Comparison of Intracellular Stress Response of NCI-H526 Small Cell Lung Cancer (SCLC) Cells to Platinum(II) Cisplatin and Platinum(IV) Oxoplatin

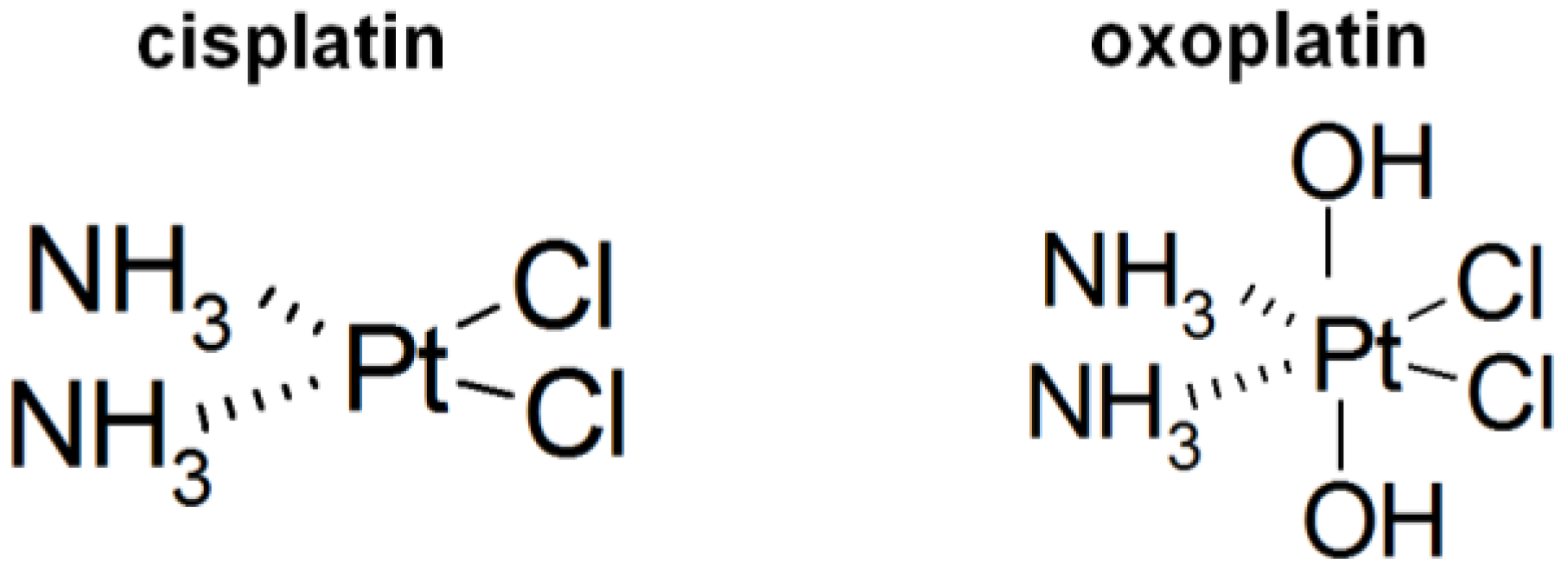{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
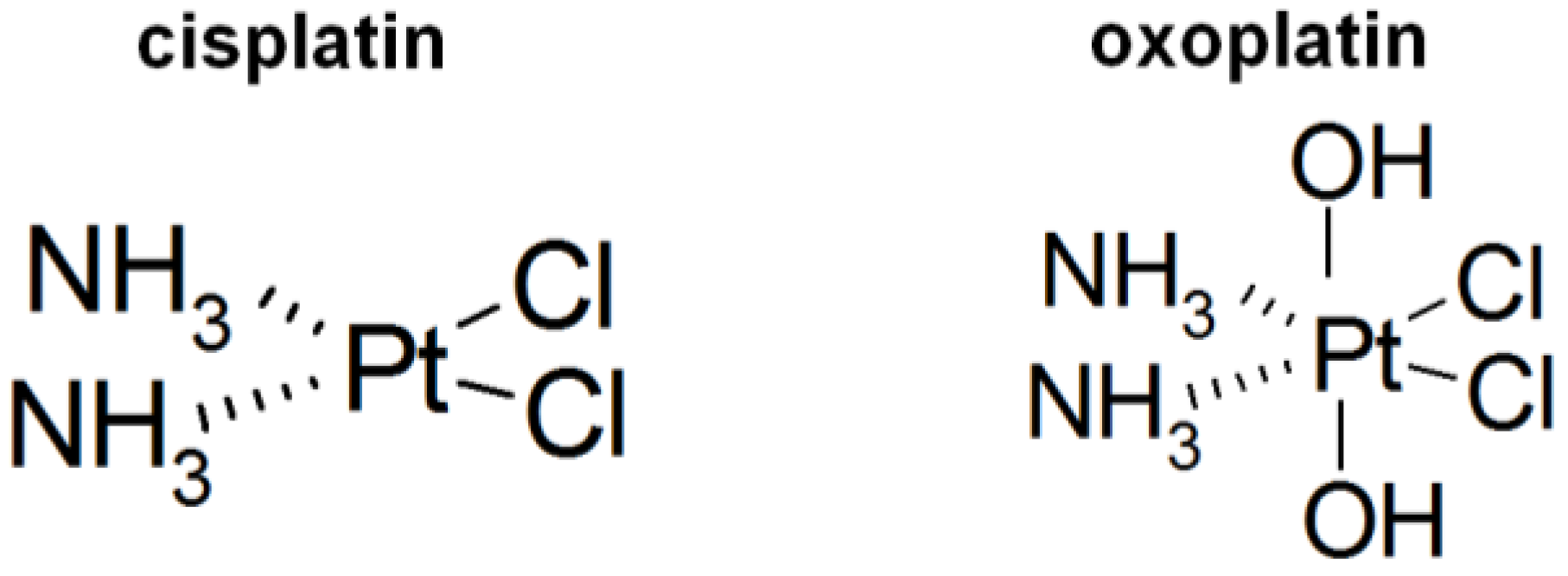
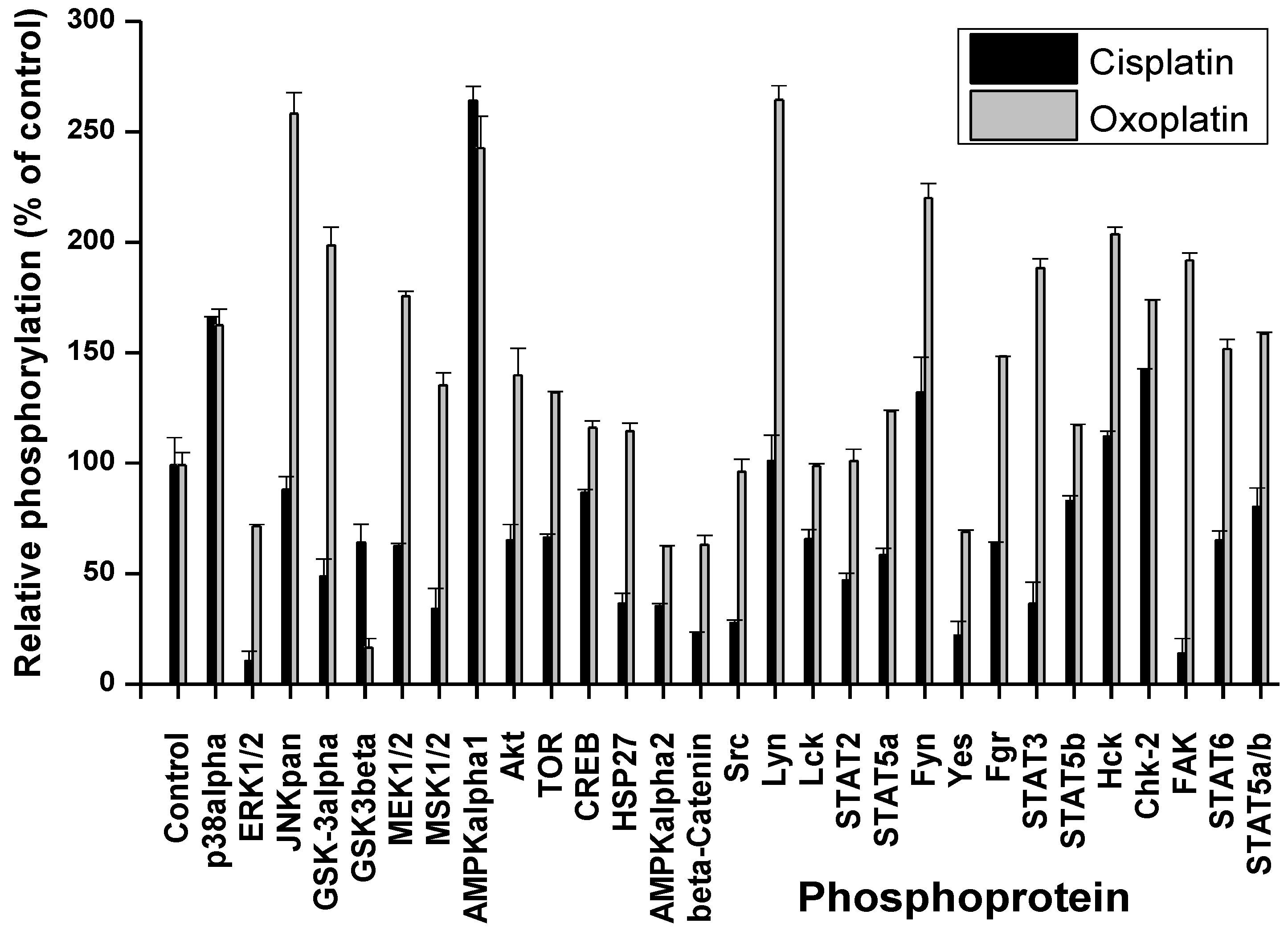
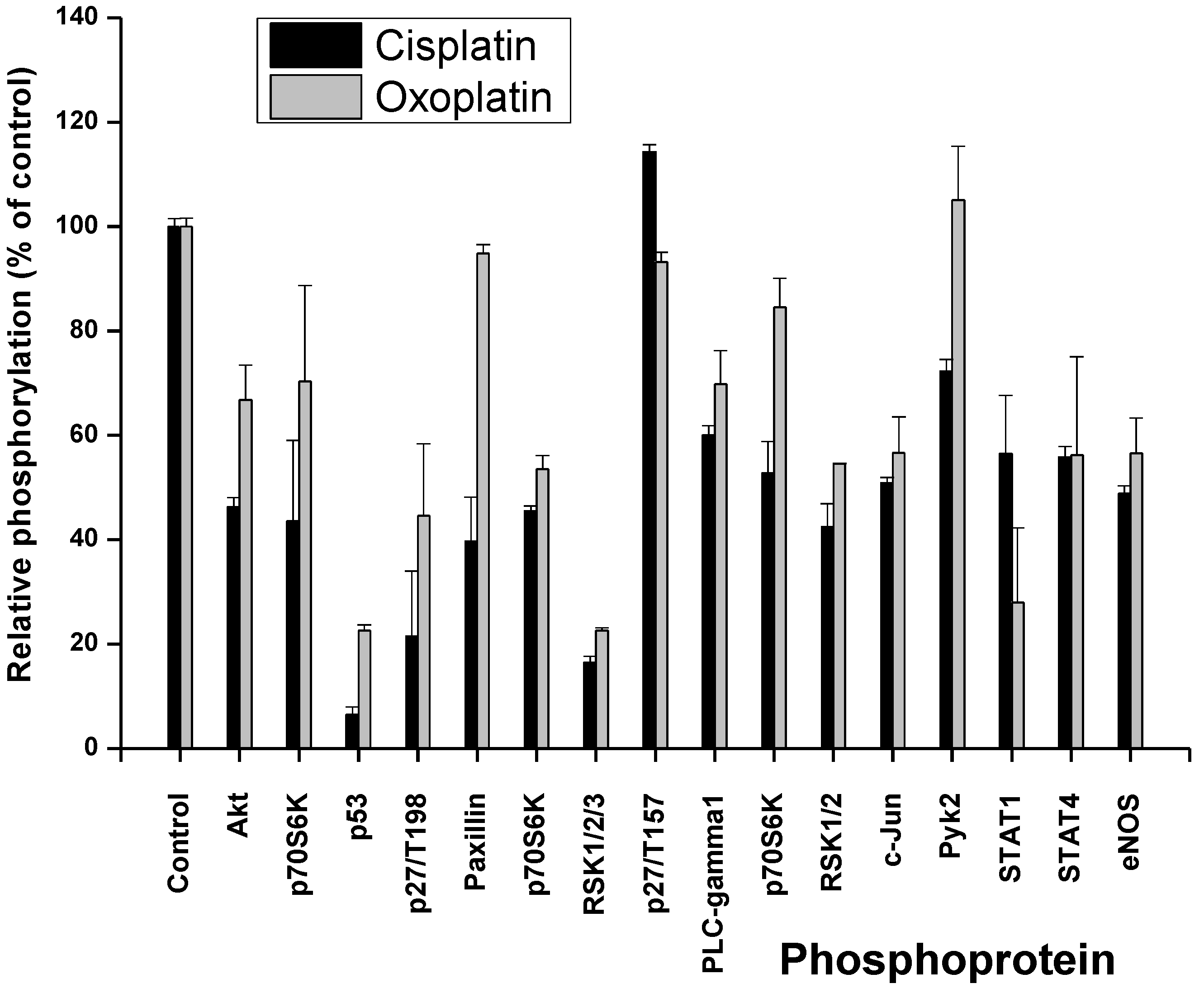
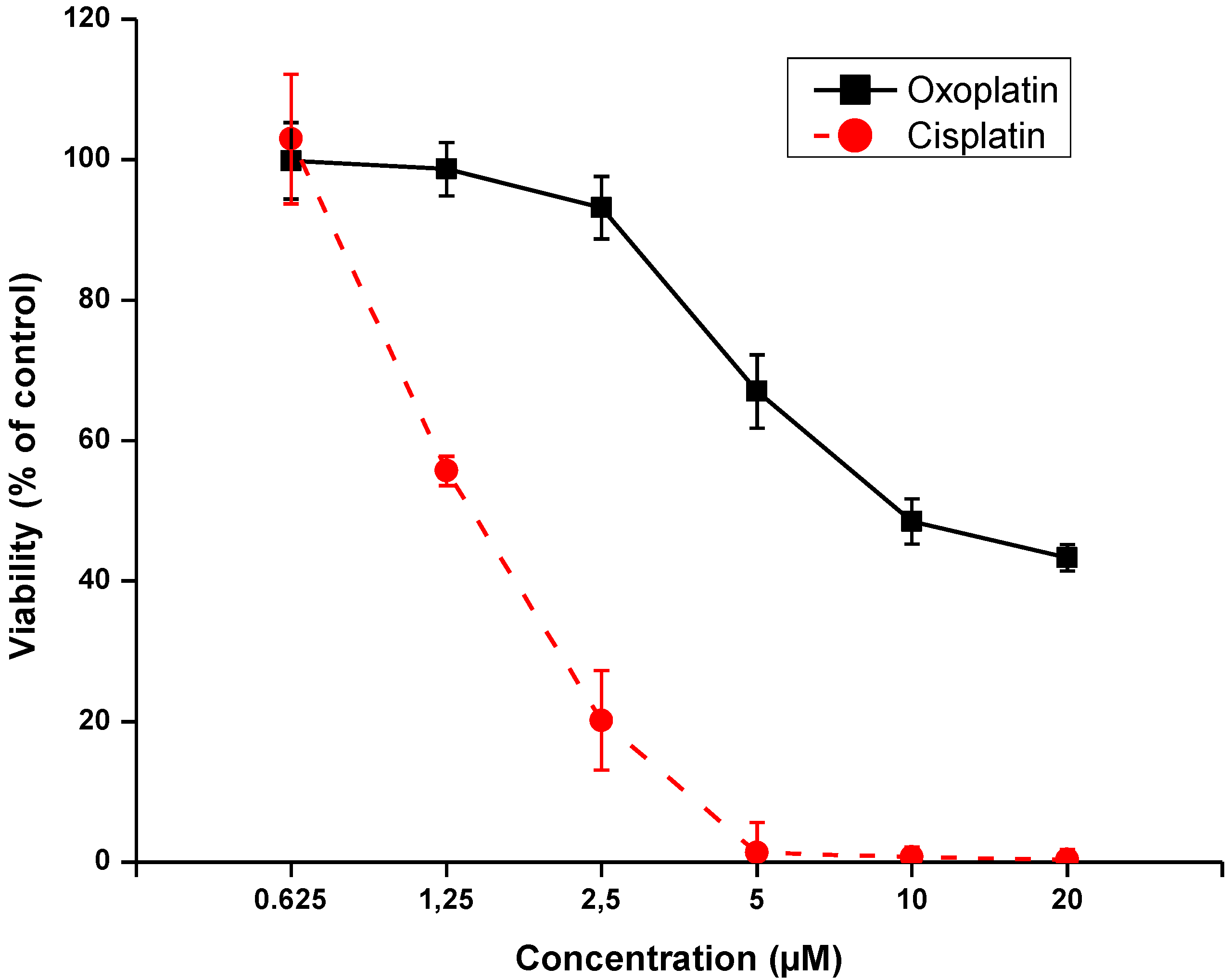
2. Results
2.1. Cisplatin- and Oxoplatin-Induced Alterations of the NCI-H526 Phosphome

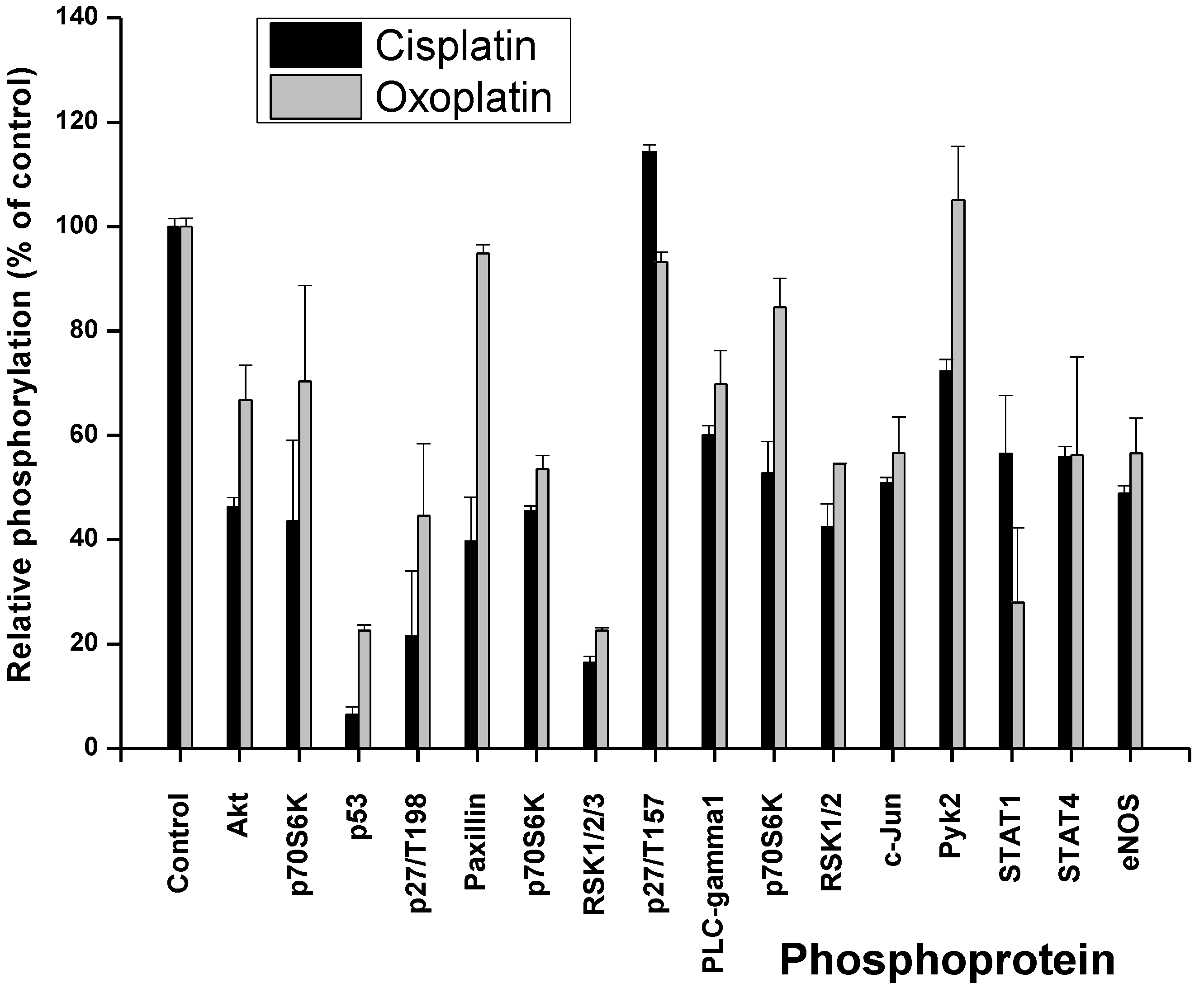
2.2. Effects of Short-Term Exposure of NCI-H526 Cells to Oxoplatin or Cisplatin on Cell Viability

3. Discussion
4. Experimental
4.1. Chemicals
4.2. Cell Culture
4.3. Phosphokinase Array
4.4. Cytotoxicity Assay
4.5. Statistics
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Youlden, D.R.; Cramb, S.M.; Baade, P.D. The international epidemiology of lung cancer: Geographical distribution and secular trends. J. Thorac. Oncol. 2008, 3, 819–831. [Google Scholar] [CrossRef]
- Califano, A.; Abidin, Z.; Peck, R.; Faivre-Finn, C.; Lorigan, P.R. Management of small cell lung cancer: Recent developments for optimal care. Drugs 2012, 72, 471–490. [Google Scholar] [CrossRef]
- William, W.N.; Glisson, B.S. Novel strategies for the treatment of small-cell lung carcinoma. Nat. Rev. Clin. Oncol. 2011, 8, 611–619. [Google Scholar] [CrossRef]
- Ali, I.; Wani, W.A.; Saleem, K.; Haque, A. Platinum compounds: A hope for future cancer chemotherapy. Anticancer Agents Med. Chem. 2013, 13, 296–306. [Google Scholar] [CrossRef]
- Olszewski, U.; Hamilton, G. A better platinum-based anticancer drug yet to come? Anticancer Agents Med. Chem. 2010, 10, 293–301. [Google Scholar] [CrossRef]
- Shen, D.W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef]
- Hamilton, G.; Olszewski, U. Picoplatin pharmacokinetics and chemotherapy of non-small cell lung cancer. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1381–1390. [Google Scholar] [CrossRef]
- Doshi, G.; Sonpavde, G.; Sternberg, C.N. Clinical and pharmacokinetic evaluation of satraplatin. Expert Opin. Drug Metab. Toxicol. 2012, 8, 103–111. [Google Scholar] [CrossRef]
- Wexselblatt, E.; Gibson, D. What do we know about the reduction of Pt(IV) pro-drugs? J. Inorg. Biochem. 2012, 117, 220–229. [Google Scholar] [CrossRef]
- Olszewski, U.; Ach, F.; Ulsperger, E.; Baumgartner, G.; Zeillinger, R.; Bednarski, P.; Hamilton, G. In vitro evaluation of oxoplatin: An oral platinum(IV) anticancer agent. Met.-Based Drugs 2009, 2009, 348916. [Google Scholar]
- Presnov, M.A.; Konovalova, A.L.; Kozlov, A.M.; Brovtsyn, V.K.; Romanova, L.F. The antitumor activity of oxoplatinum. Neoplasma 1985, 32, 73–83. [Google Scholar]
- Nowsheen, S.; Yang, E.S. The intersection between DNA damage response and cell death pathways. Exp. Oncol. 2012, 34, 243–254. [Google Scholar]
- Olszewski, U.; Deally, A.; Tacke, M.; Hamilton, G. Alterations of phosphoproteins in NCI-H526 small cell lung cancer cells involved in cytotoxicity of cisplatin and titanocene Y. Neoplasia 2012, 14, 813–822. [Google Scholar]
- Florea, A.-M.; Büsselberg, D. Cisplatin as an anti-tumor drug: Cellular mechanisms of activity, drug resistance and induced side effects. Cancers 2011, 3, 1351–1371. [Google Scholar] [CrossRef]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013, 332, 237–248. [Google Scholar] [CrossRef]
- Go, R.S.; Adjei, A.A. Review of the comparative pharmacology and clinical activity of cisplatin and carboplatin. J. Clin. Oncol. 1999, 17, 409–422. [Google Scholar]
- Olszewski, U.; Ulsperger, E.; Geissler, K.; Hamilton, G. Comparison of the effects of the oral anticancer platinum(IV) complexes oxoplatin and metabolite cis-diammine-tetrachlorido-platinum(IV) on global gene expression of NCI-H526 cells. J. Exp. Pharmacol. 2011, 3, 43–50. [Google Scholar]
- Harhaji-Trajkovic, L.; Vilimanovich, U.; Kravic-Stevovic, T.; Bumbasirevic, V.; Trajkovic, V. AMPK-mediated autophagy inhibits apoptosis in cisplatin-treated tumour cells. J. Cell. Mol. Med. 2009, 13, 3644–3654. [Google Scholar] [CrossRef]
- Levresse, V.; Marek, L.; Blumberg, D.; Heasley, L.E. Regulation of platinum-compound cytotoxicity by the c-Jun N-terminal kinase and c-Jun signaling pathway in small-cell lung cancer cells. Mol. Pharmacol. 2002, 62, 689–697. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, Z.; Zhang, X.; He, J.; Pan, Y.; Hao, F.; Xie, L.; Li, Q.; Qiu, X.; Wang, E. Inhibition of cytoplasmic GSK-3β increases cisplatin resistance through activation of Wnt/β-catenin signaling in A549/DDP cells. Cancer Lett. 2013, 336, 231–239. [Google Scholar] [CrossRef]
- Cai, G.; Wang, J.; Xin, X.; Ke, Z.; Luo, J. Phosphorylation of glycogen synthase kinase-3 beta at serine 9 confers cisplatin resistance in ovarian cancer cells. Int. J. Oncol. 2007, 31, 657–662. [Google Scholar]
- Wang, J.; Zhou, J.Y.; Wu, G.S. ERK-dependent MKP-1-mediated cisplatin resistance in human ovarian cancer cells. Cancer Res. 2007, 67, 11933–11941. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, J.Y.; Zhang, L.; Wu, G.S. Involvement of MKP-1 and Bcl-2 in acquired cisplatin resistance in ovarian cancer cells. Cell Cycle 2009, 8, 3191–3198. [Google Scholar] [CrossRef]
- Staples, C.J.; Owens, D.M.; Maier, J.V.; Cato, A.C.; Keyse, S.M. Cross-talk between the p38alpha and JNK MAPK pathways mediated by MAP kinase phosphatase-1 determines cellular sensitivity to UV radiation. J. Biol. Chem. 2010, 285, 25928–25940. [Google Scholar] [CrossRef]
- Sheppard, K.; Kinross, K.M.; Solomon, B.; Pearson, R.B.; Phillips, W.A. Targeting PI3 kinase/AKT/mTOR signaling in cancer. Crit. Rev. Oncog. 2012, 17, 69–95. [Google Scholar]
- Liu, L.Z.; Zhou, X.D.; Qian, G.; Shi, X.; Fang, J.; Jiang, B.H. AKT1 amplification regulates cisplatin resistance in human lung cancer cells through the mammalian target of rapamycin/p70S6K1 pathway. Cancer Res. 2007, 67, 6325–6332. [Google Scholar] [CrossRef]
- Peng, D.J.; Wang, J.; Zhou, J.Y.; Wu, G.S. Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells. Biochem. Biophys. Res. Commun. 2010, 394, 600–605. [Google Scholar] [CrossRef]
- Sinnberg, T.; Lasithiotakis, K.; Niessner, H.; Schittek, B.; Flaherty, K.T.; Kulms, D.; Maczey, E.; Campos, M.; Gogel, J.; Garbe, C.; et al. Inhibition of PI3K-AKT-mTOR signaling sensitizes melanoma cells to cisplatin and temozolomide. J. Investig. Dermatol. 2009, 129, 1500–1515. [Google Scholar] [CrossRef]
- Ceppi, P.; Papotti, M.; Monica, V.; Lo Iacono, M.; Saviozzi, S.; Pautasso, M.; Novello, S.; Mussino, S.; Bracco, E.; Volante, M.; et al. Effects of Src kinase inhibition induced by dasatinib in non-small cell lung cancer cell lines treated with cisplatin. Mol. Cancer Ther. 2009, 8, 3066–3074. [Google Scholar] [CrossRef]
- Levitt, J.M.; Yamashita, H.; Jian, W.; Lerner, S.P.; Sonpavde, G. Dasatinib is preclinically active against Src-overexpressing human transitional cell carcinoma of the urothelium with activated Src signaling. Mol. Cancer Ther. 2010, 9, 1128–1135. [Google Scholar] [CrossRef]
- Peterson-Roth, E.; Brdlik, C.M.; Glazer, P.M. Src-induced cisplatin resistance mediated by cell-to-cell communication. Cancer Res. 2009, 69, 3619–3624. [Google Scholar] [CrossRef]
- Yoshida, K.; Weichselbaum, R.; Kharbanda, S.; Kufe, D. Role for Lyn tyrosine kinase as a regulator of stress-activated protein kinase activity in response to DNA damage. Mol. Cell. Biol. 2000, 20, 5370–5380. [Google Scholar]
- Zhang, S.; Yu, D. Targeting Src family kinases in anti-cancer therapies: Turning promise into triumph. Trends Pharmacol. Sci. 2012, 33, 122–128. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Kunnumakkara, A.B.; Harikumar, K.B.; Gupta, S.R.; Tharakan, S.T.; Koca, C.; Dey, S.; Sung, B. Signal transducer and activator of transcription-3, inflammation, and cancer: How intimate is the relationship? Ann. NY Acad. Sci. 2009, 1171, 59–76. [Google Scholar] [CrossRef]
- Ikuta, K.; Takemura, K.; Kihara, M.; Nishimura, M.; Ueda, N.; Naito, S.; Lee, E.; Shimizu, E.; Yamauchi, A. Overexpression of constitutive signal transducer and activator of transcription 3 mRNA in cisplatin-resistant human non-small cell lung cancer cells. Oncol. Rep. 2005, 13, 217–222. [Google Scholar]
- Sheng, W.J.; Jiang, H.; Wu, D.L.; Zheng, J.H. Early responses of the STAT3 pathway to platinum drugs are associated with cisplatin resistance in epithelial ovarian cancer. Braz. J. Med. Biol. Res. 2013, 46, 650–658. [Google Scholar] [CrossRef]
- Ji, T.; Gong, D.; Han, Z.; Wei, X.; Yan, Y.; Ye, F.; Ding, W.; Wang, J.; Xia, X.; Li, F.; et al. Abrogation of constitutive Stat3 activity circumvents cisplatin resistant ovarian cancer. Cancer Lett. 2013, 341, 231–239. [Google Scholar] [CrossRef]
- Han, Z.; Feng, J.; Hong, Z.; Chen, L.; Li, W.; Liao, S.; Wang, X.; Ji, T.; Wang, S.; Ma, D.; et al. Silencing of the STAT3 signaling pathway reverses the inherent and induced chemoresistance of human ovarian cancer cells. Biochem. Biophys. Res. Commun. 2013, 435, 188–194. [Google Scholar] [CrossRef]
- Kulesza, D.W.; Carré, T.; Chouaib, S.; Kaminska, B. Silencing of the transcription factor STAT3 sensitizes lung cancer cells to DNA damaging drugs, but not to TNFα- and NK cytotoxicity. Exp. Cell Res. 2013, 319, 506–516. [Google Scholar] [CrossRef]
- Yin, Z.; Zhang, Y.; Li, Y.; Lv, T.; Liu, J.; Wang, X. Prognostic significance of STAT3 expression and its correlation with chemoresistance of non-small cell lung cancer cells. Acta Histochem. 2012, 114, 151–158. [Google Scholar] [CrossRef]
- Shi, Y.; Felley-Bosco, E.; Marti, T.M.; Orlowski, K.; Pruschy, M.; Stahel, R.A. Starvation-induced activation of ATM/Chk2/p53 signaling sensitizes cancer cells to cisplatin. BMC Cancer 2012, 12, 571. [Google Scholar] [CrossRef] [Green Version]
- Wittig, I.; Groner, B. Signal transducer and activator of transcription 5 (STAT5), a crucial regulator of immune and cancer cells. Curr. Drug Targets Immune Endocr. Metabol. Disord. 2005, 5, 449–463. [Google Scholar] [CrossRef]
- Hato, S.V.; de Vries, I.J.; Lesterhuis, W.J. STATing the importance of immune modulation by platinum chemotherapeutics. Oncoimmunology 2012, 1, 234–236. [Google Scholar] [CrossRef]
- Pabla, N.; Huang, S.; Mi, Q.S.; Daniel, R.; Dong, Z. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J. Biol. Chem. 2008, 283, 6572–6583. [Google Scholar] [CrossRef]
- Chiang, Y.Y.; Chow, K.C.; Lin, T.Y.; Chiang, I.P.; Fang, H.Y. Hepatocyte growth factor and HER2/neu downregulate expression of apoptosis-inducing factor in non-small cell lung cancer. Oncol. Rep. 2014, 31, 597–604. [Google Scholar]
- Wang, J.; Zu, J.; Xu, G.; Zhao, W.; Yan, J. Inhibition of focal adhesion kinase induces apoptosis in human osteosarcoma SAOS-2 cells. Tumor Biol. 2014, 35, 1551–1556. [Google Scholar] [CrossRef]
- Villedieu, M.; Deslandes, E.; Duval, M.; Héron, J.F.; Gauduchon, P.; Poulain, L. Acquisition of chemoresistance following discontinuous exposures to cisplatin is associated in ovarian carcinoma cells with progressive alteration of FAK, ERK and p38 activation in response to treatment. Gynecol. Oncol. 2006, 101, 507–519. [Google Scholar] [CrossRef]
- Nakahara, S.; Miyoshi, E.; Noda, K.; Ihara, S.; Gu, J.; Honke, K.; Inohara, H.; Kubo, T.; Taniguchi, N. Involvement of oligosaccharide changes in alpha5beta1 integrin in a cisplatin-resistant human squamous cell carcinoma cell line. Mol. Cancer Ther. 2003, 2, 1207–1214. [Google Scholar]
- Wu, D.W.; Wu, T.C.; Wu, J.Y.; Cheng, Y.W.; Chen, Y.C.; Lee, M.C.; Chen, C.Y.; Lee, H. Phosphorylation of paxillin confers cisplatin resistance in non-small cell lung cancer via activating ERK-mediated Bcl-2 expression. Oncogene 2013. [Google Scholar] [CrossRef]
- Geng, W.; Ng, K.T.; Sun, C.K.; Yau, W.L.; Liu, X.B.; Cheng, Q.; Poon, R.T.; Lo, C.M.; Man, K.; Fan, S.T. The role of proline rich tyrosine kinase 2 (Pyk2) on cisplatin resistance in hepatocellular carcinoma. PLoS One 2011, 6, e27362. [Google Scholar]
- Hall, M.D.; Alderden, R.A.; Zhang, M.; Beale, P.J.; Cai, Z.; Lai, B.; Stampfl, A.P.; Hambley, T.W. The fate of platinum(II) and platinum(IV) anti-cancer agents in cancer cells and tumours. J. Struct. Biol. 2006, 155, 38–44. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hamilton, G. Comparison of Intracellular Stress Response of NCI-H526 Small Cell Lung Cancer (SCLC) Cells to Platinum(II) Cisplatin and Platinum(IV) Oxoplatin. Cancers 2014, 6, 1487-1499. https://doi.org/10.3390/cancers6031487
Hamilton G. Comparison of Intracellular Stress Response of NCI-H526 Small Cell Lung Cancer (SCLC) Cells to Platinum(II) Cisplatin and Platinum(IV) Oxoplatin. Cancers. 2014; 6(3):1487-1499. https://doi.org/10.3390/cancers6031487
Chicago/Turabian StyleHamilton, Gerhard. 2014. "Comparison of Intracellular Stress Response of NCI-H526 Small Cell Lung Cancer (SCLC) Cells to Platinum(II) Cisplatin and Platinum(IV) Oxoplatin" Cancers 6, no. 3: 1487-1499. https://doi.org/10.3390/cancers6031487