
Insights into the Molecular Pathogenesis of Activated B-Cell-like Diffuse Large B-Cell Lymphoma and Its Therapeutic Implications

{kind=link}
Abstract
:1. Introduction
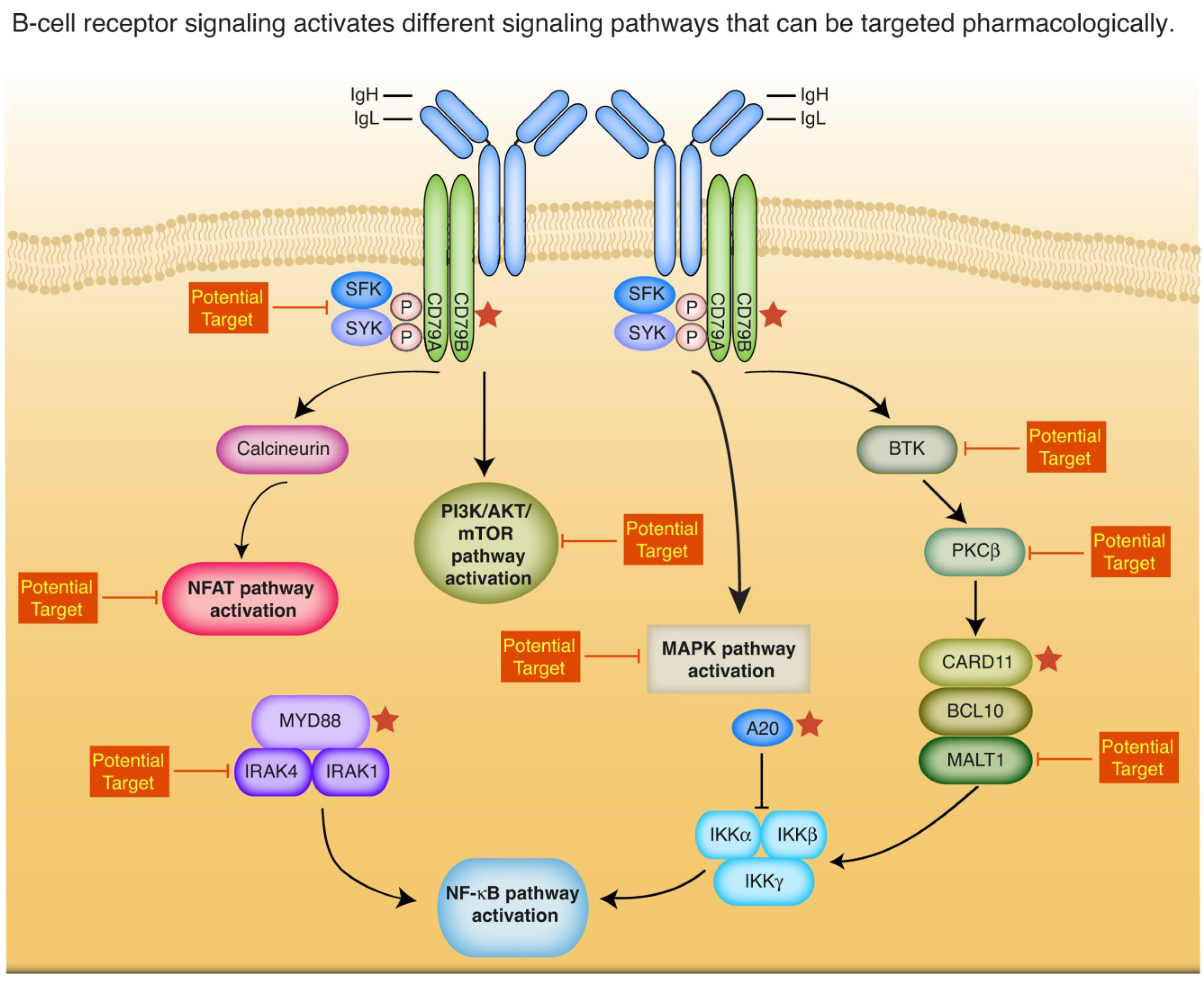
2. Insights into the Biology of ABC DLBCL

3. Potential Targets for the Treatment of GCB DLBCL Patients
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardim, J.W. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2008. [Google Scholar]
- Bernd, H.W.; Ziepert, M.; Thorns, C.; Klapper, W.; Wacker, H.H.; Hummel, M.; Stein, H.; Hansmann, M.L.; Ott, G.; Rosenwald, A.; et al. Loss of HLA-DR expression and immunoblastic morphology predict adverse outcome in diffuse large B-cell lymphoma—Analyses of cases from two prospective randomized clinical trials. Haematologica 2009, 94, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Alizadeh, A.A.; Eisen, M.B.; Davis, R.E.; Ma, C.; Lossos, I.S.; Rosenwald, A.; Boldrick, J.C.; Sabet, H.; Tran, T.; Yu, X.; et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 2000, 403, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Leroy, K.; Yu, X.; Gaulard, P.; Gascoyne, R.D.; Chan, W.C.; Zhao, T.; Haioun, C.; Greiner, T.C.; et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J. Exp. Med. 2003, 198, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.J.; Monti, S.; Kutok, J.L.; Cattoretti, G.; Neuberg, D.; De Leval, L.; Kurtin, P.; Dal Cin, P.; Ladd, C.; Feuerhake, F.; et al. The molecular signature of mediastinal large B-cell lymphoma differs from that of other diffuse large B-cell lymphomas and shares features with classical Hodgkin lymphoma. Blood 2003, 102, 3871–3879. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.; Tan, B.; Rosenwald, A.; Hurt, E.H.; Wiestner, A.; Staudt, L.M. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc. Natl. Acad. Sci. USA 2003, 100, 9991–9996. [Google Scholar] [CrossRef] [PubMed]
- Shipp, M.A.; Ross, K.N.; Tamayo, P.; Weng, A.P.; Kutok, J.L.; Aguiar, R.C.T.; Gaasenbeek, M.; Angelo, M.; Reich, M.; Pinkus, G.S.; et al. Diffuse large B-cell lymphoma outcome prediction by gene-expression profiling and supervised machine learning. Nat. Med. 2002, 8, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.; Savage, K.J.; Kutok, J.L.; Feuerhake, F.; Kurtin, P.; Mihm, M.; Wu, B.; Pasqualucci, L.; Neuberg, D.; Aguiar, R.C.; et al. Molecular profiling of diffuse large B-cell lymphoma identifies robust subtypes including one characterized by host inflammatory response. Blood 2005, 105, 1851–1861. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Wright, G.; Chan, W.C.; Connors, J.M.; Campo, E.; Fisher, R.I.; Gascoyne, R.D.; Muller-Hermelink, H.K.; Smeland, E.B.; Giltnane, J.M.; et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N. Engl. J. Med. 2002, 346, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Copie-Bergman, C.; Plonquet, A.; Alonso, M.A.; Boulland, M.L.; Marquet, J.; Divine, M.; Moller, P.; Leroy, K.; Gaulard, P. MAL expression in lymphoid cells: Further evidence for MAL as a distinct molecular marker of primary mediastinal large B-cell lymphomas. Mod. Pathol. 2002, 15, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.; Dave, S.S.; Xiao, W.; Powell, J.; Zhao, H.; Xu, W.; Tan, B.; Goldschmidt, N.; Iqbal, J.; et al. Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 2008, 359, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Pasqualucci, L.; Compagno, M.; Houldsworth, J.; Monti, S.; Grunn, A.; Nandula, S.V.; Aster, J.C.; Murty, V.V.; Shipp, M.A.; Dalla-Favera, R. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J. Exp. Med. 2006, 203, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Tam, W.; Gomez, M.; Chadburn, A.; Lee, J.W.; Chan, W.C.; Knowles, D.M. Mutational analysis of prdm1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood 2006, 107, 4090–4100. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, A.L.; Lin, K.I.; Kuo, T.C.; Yu, X.; Hurt, E.M.; Rosenwald, A.; Giltnane, J.M.; Yang, L.; Zhao, H.; Calame, K.; et al. Blimp-1 orchestrates plasma cell differentiation by extinguishing the mature B cell gene expression program. Immunity 2002, 17, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Mandelbaum, J.; Bhagat, G.; Tang, H.; Mo, T.; Brahmachary, M.; Shen, Q.; Chadburn, A.; Rajewsky, K.; Tarakhovsky, A.; Pasqualucci, L.; et al. Blimp1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large b cell lymphoma. Cancer Cell 2010, 18, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Greiner, T.C.; Patel, K.; Dave, B.J.; Smith, L.; Ji, J.; Wright, G.; Sanger, W.G.; Pickering, D.L.; Jain, S.; et al. Distinctive patterns of BCL6 molecular alterations and their functional consequences in different subgroups of diffuse large B-cell lymphoma. Leukemia 2007, 21, 2332–2343. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Wright, G.W.; Emre, N.C.; Kohlhammer, H.; Dave, S.S.; Davis, R.E.; Carty, S.; Lam, L.T.; Shaffer, A.L.; Xiao, W.; et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl. Acad. Sci. USA 2008, 105, 13520–13525. [Google Scholar] [CrossRef] [PubMed]
- Schmidlin, H.; Diehl, S.A.; Nagasawa, M.; Scheeren, F.A.; Schotte, R.; Uittenbogaart, C.H.; Spits, H.; Blom, B. Spi-B inhibits human plasma cell differentiation by repressing BLIMP1 and XBP-1 expression. Blood 2008, 112, 1804–1812. [Google Scholar] [CrossRef] [PubMed]
- Calado, D.P.; Zhang, B.; Srinivasan, L.; Sasaki, Y.; Seagal, J.; Unitt, C.; Rodig, S.; Kutok, J.; Tarakhovsky, A.; Schmidt-Supprian, M.; et al. Constitutive canonical NF-kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell 2010, 18, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Brown, K.D.; Siebenlist, U.; Staudt, L.M. Constitutive nuclear factor kappaB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2001, 194, 1861–1874. [Google Scholar] [CrossRef] [PubMed]
- Jost, P.J.; Ruland, J. Aberrant NF-kappaB signaling in lymphoma: Mechanisms, consequences, and therapeutic implications. Blood 2007, 109, 2700–2707. [Google Scholar] [PubMed]
- Hinz, M.; Arslan, S.C.; Scheidereit, C. It takes two to tango: IkappaBs, the multifunctional partners of NF-kappaB. Immunol. Rev. 2012, 246, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Loser, P.; Mathas, S.; Krappmann, D.; Dorken, B.; Scheidereit, C. Constitutive NF-kappaB maintains high expression of a characteristic gene network, including CD40, CD86, and a set of antiapoptotic genes in Hodgkin/Reed-Sternberg cells. Blood 2001, 97, 2798–2807. [Google Scholar] [CrossRef] [PubMed]
- Ruland, J.; Duncan, G.S.; Elia, A.; del Barco Barrantes, I.; Nguyen, L.; Plyte, S.; Millar, D.G.; Bouchard, D.; Wakeham, A.; Ohashi, P.S.; et al. BCL10 is a positive regulator of antigen receptor-induced activation of NF-kappaB and neural tube closure. Cell 2001, 104, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Lenz, G.; Davis, R.E.; Ngo, V.N.; Lam, L.; George, T.C.; Wright, G.W.; Dave, S.S.; Zhao, H.; Xu, W.; Rosenwald, A.; et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science 2008, 319, 1676–1679. [Google Scholar] [CrossRef] [PubMed]
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Compagno, M.; Lim, W.K.; Grunn, A.; Nandula, S.V.; Brahmachary, M.; Shen, Q.; Bertoni, F.; Ponzoni, M.; Scandurra, M.; Califano, A.; et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009, 459, 717–721. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Sanada, M.; Kato, I.; Sato, Y.; Takita, J.; Takeuchi, K.; Niwa, A.; Chen, Y.; Nakazaki, K.; Nomoto, J.; et al. Frequent inactivation of a20 in B-cell lymphomas. Nature 2009, 459, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Young, R.M.; Schmitz, R.; Jhavar, S.; Xiao, W.; Lim, K.H.; Kohlhammer, H.; Xu, W.; Yang, Y.; Zhao, H.; et al. Oncogenically active MYD88 mutations in human lymphoma. Nature 2011, 470, 115–119. [Google Scholar] [CrossRef] [PubMed]
- Staudt, L.M., II. Therapy of DLBCL based on genomics. Hematol. Oncol. 2013, 31, S26–S28. [Google Scholar] [CrossRef]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar] [CrossRef] [PubMed]
- Sander, S.; Calado, D.P.; Srinivasan, L.; Kochert, K.; Zhang, B.; Rosolowski, M.; Rodig, S.J.; Holzmann, K.; Stilgenbauer, S.; Siebert, R.; et al. Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis. Cancer Cell 2012, 22, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.T.; Davis, R.E.; Pierce, J.; Hepperle, M.; Xu, Y.; Hottelet, M.; Nong, Y.; Wen, D.; Adams, J.; Dang, L.; et al. Small molecule inhibitors of IkappaB kinase are selectively toxic for subgroups of diffuse large B-cell lymphoma defined by gene expression profiling. Clin. Cancer Res. 2005, 11, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Ngo, V.N.; Davis, R.E.; Lamy, L.; Yu, X.; Zhao, H.; Lenz, G.; Lam, L.T.; Dave, S.; Yang, L.; Powell, J.; et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature 2006, 441, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Ferch, U.; Kloo, B.; Gewies, A.; Pfander, V.; Duwel, M.; Peschel, C.; Krappmann, D.; Ruland, J. Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 2009, 206, 2313–2320. [Google Scholar] [CrossRef] [PubMed]
- Hailfinger, S.; Lenz, G.; Ngo, V.; Posvitz-Fejfar, A.; Rebeaud, F.; Guzzardi, M.; Penas, E.M.; Dierlamm, J.; Chan, W.C.; Staudt, L.M.; et al. Essential role of MALT1 protease activity in activated B cell-like diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2009, 106, 19946–19951. [Google Scholar] [CrossRef] [PubMed]
- Nagel, D.; Spranger, S.; Vincendeau, M.; Grau, M.; Raffegerst, S.; Kloo, B.; Hlahla, D.; Neuenschwander, M.; Peter von Kries, J.; Hadian, K.; et al. Pharmacologic inhibition of MALT1 protease by phenothiazines as a therapeutic approach for the treatment of aggressive ABC-DLBCl. Cancer Cell 2012, 22, 825–837. [Google Scholar] [CrossRef] [PubMed]
- Fontan, L.; Yang, C.; Kabaleeswaran, V.; Volpon, L.; Osborne, M.J.; Beltran, E.; Garcia, M.; Cerchietti, L.; Shaknovich, R.; Yang, S.N.; et al. MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell 2012, 22, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Thome, M. Multifunctional roles for MALT1 in T-cell activation. Nat. Rev. Immunol. 2008, 8, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Jaworski, M.; Marsland, B.J.; Gehrig, J.; Held, W.; Favre, S.; Luther, S.A.; Perroud, M.; Golshayan, D.; Gaide, O.; Thome, M. Malt1 protease inactivation efficiently dampens immune responses but causes spontaneous autoimmunity. EMBO J. 2014, 33, 2765–2781. [Google Scholar] [CrossRef] [PubMed]
- Coornaert, B.; Baens, M.; Heyninck, K.; Bekaert, T.; Haegman, M.; Staal, J.; Sun, L.; Chen, Z.J.; Marynen, P.; Beyaert, R. T cell antigen receptor stimulation induces Malt1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat. Immunol. 2008, 9, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Staal, J.; Driege, Y.; Bekaert, T.; Demeyer, A.; Muyllaert, D.; van Damme, P.; Gevaert, K.; Beyaert, R. T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. 2011, 30, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Hailfinger, S.; Nogai, H.; Pelzer, C.; Jaworski, M.; Cabalzar, K.; Charton, J.E.; Guzzardi, M.; Decaillet, C.; Grau, M.; Dorken, B.; et al. Malt1-dependent RelB cleavage promotes canonical NF-kappaB activation in lymphocytes and lymphoma cell lines. Proc. Natl. Acad. Sci. USA 2011, 108, 14596–14601. [Google Scholar] [CrossRef] [PubMed]
- Kloo, B.; Nagel, D.; Pfeifer, M.; Grau, M.; Duwel, M.; Vincendeau, M.; Dorken, B.; Lenz, P.; Lenz, G.; Krappmann, D. Critical role of PI3K signaling for NF-kappaB-dependent survival in a subset of activated B-cell-like diffuse large B-cell lymphoma cells. Proc. Natl. Acad. Sci. USA 2011, 108, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Naylor, T.L.; Tang, H.; Ratsch, B.A.; Enns, A.; Loo, A.; Chen, L.; Lenz, P.; Waters, N.J.; Schuler, W.; Dorken, B.; et al. Protein kinase c inhibitor sotrastaurin selectively inhibits the growth of CD79 mutant diffuse large B-cell lymphomas. Cancer Res. 2011, 71, 2643–2653. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.; Gerecitano, J.; Goy, A.; de Vos, S.; Kenkre, V.; Barr, P.; Blu, K.; Shustov, A.; Advani, R.; Lih, J.; et al. The bruton’s tyrosine kinase (BTK) inhibitor, ibrutinib (PCI-32765), has preferential activity in the ABC subtype of relapsed/refractory de novo diffuse large B-cell lymphoma (DLBCL): Interim results of a multicenter, open-label, phase 2 study. Blood 2012, 120. ASH Abstract 623. [Google Scholar]
- Mathews Griner, L.A.; Guha, R.; Shinn, P.; Young, R.M.; Keller, J.M.; Liu, D.; Goldlust, I.S.; Yasgar, A.; McKnight, C.; Boxer, M.B.; et al. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill activated B-cell-like diffuse large B-cell lymphoma cells. Proc. Natl. Acad. Sci. USA 2014, 111, 2349–2354. [Google Scholar] [CrossRef] [PubMed]
- Nogai, H.; Wenzel, S.S.; Hailfinger, S.; Grau, M.; Kaergel, E.; Seitz, V.; Wollert-Wulf, B.; Pfeifer, M.; Wolf, A.; Frick, M.; et al. IkappaB-zeta controls the constitutive NF-kappaB target gene network and survival of ABC DLBCL. Blood 2013, 122, 2242–2250. [Google Scholar] [CrossRef] [PubMed]
- Lam, L.T.; Wright, G.; Davis, R.E.; Lenz, G.; Farinha, P.; Dang, L.; Chan, J.W.; Rosenwald, A.; Gascoyne, R.D.; Staudt, L.M. Cooperative signaling through the signal transducer and activator of transcription 3 and nuclear factor-{kappa}B pathways in subtypes of diffuse large B-cell lymphoma. Blood 2008, 111, 3701–3713. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.B.; Yu, J.J.; Yu, R.Y.; Mendez, L.M.; Shaknovich, R.; Zhang, Y.; Cattoretti, G.; Ye, B.H. Constitutively activated STAT3 promotes cell proliferation and survival in the activated B-cell subtype of diffuse large B-cell lymphomas. Blood 2008, 111, 1515–1523. [Google Scholar] [CrossRef] [PubMed]
- Scuto, A.; Kujawski, M.; Kowolik, C.; Krymskaya, L.; Wang, L.; Weiss, L.M.; Digiusto, D.; Yu, H.; Forman, S.; Jove, R. STAT3 inhibition is a therapeutic strategy for ABC-like diffuse large B-cell lymphoma. Cancer Res. 2011, 71, 3182–3188. [Google Scholar] [CrossRef] [PubMed]
- Bea, S.; Zettl, A.; Wright, G.; Salaverria, I.; Jehn, P.; Moreno, V.; Burek, C.; Ott, G.; Puig, X.; Yang, L.; et al. Diffuse large B-cell lymphoma subgroups have distinct genetic profiles that influence tumor biology and improve gene-expression-based survival prediction. Blood 2005, 106, 3183–3190. [Google Scholar] [CrossRef] [PubMed]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, S.S.; Grau, M.; Mavis, C.; Hailfinger, S.; Wolf, A.; Madle, H.; Deeb, G.; Dorken, B.; Thome, M.; Lenz, P.; et al. MCL1 is deregulated in subgroups of diffuse large B-cell lymphoma. Leukemia 2013, 27, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Cerchietti, L.C.; Ghetu, A.F.; Zhu, X.; Da Silva, G.F.; Zhong, S.; Matthews, M.; Bunting, K.L.; Polo, J.M.; Fares, C.; Arrowsmith, C.H.; et al. A small-molecule inhibitor of BCL6 kills DLBCL cells in vitro and in vivo. Cancer Cell 2010, 17, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, M.; Grau, M.; Lenze, D.; Wenzel, S.S.; Wolf, A.; Wollert-Wulf, B.; Dietze, K.; Nogai, H.; Storek, B.; Madle, H.; et al. PTEN loss defines a PI3K/AKT pathway-dependent germinal center subtype of diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 12420–12425. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Monti, S.; Juszczynski, P.; Ouyang, J.; Chapuy, B.; Neuberg, D.; Doench, J.G.; Bogusz, A.M.; Habermann, T.M.; Dogan, A.; et al. SYK inhibition modulates distinct PI3K/AKT-dependent survival pathways and cholesterol biosynthesis in diffuse large B cell lymphomas. Cancer Cell 2013, 23, 826–838. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Johnson, N.A.; Severson, T.M.; Mungall, A.J.; An, J.; Goya, R.; Paul, J.E.; Boyle, M.; Woolcock, B.W.; Kuchenbauer, F.; et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat. Genet. 2010, 42, 181–185. [Google Scholar] [CrossRef] [PubMed]
- McCabe, M.T.; Ott, H.M.; Ganji, G.; Korenchuk, S.; Thompson, C.; van Aller, G.S.; Liu, Y.; Graves, A.P.; Della Pietra, A., 3rd; Diaz, E.; et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature 2012, 492, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Chan, H.; Teng, L.; Li, L.; Chuai, S.; Zhang, R.; Zeng, J.; Li, M.; Fan, H.; Lin, Y.; et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc. Nat. Acad. Sci. USA 2012, 109, 21360–21365. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W.; Wright, G.W.; Williams, P.M.; Lih, C.J.; Walsh, W.; Jaffe, E.S.; Rosenwald, A.; Campo, E.; Chan, W.C.; Connors, J.M.; et al. Determining cell-of-origin subtypes of diffuse large B-cell lymphoma using gene expression in formalin-fixed paraffin-embedded tissue. Blood 2014, 123, 1214–1217. [Google Scholar] [CrossRef] [PubMed]
- Masque-Soler, N.; Szczepanowski, M.; Kohler, C.W.; Spang, R.; Klapper, W. Molecular classification of mature aggressive B-cell lymphoma using digital multiplexed gene expression on formalin-fixed paraffin-embedded biopsy specimens. Blood 2013, 122, 1985–1986. [Google Scholar] [CrossRef] [PubMed]
- Monti, S.; Chapuy, B.; Takeyama, K.; Rodig, S.J.; Hao, Y.; Yeda, K.T.; Inguilizian, H.; Mermel, C.; Currie, T.; Dogan, A.; et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell 2012, 22, 359–372. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenz, G. Insights into the Molecular Pathogenesis of Activated B-Cell-like Diffuse Large B-Cell Lymphoma and Its Therapeutic Implications. Cancers 2015, 7, 811-822. https://doi.org/10.3390/cancers7020812
Lenz G. Insights into the Molecular Pathogenesis of Activated B-Cell-like Diffuse Large B-Cell Lymphoma and Its Therapeutic Implications. Cancers. 2015; 7(2):811-822. https://doi.org/10.3390/cancers7020812
Chicago/Turabian StyleLenz, Georg. 2015. "Insights into the Molecular Pathogenesis of Activated B-Cell-like Diffuse Large B-Cell Lymphoma and Its Therapeutic Implications" Cancers 7, no. 2: 811-822. https://doi.org/10.3390/cancers7020812