
Human BK Polyomavirus—The Potential for Head and Neck Malignancy and Disease

Abstract
:1. Polyomavirus Family

{kind=link}
{kind=link}
{kind=link}
| HPyV | Year | Source | Reference |
|---|---|---|---|
| BKPyV | 1971 | Urine, transplant patient | [2] |
| JCPyV | 1971 | Brain specimen, Hodgkin’s disease patient | [3] |
| KIPyV | 2007 | Nasopharyngeal aspirate | [7] |
| WUPyV | 2007 | Nasopharyngeal aspirate | [8] |
| MCPyV | 2008 | Merkel cell carcinoma | [4] |
| HPyV6 | 2010 | Skin swab | [10] |
| HPyV7 | 2010 | Skin swab | [10] |
| TSPyV | 2010 | Nose spicules, trichodysplasia spinulosa patient | [9] |
| HPyV9 | 2011 | Serum, kidney transplant patient | [11] |
| HPyV10 | 2012 | Stool sample, child | [12,13] |
| STLPyV | 2013 | Stool sample, child | [17] |
| HPyV12 | 2013 | Liver tissue | [19] |
| NJPyV | 2014 | Muscle specimen, pancreatic transplant patient | [20] |
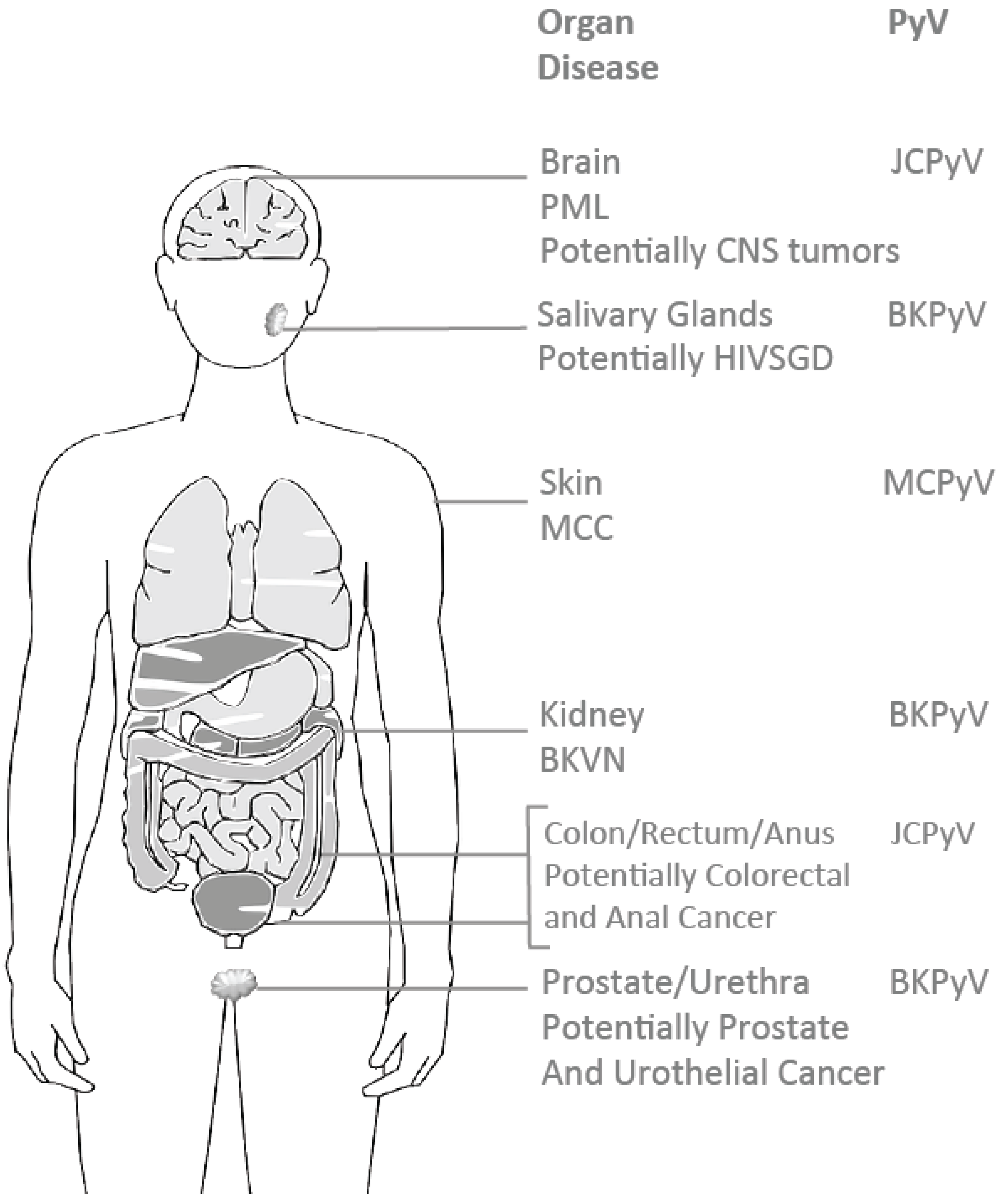
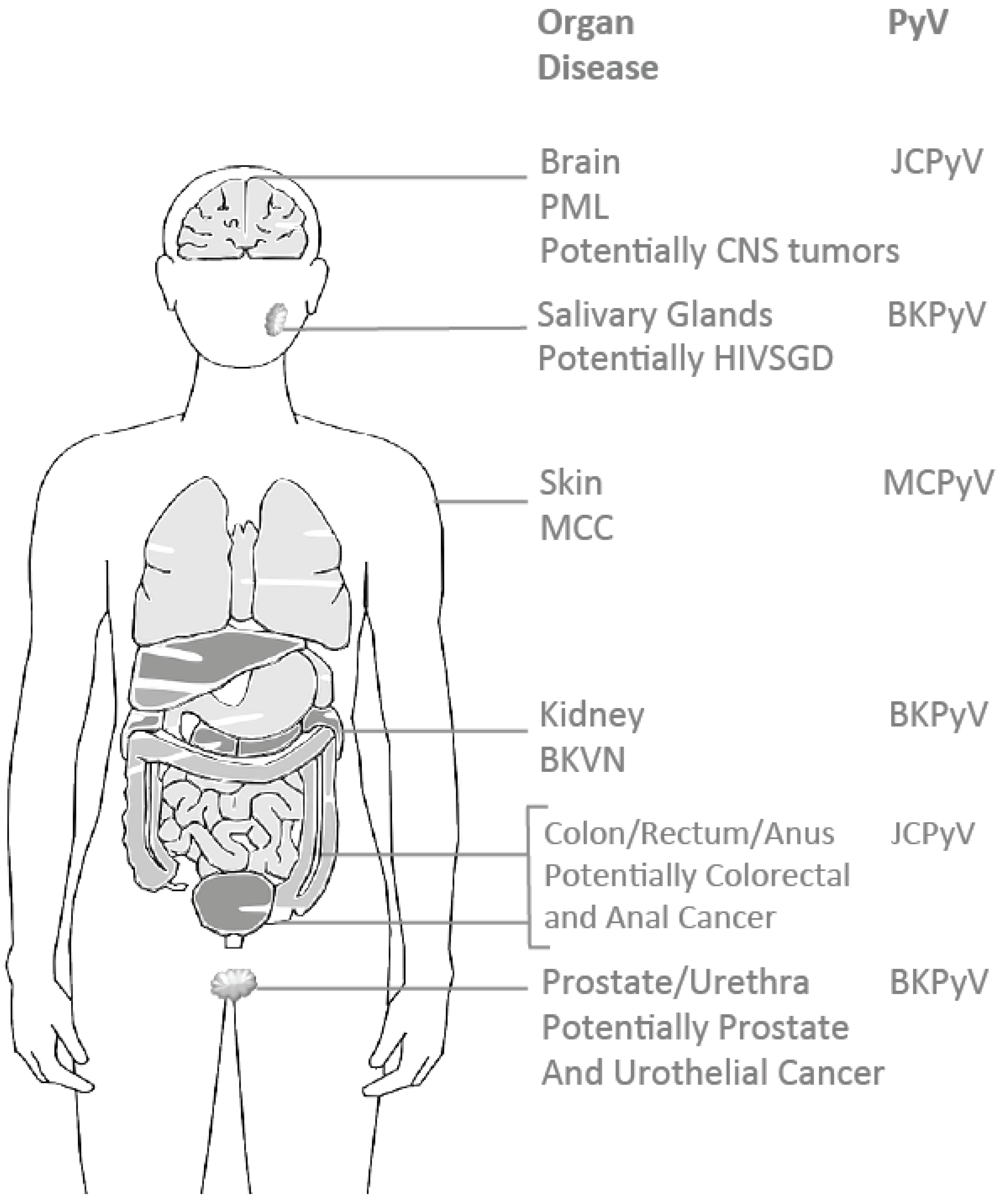
2. The Oncogenic Potential of Human Polyomaviruses
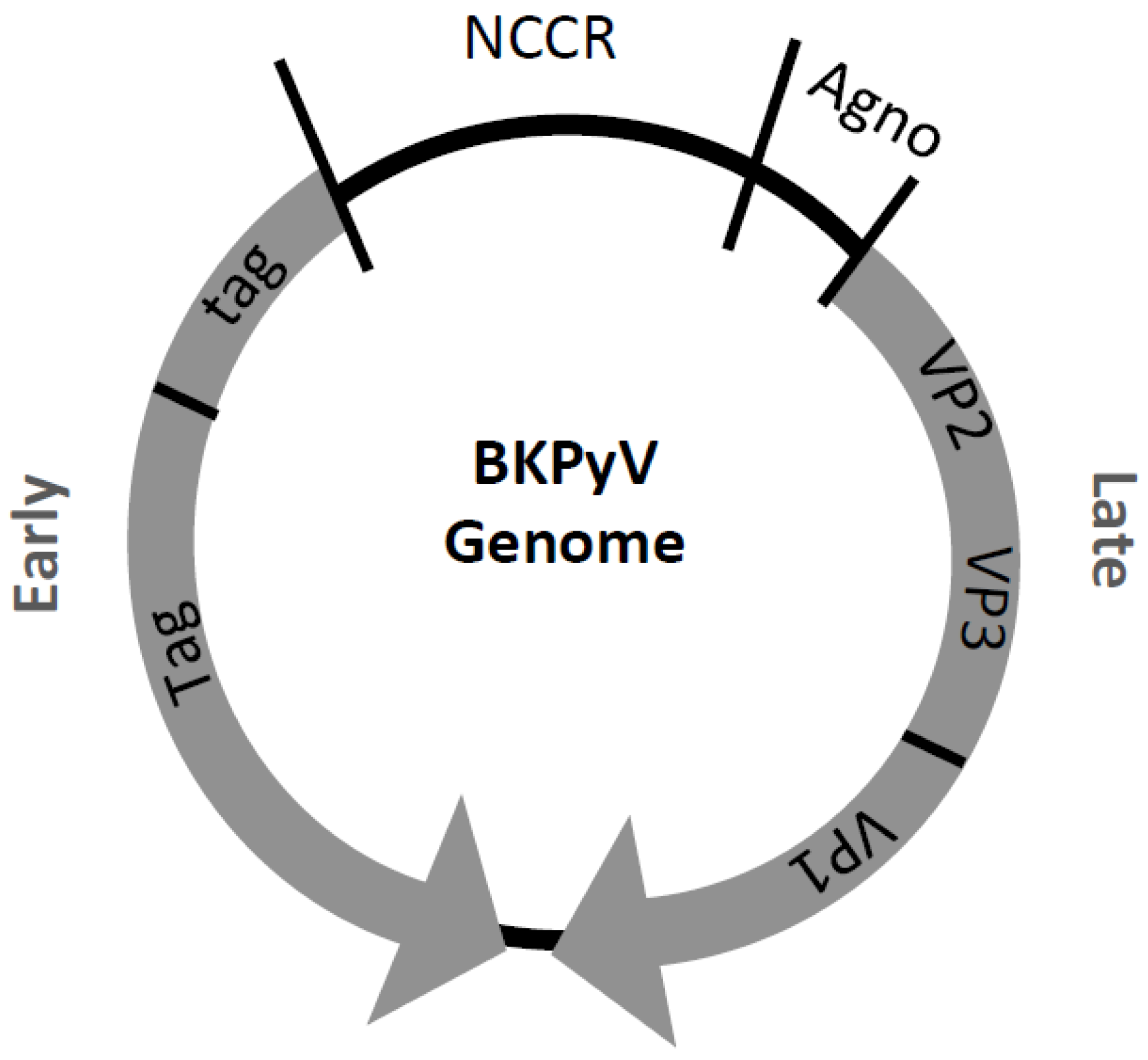
3. BK Polyomavirus (BKPyV)

4. BKPyV-Associated Renal Diseases
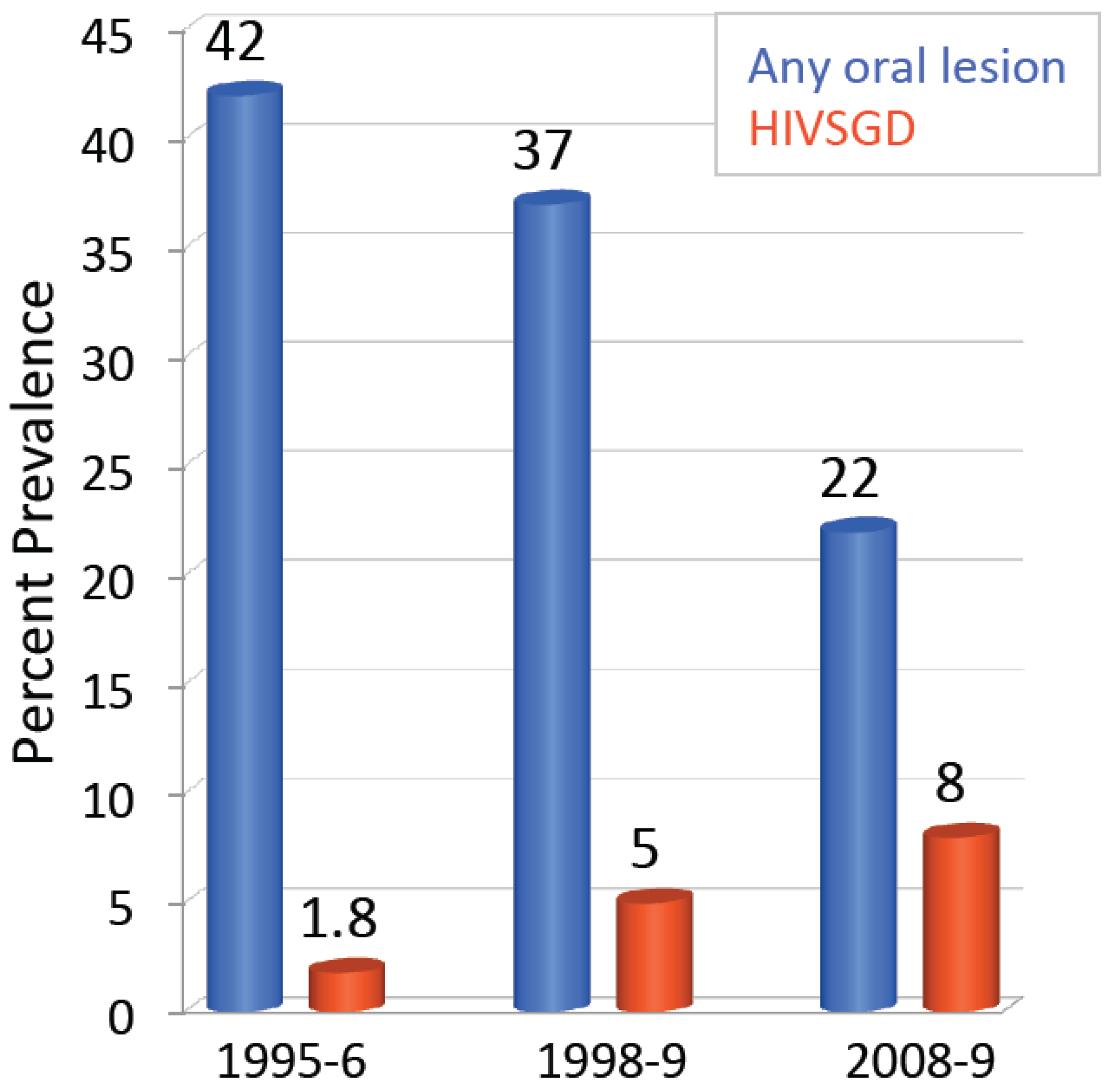
5. HIV-Associated Salivary Gland Disease (HIVSGD)

6. HIVSGD-Lymphoma Association
7. BK Polyomavirus HIVSGD Association
8. Potential Factors Allowing for BKPyV Tropism in the Head and Neck
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dalianis, T.; Hirsch, H.H. Human polyomaviruses in disease and cancer. Virology 2013, 437, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Gardner, S.D.; Field, A.M.; Coleman, D.V.; Hulme, B. New human papovavirus (BK) isolated from urine after renal transplantation. Lancet 1971, 1, 1253–1257. [Google Scholar] [CrossRef]
- Zu Rhein, G.; Chou, S.M. Particles resembling papova viruses in human cerebral demyelinating disease. Science 1965, 148, 1477–1479. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a polyomavirus in human merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Bialasiewicz, S.; Whiley, D.M.; Lambert, S.B.; Nissen, M.D.; Sloots, T.P. Detection of BK, JC, WU, or KI polyomaviruses in faecal, urine, blood, cerebrospinal fluid and respiratory samples. J. Clin. Virol. 2009, 45, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Wiedinger, K.; Bitsaktsis, C.; Chang, S. Reactivation of human polyomaviruses in immunocompromised states. J. Neurovirol. 2014, 20, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Allander, T.; Andreasson, K.; Gupta, S.; Bjerkner, A.; Bogdanovic, G.; Persson, M.A.; Dalianis, T.; Ramqvist, T.; Andersson, B. Identification of a third human polyomavirus. J. Virol. 2007, 81, 4130–4136. [Google Scholar] [CrossRef] [PubMed]
- Gaynor, A.M.; Nissen, M.D.; Whiley, D.M.; Mackay, I.M.; Lambert, S.B.; Wu, G.; Brennan, D.C.; Storch, G.A.; Sloots, T.P.; Wang, D. Identification of a novel polyomavirus from patients with acute respiratory tract infections. PLoS Pathog. 2007, 3, e64. [Google Scholar] [CrossRef] [PubMed]
- Kazem, S.; van der Meijden, E.; Feltkamp, M.C. The trichodysplasia spinulosa-associated polyomavirus: Virological background and clinical implications. Apmis 2013, 121, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Schowalter, R.M.; Pastrana, D.V.; Pumphrey, K.A.; Moyer, A.L.; Buck, C.B. Merkel cell polyomavirus and two previously unknown polyomaviruses are chronically shed from human skin. Cell Host Microbe 2010, 7, 509–515. [Google Scholar] [CrossRef] [PubMed]
- Scuda, N.; Hofmann, J.; Calvignac-Spencer, S.; Ruprecht, K.; Liman, P.; Kuhn, J.; Hengel, H.; Ehlers, B. A novel human polyomavirus closely related to the african green monkey-derived lymphotropic polyomavirus. J. Virol. 2011, 85, 4586–4590. [Google Scholar] [CrossRef] [PubMed]
- Siebrasse, E.A.; Reyes, A.; Lim, E.S.; Zhao, G.; Mkakosya, R.S.; Manary, M.J.; Gordon, J.I.; Wang, D. Identification of mw polyomavirus, a novel polyomavirus in human stool. J. Virol. 2012, 86, 10321–10326. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Phan, G.Q.; Raiji, M.T.; Murphy, P.M.; McDermott, D.H.; McBride, A.A. Complete genome sequence of a tenth human polyomavirus. J. Virol. 2012, 86, 10887. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; van Ghelue, M.; Ehlers, B. Are human polyomaviruses co-factors for cancers induced by other oncoviruses? Rev. Med. Virol. 2014, 24, 343–360. [Google Scholar] [CrossRef] [PubMed]
- Rennspiess, D.; Pujari, S.; Keijzers, M.; Abdul-Hamid, M.A.; Hochstenbag, M.; Dingemans, A.M.; Kurz, A.K.; Speel, E.J.; Haugg, A.; Pastrana, D.V.; et al. Detection of human polyomavirus 7 in human thymic epithelial tumors. J. Thorac. Oncol. 2015, 10, 360–366. [Google Scholar] [CrossRef] [PubMed]
- Wieland, U.; Silling, S.; Hellmich, M.; Potthoff, A.; Pfister, H.; Kreuter, A. Human polyomaviruses 6, 7, 9, 10 and trichodysplasia spinulosa-associated polyomavirus in hiv-infected men. J. Gen. Virol. 2014, 95, 928–932. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.S.; Reyes, A.; Antonio, M.; Saha, D.; Ikumapayi, U.N.; Adeyemi, M.; Stine, O.C.; Skelton, R.; Brennan, D.C.; Mkakosya, R.S.; et al. Discovery of STL polyomavirus, a polyomavirus of ancestral recombinant origin that encodes a unique t antigen by alternative splicing. Virology 2013, 436, 295–303. [Google Scholar] [CrossRef] [PubMed]
- DeCaprio, J.A.; Garcea, R.L. A cornucopia of human polyomaviruses. Nat. Rev. Microbiol. 2013, 11, 264–276. [Google Scholar] [CrossRef] [PubMed]
- Korup, S.; Rietscher, J.; Calvignac-Spencer, S.; Trusch, F.; Hofmann, J.; Moens, U.; Sauer, I.; Voigt, S.; Schmuck, R.; Ehlers, B. Identification of a novel human polyomavirus in organs of the gastrointestinal tract. PLoS ONE 2013, 8, e58021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, N.; Pereira, M.; Rhodes, R.H.; An, P.; Pipas, J.M.; Jain, K.; Kapoor, A.; Briese, T.; Faust, P.L.; Lipkin, W.I. Identification of a novel polyomavirus in a pancreatic transplant recipient with retinal blindness and vasculitic myopathy. J. Infect. Dis. 2014, 210, 1595–1599. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Steiger, J. Polyomavirus BK. Lancet. Infect. Dis. 2003, 3, 611–623. [Google Scholar] [CrossRef]
- Lu, M.C.; Yu, C.L.; Yin, W.Y.; Tung, C.H.; Huang, K.Y.; Liu, S.Q.; Lai, N.S. Increased prevalence of polyomavirus BK viruria that correlates with thrombocytopenia in patients with systemic lupus erythematosus on intensive immunosuppressive therapy. Autoimmunity 2009, 42, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.C.; Yin, W.Y.; Liu, S.Q.; Koo, M.; Tung, C.H.; Huang, K.Y.; Lai, N.S. Increased prevalence of JC polyomavirus viruria was associated with arthritis/arthralgia in patients with systemic lupus erythematosus. Lupus 2015, 24, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Rianthavorn, P.; Posuwan, N.; Payungporn, S.; Theamboonlers, A.; Poovorawan, Y. Polyomavirus reactivation in pediatric patients with systemic lupus erythematosus. Tohoku J. Exp. Med. 2012, 228, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Sundsfjord, A.; Osei, A.; Rosenqvist, H.; van Ghelue, M.; Silsand, Y.; Haga, H.J.; Rekvig, O.P.; Moens, U. BK and JC viruses in patients with systemic lupus erythematosus: Prevalent and persistent bk viruria, sequence stability of the viral regulatory regions, and nondetectable viremia. J. Infect. Dis. 1999, 180, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Gerits, N.; Moens, U. Agnoprotein of mammalian polyomaviruses. Virology 2012, 432, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.J.; Daugherty, M.D.; Qi, X.; Bheda-Malge, A.; Wipf, G.C.; Robinson, K.; Roman, A.; Malik, H.S.; Galloway, D.A. Identification of an overprinting gene in merkel cell polyomavirus provides evolutionary insight into the birth of viral genes. Proc. Natl. Acad. Sci. USA 2013, 110, 12744–12749. [Google Scholar] [CrossRef] [PubMed]
- Neumann, F.; Borchert, S.; Schmidt, C.; Reimer, R.; Hohenberg, H.; Fischer, N.; Grundhoff, A. Replication, gene expression and particle production by a consensus merkel cell polyomavirus (MCPyV) genome. PLoS ONE 2011, 6, e29112. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Kardas, P.; Kranz, D.; Leboeuf, C. The human jc polyomavirus (JCPyV): Virological background and clinical implications. Acta Pathol. Microbil. Immunol. Scand. 2013, 121, 685–727. [Google Scholar] [CrossRef] [PubMed]
- Sweet, B.H.; Hilleman, M.R. The vacuolating virus, SV 40. Exp. Biol. Med. 1960, 105, 420–427. [Google Scholar] [CrossRef]
- Butel, J.S.; Lednicky, J.A. Cell and molecular biology of simian virus 40: Implications for human infections and disease. J. Natl. Cancer Inst. 1999, 91, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Gross, L. A filterable agent, recovered from ak leukemic extracts, causing salivary gland carcinomas in c3h mice. Proc. Soc. Exp. Biol. Med. 1953, 83, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Qi, F.; Carbone, M.; Yang, H.; Gaudino, G. Simian virus 40 transformation, malignant mesothelioma and brain tumors. Expert Rev. Res. Med. 2011, 5, 683–697. [Google Scholar] [CrossRef] [PubMed]
- Colvin, E.K.; Weir, C.; Ikin, R.J.; Hudson, A.L. SV40 tag mouse models of cancer. Semin. Cell Dev. Biol. 2014, 27, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef] [PubMed]
- Giraud, G.; Ramqvist, T.; Ragnarsson-Olding, B.; Dalianis, T. DNA from BK virus and JC virus and from KI, WU, and MC polyomaviruses as well as from simian virus 40 is not detected in non-UV-light-associated primary malignant melanomas of mucous membranes. J. Clin. Microbiol. 2008, 46, 3595–3598. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; DeCaprio, J.A.; Fluck, M.M.; Schaffhausen, B.S. Cellular transformation by simian virus 40 and murine polyoma virus T antigens. Semin. Cancer Biol. 2009, 19, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Tognon, M.; Corallini, A.; Martini, F.; Negrini, M.; Barbanti-Brodano, G. Oncogenic transformation by BK virus and association with human tumors. Oncogene 2003, 22, 5192–5200. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.; Corallini, A.; Grossi, M.P.; Carra, L.; Balboni, P.G.; Negrini, M.; Milanesi, G.; Federspil, G.; Barbanti-Brodano, G. Episomal DNA of a BK virus variant in a human insulinoma. J. Med. Virol. 1983, 12, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Corallini, A.; Altavilla, G.; Cecchetti, M.G.; Fabris, G.; Grossi, M.P.; Balboni, P.G.; Lanza, G.; Barbanti-Brodano, G. Ependymomas, malignant tumors of pancreatic islets, and osteosarcomas induced in hamsters by BK virus, a human papovavirus. J. Natl. Cancer Inst. 1978, 61, 875–883. [Google Scholar] [PubMed]
- Verhaegen, M.E.; Mangelberger, D.; Harms, P.W.; Vozheiko, T.D.; Weick, J.W.; Wilbert, D.M.; Saunders, T.L.; Ermilov, A.N.; Bichakjian, C.K.; Johnson, T.M.; et al. Merkel cell polyomavirus small T antigen is oncogenic in transgenic mice. J. Investig. Dermatol. 2015, 135, 1415–1424. [Google Scholar] [CrossRef] [PubMed]
- Spurgeon, M.E.; Cheng, J.; Bronson, R.T.; Lambert, P.F.; DeCaprio, J.A. Tumorigenic activity of merkel cell polyomavirus T antigens expressed in the stratified epithelium of mice. Cancer Res. 2015, 75, 1068–1079. [Google Scholar] [CrossRef] [PubMed]
- Reiss, K.; Khalili, K. Viruses and cancer: Lessons from the human polyomavirus, JCV. Oncogene 2003, 22, 6517–6523. [Google Scholar] [CrossRef] [PubMed]
- Frisque, R.J.; Rifkin, D.B.; Walker, D.L. Transformation of primary hamster brain cells with JC virus and its DNA. J. Virol. 1980, 35, 265–269. [Google Scholar] [PubMed]
- Kang, S.; Folk, W.R. Lymphotropic papovavirus transforms hamster cells without altering the amount or stability of p53. Virology 1992, 191, 754–764. [Google Scholar] [CrossRef]
- Hatfield, J.M.; Walker, P.M. Satellite DNA replication in baby mouse kidney cells infected with polyoma virus. Nat. New Biol. 1973, 242, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Varakis, J.; ZuRhein, G.M.; Padgett, B.L.; Walker, D.L. Induction of peripheral neuroblastomas in syrian hamsters after injection as neonates with JC virus, a human polyoma virus. Cancer Res. 1978, 38, 1718–1722. [Google Scholar] [PubMed]
- Zu Rhein, G.M.; Varakis, J.N. Perinatal induction of medulloblastomas in syrian golden hamsters by a human polyoma virus (JC). Natl. Cancer Inst. Monogr. 1979, 205–208. [Google Scholar]
- Delbue, S.; Ferrante, P.; Provenzano, M. Polyomavirus BK and prostate cancer: An unworthy scientific effort? Oncoscience 2014, 1, 296–303. [Google Scholar] [PubMed]
- Justice, J.L.; Verhalen, B.; Jiang, M. Polyomavirus interaction with the DNA damage response. Virol. Sin. 2015, 30, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Coursaget, P.; Samimi, M.; Nicol, J.T.; Gardair, C.; Touze, A. Human merkel cell polyomavirus: Virological background and clinical implications. Acta Pathol. Microbil. Immunol. Scand. 2013, 121, 755–769. [Google Scholar] [CrossRef] [PubMed]
- Triozzi, P.L.; Fernandez, A.P. The role of the immune response in merkel cell carcinoma. Cancers 2013, 5, 234–254. [Google Scholar] [CrossRef] [PubMed]
- Pastrana, D.V.; Tolstov, Y.L.; Becker, J.C.; Moore, P.S.; Chang, Y.; Buck, C.B. Quantitation of human seroresponsiveness to merkel cell polyomavirus. PLoS Pathog. 2009, 5, e1000578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Touze, A.; Le Bidre, E.; Laude, H.; Fleury, M.J.; Cazal, R.; Arnold, F.; Carlotti, A.; Maubec, E.; Aubin, F.; Avril, M.F.; et al. High levels of antibodies against merkel cell polyomavirus identify a subset of patients with merkel cell carcinoma with better clinical outcome. J. Clin. Oncol. 2011, 29, 1612–1619. [Google Scholar] [CrossRef] [PubMed]
- Kassem, A.; Technau, K.; Kurz, A.K.; Pantulu, D.; Loning, M.; Kayser, G.; Stickeler, E.; Weyers, W.; Diaz, C.; Werner, M.; et al. Merkel cell polyomavirus sequences are frequently detected in nonmelanoma skin cancer of immunosuppressed patients. Int. J. Cancer 2009, 125, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Wetzels, C.T.; Hoefnagel, J.G.; Bakkers, J.M.; Dijkman, H.B.; Blokx, W.A.; Melchers, W.J. Ultrastructural proof of polyomavirus in merkel cell carcinoma tumour cells and its absence in small cell carcinoma of the lung. PLoS ONE 2009, 4, e4958. [Google Scholar] [CrossRef] [PubMed]
- Bluemn, E.G.; Paulson, K.G.; Higgins, E.E.; Sun, Y.; Nghiem, P.; Nelson, P.S. Merkel cell polyomavirus is not detected in prostate cancers, surrounding stroma, or benign prostate controls. J. Clin. Virol. 2009, 44, 164–166. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, B.; Honkaniemi, E.; Goh, S.; Giraud, G.; Forestier, E.; von Dobeln, U.; Allander, T.; Dalianis, T.; Bogdanovic, G. KI, WU, and merkel cell polyomavirus DNA was not detected in guthrie cards of children who later developed acute lymphoblastic leukemia. J. Pediatr. Hematol. Oncol. 2012, 34, 364–367. [Google Scholar] [CrossRef] [PubMed]
- Sastre-Garau, X.; Peter, M.; Avril, M.F.; Laude, H.; Couturier, J.; Rozenberg, F.; Almeida, A.; Boitier, F.; Carlotti, A.; Couturaud, B.; et al. Merkel cell carcinoma of the skin: Pathological and molecular evidence for a causative role of MCV in oncogenesis. J. Pathol. 2009, 218, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Mogha, A.; Fautrel, A.; Mouchet, N.; Guo, N.; Corre, S.; Adamski, H.; Watier, E.; Misery, L.; Galibert, M.D. Merkel cell polyomavirus small T antigen mrna level is increased following in vivo UV-radiation. PLoS ONE 2010, 5, e11423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadeghi, F.; Salehi-Vaziri, M.; Alizadeh, A.; Ghodsi, S.M.; Bokharaei-Salim, F.; Fateh, A.; Monavari, S.H.; Keyvani, H. Detection of merkel cell polyomavirus large T-antigen sequences in human central nervous system tumors. J. Med. Virol. 2015, 87, 1241–1247. [Google Scholar] [CrossRef] [PubMed]
- Koralnik, I.J.; Schellingerhout, D.; Frosch, M.P. Case records of the massachusetts general hospital. Weekly clinicopathological exercises. Case 14-2004. A 66-year-old man with progressive neurologic deficits. New Engl. J. Med. 2004, 350, 1882–1893. [Google Scholar] [PubMed]
- Power, C.; Gladden, J.G.; Halliday, W.; Del Bigio, M.R.; Nath, A.; Ni, W.; Major, E.O.; Blanchard, J.; Mowat, M. Aids- and non-AIDS-related pml association with distinct p53 polymorphism. Neurology 2000, 54, 743–746. [Google Scholar] [CrossRef] [PubMed]
- Berger, J.R.; Kaszovitz, B.; Post, M.J.; Dickinson, G. Progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. A review of the literature with a report of sixteen cases. Ann. Intern. Med. 1987, 107, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Dobson, S. BK and JC virus: A review. J. Infect. 2014, 68, S2–S8. [Google Scholar] [CrossRef] [PubMed]
- Coelho, T.R.; Almeida, L.; Lazo, P.A. JC virus in the pathogenesis of colorectal cancer, an etiological agent or another component in a multistep process? Virol. J. 2010, 7, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization. Cancer Fact Sheet; WHO: Geneva, Switzerland, 2014. [Google Scholar]
- Ricciardiello, L.; Chang, D.K.; Laghi, L.; Goel, A.; Chang, C.L.; Boland, C.R. Mad-1 is the exclusive JC virus strain present in the human colon, and its transcriptional control region has a deleted 98-base-pair sequence in colon cancer tissues. J. Virol. 2001, 75, 1996–2001. [Google Scholar] [CrossRef] [PubMed]
- Haggerty, S.; Walker, D.L.; Frisque, R.J. JC virus-simian virus 40 genomes containing heterologous regulatory signals and chimeric early regions: Identification of regions restricting transformation by JC virus. J. Virol. 1989, 63, 2180–2190. [Google Scholar] [PubMed]
- Yin, W.Y.; Lee, M.C.; Lai, N.S.; Lu, M.C. BK virus as a potential oncovirus for bladder cancer in a renal transplant patient. J. Formos. Med. Assoc. 2015, 114, 373–374. [Google Scholar] [CrossRef] [PubMed]
- Mischitelli, M.; Bellizzi, A.; Anzivino, E.; Rodio, D.M.; Sciarra, A.; Gentile, V.; Pietropaolo, V. Results, questions, perspectives of a study on human polyomavirus BK and molecular actors in prostate cancer development. Cancer Genom. Proteom. 2015, 12, 57–65. [Google Scholar]
- Tang, W.; Duan, J.; Zhang, J.G.; Wang, Y.P. Subtyping glioblastoma by combining mirna and mrna expression data using compressed sensing-based approach. EURASIP J. Bioinf. Syst. Biol. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Shah, R.B.; Imperiale, M.J. Detection and expression of human BK virus sequences in neoplastic prostate tissues. Oncogene 2004, 23, 7031–7046. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Wojno, K.; Imperiale, M.J. BK virus as a cofactor in the etiology of prostate cancer in its early stages. J. Virol. 2008, 82, 2705–2714. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, L.K.; Madden, V.; Webster-Cyriaque, J. BK virus has tropism for human salivary gland cells in vitro: Implications for transmission. Virology 2009, 394, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, L.; Webster-Cyriaque, J.Y. Viruses and salivary gland disease (SGD): Lessons from HIV SGD. Adv. Dent. Res. 2011, 23, 79–83. [Google Scholar] [CrossRef] [PubMed]
- Jeffers, L.K.; Burger-Calderon, R.; Webster-Cyriaque, J. Effect of leflunomide, cidofovir and ciprofloxacin on replication of BKPyV in a salivary gland in vitro culture system. Antivir. Res. 2015, 118, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Burger-Calderon, R.; Madden, V.; Hallett, R.A.; Gingerich, A.D.; Nickeleit, V.; Webster-Cyriaque, J. Replication of oral BK virus in human salivary gland cells. J. Virol. 2014, 88, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Polz, D.; Morshed, K.; Stec, A.; Podsiadlo, L.; Polz-Dacewicz, M. Do polyomavirus hominis strains BK and JC play a role in oral squamous cell carcinoma? Ann. Agric. Environ. Med. 2015, 22, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M.C.; Jensen, P.N.; Hou, J.; Durham, L.C.; Major, E.O. Detection of JC virus DNA in human tonsil tissue: Evidence for site of initial viral infection. J. Virol. 1998, 72, 9918–9923. [Google Scholar] [PubMed]
- Kato, A.; Kitamura, T.; Takasaka, T.; Tominaga, T.; Ishikawa, A.; Zheng, H.Y.; Yogo, Y. Detection of the archetypal regulatory region of JC virus from the tonsil tissue of patients with tonsillitis and tonsilar hypertrophy. J. Neurovirol. 2004, 10, 244–249. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, A.; Carinci, F.; Martinelli, M.; Spinelli, G.; Lo Muzio, L.; Rubini, C.; Scapoli, L. Absence of simian virus 40, BK, and JC polyomavirus DNA in squamous cell carcinoma limited to the oral cavity. Head Neck 2010, 32, 375–380. [Google Scholar] [CrossRef] [PubMed]
- Imperiale, M.J. The human polyomaviruses, BKV and JCV: Molecular pathogenesis of acute disease and potential role in cancer. Virology 2000, 267, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Boldorini, R.; Allegrini, S.; Miglio, U.; Paganotti, A.; Cocca, N.; Zaffaroni, M.; Riboni, F.; Monga, G.; Viscidi, R. Serological evidence of vertical transmission of JC and BK polyomaviruses in humans. J. Gen. Virol. 2011, 92, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Hashida, Y.; Gaffney, P.C.; Yunis, E.J. Acute hemorrhagic cystitis of childhood and papovavirus-like particles. J. Pediatr. 1976, 89, 85–87. [Google Scholar] [CrossRef]
- Weinberg, G.A.; Mian, A.N. BK virus nephropathy and other polyoma virus infections. Pediatr. Infect. Dis. J. 2010, 29, 257–260. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.F.; Arvin, A.M.; Salter, L.; Bailey, J.R.; Berk, A.J.; Berns, K.I.; Braciale, T.J.; Beirne, B.; Broder, C.C.; Buchmeier, M.J.; et al. Fields’ Virology, 5th ed.; Knipe, D.M., Howley, P.M., Eds.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Rinaldo, C.H.; Tylden, G.D.; Sharma, B.N. The human polyomavirus BK (BKPyV): Virological background and clinical implications. Acta Pathol. Microbil. Immunol. Scand. 2013, 121, 728–745. [Google Scholar] [CrossRef] [PubMed]
- Broekema, N.M.; Abend, J.R.; Bennett, S.M.; Butel, J.S.; Vanchiere, J.A.; Imperiale, M.J. A system for the analysis of BKV non-coding control regions: Application to clinical isolates from an HIV/AIDS patient. Virology 2010, 407, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, J.I.; Seternes, O.M.; Johansen, T.; Moens, U.; Mantyjarvi, R.; Traavik, T. Subpopulations of non-coding control region variants within a cell culture-passaged stock of BK virus: Sequence comparisons and biological characteristics. J. Gen. Virol. 1995, 76 Pt 7, 1571–1581. [Google Scholar] [CrossRef] [PubMed]
- Gorrill, T.S.; Khalili, K. Cooperative interaction of p65 and C/EBPbeta modulates transcription of BKV early promoter. Virology 2005, 335, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, R.B.; Dynan, W.S. Binding of cellular proteins to the regulatory region of BK virus DNA. J. Virol. 1988, 62, 3388–3398. [Google Scholar] [PubMed]
- Cho, S.; Tian, Y.; Benjamin, T.L. Binding of p300/CBP co-activators by polyoma large T antigen. J. Biol. Chem. 2001, 276, 33533–33539. [Google Scholar] [CrossRef] [PubMed]
- Poulin, D.L.; Kung, A.L.; DeCaprio, J.A. p53 targets simian virus 40 large T antigen for acetylation by CBP. J. Virol. 2004, 78, 8245–8253. [Google Scholar] [CrossRef] [PubMed]
- Liang, B.; Tikhanovich, I.; Nasheuer, H.P.; Folk, W.R. Stimulation of BK virus DNA replication by NFI family transcription factors. J. Virol. 2012, 86, 3264–3275. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.J.; Shadley, L.; Mertz, J.E. Nuclear factor 1 family members mediate repression of the BK virus late promoter. Virology 2001, 287, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Pipas, J.M. Common and unique features of T antigens encoded by the polyomavirus group. J. Virol. 1992, 66, 3979–3985. [Google Scholar] [PubMed]
- Daniels, R.; Sadowicz, D.; Hebert, D.N. A very late viral protein triggers the lytic release of SV40. PLoS Pathog. 2007, 3, e98. [Google Scholar] [CrossRef] [PubMed]
- Cubukcu-Dimopulo, O.; Greco, A.; Kumar, A.; Karluk, D.; Mittal, K.; Jagirdar, J. BK virus infection in aids. Am. J. Surg. Pathol. 2000, 24, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Vidal, J.E.; Fink, M.C.; Cedeno-Laurent, F.; Delbue, S.; Ferrante, P.; Dauar, R.F.; Filho, F.B.; Nogueira, R.S.; Calore, E.E.; Pannuti, C.S.; et al. BK virus associated meningoencephalitis in an AIDS patient treated with HAART. AIDS Res. Ther. 2007, 4, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Abend, J.R.; Johnson, S.F.; Imperiale, M.J. The role of polyomaviruses in human disease. Virology 2009, 384, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Nickeleit, V.; Mihatsch, M.J. Polyomavirus nephropathy in native kidneys and renal allografts: An update on an escalating threat. Transpl. Int. 2006, 19, 960–973. [Google Scholar] [CrossRef] [PubMed]
- Shanti, R.M.; Aziz, S.R. HIV-associated salivary gland disease. Oral. Maxillofac. Surg. Clin. North. Am. 2009, 21, 339–343. [Google Scholar] [CrossRef] [PubMed]
- McArthur, C.P.; Subtil-DeOliveira, A.; Palmer, D.; Fiorella, R.M.; Gustafson, S.; Tira, D.; Miranda, R.N. Characteristics of salivary diffuse infiltrative lymphocytosis syndrome in west africa. Arch. Pathol. Lab. Med. 2000, 124, 1773–1779. [Google Scholar] [PubMed]
- Ioachim, H.L.; Ryan, J.R. Salivary gland lymphadenopathies associated with aids. Hum. Pathol. 1988, 19, 616–617. [Google Scholar] [CrossRef]
- Ioachim, H.L.; Ryan, J.R.; Blaugrund, S.M. Salivary gland lymph nodes. The site of lymphadenopathies and lymphomas associated with human immunodeficiency virus infection. Arch. Pathol. Lab. Med. 1988, 112, 1224–1228. [Google Scholar] [PubMed]
- DiGiuseppe, J.A.; Corio, R.L.; Westra, W.H. Lymphoid infiltrates of the salivary glands: Pathology, biology and clinical significance. Curr. Opin. Oncol. 1996, 8, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Schiodt, M.; Greenspan, D.; Levy, J.A.; Nelson, J.A.; Chernoff, D.; Hollander, H.; Greenspan, J.S. Does HIV cause salivary gland disease? Aids 1989, 3, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Program, R.W.H. Oral health release. Available online: http://hab.hrsa.gov/abouthab/files/oral_health_fact_sheet.pdf (accessed on 22 January 2014).
- Patton, L.L.; McKaig, R.; Strauss, R.; Rogers, D.; Eron, J.J., Jr. Changing prevalence of oral manifestations of human immuno-deficiency virus in the era of protease inhibitor therapy. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2000, 89, 299–304. [Google Scholar] [CrossRef]
- Greenspan, D.; Canchola, A.J.; MacPhail, L.A.; Cheikh, B.; Greenspan, J.S. Effect of highly active antiretroviral therapy on frequency of oral warts. Lancet 2001, 357, 1411–1412. [Google Scholar] [CrossRef]
- Ellis, G.L. Lymphoid lesions of salivary glands: Malignant and benign. Med. Oral Patol. Oral y Cirugía Bucal 2007, 12, E479–E485. [Google Scholar]
- Chadburn, A.; Abdul-Nabi, A.M.; Teruya, B.S.; Lo, A.A. Lymphoid proliferations associated with human immunodeficiency virus infection. Arch. Pathol. Lab. Med. 2013, 137, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Greaves, W.O.; Wang, S.A. Selected topics on lymphoid lesions in the head and neck regions. Head Neck Pathol. 2011, 5, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Ioachim, H.L.; Antonescu, C.; Giancotti, F.; Dorsett, B. EBV-associated primary lymphomas in salivary glands of HIV-infected patients. Pathol. Res. Pract. 1998, 194, 87–95. [Google Scholar] [CrossRef]
- Dovigi, D.A. HIV Salivary Gland Disease: A Role for Viral Infection. Master of Science Thesis, University of North Carolina at Chapel Hill, Chapel Hill, NC, USA, 2005. [Google Scholar]
- Goudsmit, J.; Baak, M.L.; Sleterus, K.W.; van der Noordaa, J. Human papovavirus isolated from urine of a child with acute tonsillitis. Br. Med. J. 1981, 283, 1363–1364. [Google Scholar] [CrossRef]
- Goudsmit, J.; Wertheim-Van Dillen, P.; van Strien, A.; van der Noordaa, J. The role of BK virus in acute respiratory tract disease and the presence of BKV DNA in tonsils. J. Med. Virol. 1982, 10, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Nickeleit, V.; Hirsch, H.H.; Binet, I.F.; Gudat, F.; Prince, O.; Dalquen, P.; Thiel, G.; Mihatsch, M.J. Polyomavirus infection of renal allograft recipients: From latent infection to manifest disease. J. Am. Soc. Nephrol. 1999, 10, 1080–1089. [Google Scholar] [PubMed]
- Gosert, R.; Rinaldo, C.H.; Funk, G.A.; Egli, A.; Ramos, E.; Drachenberg, C.B.; Hirsch, H.H. Polyomavirus BK with rearranged noncoding control region emerge in vivo in renal transplant patients and increase viral replication and cytopathology. J. Exp. Med. 2008, 205, 841–852. [Google Scholar] [CrossRef] [PubMed]
- Funk, G.A.; Gosert, R.; Hirsch, H.H. Viral dynamics in transplant patients: Implications for disease. Lancet Infect. Dis. 2007, 7, 460–472. [Google Scholar] [CrossRef]
- Robaina, T.F.; Mendes, G.S.; Benati, F.J.; Pena, G.A.; Silva, R.C.; Montes, M.A.; Janini, M.E.; Camara, F.P.; Santos, N. Shedding of polyomavirus in the saliva of immunocompetent individuals. J. Med. Virol. 2013, 85, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Ortega, K.L.; Ceballos-Salobrena, A.; Gaitan-Cepeda, L.A.; Magalhaes, M.G. Oral manifestations after immune reconstitution in HIV patients on haart. Int. J. STD AIDS 2008, 19, 305–308. [Google Scholar] [CrossRef] [PubMed]
- Shelburne, S.A.; Visnegarwala, F.; Darcourt, J.; Graviss, E.A.; Giordano, T.P.; White, A.C., Jr.; Hamill, R.J. Incidence and risk factors for immune reconstitution inflammatory syndrome during highly active antiretroviral therapy. Aids 2005, 19, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Williams, F.M.; Ahn, C.W.; Reveille, J.D. Changing spectrum of the diffuse infiltrative lymphocytosis syndrome. Arthritis Rheum. 2006, 55, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Itescu, S.; Brancato, L.J.; Winchester, R. A sicca syndrome in HIV infection: Association with hla-dr5 and cd8 lymphocytosis. Lancet 1989, 2, 466–468. [Google Scholar] [CrossRef]
- Ashok, A.; Atwood, W.J. Virus receptors and tropism. Adv. Exp. Med. Biol. 2006, 577, 60–72. [Google Scholar] [PubMed]
- Neu, U.; Allen, S.A.; Blaum, B.S.; Liu, Y.; Frank, M.; Palma, A.S.; Stroh, L.J.; Feizi, T.; Peters, T.; Atwood, W.J.; et al. A structure-guided mutation in the major capsid protein retargets BK polyomavirus. PLoS Pathog. 2013, 9, e1003688. [Google Scholar] [CrossRef] [PubMed]
- Maraldi, N.M.; Barbanti-Brodano, G.; Portolani, M.; la Placa, M. Ultrastructural aspects of BK virus uptake and replication in human fibroblasts. J. Gen. Virol. 1975, 27, 71–80. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Johansen, T.; Johnsen, J.I.; Seternes, O.M.; Traavik, T. Noncoding control region of naturally occurring BK virus variants: Sequence comparison and functional analysis. Virus Genes 1995, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, R.; Schoonakker, B.C.; Harley, E.H. Recurring theme of changes in the transcriptional control region of BK virus during adaptation to cell culture. J. Virol. 1991, 65, 1600–1604. [Google Scholar] [PubMed]
- Sundsfjord, A.; Johansen, T.; Flaegstad, T.; Moens, U.; Villand, P.; Subramani, S.; Traavik, T. At least two types of control regions can be found among naturally occurring BK virus strains. J. Virol. 1990, 64, 3864–3871. [Google Scholar] [PubMed]
- Sundsfjord, A.; Flaegstad, T.; Flo, R.; Spein, A.R.; Pedersen, M.; Permin, H.; Julsrud, J.; Traavik, T. BK and JC viruses in human immunodeficiency virus type 1-infected persons: Prevalence, excretion, viremia, and viral regulatory regions. J. Infect. Dis. 1994, 169, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, C.H.; Hansen, H.; Traavik, T. Human endothelial cells allow passage of an archetypal BK virus (BKV) strain-a tool for cultivation and functional studies of natural bkv strains. Arch. Virol. 2005, 150, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Bennett, S.M.; Broekema, N.M.; Imperiale, M.J. BK polyomavirus: Emerging pathogen. Microbes Infect. 2012, 14, 672–683. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, M.; Weyandt, T.B.; Frisque, R.J. Identification of archetype and rearranged forms of BK virus in leukocytes from healthy individuals. J. Med. Virol. 2000, 60, 353–362. [Google Scholar] [CrossRef]
- Olsen, G.H.; Andresen, P.A.; Hilmarsen, H.T.; Bjorang, O.; Scott, H.; Midtvedt, K.; Rinaldo, C.H. Genetic variability in BK virus regulatory regions in urine and kidney biopsies from renal-transplant patients. J. Med. Virol. 2006, 78, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Perets, T.T.; Silberstein, I.; Rubinov, J.; Sarid, R.; Mendelson, E.; Shulman, L.M. High frequency and diversity of rearrangements in polyomavirus BK noncoding regulatory regions cloned from urine and plasma of Israeli renal transplant patients and evidence for a new genetic subtype. J. Clin. Microbiol. 2009, 47, 1402–1411. [Google Scholar] [CrossRef] [PubMed]
- Barcena-Panero, A.; Echevarria, J.E.; van Ghelue, M.; Fedele, G.; Royuela, E.; Gerits, N.; Moens, U. BK polyomavirus with archetypal and rearranged non-coding control regions is present in cerebrospinal fluids from patients with neurological complications. J. Gen. Virol. 2012, 93, 1780–1794. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; van Ghelue, M. Polymorphism in the genome of non-passaged human polyomavirus BK: Implications for cell tropism and the pathological role of the virus. Virology 2005, 331, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Broekema, N.M.; Imperiale, M.J. Efficient propagation of archetype BK and JC polyomaviruses. Virology 2012, 422, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Olsen, G.H.; Hirsch, H.H.; Rinaldo, C.H. Functional analysis of polyomavirus BK non-coding control region quasispecies from kidney transplant recipients. J. Med. Virol. 2009, 81, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Martin, J.D.; King, D.M.; Slauch, J.M.; Frisque, R.J. Differences in regulatory sequences of naturally occurring JC virus variants. J. Virol. 1985, 53, 306–311. [Google Scholar] [PubMed]
- White, F.A., 3rd; Ishaq, M.; Stoner, G.L.; Frisque, R.J. JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J. Virol. 1992, 66, 5726–5734. [Google Scholar] [PubMed]
- Kato, K.; Guo, J.; Taguchi, F.; Daimaru, O.; Tajima, M.; Haibara, H.; Matsuda, J.; Sumiya, M.; Yogo, Y. Phylogenetic comparison between archetypal and disease-associated JC virus isolates in japan. Jpn. J. Med. Sci. Biol. 1994, 47, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Ciappi, S.; Azzi, A.; de Santis, R.; Leoncini, F.; Sterrantino, G.; Mazzotta, F.; Mecocci, L. Archetypal and rearranged sequences of human polyomavirus JC transcription control region in peripheral blood leukocytes and in cerebrospinal fluid. J. Gen. Virol. 1999, 80, 1017–1023. [Google Scholar] [PubMed]
- Newman, J.T.; Frisque, R.J. Identification of JC virus variants in multiple tissues of pediatric and adult pml patients. J. Med. Virol. 1999, 58, 79–86. [Google Scholar] [CrossRef]
- Daniel, A.M.; Swenson, J.J.; Mayreddy, R.P.; Khalili, K.; Frisque, R.J. Sequences within the early and late promoters of archetype JC virus restrict viral DNA replication and infectivity. Virology 1996, 216, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Inoue, T.; Nagata, K.; Mikoshiba, K. Enhancer of human polyoma jc virus contains nuclear factor i-binding sequences; analysis using mouse brain nuclear extracts. Biochem. Biophys. Res. Commun. 1988, 157, 419–425. [Google Scholar] [CrossRef]
- Khalili, K.; Rappaport, J.; Khoury, G. Nuclear factors in human brain cells bind specifically to the jcv regulatory region. Embo. J. 1988, 7, 1205–1210. [Google Scholar] [PubMed]
- Feigenbaum, L.; Hinrichs, S.H.; Jay, G. JC virus and simian virus 40 enhancers and transforming proteins: Role in determining tissue specificity and pathogenicity in transgenic mice. J. Virol. 1992, 66, 1176–1182. [Google Scholar] [PubMed]
- Kerr, D.; Chang, C.F.; Chen, N.; Gallia, G.; Raj, G.; Schwartz, B.; Khalili, K. Transcription of a human neurotropic virus promoter in glial cells: Effect of YB-1 on expression of the JC virus late gene. J. Virol. 1994, 68, 7637–7643. [Google Scholar] [PubMed]
- Chen, N.N.; Khalili, K. Transcriptional regulation of human JC polyomavirus promoters by cellular proteins YB-1 and pur alpha in glial cells. J. Virol. 1995, 69, 5843–5848. [Google Scholar] [PubMed]
- Chen, N.N.; Chang, C.F.; Gallia, G.L.; Kerr, D.A.; Johnson, E.M.; Krachmarov, C.P.; Barr, S.M.; Frisque, R.J.; Bollag, B.; Khalili, K. Cooperative action of cellular proteins YB-1 and pur alpha with the tumor antigen of the human JC polyomavirus determines their interaction with the viral lytic control element. Proc. Natl. Acad. Sci. USA 1995, 92, 1087–1091. [Google Scholar] [CrossRef] [PubMed]
- Safak, M.; Gallia, G.L.; Khalili, K. A 23-bp sequence element from human neurotropic JC virus is responsive to NF-kappa B subunits. Virology 1999, 262, 178–189. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.M.; Gupta, G.; Vats, A.; Shapiro, R.; Randhawa, P.S. Polyomavirus BK non-coding control region rearrangements in health and disease. J. Med. Virol. 2007, 79, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Boldorini, R.; Veggiani, C.; Turello, E.; Barco, D.; Monga, G. Are sequence variations in the BK virus control region essential for the development of polyomavirus nephropathy? Am. J. Clin. Pathol. 2005, 124, 303–312. [Google Scholar] [CrossRef] [PubMed]
- Elsner, C.; Dorries, K. Human polyomavirus jc control region variants in persistently infected CNS and kidney tissue. J. Gen. Virol. 1998, 79 Pt 4, 789–799. [Google Scholar] [PubMed]
- White, M.K.; Pagano, J.S.; Khalili, K. Viruses and human cancers: A long road of discovery of molecular paradigms. Clin. Microbiol. Rev. 2014, 27, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Nickeleit, V. Animal models of polyomavirus nephropathy: Hope and reality. Am. J. Transplant. 2006, 6, 1507–1509. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burger-Calderon, R.; Webster-Cyriaque, J. Human BK Polyomavirus—The Potential for Head and Neck Malignancy and Disease. Cancers 2015, 7, 1244-1270. https://doi.org/10.3390/cancers7030835
Burger-Calderon R, Webster-Cyriaque J. Human BK Polyomavirus—The Potential for Head and Neck Malignancy and Disease. Cancers. 2015; 7(3):1244-1270. https://doi.org/10.3390/cancers7030835
Chicago/Turabian StyleBurger-Calderon, Raquel, and Jennifer Webster-Cyriaque. 2015. "Human BK Polyomavirus—The Potential for Head and Neck Malignancy and Disease" Cancers 7, no. 3: 1244-1270. https://doi.org/10.3390/cancers7030835