N.; Sakamoto, K.M.; et al. Self-renewal of acute lymphocytic leukemia cells is limited by the Hedgehog pathway inhibitors cyclopamine and IPI-926. PLoS ONE 2010, 5, e15262. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Badura, S.; Ruthardt, M.; Rieger, M.A.; Ottmann, O.G. Modulation of leukemic stem cell self-renewal and cell fate decisions by inhibition of hedgehog signalling in human acute lymphoblastic leukemia. Blood 2012, 120, s2578. [Google Scholar]
- Bar, E.E.; Chaudhry, A.; Lin, A.; Fan, X.; Schreck, K.; Matsui, W.; Piccirillo, S.; Vescovi, A.L.; DiMeco, F.; Olivi, A.; et al. Cyclopamine-mediated Hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells 2007, 25, 2524–2533. [Google Scholar] [CrossRef] [PubMed]
- Ehtesham, M.; Sarangi, A.; Valadez, J.G.; Chanthaphaychith, S.; Becher, M.W.; Abel, T.W.; Thompson, R.C.; Cooper, M.K. Ligand-dependent activation of the hedgehog pathway in glioma progenitor cells. Oncogene 2007, 26, 5752–5761. [Google Scholar] [CrossRef] [PubMed]
- Morgenroth, A.; Vogg, A.T.J.; Ermert, K.; Zlatopolskiy, B.; Mottaghy, F.M. Hedgehog signaling sensitizes Glioma stem cells to endogenous nano-irradiation. Oncotarget 2014, 5, 5483–5493. [Google Scholar] [PubMed]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef] [PubMed]
- Memmi, E.M.; Sanarico, A.G.; Giacobbe, A.; Peschiaroli, A.; Frezza, V.; Cicalese, A.; Pisati, F.; Tosoni, D.; Zhou, H.; Tonon, G.; et al. p63 sustains self-renewal of mammary cancer stem cells through regulation of Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2015, 112, 3499–3504. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, N.; Huo, Q.; Sun, M.; Dong, L.; Zhang, Y.; Xu, G.; Yang, Q. Huaier aqueous extract inhibits stem-like characteristics of MCF7 breast cancer cells via inactivation of hedgehog pathway. Tumour Biol. 2014, 35, 10805–10813. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Martelotto, L.G.; Peifer, M.; Sos, M.L.; Karnezis, A.N.; Mahjoub, M.R.; Bernard, K.; Conklin, J.F.; Szczepny, A.; Yuan, J.; et al. A crucial requirement for Hedgehog signaling in small cell lung cancer. Nat. Med. 2011, 17, 1504–1508. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Mysliwietz, J.; Ellwart, J.; Gamarra, F.; Huber, R.M.; Bergner, A. Effects of the Hedgehog pathway inhibitor GDC-0449 on lung cancer cell lines are mediated by side populations. Clin. Exp. Med. 2012, 12, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Maitah, M.Y.; Ginnebaugh, K.R.; Li, Y.; Bao, B.; Gadgeel, S.M.; Sarkar, F.H. Inhibition of Hedgehog signaling sensitizes NSCLC cells to standard therapies through modulation of EMT-regulating miRNAs. J. Hematol. Oncol. 2013, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.; Park, D.J.; Schmidt, B.; Thomas, N.J.; Lee, H.J.; Kim, T.S.; Janjigian, Y.Y.; Cohen, D.J.; Yoon, S.S. CD44 expression denotes a subpopulation of gastric cancer cells in which Hedgehog signaling promotes chemotherapy resistance. Clin. Cancer Res. 2014, 20, 3974–3988. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Yue, W.; Wei, B.; Wang, N.; Li, T.; Guan, L.; Shi, S.; Zeng, Q.; Pei, X.; Chen, L. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PLoS ONE 2011, 6, e17687. [Google Scholar] [CrossRef] [PubMed]
- Rodova, M.; Fu, J.; Watkins, D.N.; Srivastava, R.K.; Shankar, S. Sonic hedgehog signaling inhibition provides opportunities for targeted therapy by sulforaphane in regulating pancreatic cancer stem cell self-renewal. PLoS ONE 2012, 7, e46083. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.T.; Zhuan-Sun, Y.X.; Zhuang, Y.Y.; Wei, S.L.; Tang, J.; Chen, W.B.; Zhang, S.N. Inhibition of hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Int. J. Oncol. 2012, 41, 1707–1714. [Google Scholar] [PubMed]
- Gu, D.; Liu, H.; Su, G.H.; Zhang, X.; Chin-Sinex, H.; Hanenberg, H.; Mendonca, M.S.; Shannon, H.E.; Chiorean, E.G.; Xie, J. Combining hedgehog signaling inhibition with focal irradiation on reduction of pancreatic cancer metastasis. Mol. Cancer Ther. 2013, 12, 1038–1048. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: Molecular mechanisms. PLoS ONE 2011, 6, e27306. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; An, Y.; Wei, J.S.; Ji, Z.L.; Lu, Z.P.; Wu, J.L.; Jiang, K.R.; Chen, P.; Xu, Z.K.; Miao, Y. Cyclopamine reverts acquired chemoresistance and down-regulates cancer stem cell markers in pancreatic cancer cell lines. Swiss Medical Weekly 2011, 141, w13208. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.N.; Fu, J.; Nall, D.; Rodova, M.; Shankar, S.; Srivastava, R.K. Inhibition of sonic hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int. J. Cancer 2012, 131, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Han, J.B.; Sang, F.; Chang, J.J.; Hua, Y.Q.; Shi, W.D.; Tang, L.H.; Liu, L.M. Arsenic trioxide inhibits viability of pancreatic cancer stem cells in culture and in a xenograft model via binding to SHH-Gli. OncoTargets Ther. 2013, 6, 1129–1138. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Li, L.; Jiao, M.; Wu, D.; Wu, K.; Li, X.; Zhu, G.; Yang, L.; Wang, X.; Hsieh, J.T.; et al. Genistein inhibits the stemness properties of prostate cancer cells through targeting Hedgehog-Gli1 pathway. Cancer Lett. 2012, 323, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Chitkara, D.; Mehrazin, R.; Behrman, S.W.; Wake, R.W.; Mahato, R.I. Chemoresistance in prostate cancer cells is regulated by miRNAs and Hedgehog pathway. PLoS ONE 2012, 7, e40021. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Domenech, J.; Vidal, S.J.; Rodriguez-Bravo, V.; Castillo-Martin, M.; Quinn, S.; Rodriguez-Barrueco, R.; Bonal, D.; Charytonowicz, E.; Gladoun, N.; de la Iglesia-Vicente, J.; et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell 2012, 22, 373–388. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Chen, B.Y.; Wu, C.Y.; Tsao, Z.J.; Chen, Y.Y.; Chang, C.P.; Yang, C.R.; Lin, D.P.C. Hedgehog overexpression leads to the formation of prostate cancer stem cells with metastatic property irrespective of androgen receptor expression in the mouse model. J. Biomed. Sci. 2011, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Santini, R.; Vinci, M.C.; Pandolfi, S.; Penachioni, J.Y.; Montagnani, V.; Olivito, B.; Gattai, R.; Pimpinelli, N.; Gerlini, G.; Borgognoni, L.; et al. HEDGEHOG-GLI signaling drives self-renewal and tumorigenicity of human melanoma-initiating cells. Stem Cells 2012, 30, 1808–1818. [Google Scholar] [CrossRef] [PubMed]
- Santini, R.; Pietrobono, S.; Pandolfi, S.; Montagnani, V.; D'Amico, M.; Penachioni, J.Y.; Vinci, M.C.; Borgognoni, L.; Stecca, B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene 2014, 33, 4697–4708. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, S.; Montagnani, V.; Penachioni, J.Y.; Vinci, M.C.; Olivito, B.; Borgognoni, L.; Stecca, B. WIP1 phosphatase modulates the Hedgehog signaling by enhancing GLI1 function. Oncogene 2013, 32, 4737–4747. [Google Scholar] [CrossRef] [PubMed]
- Riether, C.; Schurch, C.M.; Ochsenbein, A.F. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ 2015, 22, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Testa, U. Leukemia stem cells. Ann. Hematol. 2011, 90, 245–271. [Google Scholar] [CrossRef] [PubMed]
- Huntly, B.J.P.; Gilliland, D.G. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat. Rev. Cancer 2005, 5, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Goardon, N.; Marchi, E.; Atzberger, A.; Quek, L.; Schuh, A.; Soneji, S.; Woll, P.; Mead, A.; Alford, K.A.; Rout, R.; et al. Coexistence of LMPP-like and GMP-like leukemia stem cells in acute myeloid leukemia. Cancer Cell 2011, 19, 138–152. [Google Scholar] [CrossRef] [PubMed]
- Deininger, M.W.N.; Goldman, J.M.; Melo, J.V. The molecular biology of chronic myeloid leukemia. Blood 2000, 96, 3343–3356. [Google Scholar] [PubMed]
- Mehrotra, B.; George, T.I.; Kavanau, K.; Avet-Loiseau, H.; Moore Ii, D.; Willman, C.L.; Slovak, M.L.; Atwater, S.; Head, D.R.; Pallavicini, M.G. Cytogenetically aberrant cells in the stem cell compartment (CD34+lin-) in acute myeloid leukemia. Blood 1995, 86, 1139–1147. [Google Scholar] [PubMed]
- Haase, D.; Feuring-Buske, M.; Konemann, S.; Fonatsch, C.; Troff, C.; Verbeek, W.; Pekrun, A.; Hiddemann, W.; Wormann, B. Evidence for malignant transformation in acute myeloid leukemia at the level of early hematopoietic stem cells by cytogenetic analysis of CD34+ subpopulations. Blood 1995, 86, 2906–2912. [Google Scholar] [PubMed]
- Quijano, C.A.; Moore Ii, D.; Arthur, D.; Feusner, J.; Winter, S.S.; Pallavicini, M.G. Cytogenetically aberrant cells are present in the CD34+CD33-38-19-marrow compartment in children with acute lymphoblastic leukemia. Leukemia 1997, 11, 1508–1515. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.J.; Najfeld, V.; Hansen, J.A.; Penfold, G.K.; Jacobson, R.J.; Fialkow, P.J. Involvement of the B-lymphoid system in chronic myelogenous leukaemia. Nature 1980, 287, 49–50. [Google Scholar] [CrossRef] [PubMed]
- Passegué, E.; Wagner, E.F.; Weissman, I.L. JunB deficiency leads to a myeloproliferative disorder arising from hematopoietic stem cells. Cell 2004, 119, 431–443. [Google Scholar] [CrossRef] [PubMed]
- Taussig, D.C.; Vargaftig, J.; Miraki-Moud, F.; Griessinger, E.; Sharrock, K.; Luke, T.; Lillington, D.; Oakervee, H.; Cavenagh, J.; Agrawal, S.G.; et al. Leukemia-initiating cells from some acute myeloid leukemia patients with mutated nucleophosmin reside in the CD34-fraction. Blood 2010, 115, 1976–1984. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, K.; Ayton, P.M.; Carapeti, M.; Kutok, J.L.; Snyder, C.S.; Williams, I.R.; Cross, N.C.P.; Glass, C.K.; Cleary, M.L.; Gilliland, D.G. MOZ-TIF2-induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2-mediated recruitment of CBP. Cancer Cell 2003, 3, 259–271. [Google Scholar] [CrossRef]
- Schreiner, S.; Birke, M.; García-Cuéllar, M.P.; Zilles, O.; Greil, J.; Slany, R.K. MLL-ENL causes a reversible and myc-dependent block of myelomonocytic cell differentiation. Cancer Res. 2001, 61, 6480–6486. [Google Scholar] [PubMed]
- Jørgensen, H.G.; Allan, E.K.; Jordanides, N.E.; Mountford, J.C.; Holyoake, T.L. Nilotinib exerts equipotent antiproliferative effects to imatinib and does not induce apoptosis in CD34+ CML cells. Blood 2007, 109, 4016–4019. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Zhu, H.; Zhu, C.; Liu, T.; Meng, W. Activation of the hedgehog pathway in chronic myelogeneous leukemia patients. J. Exp. Clin. Cancer Res.: CR 2011, 30, 8. [Google Scholar] [CrossRef] [PubMed]
- van Rhenen, A.; Moshaver, B.; Kelder, A.; Feller, N.; Nieuwint, A.W.M.; Zweegman, S.; Ossenkoppele, G.J.; Schuurhuis, G.J. Aberrant marker expression patterns on the CD34+CD38-stem cell compartment in acute myeloid leukemia allows to distinguish the malignant from the normal stem cell compartment both at diagnosis and in remission. Leukemia 2007, 21, 1700–1707. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.Y.; Chiu, C.F.; Lin, C.W.; Hsu, N.Y.; Lin, C.L.; Lo, W.J.; Kao, M.C. Differential expression of Sonic hedgehog and Gli1 in hematological malignancies. Leukemia 2007, 22, 226–228. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Graves, S.; Koch, U.; Liu, S.; Jankovic, V.; Buonamici, S.; El Andaloussi, A.; Nimer, S.D.; Kee, B.L.; Taichman, R.; et al. Hedgehog Signaling Is Dispensable for Adult Hematopoietic Stem Cell Function. Cell Stem Cell 2009, 4, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, I.; Stover, E.H.; Cullen, D.E.; Mao, J.; Morgan, K.J.; Lee, B.H.; Kharas, M.G.; Miller, P.G.; Cornejo, M.G.; Okabe, R.; et al. Hedgehog signaling is dispensable for adult murine hematopoietic stem cell function and hematopoiesis. Cell Stem Cell 2009, 4, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Campbell, V.; Copland, M. Hedgehog signaling in cancer stem cells: A focus on hematological cancers. Stem Cells Cloning 2015, 8, 27–38. [Google Scholar] [PubMed]
- Ji, Z.; Mei, F.C.; Johnson, B.H.; Thompson, E.B.; Cheng, X. Protein kinase A, not Epac, suppresses hedgehog activity and regulates glucocorticoid sensitivity in acute lymphoblastic leukemia cells. J. Biol. Chem. 2007, 282, 37370–37377. [Google Scholar] [CrossRef] [PubMed]
- Matsui, W.; Huff, C.A.; Wang, Q.; Malehorn, M.T.; Barber, J.; Tanhehco, Y.; Smith, B.D.; Civin, C.I.; Jones, R.J. Characterization of clonogenic multiple myeloma cells. Blood 2003, 103, 2332–2336. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xu, J.; He, J.; Zheng, Y.; Li, H.; Lu, Y.; Qian, J.; Lin, P.; Weber, D.M.; Yang, J.; et al. A critical role of autocrine sonic hedgehog signaling in human CD138+ myeloma cell survival and drug resistance. Blood 2014, 124, 2061–2071. [Google Scholar] [CrossRef] [PubMed]
- Merchant, A.A.; Matsui, W. Targeting hedgehog—a cancer stem cell pathway. Clin. Cancer Res. 2010, 16, 3130–3140. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, X.; Chen, L.; Du, W.; Cui, Y.; Piao, X.; Li, Y.; Jiang, C. Targeting glioma stem cells via the Hedgehog signaling pathway. Neuroimmunol. Neuroinflamm. 2014, 1, 51–59. [Google Scholar]
- Hermann, P.C.; Bhaskar, S.; Cioffi, M.; Heeschen, C. Cancer stem cells in solid tumors. Semin. Cancer Biol. 2010, 20, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Lewis, M.T.; Ross, S.; Strickland, P.A.; Sugnet, C.W.; Jimenez, E.; Scott, M.P.; Daniel, C.W. Defects in mouse mammary gland development caused by conditional haploinsufficiency of Patched-1. Development 1999, 126, 5181–5193. [Google Scholar] [PubMed]
- Moraes, R.C.; Zhang, X.; Harrington, N.; Fung, J.Y.; Wu, M.F.; Hilsenbeck, S.G.; Allred, D.C.; Lewis, M.T. Constitutive activation of smoothened (SMO) in mammary glands of transgenic mice leads to increased proliferation, altered differentiation and ductal dysplasia. Development 2007, 134, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Jaks, V.; Fiaschi, M.; Toftgård, R. Hedgehog signalling in breast cancer. Carcinogenesis 2009, 30, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, C.; He, F.; Cai, Y.; Yang, H. Identification of CD44+CD24+ gastric cancer stem cells. J. Cancer Res. Clin. Oncol. 2011, 137, 1679–1686. [Google Scholar] [CrossRef] [PubMed]
- Humphries, A.; Wright, N.A. Colonic crypt organization and tumorigenesis. Nat. Rev. Cancer 2008, 8, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Lee, C.J.; Simeone, D.M. Identification of human pancreatic cancer stem cells. Methods Mol. Biol. 2009, 568, 161–173. [Google Scholar] [PubMed]
- Kelleher, F.C. Hedgehog signaling and therapeutics in pancreatic cancer. Carcinogenesis 2011, 32, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Onishi, H.; Katano, M. Hedgehog signaling pathway as a new therapeutic target in pancreatic cancer. World J. Gastroenterol. 2014, 20, 2335–2342. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; Roy, I.; Anchoori, R.K.; Fazli, S.; Maitra, A.; Beachy, P.A.; Khan, S.R. Targeted inhibition of hedgehog signaling by cyclopamine prodrugs for advanced prostate cancer. Bioorg. Med. Chem. 2008, 16, 2764–2768. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Cozzi, P.; Hao, J.; Duan, W.; Graham, P.; Kearsley, J.; Li, Y. Cancer stem cells in prostate cancer chemoresistance. Curr. Cancer Drug Targets 2014, 14, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.N.; Berman, D.M.; Baylin, S.B. Hedgehog signaling: progenitor phenotype in small-cell lung cancer. Cell cycle 2003, 2, 196–198. [Google Scholar] [CrossRef] [PubMed]
- Daniel, V.C.; Peacock, C.D.; Watkins, D.N. Developmental signalling pathways in lung cancer. Respirology 2006, 11, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Justilien, V.; Fields, A.P. Molecular pathways: Novel approaches for improved therapeutic targeting of hedgehog signaling in cancer stem cells. Clin. Cancer Res. 2015, 21, 505–513. [Google Scholar] [PubMed]
- Schatton, T.; Frank, M.H. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res. 2008, 21, 39–55. [Google Scholar] [PubMed]
- O’Reilly, K.E.; de Miera, E.V.S.; Segura, M.F.; Friedman, E.; Poliseno, L.; Han, S.W.; Zhong, J.; Zavadil, J.; Pavlick, A.; Hernando, E.; et al. Hedgehog pathway blockade inhibits melanoma cell growth in vitro and in vivo. Pharmaceuticals 2013, 6, 1429–1450. [Google Scholar] [PubMed]
- Sun, G.G.; Wang, Y.D.; Liu, Q.; Hu, W.N. Expression of Wip1 in kidney carcinoma and its correlation with tumor metastasis and clinical significance. Pathol. Oncol. Res. 2015, 21, 219–224. [Google Scholar] [PubMed]
- Yang, D.H.; He, J.A.; Li, J.; Ma, W.F.; Hu, X.H.; Xin, S.J.; Duan, Z.Q. Expression of proto-oncogene Wip1 in breast cancer and its clinical significance. Natl. Med. J. China 2010, 90, 519–522. [Google Scholar]
- Harrison, M.; Li, J.; Degenhardt, Y.; Hoey, T.; Powers, S. Wip1-deficient mice are resistant to common cancer genes. Trends Mol. Med. 2004, 10, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.T.; Zhang, L.; Gao, X.Z.; Jiang, X.H.; Sun, L.Q. Expression and significance of the Wip1 proto-oncogene in colorectal cancer. Asian Pac. J. Cancer Prev. 2013, 14, 1975–1979. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Qi, L.; Han, W.; Wan, X.; Jiang, S.; Li, Y.; Xie, Y.; Liu, L.; Zeng, F.; Liu, Z.; et al. Overexpression of Wip1 is associated with biologic behavior in human clear cell renal cell carcinoma. PLoS ONE 2014, 9, e110218. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cochrane, C.R.; Szczepny, A.; Watkins, D.N.; Cain, J.E. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers 2015, 7, 1554-1585. https://doi.org/10.3390/cancers7030851
Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog Signaling in the Maintenance of Cancer Stem Cells. Cancers. 2015; 7(3):1554-1585. https://doi.org/10.3390/cancers7030851
Chicago/Turabian StyleCochrane, Catherine R., Anette Szczepny, D. Neil Watkins, and Jason E. Cain. 2015. "Hedgehog Signaling in the Maintenance of Cancer Stem Cells" Cancers 7, no. 3: 1554-1585. https://doi.org/10.3390/cancers7030851