
Chromatinization of the KSHV Genome During the KSHV Life Cycle


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Clinical Diseases Associated with KSHV Infection
1.1.1. Kaposi’s Sarcoma (KS)
1.1.2. Primary Effusion Lymphoma (PEL)
1.1.3. Multicentric Castleman’s Disease
1.2. Epidemiology and KSHV Transmission
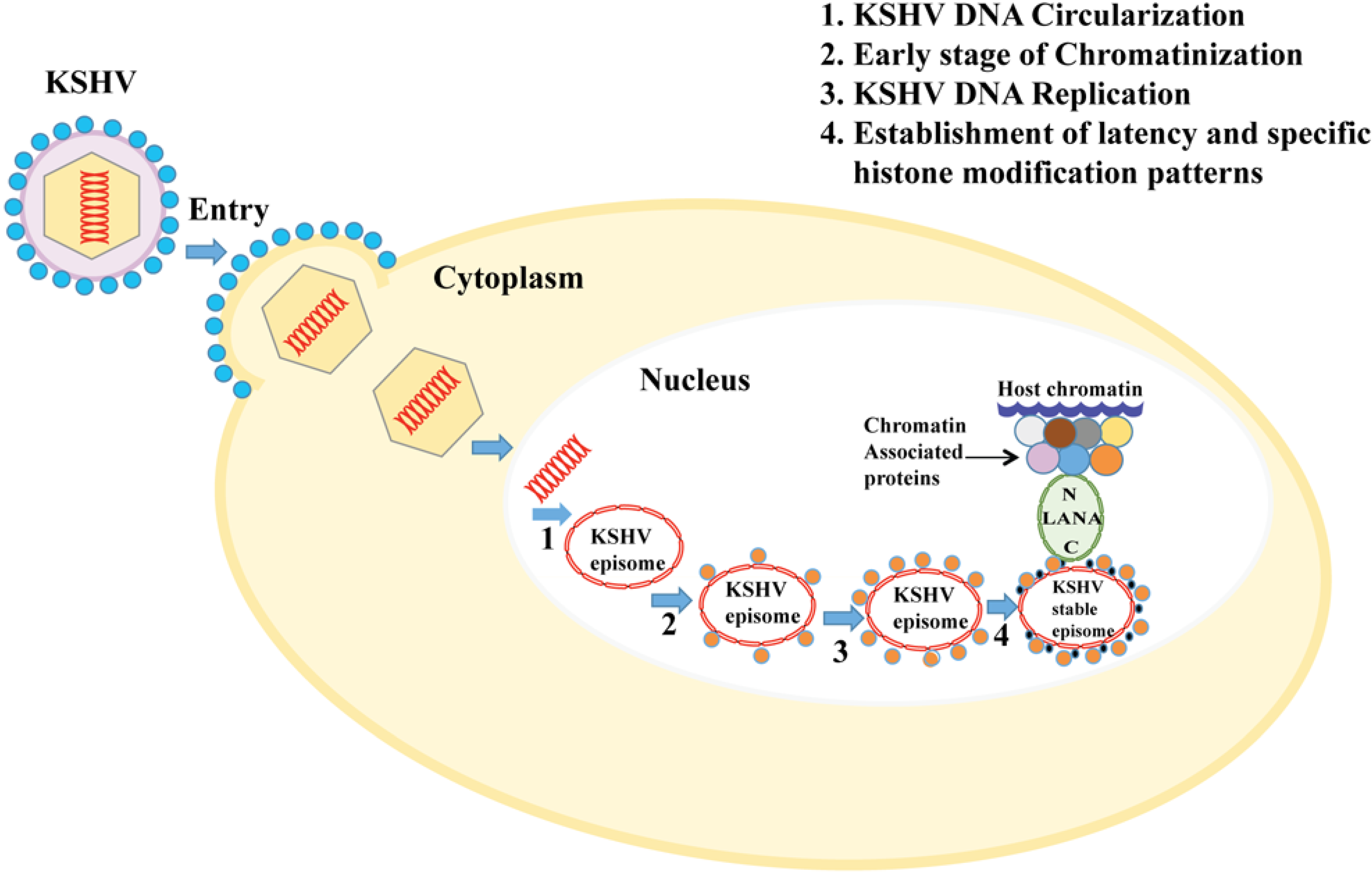
1.3. KSHV Genome and Its Chromatinization
2. Chromatin Regulation and Gene Expression Pattern
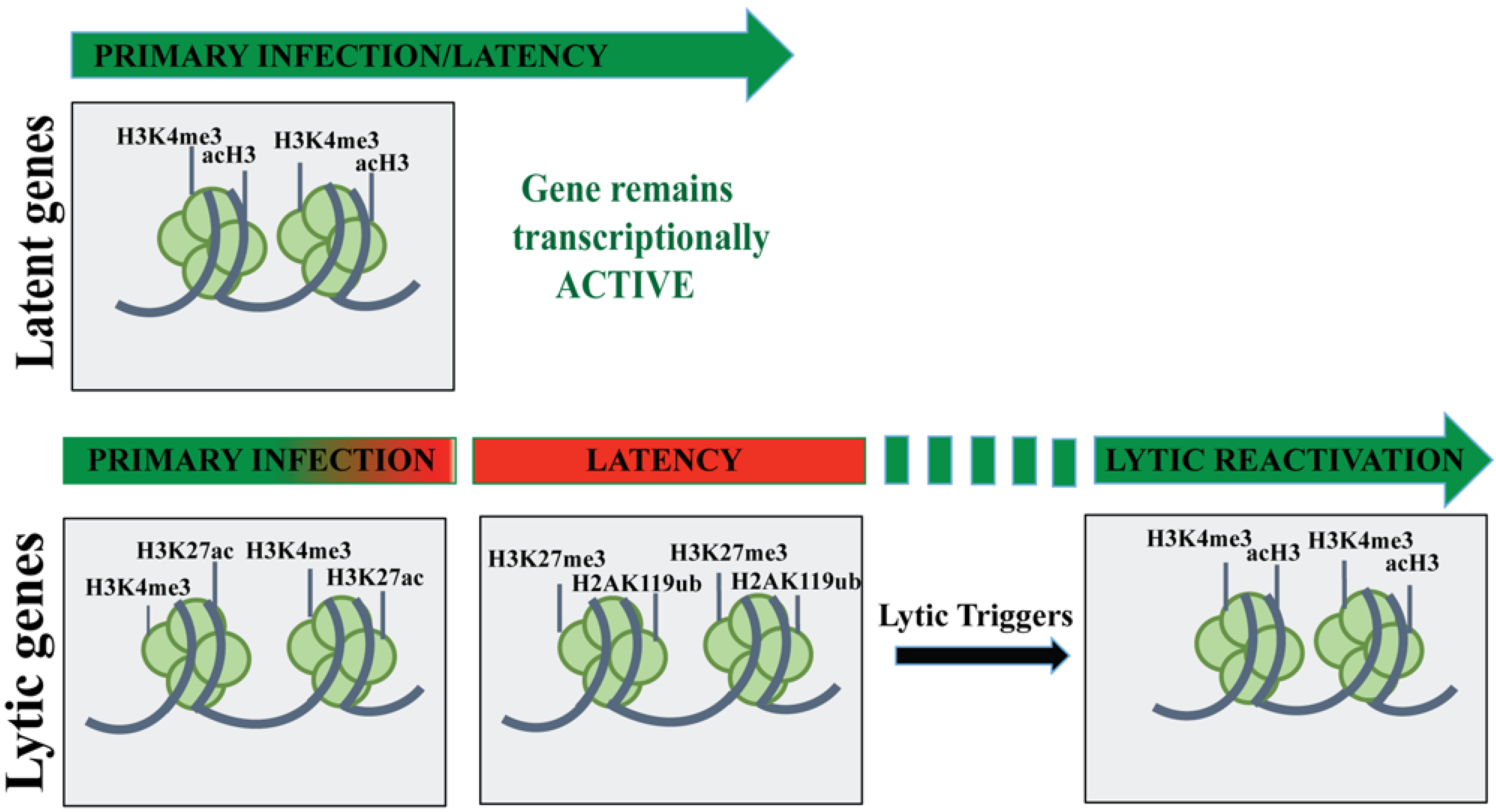
3. KSHV Latent Gene Expression Profile
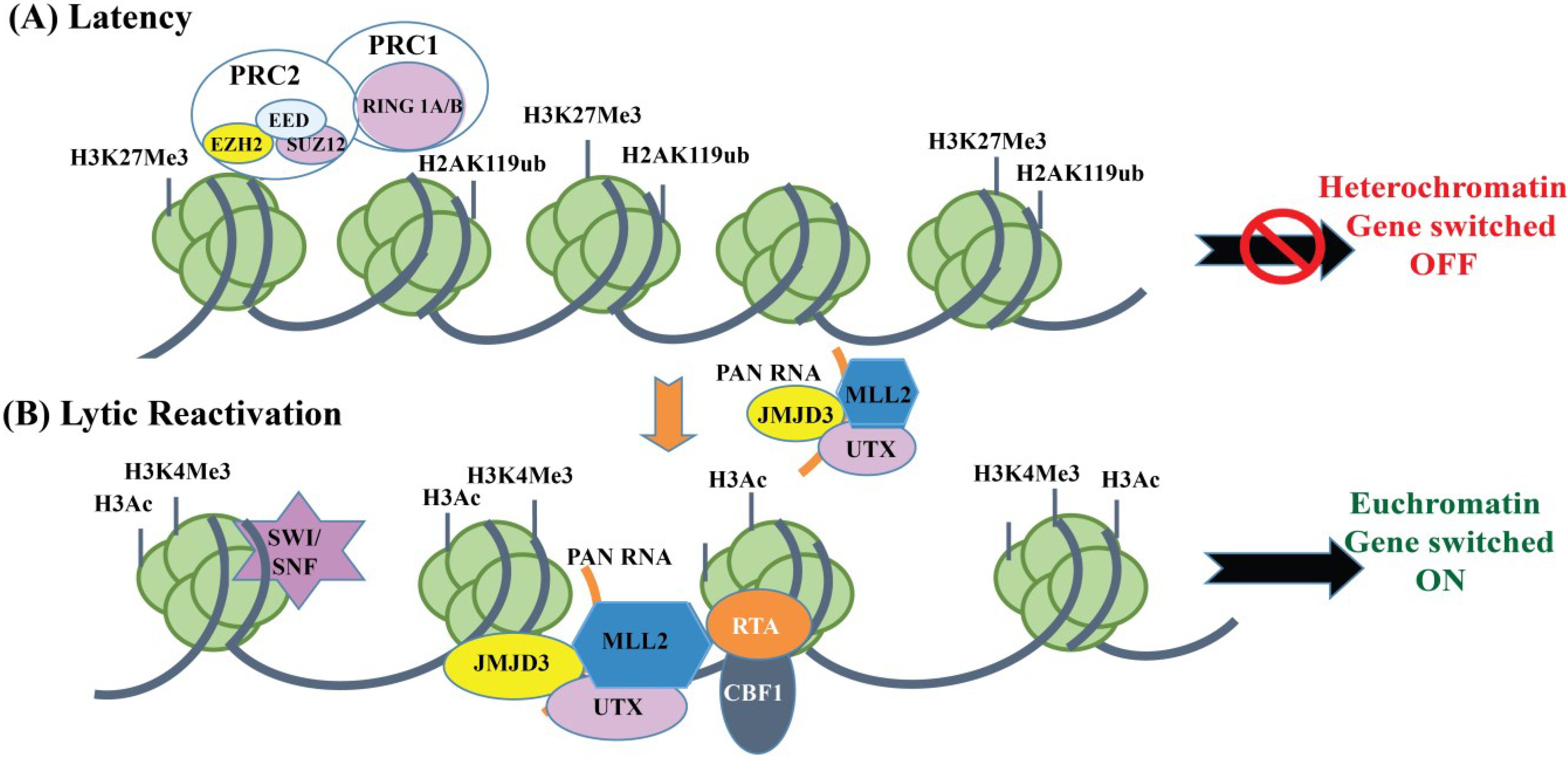
Chromatin Organization of KSHV Genome during Latent Infection
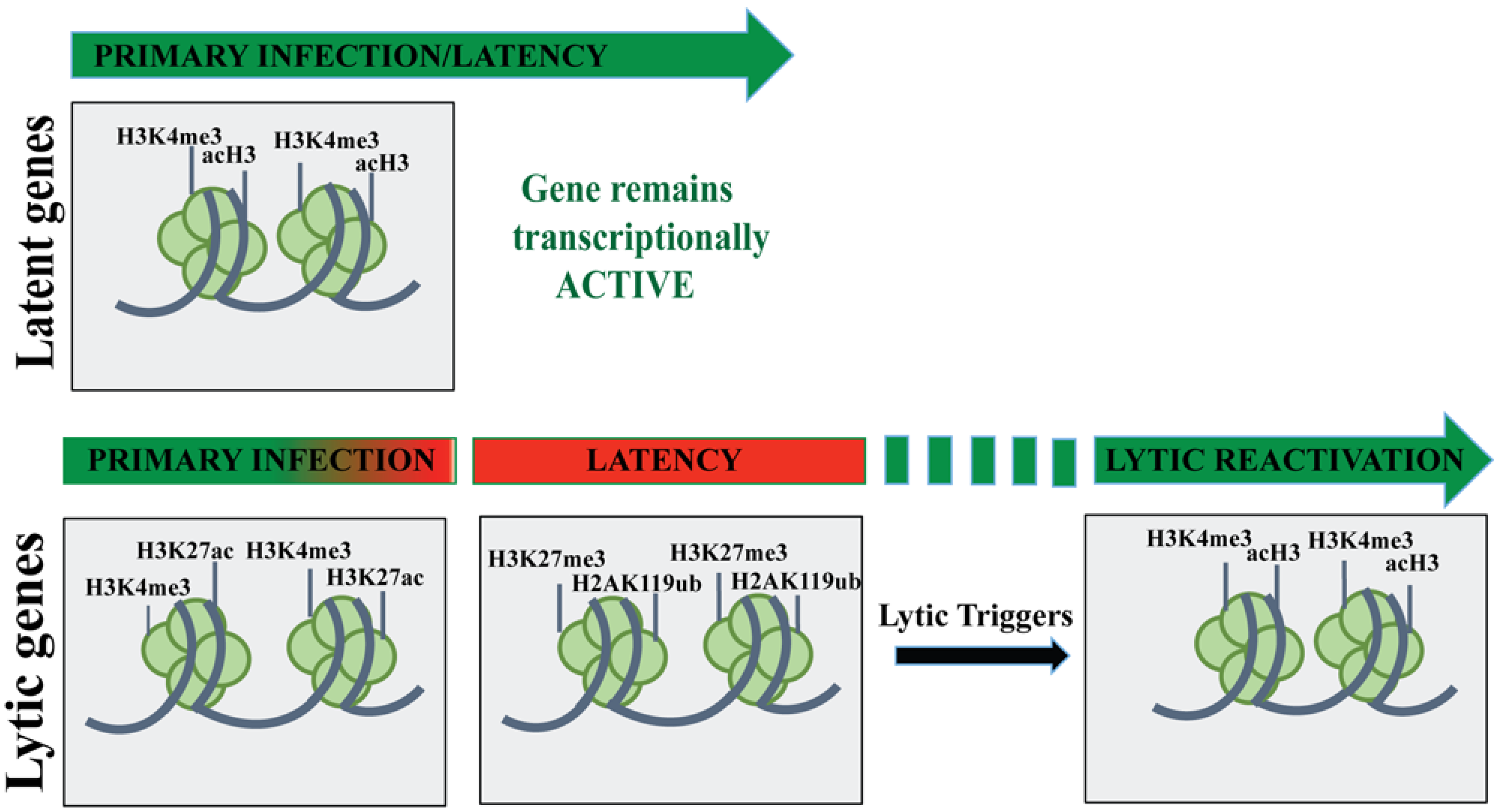
4. KSHV Lytic Gene Expression Profile
4.1. Chromatin Organization of the KSHV Genome during Lytic Reactivation

4.2. Chromatin Organization of KSHV Genome during de Novo Infection
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in aids-associated Kaposiʼs sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposiʼs sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; dʼAgay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposiʼs sarcoma-associated herpesvirus-like DNA sequences in multicentric castlemanʼs disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Deloose, S.T.; Smit, L.A.; Pals, F.T.; Kersten, M.J.; van Noesel, C.J.; Pals, S.T. High incidence of Kaposi sarcoma-associated herpesvirus infection in HIV-related solid immunoblastic/plasmablastic diffuse large B-cell lymphoma. Leukemia 2005, 19, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Cool, C.D.; Rai, P.R.; Yeager, M.E.; Hernandez-Saavedra, D.; Serls, A.E.; Bull, T.M.; Geraci, M.W.; Brown, K.K.; Routes, J.M.; Tuder, R.M.; et al. Expression of human herpesvirus 8 in primary pulmonary hypertension. N. Engl. J. Med. 2003, 349, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Gloghini, A.; Vaccher, E.; Cerri, M.; Gaidano, G.; Dalla-Favera, R.; Tirelli, U. Kaposiʼs sarcoma-associated herpesvirus/human herpesvirus type 8-positive solid lymphomas: A tissue-based variant of primary effusion lymphoma. J. Mol. Diagn. 2005, 7, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Du, M.Q.; Diss, T.C.; Liu, H.; Ye, H.; Hamoudi, R.A.; Cabecadas, J.; Dong, H.Y.; Harris, N.L.; Chan, J.K.; Rees, J.W.; et al. KSHV- and EBV-associated germinotropic lymphoproliferative disorder. Blood 2002, 100, 3415–3418. [Google Scholar] [CrossRef] [PubMed]
- Gbabe, O.F.; Okwundu, C.I.; Dedicoat, M.; Freeman, E.E. Treatment of severe or progressive Kaposiʼs sarcoma in HIV-infected adults. Cochrane Database Syst. Rev. 2014, 8, CD003256. [Google Scholar] [PubMed]
- Cornali, E.; Zietz, C.; Benelli, R.; Weninger, W.; Masiello, L.; Breier, G.; Tschachler, E.; Albini, A.; Sturzl, M. Vascular endothelial growth factor regulates angiogenesis and vascular permeability in Kaposiʼs sarcoma. Am. J. Pathol. 1996, 149, 1851–1869. [Google Scholar] [PubMed]
- Gessain, A.; Duprez, R. Spindle cells and their role in Kaposiʼs sarcoma. Int. J. Biochem. Cell Biol. 2005, 37, 2457–2465. [Google Scholar] [CrossRef] [PubMed]
- Gasperini, P.; Espigol-Frigole, G.; McCormick, P.J.; Salvucci, O.; Maric, D.; Uldrick, T.S.; Polizzotto, M.N.; Yarchoan, R.; Tosato, G. Kaposi sarcoma herpesvirus promotes endothelial-to-mesenchymal transition through notch-dependent signaling. Cancer Res. 2012, 72, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.W.; Trotter, M.W.; Lagos, D.; Bourboulia, D.; Henderson, S.; Makinen, T.; Elliman, S.; Flanagan, A.M.; Alitalo, K.; Boshoff, C. Kaposi sarcoma herpesvirus-induced cellular reprogramming contributes to the lymphatic endothelial gene expression in Kaposi sarcoma. Nat. Genet. 2004, 36, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.K.; Foreman, K.; Shin, J.W.; Hirakawa, S.; Curry, C.L.; Sage, D.R.; Libermann, T.; Dezube, B.J.; Fingeroth, J.D.; Detmar, M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma-associated herpesvirus. Nat. Genet. 2004, 36, 683–685. [Google Scholar] [CrossRef] [PubMed]
- Wen, K.W.; Damania, B. Kaposi sarcoma-associated herpesvirus (KSHV): Molecular biology and oncogenesis. Cancer Lett. 2010, 289, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Dourmishev, L.A.; Dourmishev, A.L.; Palmeri, D.; Schwartz, R.A.; Lukac, D.M. Molecular genetics of Kaposiʼs sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol Mol. Biol. Rev. 2003, 67, 175–212. [Google Scholar] [CrossRef] [PubMed]
- Pantanowitz, L.; Dezube, B.J. Kaposi sarcoma in unusual locations. BMC cancer 2008, 8, 190. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T.; Lawrence, T.S.; Rosenberg, S.A. Cancer: Principles and Practice of Oncology, 8th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2008; Volume 2, p. 3151. [Google Scholar]
- Wool, G.M. Aids-related malignancies. Oncologist 1998, 3, 279–283. [Google Scholar] [PubMed]
- Ablashi, D.V.; Chatlynne, L.G.; Whitman, J.E., Jr.; Cesarman, E. Spectrum of Kaposiʼs sarcoma-associated herpesvirus, or human herpesvirus 8, diseases. Clin. Microbiol. Rev. 2002, 15, 439–464. [Google Scholar] [CrossRef] [PubMed]
- Nador, R.G.; Cesarman, E.; Chadburn, A.; Dawson, D.B.; Ansari, M.Q.; Sald, J.; Knowles, D.M. Primary effusion lymphoma: A distinct clinicopathologic entity associated with the Kaposiʼs sarcoma-associated herpes virus. Blood 1996, 88, 645–656. [Google Scholar] [PubMed]
- Edelman, D.C. Human herpesvirus 8—A novel human pathogen. Virol. J. 2005, 2, 78. [Google Scholar] [CrossRef] [PubMed]
- Haecker, I.; Gay, L.A.; Yang, Y.; Hu, J.; Morse, A.M.; McIntyre, L.M.; Renne, R. Ago HITS-clip expands understanding of Kaposiʼs sarcoma-associated herpesvirus miRNA function in primary effusion lymphomas. PLOS Pathog. 2012, 8, e1002884. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Bubman, D.; Chadburn, A.; Harrington, W.J., Jr.; Cesarman, E.; Knowles, D.M. Distinct subsets of primary effusion lymphoma can be identified based on their cellular gene expression profile and viral association. J. Virol. 2005, 79, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Ohsaki, E.; Nakano, K.; Zheng, X. Characterization of Kaposiʼs sarcoma-associated herpesvirus-related lymphomas by DNA microarray analysis. Leuk. Res. Treat. 2011, 2011, 726964. [Google Scholar]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—An update. Curr. Opin. Virol. 2013, 3, 238–244. [Google Scholar] [CrossRef] [PubMed]
- Curioni, S.; DʼAmico, M.; Quartagno, R.; Martino, S.; DellʼAntonio, G.; Cusi, D. Castlemanʼs disease with nephrotic syndrome, amyloidosis and autoimmune manifestations. Nephrol. Dial. Transplant. 2001, 16, 1475–1478. [Google Scholar] [CrossRef] [PubMed]
- Dupin, N.; Fisher, C.; Kellam, P.; Ariad, S.; Tulliez, M.; Franck, N.; van Marck, E.; Salmon, D.; Gorin, I.; Escande, J.P.; et al. Distribution of human herpesvirus-8 latently infected cells in kaposiʼs sarcoma, multicentric castlemanʼs disease, and primary effusion lymphoma. Proc. Natl. Acad. Sci. USA 1999, 96, 4546–4551. [Google Scholar] [CrossRef] [PubMed]
- Carbone, A.; Cesarman, E.; Spina, M.; Gloghini, A.; Schulz, T.F. HIV-associated lymphomas and gamma-herpesviruses. Blood 2009, 113, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Nishi, J.; Arimura, K.; Utsunomiya, A.; Yonezawa, S.; Kawakami, K.; Maeno, N.; Ijichi, O.; Ikarimoto, N.; Nakata, M.; Kitajima, I.; et al. Expression of vascular endothelial growth factor in sera and lymph nodes of the plasma cell type of castlemanʼs disease. Br. J. Haematol. 1999, 104, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.C.; Kajumbula, H.; Boto, W.; Hayward, G.S. Evaluation of global clustering patterns and strain variation over an extended ORF26 gene locus from Kaposiʼs sarcoma herpesvirus. J. Clin. Virol. 2007, 40, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Hayward, G.S. KSHV strains: The origins and global spread of the virus. Semin. Cancer Biol. 1999, 9, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Hayward, G.S.; Zong, J.C. Modern evolutionary history of the human KSHV genome. Curr. Top. Microbiol. Immunol. 2007, 312, 1–42. [Google Scholar] [PubMed]
- Stebbing, J.; Bourboulia, D.; Johnson, M.; Henderson, S.; Williams, I.; Wilder, N.; Tyrer, M.; Youle, M.; Imami, N.; Kobu, T.; et al. Kaposiʼs sarcoma-associated herpesvirus cytotoxic T lymphocytes recognize and target darwinian positively selected autologous K1 epitopes. J. Virol. 2003, 77, 4306–4314. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.J.; Zong, J.C.; Ciufo, D.M.; Alcendor, D.J.; Cannon, J.S.; Ambinder, R.; Orenstein, J.M.; Reitz, M.S.; Hayward, G.S. Comparison of genetic variability at multiple loci across the genomes of the major subtypes of Kaposiʼs sarcoma-associated herpesvirus reveals evidence for recombination and for two distinct types of open reading frame K15 alleles at the right-hand end. J. Virol. 1999, 73, 6646–6660. [Google Scholar] [PubMed]
- Zong, J.; Ciufo, D.M.; Viscidi, R.; Alagiozoglou, L.; Tyring, S.; Rady, P.; Orenstein, J.; Boto, W.; Kalumbuja, H.; Romano, N.; et al. Genotypic analysis at multiple loci across Kaposiʼs sarcoma herpesvirus (KSHV) DNA molecules: Clustering patterns, novel variants and chimerism. J. Clin. Virol. 2002, 23, 119–148. [Google Scholar] [CrossRef] [PubMed]
- Cook, P.M.; Whitby, D.; Calabro, M.L.; Luppi, M.; Kakoola, D.N.; Hjalgrim, H.; Ariyoshi, K.; Ensoli, B.; Davison, A.J.; Schulz, T.F. Variability and evolution of Kaposiʼs sarcoma-associated herpesvirus in Europe and Africa. International collaborative group. AIDS 1999, 13, 1165–1176. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.X.; Spira, T.J.; Bhat, G.J.; Birch, C.J.; Druce, J.D.; Edlin, B.R.; Edwards, R.; Gunthel, C.; Newton, R.; Stamey, F.R.; et al. Individuals from North America, Australasia, and Africa are infected with four different genotypes of human herpesvirus 8. Virology 1999, 261, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Biggar, R.J.; Whitby, D.; Marshall, V.; Linhares, A.C.; Black, F. Human herpesvirus 8 in Brazilian amerindians: A hyperendemic population with a new subtype. J. Infect. Dis. 2000, 181, 1562–1568. [Google Scholar] [CrossRef] [PubMed]
- Engels, E.A.; Sinclair, M.D.; Biggar, R.J.; Whitby, D.; Ebbesen, P.; Goedert, J.J.; Gastwirth, J.L. Latent class analysis of human herpesvirus 8 assay performance and infection prevalence in sub-Saharan Africa and Malta. Int. J. Cancer 2000, 88, 1003–1008. [Google Scholar] [CrossRef] [PubMed]
- Serraino, D.; Toma, L.; Andreoni, M.; Butto, S.; Tchangmena, O.; Sarmati, L.; Monini, P.; Franceschi, S.; Ensoli, B.; Rezza, G. A seroprevalence study of human herpesvirus type 8 (HHV8) in Eastern and Central Africa and in the mediterranean area. Eur. J. Epidemiol. 2001, 17, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Pellett, P.E.; Wright, D.J.; Engels, E.A.; Ablashi, D.V.; Dollard, S.C.; Forghani, B.; Glynn, S.A.; Goedert, J.J.; Jenkins, F.J.; Lee, T.H.; et al. Multicenter comparison of serologic assays and estimation of human herpesvirus 8 seroprevalence among us blood donors. Transfusion 2003, 43, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Vitale, F.; Viviano, E.; Perna, A.M.; Bonura, F.; Mazzola, G.; Ajello, F.; Romano, N. Serological and virological evidence of non-sexual transmission of human herpesvirus type 8 (HHV8). Epidemiol. Infect. 2000, 125, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Mbulaiteye, S.; Marshall, V.; Bagni, R.K.; Wang, C.D.; Mbisa, G.; Bakaki, P.M.; Owor, A.M.; Ndugwa, C.M.; Engels, E.A.; Katongole-Mbidde, E.; et al. Molecular evidence for mother-to-child transmission of Kaposi sarcoma-associated herpesvirus in uganda and K1 gene evolution within the host. J. Infect. Dis. 2006, 193, 1250–1257. [Google Scholar] [CrossRef] [PubMed]
- Mesri, E.A.; Cesarman, E.; Boshoff, C. Kaposiʼs sarcoma and its associated herpesvirus. Nat. Rev. Cancer 2010, 10, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Coluzzi, M.; Manno, D.; Guzzinati, S.; Tognazzo, S.; Zambon, P.; Arca, B.; Costantini, C.; Ascoli, V. The bloodsucking arthropod bite as possible cofactor in the transmission of human herpesvirus-8 infection and in the expression of Kaposiʼs sarcoma disease. Parassitologia 2002, 44, 123–129. [Google Scholar] [PubMed]
- Ascoli, V.; Facchinelli, L.; Valerio, L.; Manno, D.; Coluzzi, M. Kaposiʼs sarcoma, human herpesvirus 8 infection and the potential role of promoter-arthropod bites in Northern Sweden. J. Med. Virol. 2006, 78, 1452–1455. [Google Scholar] [CrossRef] [PubMed]
- Mbulaiteye, S.M.; Biggar, R.J.; Pfeiffer, R.M.; Bakaki, P.M.; Gamache, C.; Owor, A.M.; Katongole-Mbidde, E.; Ndugwa, C.M.; Goedert, J.J.; Whitby, D.; et al. Water, socioeconomic factors, and human herpesvirus 8 infection in ugandan children and their mothers. J. Acquir. Immune Defic. Syndr. 2005, 38, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Lagunoff, M.; Zhong, W.; Ganem, D. The size and conformation of Kaposiʼs sarcoma-associated herpesvirus (human herpesvirus 8) DNA in infected cells and virions. J. Virol. 1996, 70, 8151–8154. [Google Scholar] [PubMed]
- Russo, J.J.; Bohenzky, R.A.; Chien, M.C.; Chen, J.; Yan, M.; Maddalena, D.; Parry, J.P.; Peruzzi, D.; Edelman, I.S.; Chang, Y.; et al. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. USA 1996, 93, 14862–14867. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.L.; Damania, B. Linking KSHV to human cancer. Curr. Oncol. Rep. 2005, 7, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.X.; Chong, J.M.; Wu, L.; Yuan, Y. Virion proteins of Kaposiʼs sarcoma-associated herpesvirus. J. Virol. 2005, 79, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Spear, P.G.; Longnecker, R. Herpesvirus entry: An update. J. Virol. 2003, 77, 10179–10185. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Keeping it quiet: Chromatin control of gammaherpesvirus latency. Nat. Rev. Microbiol. 2013, 11, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Chromatin regulation of virus infection. Trends Microbiol. 2006, 14, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Luger, K.; Mader, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 a resolution. Nature 1997, 389, 251–260. [Google Scholar] [CrossRef]
- Choudhuri, S.; Cui, Y.; Klaassen, C.D. Molecular targets of epigenetic regulation and effectors of environmental influences. Toxicol. Appl. Pharmacol. 2010, 245, 378–393. [Google Scholar] [CrossRef] [PubMed]
- Campos, E.I.; Reinberg, D. Histones: Annotating chromatin. Annu. Rev. Genet. 2009, 43, 559–599. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Karlic, R.; Chung, H.R.; Lasserre, J.; Vlahovicek, K.; Vingron, M. Histone modification levels are predictive for gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 2926–2931. [Google Scholar] [CrossRef] [PubMed]
- Hirst, M.; Marra, M.A. Next generation sequencing based approaches to epigenomics. Brief. Funct. Genomics 2010, 9, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Sarda, S.; Hannenhalli, S. Next-generation sequencing and epigenomics research: A hammer in search of nails. Genomics Inf. 2014, 12, 2–11. [Google Scholar] [CrossRef]
- Chen, H.S.; Wikramasinghe, P.; Showe, L.; Lieberman, P.M. Cohesins repress Kaposiʼs sarcoma-associated herpesvirus immediate early gene transcription during latency. J. Virol. 2012, 86, 9454–9464. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Cho, H.; Sung, G.H.; Lieberman, P.M. CTCF regulates Kaposiʼs sarcoma-associated herpesvirus latency transcription by nucleosome displacement and RNA polymerase programming. J. Virol. 2013, 87, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Lieberman, P.M. Cell cycle control of Kaposiʼs sarcoma-associated herpesvirus latency transcription by CTCF-cohesin interactions. J. Virol. 2009, 83, 6199–6210. [Google Scholar] [CrossRef] [PubMed]
- Hilton, I.B.; Simon, J.M.; Lieb, J.D.; Davis, I.J.; Damania, B.; Dittmer, D.P. The open chromatin landscape of Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2013, 87, 11831–11842. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.; Kuhne, K.; Ye, F.; Chen, J.; Zhou, F.; Lei, X.; Gao, S.J. Molecular biology of KSHV in relation to AIDS-associated oncogenesis. Cancer Treat. Res. 2007, 133, 69–127. [Google Scholar] [PubMed]
- Toth, Z.; Brulois, K.; Jung, J.U. The chromatin landscape of Kaposi’s sarcoma-associated herpesvirus. Viruses 2013, 5, 1346–1373. [Google Scholar] [CrossRef] [PubMed]
- Dittmer, D.; Lagunoff, M.; Renne, R.; Staskus, K.; Haase, A.; Ganem, D. A cluster of latently expressed genes in Kaposiʼs sarcoma-associated herpesvirus. J. Virol. 1998, 72, 8309–8315. [Google Scholar] [PubMed]
- Sturzl, M.; Hohenadl, C.; Zietz, C.; Castanos-Velez, E.; Wunderlich, A.; Ascherl, G.; Biberfeld, P.; Monini, P.; Browning, P.J.; Ensoli, B. Expression of K13/V-FLIP gene of human herpesvirus 8 and apoptosis in Kaposiʼs sarcoma spindle cells. J. Natl. Cancer Inst. 1999, 91, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.A.; Sturzl, M.A.; Blasig, C.; Schreier, A.; Guo, H.G.; Reitz, M.; Opalenik, S.R.; Browning, P.J. Expression of human herpesvirus 8-encoded cyclin D in Kaposiʼs sarcoma spindle cells. J. Natl. Cancer Inst. 1997, 89, 1868–1874. [Google Scholar] [CrossRef] [PubMed]
- Muralidhar, S.; Veytsmann, G.; Chandran, B.; Ablashi, D.; Doniger, J.; Rosenthal, L.J. Characterization of the human herpesvirus 8 (Kaposiʼs sarcoma-associated herpesvirus) oncogene, kaposin (ORF K12). J. Clin. Virol. 2000, 16, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Lu, S.; Zhang, Z.; Gonzalez, C.M.; Damania, B.; Cullen, B.R. Kaposiʼs sarcoma-associated herpesvirus expresses an array of viral micrornas in latently infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 5570–5575. [Google Scholar] [CrossRef] [PubMed]
- Ballestas, M.E.; Chatis, P.A.; Kaye, K.M. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 1999, 284, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.C.; Zhou, F.C.; Xie, J.P.; Kang, T.; Greene, W.; Kuhne, K.; Lei, X.F.; Li, Q.H.; Gao, S.J. Kaposiʼs sarcoma-associated herpesvirus latent gene vFLIP inhibits viral lytic replication through NF-kappaB-mediated suppression of the AP-1 pathway: A novel mechanism of virus control of latency. J. Virol. 2008, 82, 4235–4249. [Google Scholar] [CrossRef] [PubMed]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.L.; Schroter, M.; et al. Viral flice-inhibitory proteins (FLIPS) prevent apoptosis induced by death receptors. Nature 1997, 386, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Godden-Kent, D.; Talbot, S.J.; Boshoff, C.; Chang, Y.; Moore, P.; Weiss, R.A.; Mittnacht, S. The cyclin encoded by Kaposiʼs sarcoma-associated herpesvirus stimulates CDK6 to phosphorylate the retinoblastoma protein and histone H1. J. Virol. 1997, 71, 4193–4198. [Google Scholar] [PubMed]
- Li, H.; Komatsu, T.; Dezube, B.J.; Kaye, K.M. The Kaposiʼs sarcoma-associated herpesvirus K12 transcript from a primary effusion lymphoma contains complex repeat elements, is spliced, and initiates from a novel promoter. J. Virol. 2002, 76, 11880–11888. [Google Scholar] [CrossRef] [PubMed]
- Bieleski, L.; Talbot, S.J. Kaposiʼs sarcoma-associated herpesvirus vcyclin open reading frame contains an internal ribosome entry site. J. Virol. 2001, 75, 1864–1869. [Google Scholar] [CrossRef] [PubMed]
- Tempera, I.; Lieberman, P.M. Chromatin organization of gammaherpesvirus latent genomes. Biochim. Biophys. Acta 2010, 1799, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, K.L.; Frappier, L. Viral proteomics. Microbiol. Mol. Biol. Rev. 2007, 71, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Cotter, M.A., 2nd; Robertson, E.S. The latency-associated nuclear antigen tethers the Kaposiʼs sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology 1999, 264, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Si, H.; Verma, S.C.; Lampson, M.A.; Cai, Q.; Robertson, E.S. Kaposiʼs sarcoma-associated herpesvirus-encoded LANA can interact with the nuclear mitotic apparatus protein to regulate genome maintenance and segregation. J. Virol. 2008, 82, 6734–6746. [Google Scholar] [CrossRef] [PubMed]
- Kelley-Clarke, B.; de Leon-Vazquez, E.; Slain, K.; Barbera, A.J.; Kaye, K.M. Role of Kaposiʼs sarcoma-associated herpesvirus C-terminal LANA chromosome binding in episome persistence. J. Virol. 2009, 83, 4326–4337. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Hayward, S.D. The latency-associated nuclear antigen of Kaposiʼs sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3beta. J. Virol. 2003, 77, 8019–8030. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Wu, F.Y.; ApRhys, C.; Kajumbula, H.; Young, D.B.; Hayward, G.S.; Hayward, S.D. A novel viral mechanism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency. Nat. Med. 2003, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Lan, K.; Verma, S.C.; Si, H.; Lin, D.; Robertson, E.S. Kaposiʼs sarcoma-associated herpesvirus latent protein LANA interacts with HIF-1 alpha to upregulate RTA expression during hypoxia: Latency control under low oxygen conditions. J. Virol. 2006, 80, 7965–7975. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Martin, H.J.; Liao, G.; Hayward, S.D. The Kaposiʼs sarcoma-associated herpesvirus LANA protein stabilizes and activates C-myc. J. Virol. 2007, 81, 10451–10459. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Murakami, M.; Si, H.; Robertson, E.S. A potential alpha-helix motif in the amino terminus of LANA encoded by Kaposiʼs sarcoma-associated herpesvirus is critical for nuclear accumulation of HIF-1alpha in normoxia. J. Virol. 2007, 81, 10413–10423. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.L.; Knight, J.S.; Verma, S.C.; Zald, P.; Robertson, E.S. Ec5s ubiquitin complex is recruited by KSHV latent antigen LANA for degradation of the VHL and P53 tumor suppressors. PLOS Pathog. 2006, 2, e116. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.; Yamboliev, I.; Pari, G.S. Kaposiʼs sarcoma-associated herpesvirus/human herpesvirus 8 K-BZIP modulates latency-associated nuclear protein-mediated suppression of lytic origin-dependent DNA synthesis. J. Virol. 2009, 83, 8492–8501. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Verma, S.C.; Murakami, M.; Cai, Q.; Kumar, P.; Xiao, B.; Robertson, E.S. Latency-associated nuclear antigen of Kaposiʼs sarcoma-associated herpesvirus (KSHV) upregulates survivin expression in KSHV-associated B-lymphoma cells and contributes to their proliferation. J. Virol. 2009, 83, 7129–7141. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Verma, S.C.; Cai, Q.; Kaul, R.; Lu, J.; Saha, A.; Robertson, E.S. BUB1 and CENP-F can contribute to Kaposiʼs sarcoma-associated herpesvirus genome persistence by targeting LANA to kinetochores. J. Virol. 2010, 84, 9718–9732. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.C.; Choi, J.Y.; Ma, M.; Robertson, E.S. Kaposiʼs sarcoma-associated herpesvirus inhibits interleukin-4-mediated STAT6 phosphorylation to regulate apoptosis and maintain latency. J. Virol. 2010, 84, 11134–11144. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.C.; Shu, M.A.; Hu, J.; Renne, R. DNA binding and modulation of gene expression by the latency-associated nuclear antigen of Kaposiʼs sarcoma-associated herpesvirus. J. Virol. 2001, 75, 7882–7892. [Google Scholar] [CrossRef] [PubMed]
- Cotter, M.A., 2nd; Subramanian, C.; Robertson, E.S. The Kaposiʼs sarcoma-associated herpesvirus latency-associated nuclear antigen binds to specific sequences at the left end of the viral genome through its carboxy-terminus. Virology 2001, 291, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.C.; Hu, J.; Renne, R. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem. 2002, 277, 27401–27411. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.Y.; Wilson, A.C. Kaposiʼs sarcoma-associated herpesvirus latency-associated nuclear antigen induces a strong bend on binding to terminal repeat DNA. J. Virol. 2005, 79, 13829–13836. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, T.; Ballestas, M.E.; Barbera, A.J.; Kelley-Clarke, B.; Kaye, K.M. KSHV LANA1 binds DNA as an oligomer and residues N-terminal to the oligomerization domain are essential for DNA binding, replication, and episome persistence. Virology 2004, 319, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Lee, D.; Seo, T.; Choi, C.; Choe, J. Latency-associated nuclear antigen of Kaposiʼs sarcoma-associated herpesvirus functionally interacts with heterochromatin protein 1. J. Biol. Chem. 2003, 278, 7397–7405. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Cai, Q.; Kreider, E.; Lu, J.; Robertson, E.S. Comprehensive analysis of LANA interacting proteins essential for viral genome tethering and persistence. PLOS ONE 2013, 8, e74662. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.C.; Lu, J.; Robertson, E.S. Molecular biology of Kaposiʼs sarcoma-associated herpesvirus and related oncogenesis. Adv. Virus Res. 2010, 78, 87–142. [Google Scholar] [PubMed]
- Hu, J.; Garber, A.C.; Renne, R. The latency-associated nuclear antigen of Kaposiʼs sarcoma-associated herpesvirus supports latent DNA replication in dividing cells. J. Virol. 2002, 76, 11677–11687. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Lan, K.; Robertson, E. Structure and function of latency-associated nuclear antigen. Curr. Top. Microbiol. Immunol. 2007, 312, 101–136. [Google Scholar] [PubMed]
- Stedman, W.; Deng, Z.; Lu, F.; Lieberman, P.M. Orc, mcm, and histone hyperacetylation at the Kaposi’s sarcoma-associated herpesvirus latent replication origin. J. Virol. 2004, 78, 12566–12575. [Google Scholar] [CrossRef] [PubMed]
- Purushothaman, P.; McDowell, M.E.; McGuinness, J.; Salas, R.; Rumjahn, S.M.; Verma, S.C. Kaposiʼs sarcoma-associated herpesvirus-encoded LANA recruits topoisomerase iibeta for latent DNA replication of the terminal repeats. J. Virol. 2012, 86, 9983–9994. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Choudhuri, T.; Kaul, R.; Robertson, E.S. Latency-associated nuclear antigen (LANA) of Kaposi’s sarcoma-associated herpesvirus interacts with origin recognition complexes at the LANA binding sequence within the terminal repeats. J. Virol. 2006, 80, 2243–2256. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Choudhuri, T.; Robertson, E.S. The minimal replicator element of the Kaposiʼs sarcoma-associated herpesvirus terminal repeat supports replication in a semiconservative and cell-cycle-dependent manner. J. Virol. 2007, 81, 3402–3413. [Google Scholar] [CrossRef] [PubMed]
- Nishitani, H.; Lygerou, Z. Control of DNA replication licensing in a cell cycle. Genes Cells 2002, 7, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Bell, S.P.; Dutta, A. DNA replication in eukaryotic cells. Annu. Rev. Biochem. 2002, 71, 333–374. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Tsurimoto, T.; Juillard, F.; Li, L.; Li, S.; de Leon Vazquez, E.; Chen, S.; Kaye, K. Kaposiʼs sarcoma-associated herpesvirus LANA recruits the DNA polymerase clamp loader to mediate efficient replication and virus persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 11816–11821. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.C.; Borah, S.; Robertson, E.S. Latency-associated nuclear antigen of Kaposiʼs sarcoma-associated herpesvirus up-regulates transcription of human telomerase reverse transcriptase promoter through interaction with transcription factor SP1. J. Virol. 2004, 78, 10348–10359. [Google Scholar] [CrossRef] [PubMed]
- Friborg, J., Jr.; Kong, W.; Hottiger, M.O.; Nabel, G.J. P53 inhibition by the LANA protein of KSHV protects against cell death. Nature 1999, 402, 889–894. [Google Scholar] [PubMed]
- Radkov, S.A.; Kellam, P.; Boshoff, C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene HRAS transforms primary rat cells. Nat. Med. 2000, 6, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Van Dross, R.; Yao, S.; Asad, S.; Westlake, G.; Mays, D.J.; Barquero, L.; Duell, S.; Pietenpol, J.A.; Browning, P.J. Constitutively active K-cyclin/CDK6 kinase in Kaposi sarcoma-associated herpesvirus-infected cells. J. Natl. Cancer Inst. 2005, 97, 656–666. [Google Scholar] [CrossRef] [PubMed]
- Direkze, S.; Laman, H. Regulation of growth signalling and cell cycle by Kaposiʼs sarcoma-associated herpesvirus genes. Int. J. Exp. Pathol. 2004, 85, 305–319. [Google Scholar] [CrossRef] [PubMed]
- Sarek, G.; Jarviluoma, A.; Moore, H.M.; Tojkander, S.; Vartia, S.; Biberfeld, P.; Laiho, M.; Ojala, P.M. Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency. PLOS Pathog. 2010, 6, e1000818. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Paden, C.R.; Morales, F.M.; Powers, R.P.; Jacob, J.; Speck, S.H. Murine gamma-herpesvirus immortalization of fetal liver-derived B cells requires both the viral cyclin D homolog and latency-associated nuclear antigen. PLOS Pathog. 2011, 7, e1002220. [Google Scholar] [CrossRef] [PubMed]
- Guasparri, I.; Keller, S.A.; Cesarman, E. KSHV vFLIP is essential for the survival of infected lymphoma cells. J. Exp. Med. 2004, 199, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Israel, A. The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harbor Perspect. Biol. 2010, 2, a000158. [Google Scholar] [CrossRef]
- Field, N.; Low, W.; Daniels, M.; Howell, S.; Daviet, L.; Boshoff, C.; Collins, M. KSHV vFLIP binds to IKK-gamma to activate IKK. J. Cell Sci. 2003, 116, 3721–3728. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Lei, X.; Gao, S.J. Mechanisms of Kaposiʼs sarcoma-associated herpesvirus latency and reactivation. Adv. Virol. 2011, 2011. Article ID 193860. [Google Scholar] [PubMed]
- Sadler, R.; Wu, L.; Forghani, B.; Renne, R.; Zhong, W.; Herndier, B.; Ganem, D. A complex translational program generates multiple novel proteins from the latently expressed kaposin (K12) locus of Kaposiʼs sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5722–5730. [Google Scholar] [PubMed]
- McCormick, C.; Ganem, D. The kaposin B protein of KSHV activates the P38/MK2 pathway and stabilizes cytokine mrnas. Science 2005, 307, 739–741. [Google Scholar] [CrossRef] [PubMed]
- Kanno, T.; Sato, Y.; Sata, T.; Katano, H. Expression of Kaposiʼs sarcoma-associated herpesvirus-encoded K10/10.1 protein in tissues and its interaction with poly(A)-binding protein. Virology 2006, 352, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Esteban, M.; Garcia, M.A.; Domingo-Gil, E.; Arroyo, J.; Nombela, C.; Rivas, C. The latency protein LANA2 from Kaposiʼs sarcoma-associated herpesvirus inhibits apoptosis induced by dsRNA-activated protein kinase but not RNase l activation. J. Gen. Virol. 2003, 84, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Rivas, C.; Thlick, A.E.; Parravicini, C.; Moore, P.S.; Chang, Y. Kaposiʼs sarcoma-associated herpesvirus LANA2 is a B-cell-specific latent viral protein that inhibits p53. J. Virol. 2001, 75, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.T.; Kincaid, R.P.; Arasappan, D.; Dowd, S.E.; Hunicke-Smith, S.P.; Sullivan, C.S. Small RNA profiling reveals antisense transcription throughout the KSHV genome and novel small RNAs. RNA 2010, 16, 1540–1558. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Cullen, B.R. Transcriptional origin of Kaposiʼs sarcoma-associated herpesvirus micrornas. J. Virol. 2006, 80, 2234–2242. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Sullivan, C.S.; Ganem, D. A combined computational and microarray-based approach identifies novel micrornas encoded by human gamma-herpesviruses. RNA 2006, 12, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Sewer, A.; Lagos-Quintana, M.; Sheridan, R.; Sander, C.; Grasser, F.A.; van Dyk, L.F.; Ho, C.K.; Shuman, S.; Chien, M.; et al. Identification of micrornas of the herpesvirus family. Nat. Methods 2005, 2, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Samols, M.A.; Hu, J.; Skalsky, R.L.; Renne, R. Cloning and identification of a microrna cluster within the latency-associated region of Kaposiʼs sarcoma-associated herpesvirus. J. Virol. 2005, 79, 9301–9305. [Google Scholar] [CrossRef] [PubMed]
- Feldman, E.R.; Kara, M.; Coleman, C.B.; Grau, K.R.; Oko, L.M.; Krueger, B.J.; Renne, R.; van Dyk, L.F.; Tibbetts, S.A. Virus-encoded micrornas facilitate gammaherpesvirus latency and pathogenesis in vivo. mBio 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Bai, Z.; Ye, F.; Huang, Y.; Gao, S.J. Regulation of herpesvirus lifecycle by viral micrornas. Virulence 2010, 1, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.C.; Li, Z.; Chu, C.Y.; Feng, J.; Feng, J.; Sun, R.; Rana, T.M. Micrornas encoded by Kaposiʼs sarcoma-associated herpesvirus regulate viral life cycle. EMBO Rep. 2010, 11, 784–790. [Google Scholar] [CrossRef] [PubMed]
- Plaisance-Bonstaff, K.; Choi, H.S.; Beals, T.; Krueger, B.J.; Boss, I.W.; Gay, L.A.; Haecker, I.; Hu, J.; Renne, R. Kshv mirnas decrease expression of lytic genes in latently infected PEL and endothelial cells by targeting host transcription factors. Viruses 2014, 6, 4005–4023. [Google Scholar] [CrossRef] [PubMed]
- Moody, R.; Zhu, Y.; Huang, Y.; Cui, X.; Jones, T.; Bedolla, R.; Lei, X.; Bai, Z.; Gao, S.J. KSHV micrornas mediate cellular transformation and tumorigenesis by redundantly targeting cell growth and survival pathways. PLOS Pathog. 2013, 9, e1003857. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Bai, Z.; Ye, F.; Xie, J.; Kim, C.G.; Huang, Y.; Gao, S.J. Regulation of NF-kappaB inhibitor ikappabalpha and viral replication by a KSHV microrna. Nat. Cell Biol. 2010, 12, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Suffert, G.; Malterer, G.; Hausser, J.; Viiliainen, J.; Fender, A.; Contrant, M.; Ivacevic, T.; Benes, V.; Gros, F.; Voinnet, O.; et al. Kaposiʼs sarcoma herpesvirus micrornas target caspase 3 and regulate apoptosis. PLOS Pathog. 2011, 7, e1002405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, Z.; Jakymiw, A.; Findlay, V.; Parsons, C. KSHV-encoded micrornas: Lessons for viral cancer pathogenesis and emerging concepts. Int. J. Cell Biol. 2012, 2012, 603961. [Google Scholar] [CrossRef] [PubMed]
- Ballestas, M.E.; Kaye, K.M. Kaposiʼs sarcoma-associated herpesvirus latency-associated nuclear antigen 1 mediates episome persistence through CIS-acting terminal repeat (TR) sequence and specifically binds TR DNA. J. Virol. 2001, 75, 3250–3258. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Renne, R. Characterization of the minimal replicator of Kaposiʼs sarcoma-associated herpesvirus latent origin. J. Virol. 2005, 79, 2637–2642. [Google Scholar] [CrossRef] [PubMed]
- Skalsky, R.L.; Hu, J.; Renne, R. Analysis of viral cis elements conferring Kaposiʼs sarcoma-associated herpesvirus episome partitioning and maintenance. J. Virol. 2007, 81, 9825–9837. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.R.; Wong, L.Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; et al. Epigenetic analysis of KSHV latent and lytic genomes. PLOS Pathog. 2010, 6, e1001013. [Google Scholar] [CrossRef] [PubMed]
- Gunther, T.; Grundhoff, A. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLOS Pathog. 2010, 6, e1000935. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhang, W.; Bakken, T.; Schutten, M.; Toth, Z.; Jung, J.U.; Gill, P.; Cannon, M.; Gao, S.J. Cancer angiogenesis induced by Kaposi sarcoma-associated herpesvirus is mediated by EZH2. Cancer Res. 2012, 72, 3582–3592. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Fitzgerald, L.D.; Hsia, D.A.; Izumiya, Y.; Wu, C.Y.; Hsieh, W.P.; Lin, S.F.; Campbell, M.; Lam, K.S.; Luciw, P.A.; et al. Histone demethylase JMJD2A regulates Kaposiʼs sarcoma-associated herpesvirus replication and is targeted by a viral transcriptional factor. J. Virol. 2011, 85, 3283–3293. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Allday, M.J. Epigenetic reprogramming of host genes in viral and microbial pathogenesis. Trends Microbiol. 2010, 18, 439–447. [Google Scholar] [CrossRef] [PubMed]
- Gunther, T.; Schreiner, S.; Dobner, T.; Tessmer, U.; Grundhoff, A. Influence of ND10 components on epigenetic determinants of early KSHV latency establishment. PLOS Pathog. 2014, 10, e1004274. [Google Scholar] [CrossRef] [PubMed]
- Merkenschlager, M.; Odom, D.T. CTCF and Cohesin: Linking gene regulatory elements with their targets. Cell 2013, 152, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Wiedmer, A.; Yuan, Y.; Robertson, E.; Lieberman, P.M. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLOS Pathog. 2011, 7, e1002140. [Google Scholar] [CrossRef] [PubMed]
- Stedman, W.; Kang, H.; Lin, S.; Kissil, J.L.; Bartolomei, M.S.; Lieberman, P.M. Cohesins localize with CTCF at the KSHV latency control region and at cellular C-myc and H19/IGF2 insulators. EMBO J. 2008, 27, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Li, D.J.; Verma, D.; Mosbruger, T.; Swaminathan, S. CTCF and RAD21 act as host cell restriction factors for Kaposiʼs sarcoma-associated herpesvirus (KSHV) lytic replication by modulating viral gene transcription. PLOS Pathog. 2014, 10, e1003880. [Google Scholar] [CrossRef] [PubMed]
- Losada, A. Cohesin in cancer: Chromosome segregation and beyond. Nat. Rev. Cancer 2014, 14, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.S.; Lu, F.; Lieberman, P.M. Epigenetic regulation of EBV and KSHV latency. Curr. Opin. Virol. 2013, 3, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Darst, R.P.; Haecker, I.; Pardo, C.E.; Renne, R.; Kladde, M.P. Epigenetic diversity of Kaposiʼs sarcoma-associated herpesvirus. Nucl. Acids Res. 2013, 41, 2993–3009. [Google Scholar] [CrossRef] [PubMed]
- Darst, R.P.; Nabilsi, N.H.; Pardo, C.E.; Riva, A.; Kladde, M.P. DNA methyltransferase accessibility protocol for individual templates by deep sequencing. Methods Enzymol. 2012, 513, 185–204. [Google Scholar] [PubMed]
- Staskus, K.A.; Zhong, W.; Gebhard, K.; Herndier, B.; Wang, H.; Renne, R.; Beneke, J.; Pudney, J.; Anderson, D.J.; Ganem, D.; et al. Kaposiʼs sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 1997, 71, 715–719. [Google Scholar] [PubMed]
- Zhong, W.; Wang, H.; Herndier, B.; Ganem, D. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc. Natl. Acad. Sci. USA 1996, 93, 6641–6646. [Google Scholar] [CrossRef] [PubMed]
- Cavallin, L.E.; Goldschmidt-Clermont, P.; Mesri, E.A. Molecular and cellular mechanisms of KSHV oncogenesis of Kaposiʼs sarcoma associated with HIV/AIDS. PLOS Pathog. 2014, 10, e1004154. [Google Scholar] [CrossRef] [PubMed]
- Honess, R.W.; Roizman, B. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 1974, 14, 8–19. [Google Scholar] [PubMed]
- Sun, R.; Lin, S.F.; Staskus, K.; Gradoville, L.; Grogan, E.; Haase, A.; Miller, G. Kinetics of Kaposiʼs sarcoma-associated herpesvirus gene expression. J. Virol. 1999, 73, 2232–2242. [Google Scholar] [PubMed]
- Zhu, F.X.; Cusano, T.; Yuan, Y. Identification of the immediate-early transcripts of Kaposiʼs sarcoma-associated herpesvirus. J. Virol. 1999, 73, 5556–5567. [Google Scholar] [PubMed]
- Jenner, R.G.; Alba, M.M.; Boshoff, C.; Kellam, P. Kaposiʼs sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 2001, 75, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Lukac, D.M.; Kirshner, J.R.; Ganem, D. Transcriptional activation by the product of open reading frame 50 of Kaposiʼs sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 1999, 73, 9348–9361. [Google Scholar] [PubMed]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of Kaposiʼs sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Lin, S.F.; Gradoville, L.; Yuan, Y.; Zhu, F.; Miller, G. A viral gene that activates lytic cycle expression of Kaposiʼs sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 1998, 95, 10866–10871. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; AuCoin, D.P.; Huete, A.R.; Cei, S.A.; Hanson, L.J.; Pari, G.S. A Kaposiʼs sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 2005, 79, 3479–3487. [Google Scholar] [CrossRef] [PubMed]
- Glaunsinger, B.; Ganem, D. Lytic KSHV infection inhibits host gene expression by accelerating global mrna turnover. Mol. Cell 2004, 13, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Yamanegi, K.; Zheng, Z.M. Gene structure and expression of Kaposiʼs sarcoma-associated herpesvirus ORF56, ORF57, ORF58, and ORF59. J. Virol. 2006, 80, 11968–11981. [Google Scholar] [CrossRef] [PubMed]
- Goto, E.; Ishido, S.; Sato, Y.; Ohgimoto, S.; Ohgimoto, K.; Nagano-Fujii, M.; Hotta, H. C-mir, a human E3 ubiquitin ligase, is a functional homolog of herpesvirus proteins MIR1 and MIR2 and has similar activity. J. Biol. Chem. 2003, 278, 14657–14668. [Google Scholar] [CrossRef] [PubMed]
- Arias, C.; Weisburd, B.; Stern-Ginossar, N.; Mercier, A.; Madrid, A.S.; Bellare, P.; Holdorf, M.; Weissman, J.S.; Ganem, D. KSHV 2.0: A comprehensive annotation of the Kaposiʼs sarcoma-associated herpesvirus genome using next-generation sequencing reveals novel genomic and functional features. PLOS Pathog. 2014, 10, e1003847. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.Y.; Townes, T.M. Molecular mechanism for silencing virally transduced genes involves histone deacetylation and chromatin condensation. Proc. Natl. Acad. Sci. USA 2000, 97, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ueda, K.; Sakakibara, S.; Okuno, T.; Parravicini, C.; Corbellino, M.; Yamanishi, K. Activation of latent Kaposiʼs sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc. Natl. Acad. Sci. USA 2001, 98, 4119–4124. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Zhou, J.; Wiedmer, A.; Madden, K.; Yuan, Y.; Lieberman, P.M. Chromatin remodeling of the Kaposiʼs sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 2003, 77, 11425–11435. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.L.; Arnold, R.R.; Webster-Cyriaque, J. Signaling cascades triggered by bacterial metabolic end products during reactivation of Kaposiʼs sarcoma-associated herpesvirus. J. Virol. 2007, 81, 6032–6042. [Google Scholar] [CrossRef] [PubMed]
- Pantry, S.N.; Medveczky, P.G. Epigenetic regulation of Kaposiʼs sarcoma-associated herpesvirus replication. Semin. Cancer Biol. 2009, 19, 153–157. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Pari, G. KSHV PAN RNA associates with demethylases UTX and JMJD3 to activate lytic replication through a physical interaction with the virus genome. PLOS Pathog. 2012, 8, e1002680. [Google Scholar] [CrossRef] [PubMed]
- Groen, J.N.; Morris, K.V. Chromatin, non-coding RNAs, and the expression of HIV. Viruses 2013, 5, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Majerciak, V.; Ni, T.; Yang, W.; Meng, B.; Zhu, J.; Zheng, Z.M. A viral genome landscape of RNA polyadenylation from KSHV latent to lytic infection. PLOS Pathog. 2013, 9, e1003749. [Google Scholar] [CrossRef] [PubMed]
- Steitz, J.; Borah, S.; Cazalla, D.; Fok, V.; Lytle, R.; Mitton-Fry, R.; Riley, K.; Samji, T. Noncoding RNPs of viral origin. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Song, M.J.; Brown, H.J.; Wu, T.T.; Sun, R. Transcription activation of polyadenylated nuclear rna by RTA in human herpesvirus 8/Kaposiʼs sarcoma-associated herpesvirus. J. Virol. 2001, 75, 3129–3140. [Google Scholar] [CrossRef] [PubMed]
- Sahin, B.B.; Patel, D.; Conrad, N.K. Kaposiʼs sarcoma-associated herpesvirus ORF57 protein binds and protects a nuclear noncoding RNA from cellular RNA decay pathways. PLOS Pathog. 2010, 6, e1000799. [Google Scholar] [CrossRef] [PubMed]
- Massimelli, M.J.; Majerciak, V.; Kruhlak, M.; Zheng, Z.M. Interplay between polyadenylate-binding protein 1 and Kaposiʼs sarcoma-associated herpesvirus ORF57 in accumulation of polyadenylated nuclear RNA, a viral long noncoding RNA. J. Virol. 2013, 87, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Blauvelt, A.; Herndier, B.G.; Orenstein, J.M. Propagation of a human herpesvirus from AIDS-associated Kaposi’s sarcoma. N. Engl. J. Med. 1997, 336, 1837–1839. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Blackbourn, D.; Whitby, D.; Levy, J.; Ganem, D. Limited transmission of Kaposiʼs sarcoma-associated herpesvirus in cultured cells. J. Virol. 1998, 72, 5182–5188. [Google Scholar] [PubMed]
- Veettil, M.V.; Bandyopadhyay, C.; Dutta, D.; Chandran, B. Interaction of KSHV with host cell surface receptors and cell entry. Viruses 2014, 6, 4024–4046. [Google Scholar] [CrossRef] [PubMed]
- Luna, R.E.; Zhou, F.; Baghian, A.; Chouljenko, V.; Forghani, B.; Gao, S.J.; Kousoulas, K.G. Kaposiʼs sarcoma-associated herpesvirus glycoprotein K8.1 is dispensable for virus entry. J. Virol. 2004, 78, 6389–6398. [Google Scholar] [CrossRef] [PubMed]
- Chandran, B.; Bloomer, C.; Chan, S.R.; Zhu, L.; Goldstein, E.; Horvat, R. Human herpesvirus-8 ORF K8.1 gene encodes immunogenic glycoproteins generated by spliced transcripts. Virology 1998, 249, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Koyano, S.; Mar, E.C.; Stamey, F.R.; Inoue, N. Glycoproteins M and N of human herpesvirus 8 form a complex and inhibit cell fusion. J. Gen. Virol. 2003, 84, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Full, F.; Jungnickl, D.; Reuter, N.; Bogner, E.; Brulois, K.; Scholz, B.; Sturzl, M.; Myoung, J.; Jung, J.U.; Stamminger, T.; et al. Kaposiʼs sarcoma associated herpesvirus tegument protein ORF75 is essential for viral lytic replication and plays a critical role in the antagonization of ND10-instituted intrinsic immunity. PLOS Pathog. 2014, 10, e1003863. [Google Scholar] [CrossRef] [PubMed]
- Chandran, B. Early events in Kaposiʼs sarcoma-associated herpesvirus infection of target cells. J. Virol. 2010, 84, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- Toth, Z.; Brulois, K.; Lee, H.R.; Izumiya, Y.; Tepper, C.; Kung, H.J.; Jung, J.U. Biphasic euchromatin-to-heterochromatin transition on the KSHV genome following de novo infection. PLOS Pathog. 2013, 9, e1003813. [Google Scholar] [CrossRef] [PubMed]
- Morey, L.; Helin, K. Polycomb group protein-mediated repression of transcription. Trends Biochem. Sci. 2010, 35, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Lallemand-Breitenbach, V.; Zhu, J.; Puvion, F.; Koken, M.; Honore, N.; Doubeikovsky, A.; Duprez, E.; Pandolfi, P.P.; Puvion, E.; Freemont, P.; et al. Role of promyelocytic leukemia (PML) sumolation in nuclear body formation, 11s proteasome recruitment, and AS2O3-induced PML or PML/retinoic acid receptor alpha degradation. J. Exp. Med. 2001, 193, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.Y.; Li, L.; Fan, Y.H.; Mu, Z.M.; Zhang, W.W.; Chang, K.S. Cell-cycle regulation of DNA damage-induced expression of the suppressor gene PML. Biochem. Biophys. Res. Commun. 1997, 240, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Ishov, A.M.; Sotnikov, A.G.; Negorev, D.; Vladimirova, O.V.; Neff, N.; Kamitani, T.; Yeh, E.T.; Strauss, J.F., 3rd; Maul, G.G. PML is critical for ND10 formation and recruits the PML-interacting protein DAXX to this nuclear structure when modified by SUMO-1. J. Cell Biol. 1999, 147, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Kyratsous, C.A.; Silverstein, S.J. Components of nuclear domain 10 bodies regulate varicella-zoster virus replication. J. Virol. 2009, 83, 4262–4274. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.M.; Davis, B.K.; West, J.A.; Taxman, D.J.; Matsuzawa, S.; Reed, J.C.; Ting, J.P.; Damania, B. Discovery of a viral NLR homolog that inhibits the inflammasome. Science 2011, 331, 330–334. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uppal, T.; Jha, H.C.; Verma, S.C.; Robertson, E.S. Chromatinization of the KSHV Genome During the KSHV Life Cycle. Cancers 2015, 7, 112-142. https://doi.org/10.3390/cancers7010112
Uppal T, Jha HC, Verma SC, Robertson ES. Chromatinization of the KSHV Genome During the KSHV Life Cycle. Cancers. 2015; 7(1):112-142. https://doi.org/10.3390/cancers7010112
Chicago/Turabian StyleUppal, Timsy, Hem C. Jha, Subhash C. Verma, and Erle S. Robertson. 2015. "Chromatinization of the KSHV Genome During the KSHV Life Cycle" Cancers 7, no. 1: 112-142. https://doi.org/10.3390/cancers7010112