The Role of PI3K/Akt/mTOR Signaling in Gastric Carcinoma

1
Department of Surgical Oncology, Osaka City University Graduate School of Medicine, 1-4-3 Asahi-machi, Abeno-ku, Osaka 545-8585, Japan
2
Oncology Institute of Geriatrics and Medical Science, Osaka City University Graduate School of Medicine, 1-4-3 Asahi-machi, Abeno-ku, Osaka 545-8585, Japan
*
Author to whom correspondence should be addressed.
Cancers 2014, 6(3), 1441-1463; https://doi.org/10.3390/cancers6031441
Submission received: 14 April 2014
/
Revised: 24 June 2014
/
Accepted: 26 June 2014
/
Published: 7 July 2014
(This article belongs to the Special Issue Kinases and Cancer)
Abstract
:The phosphatidylinositol 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) pathway is one of the key signaling pathways induced by various receptor-tyrosine kinases. Accumulating evidence shows that this pathway is an important promoter of cell growth, metabolism, survival, metastasis, and resistance to chemotherapy. Genetic alterations in the PI3K/Akt/mTOR pathway in gastric carcinoma have often been demonstrated. Many kinds of molecular targeting therapies are currently undergoing clinical testing in patients with solid tumors. However, with the exception of the ErbB2-targeting antibody, targeting agents, including PI3K/Akt/mTOR inhibitors, have not been approved for treatment of patients with gastric carcinoma. This review summarizes the current knowledge on PI3K/Akt/mTOR signaling in the pathogenesis of gastric carcinoma and the possible therapeutic targets for gastric carcinoma. Improved knowledge of the PI3K/Akt/mTOR pathway in gastric carcinoma will be useful in understanding the mechanisms of tumor development and for identifying ideal targets of anticancer therapy for gastric carcinoma.
1. Introduction
Gastric carcinoma remains the second most common cause of cancer-related deaths in the world. Although the outcomes of gastric carcinoma have recently improved, highly malignant type of gastric carcinoma, which is characterized by rapid progression and resistance to chemotherapy, continue to have poor prognosis [1].
To date, progress in the understanding of molecular pathways involved in a variety of cancers has led to the discovery of new targeted therapies, including therapies for gastric carcinoma. Trastuzumab has already been approved as a standard therapy for human epidermal growth factor receptor 2 (HER2)-positive gastric carcinoma patients, on the basis of results of the Trastuzumab for Gastric Cancer clinical trial [2]. Further candidate molecules related to cell growth, invasion, apoptosis, angiogenesis, and metastasis are emerging as a new strategy for the treatment of gastric carcinoma. Agents targeting molecules such as epidermal growth factor receptor (EGFR), vascular endothelial growth factor (VEGF) receptor, and fibroblast growth factor receptor (FGFR), as well as pathways such as c-Met pathways, are promising candidates for targeted therapy for gastric carcinoma and are now in clinical development [3,4].
It is known that accumulation of genetic alterations results in the development of gastric carcinoma [5]. Most of the genetic mutations in gastric carcinoma correlate with changes in biological signals, such as those in the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin pathway (PI3K/Akt/mTOR pathway) [6]. PI3K signaling is a crucial regulator of many essential cellular processes, including cell growth, metabolism, survival, metastasis, and resistance to chemotherapy [7]. Although there is substantial evidence that the PI3K/Akt/mTOR pathway is frequently altered in gastric carcinoma, its precise function remains to be determined [7,8,9]. Understanding the biological pathways leading to the development of gastric carcinoma will provide an opportunity to enhance targeted therapies. In this context, we review the role of PI3K signaling in gastric carcinoma, especially on carcinogenesis, growth, adhesion, metastasis, apoptosis, and sensitivity to therapy. Novel therapies targeting the PI3K/AkT/mTOR pathway as well as its downstream substrates are summarized with the literatures in these several decade.
2. The PI3K/Akt/mTOR Pathway
The PI3K pathway (Figure 1) has many different upstream factors, including the binding of receptor tyrosine kinase (RTK), G-protein-coupled receptors (GPCR), and GTP-binding proteins to adaptor proteins [7,10]. PI3K belongs to a lipid kinase family characterized by their ability to phosphorylate the 3'-OH group of the inositol ring in inositol phospholipids [10]. They are categorized into three classes, namely, classes I, II, and III. Class I PI3Ks are further divided into class IA and IB. The catalytic subunit of class IA PI3K, p110, has four isoforms (p110α, p110β, p110γ, and p110δ), whereas its p85 regulatory subunit has three variants (p85α, p85β, and p55γ). The isoforms of p110 are encoded by the PIK3CA, PIK3CB, and PIK3CD genes. Deregulation of the PI3K/Akt/mTOR pathway can occur subsequent to oncogenic mutations of PIK3CA [11]. p110α is the most well-understood isoform in terms of signal transduction and physiological association and the most often mutated or amplified in solid tumors [12]. PI3K activation usually occurs through growth factor stimulation by phosphotyrosine kinases such as EGFR, platelet-derived factor receptor, insulin growth factor receptor, or c-Met. Activated PI3K associates with the receptor through one or two Src homology 2 domains in the regulatory subunit, which leads to the activation of the catalytic subunit. Activation of the PI3K pathway leads to the phosphorylation of the inositol ring of lipids in the plasma membrane and converts phosphatidylinositol 3-phosphate (PI) and phosphatidylinositol 4,5-bisphosphate (PIP2), the lipid substrates for class I PI3Ks, to phosphatidylinositol 3,4,5-trisphosphate (PIP3). PIP2 and PIP3 interact with pleckstrin homology (PH) domain-containing proteins on the inner surface of the plasma membrane, resulting in conformational changes of these proteins.
PH domains are found in many proteins, including Akt, which is also known as protein kinase B [13]. Akt is a serine–threonine kinase that normally exists in the cytoplasm. Recently, three members of the Akt family, namely, Akt1, Akt2, and Akt3, have been isolated. These are products of three distinct genes that share up to 80% homology at the amino acid level. Upon activation of PI3K, Akt transfers to the cell membrane, resulting in its conformational change. Akt contains a central kinase domain with a threonine residue (T308) that binds to the phosphoinositide-dependent protein kinase 1 (PDK1) and a C-terminal tail domain (S473) that binds to the second mTOR complex 2 (mTOR2). Phosphorylated Akt (p-Akt) has been shown to promote molecular functions within the cell, such as cell cycle progression and angiogenesis, as well as prevent apoptosis through a number of downstream effectors [14]. Glycogen synthase kinase 3 (GSK3), the first identified Akt substrate, is believed to be an essential metabolic enzyme and an important factor in other signaling cascades. It phosphorylates a host of downstream substrates such as p21, p27, caspase 9, FKHR, IKKα, and BAD, thereby mediating a number of effects [15]. PI3K activity is regulated by the lipid phosphatase and tensin homolog (PTEN), a tumor suppressor gene that encodes a lipid phosphatase that downregulates the PI3K signal by converting PIP3 back to PIP2 [16]. Loss of PTEN results in constitutive activation of Akt and in alteration of downstream factors in Akt signaling.
mTOR is a highly conserved protein kinase that participates as an effector in the PI3K/Akt pathway. mTOR comprises two protein complexes, mTORC1 (mTOR, mLST8, and raptor) and mTORC2 (mTOR, mLST8, mSIN1, and Rictor). mTOR1, a complex that is also modulated by extracellular-signal-regulated kinase, induces protein synthesis and cell growth by regulating ribosomal p70S6 kinase 1 (S6K1) and eukaryotic translation factor 4E-binding protein 1 (4EBP1) [17]. Activated S6K1 participates in negative feedback, thereby attenuating activation of the PI3K pathway through phosphorylation and subsequently inhibiting adaptor molecule insulin receptor substrate 1, which interrupts the signaling between IGF-1 and PI3K. mTORC2 phosphorylates Akt and SGK1 at the C-terminal and regulates the remodeling of the actin cytoskeleton, but the biological significance of these activities is still largely unknown [18]. mTOR plays a critical role in the regulation of tumor cell motility and cancer metastasis [19]. However, the underlying mechanism of mTOR regulation of cell motility and mTOR inhibitors inhibiting tumor cell motility is controversial [20].
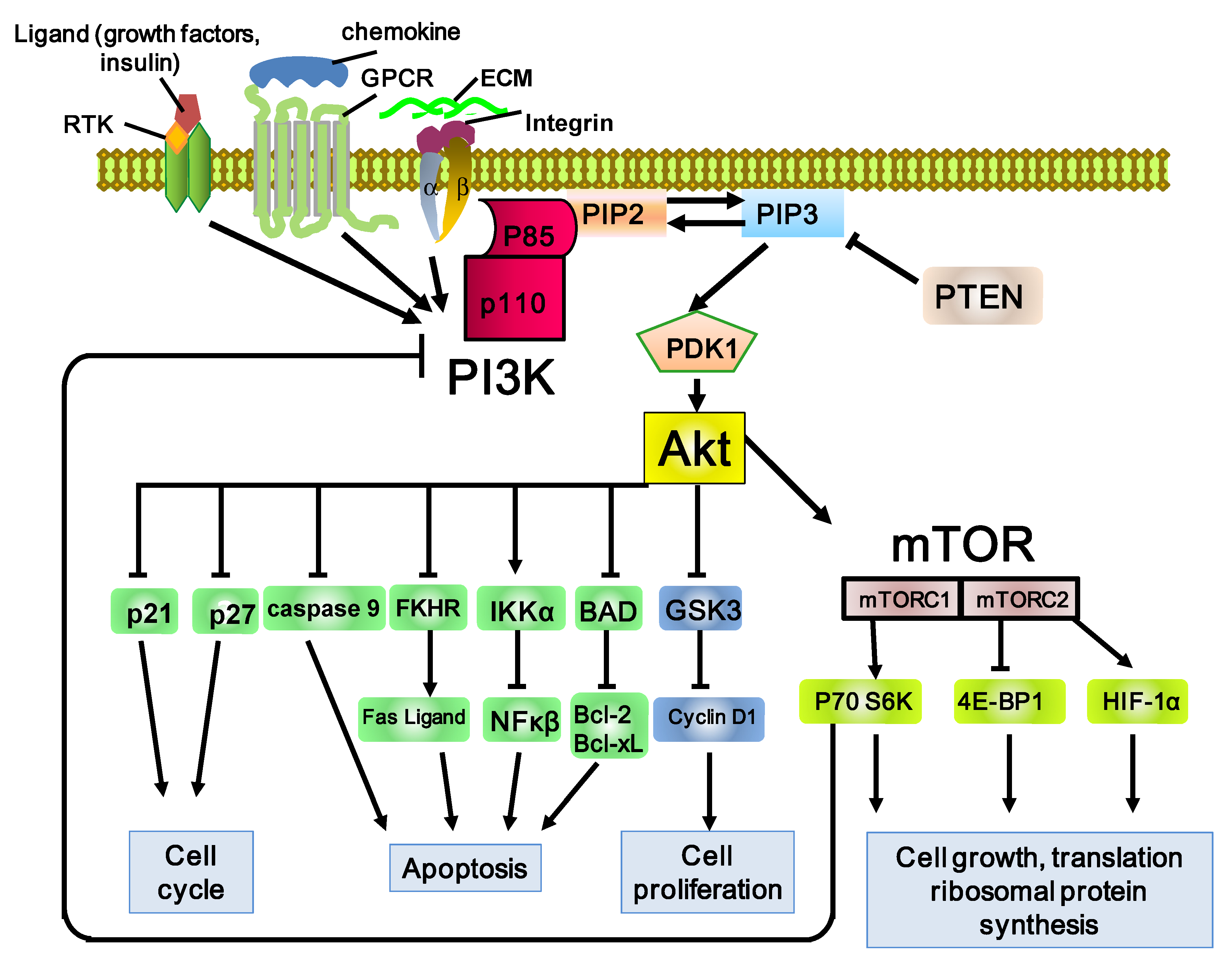
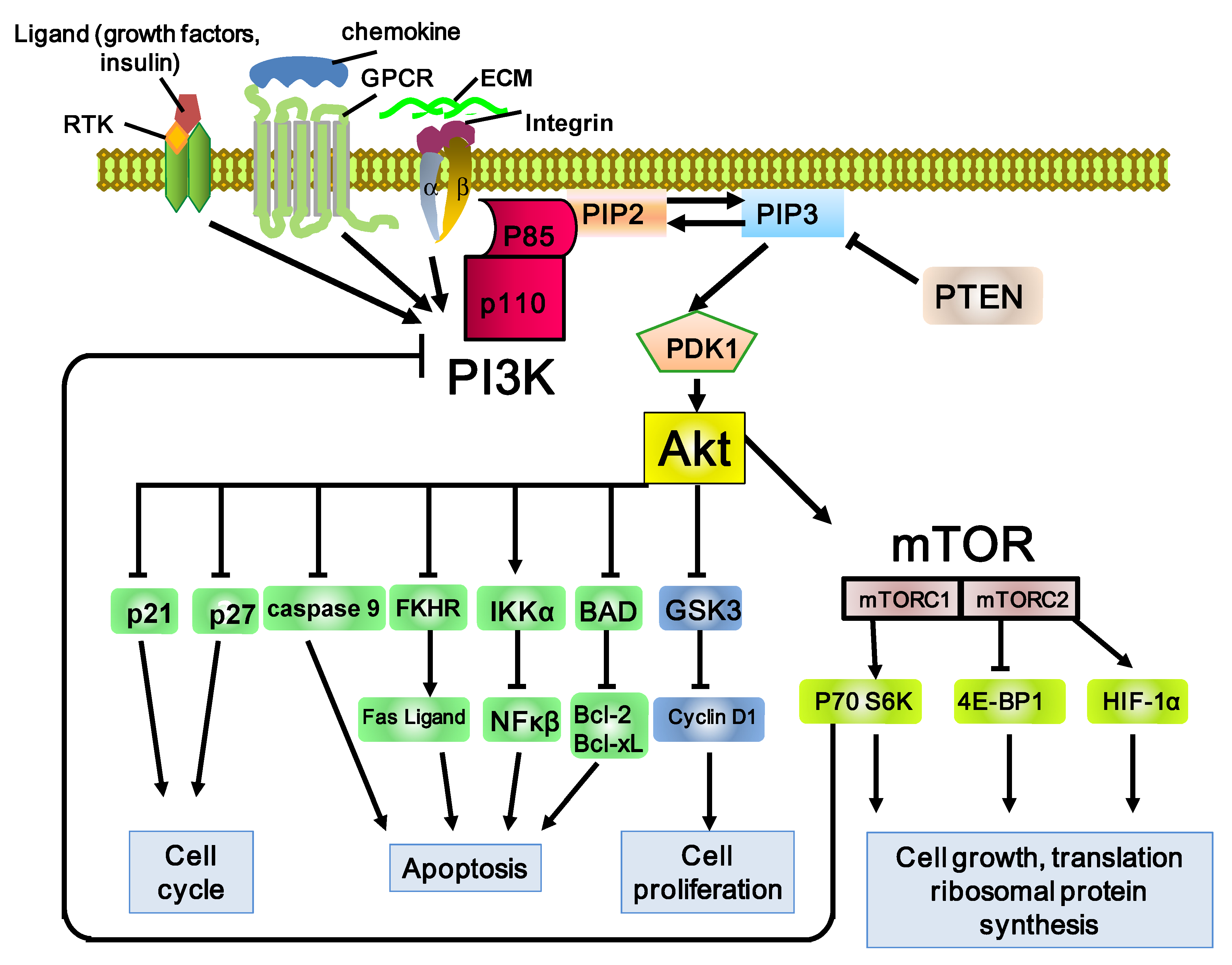
Figure 1.
Schematic representation of the PI3K/Akt/mTOR pathway. The PI3K pathway involves many factors, including the binding of receptor tyrosine kinase (RTK), G-protein-coupled receptors (GPCR), and GTP-binding proteins to adaptor proteins. PI3K consists of the catalytic subunit, p110, and the regulatory subunit, p85. PI3K phosphorylates PIP2 (phosphatidylinositol 3,4-bisphosphate) and produces PIP3 (phosphatidylinositol 3,4,5-trisphosphate). PIP3 then activates 3-phosphoinositide-dependent kinase 1 (PDK1) and its major downstream effector, Akt. Phosphorylation of Akt promotes cell proliferation, survival, migration, and differentiation. Phosphatase and tensin homolog (PTEN) dephosphorylates PIP3 and inhibits activation of Akt by PIP3. Phosphorylation of Akt induces the activation of one of the major downstream effectors, mTOR (mammalian target of rapamycin). mTOR phosphorylates S6K1 and 4EBP1, directly leading to increased translation and synthesis of cell-cycle-regulating and ribosomal proteins. Stimulatory events are indicated by arrows and inhibitory events are indicated by lines ending in flat lines.
Figure 1.
Schematic representation of the PI3K/Akt/mTOR pathway. The PI3K pathway involves many factors, including the binding of receptor tyrosine kinase (RTK), G-protein-coupled receptors (GPCR), and GTP-binding proteins to adaptor proteins. PI3K consists of the catalytic subunit, p110, and the regulatory subunit, p85. PI3K phosphorylates PIP2 (phosphatidylinositol 3,4-bisphosphate) and produces PIP3 (phosphatidylinositol 3,4,5-trisphosphate). PIP3 then activates 3-phosphoinositide-dependent kinase 1 (PDK1) and its major downstream effector, Akt. Phosphorylation of Akt promotes cell proliferation, survival, migration, and differentiation. Phosphatase and tensin homolog (PTEN) dephosphorylates PIP3 and inhibits activation of Akt by PIP3. Phosphorylation of Akt induces the activation of one of the major downstream effectors, mTOR (mammalian target of rapamycin). mTOR phosphorylates S6K1 and 4EBP1, directly leading to increased translation and synthesis of cell-cycle-regulating and ribosomal proteins. Stimulatory events are indicated by arrows and inhibitory events are indicated by lines ending in flat lines.
3. Gene Mutation and Activation of PI3K/Akt/mTOR Pathway in Gastric Carcinoma
Genetic alterations in the PI3K/Akt/mTOR pathway in gastric carcinoma have been demonstrated frequently. The alterations and activity founded in gastric carcinoma are summarized in Table 1. These events are due to several mechanisms, including mutation and amplification of genes encoding crucial components of the signaling cascade.
The catalytic subunit PIK3CA gene is mutated at a high frequency in gastric carcinoma cell lines and tumor tissues [11]. Several studies have demonstrated that somatic mutations in PI3KCA are present in 4%–25% of patients with gastric carcinoma [21,22,23,24,25]. The mutation incidence is high (21.4%) in T4 cancers and low (6.4%) in T2 cancers. Therefore, PIK3CA mutations are likely to be late events in gastric carcinogenesis [24]. Although PIK3CA mutations preferentially occur together with activating KRAS-BRAF mutations in colorectal carcinoma, PIK3CA mutations tend to occur as isolated events, such as those in mismatch repair deficiency in gastric carcinoma [21]. In contrast, another study showed that PIK3CA mutations are rare, but their amplification is very common in gastric carcinoma. Therefore, amplification could be a major mechanism in activating the PI3K/Akt pathway in this type of malignancy [26]. The PIK3R3 gene, which encodes the PI3K regulatory subunit p55γ, was found to be significantly upregulated in gastric tumor specimens. Silencing PIK3R3 resulted in reduction of the growth of gastric carcinoma cells, induction of G0/G1 cell-cycle arrest, as well as attenuation of retinoblastoma protein phosphorylation and of cyclin D1 and PCNA expression [9].
High expression levels of Akt and p-Akt were observed in 74% and 78% of gastric tumors, respectively [27]. p-Akt expression was significantly correlated with the depth of invasion, number of lymph nodes, and poor prognosis in gastric carcinoma with respect to tumor angiogenesis [28,29]. On the other hand, p-Akt expression was also significantly associated with HER2 overexpression but not with PIK3CA mutations [30]. A survey of 225 human tumors for changes involving AKT1 led to the discovery of a 20-fold amplification of this gene in gastrointestinal tumors [31]. Additionally, high activity of GSK3β was found to be frequently present in early-stage gastric carcinoma and was positively associated with good prognosis. Thus, GSK3β could be a useful prognostic marker for gastric carcinoma [32].
The tumor suppressor gene PTEN located on chromosome 10q23.3 acts as a plasma membrane lipid phosphatase. Several studies revealed that inactivation of PTEN is closely correlated with the initiation and development of gastric carcinoma. There are reports that PTEN could be a prognostic biomarker for this type of malignancy. A direct-sequencing study indicated 27 cases with mutations among 144 patients (18.8%) consisting of 15 cases of missense mutations, nine nonsense mutations, two 1-bp deletions, and a mutation in intron 6. The mutation status of PTEN was significantly related to pTNM staging and to the degree of cell differentiation [33]. The importance of the loss of function of PTEN in metastatic gastric carcinomas is well described and its role includes homozygous deletions, loss of heterozygosity, and inactivation of mutations [34]. Interestingly, the lack of PTEN in gastric carcinoma is more often found in the cardia than in other areas of the stomach [34]. In addition to amplification of the Akt1 gene locus, frequent monoallelic deletions of PTEN phosphatase antagonism of PI3K/Akt in 47% of cases [35] or of promoter methylation in 39% of cases have been described [36]. An immunohistochemical study showed positive correlations between mTOR expression in gastric carcinomas and pathological parameters such as invasive depth, differentiation, and lymph node metastasis. The positive rate of mTOR expression was found to be much higher in tumor cells, whereas little or no expression in normal gastric tissues was observed [37]. This report also showed that the expression levels of mTOR and PTEN were negatively correlated in the PI3K/Akt/mTOR signaling pathway.
Several other well-known upstream oncogenes such as those that activate mutations in RAS family members, as well as mutations and amplifications of RTKs (e.g., EGFR and HER2/neu) mediate their effect in part through activation of PI3K signaling. Interestingly, the prevalence of these events are depending on the carcinoma type. As there is evidence that various mutations are non-equivalent in their functional dependencies, clarifying the specific oncogenic events that are present in specific cancers is critical [38].

{kind=link}
{kind=link}
{kind=link}
| Study [Ref#] | PMID | Sample | Main Results |
|---|---|---|---|
| PI3K | |||
| Samuels et al. [11] | 15016963 | Tumor specimens | Mutations in PIK3CA were identified in 3 of 12 gastric cancers (25%). |
| Velho et al. [21] | 15994075 | Tumor specimens | PIK3CA mutations in exons 9 and 20 were present in 10.6% of gastric carcinomas. |
| Barbi et al. [23] | 20398348 | Tumor specimens | PIK3CA mutations were present in 16% of gastric carcinomas. No other association between PI3KCA mutations and their clinical pathological covariates was found. |
| Sukawa et al. [24] | 24458107 | Tumor specimens | The mutation incidence is high (21.4%) in T4 cancers and low (6.4%) in T2 cancers. |
| Corso et al. [25] | 20937558 | Tumor specimens | Mutations in PIK3CA gene occurred in 14.3% of the MSI gastric cancers. |
| Shi et al. [26] | 22292935 | Tumor specimens | PIK3CA mutations are rare, but their amplification is very common in gastric carcinoma. |
| Zhou et al. [9] | 22876838 | Cell lines/tumor specimens | PIK3R3 was significantly up-regulated in gastric cancer specimens, and 9.5% to 15% tumors showed more than 2 fold increase compare to the paired mucosa tissues. |
| Akt | |||
| Num et al. [27] | 14678019 | Tumor specimens | Akt expression was detected in 74% of the tumors and pAkt expression in 78%. |
| Cinti et al. [28] | 18841391 | Tumor specimens | There was a statistically significant correlation between pAkt expression and depth of infiltration of the tumor, number of infiltrated lymph nodes and p34/cdc2 expression. |
| Kobayashi et al. [29] | 16785763 | Cell lines/tumor specimens | pAkt expression was detected in 57% of the tumors, which was correlated with high clinicopathological parameters as well as a poor outcome. |
| Sukawa et al. [30] | 23236232 | Tumor specimens | pAkt expression was also significantly associated with HER2 overexpression but not with PIK3CA mutations |
| Staal et al. [31] | 3037531 | Cell lines/tumor specimens | A survey of 225 human tumors for changes involving AKT1 led to the discovery of a 20-fold amplification of this gene in one of the five gastric adenocarcinomas tested. |
| Cho et al. [32] | 20704706 | Cell lines/tumor specimens | High activity of GSK3β was found to be frequently present in early-stage gastric carcinoma and was positively associated with good prognosis. |
| PTEN | |||
| Wen et al. [33] | 20514448 | Tumor specimens | PTEN mutations were present in 55.6% of missense mutation, 33.3% of nonsense mutation, 7.4% of 1-bp deletion and 3.7% of a mutation within intron. |
| Mina et al. [34] | 22639407 | Tumor specimens | 4.4% of primary gastric cancer spots showed PTEN deletions. PTEN deletion was correlated with nodal and distant metastases. |
| Byun et al. [35] | 12569555 | Cell lines/tumor specimens | Frequent monoallelic deletions of PTEN phosphatase antagonism of PI3K/Akt in 47% of cases. |
| Kang et al. [36] | 11896207 | Tumor specimens | The promoter methylation frequency of PTEN was found to be present in 39% of cases examined, and 73% of gastric cancer tissues showing promoter methylation exhibited the loss of PTEN expression. |
| mTOR | |||
| Li et al. [37] | 23205120 | Tumor specimens | The expression levels of mTOR and PTEN were negatively correlated in the PI3K-AKT-mTOR signaling pathway. |
4. The Role of the PI3K/Akt/mTOR Pathway in the Biological Properties of Gastric Carcinoma
4.1. Apoptosis
The anti-apoptotic activities of the PI3K/Akt/mTOR pathway are closely related to the resistance of the malignant cells to a spectrum of apoptosis-stimulating factors (Figure 2). Several studies showed that the use of PI3K inhibitors substantially facilitates apoptosis [39]. A recent investigation demonstrated that PI3K inhibition induces mitochondrial function and upregulates p53 and PUMA levels [40]. A synthetic small-molecule pan-PI3K inhibitor, LY294002, significantly inhibits the growth of gastric carcinoma while promoting apoptosis, resulting in the downregulation of MMP-2, MMP-9, and VEGF [41]. Impairment of PI3K/Akt activity attenuates the activity of κ-light-chain enhancer of activated B cells (NF-κB) and increases apoptosis in gastric carcinoma cells. This effect induced by blocking PI3K/Akt was ascribed to the inhibition of NF-κB activity through IKK/IκB [42]. Furthermore, the NF-κB inhibitor SN50 leads to an increase in the effect of LY294002 on inducing death of human gastric carcinoma cells through upregulated expression of p53, PUMA, and Beclin1 [43]. Several factors have been reported to induce apoptosis via the PI3K/Akt pathway in gastric carcinoma [44,45,46,47,48]. Apoptosis of gastric carcinoma cells induced by isoalantolactone was associated with the dissipation of mitochondrial membrane potential due to downregulation of Bcl-2 and upregulation of Bax [49]. Some studies have shown that PTEN could enhance Fas/FasL or cytochrome c-mediated apoptosis that involves activation of caspase-3, suggesting that decreased PTEN expression could downregulate caspase-3, thus disrupting the apoptotic pathway in gastric carcinoma [50].
4.2. Metastasis
More than half of patients with advanced gastric carcinoma have distant metastasis upon its diagnosis [1]. To date, no molecular indicator is available for predicting metastasis in this disease. However, recent studies have shown that p-Akt is highly expressed in the lymph node and in distant metastases compared with that in the primary gastric tumor, suggesting that Akt activity plays a role in promoting metastasis in gastric carcinoma patients [51]. p-Akt positivity also correlates with microvessel density and VEGF, suggesting that p-Akt and VEGF have a significant role in angiogenesis in gastric adenocarcinoma [52]. Dominant-negative Akt inhibits the proliferation of gastric carcinoma cells and induces G1 cell cycle arrest, whereas upregulation of Akt increases cell proliferation [53]. On the other hand, gastric carcinoma tissues strongly express PI3K p85α and p-AkT, and targeted blockade of the PI3K pathway inhibits gastric carcinoma growth and metastasis through downregulation of Ki-67 and MMP-2 expression [54]. Combrestatin A4, an anti-angiogenic compound, reduces the growth of a cell line with high Akt expression. Downregulated Akt suppresses tumor formation and metastasis by reducing cell attachment, migration, and invasion [55]. The PI3K/Akt pathway also enhances the adhesiveness of metastatic gastric carcinoma cells. Our previous data reveal that attachment of OCUM-2MD3 cells, an extensively peritoneal-seeding human scirrhous gastric cancer cell line, to type IV collagen increased Akt activation; whereas PI3K/Akt inhibition decreased adhesion of in a dose-dependent manner. Immunoprecipitation studies indicated that the PI3K/Akt pathway is associated with integrin signaling through Src and vinculin, leading to cytoskeletal reorganization [56].
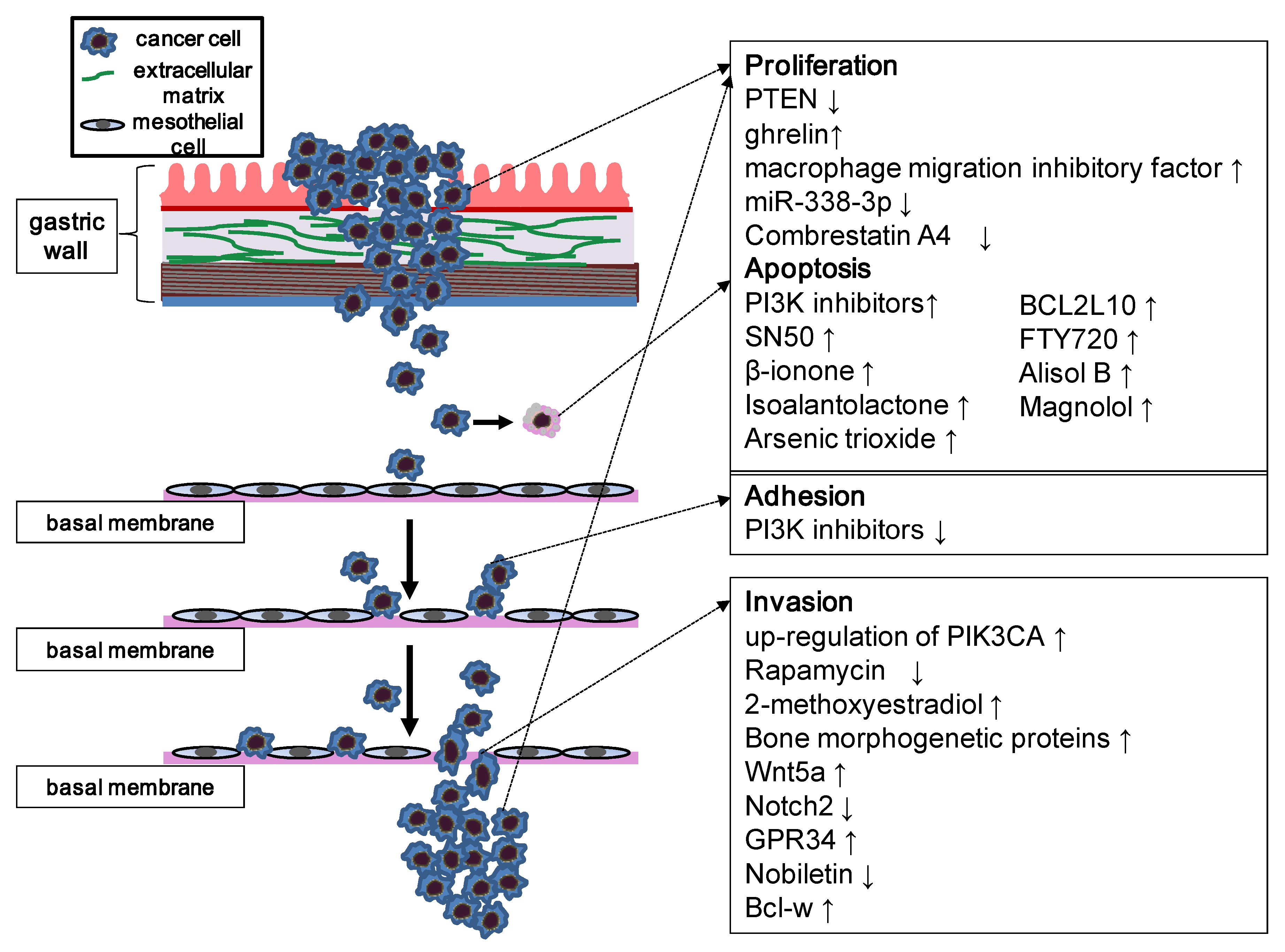
Figure 2.
Schematic representation of the PI3K/Akt/mTOR pathway involved in carcinoma metastasis. The figure focuses on the biological process of peritoneal metastasis, which is one of the most common metastatic types of gastric carcinoma, although these principles apply to most patterns of metastases. The PI3K/Akt/mTOR pathway is involved in each step of metastasis; thus, numerous inhibitors and agents can affect these processes by modulation of PI3K signaling. Stimulatory events are indicated by upward arrows and inhibitory events are indicated by downward arrows.
Figure 2.
Schematic representation of the PI3K/Akt/mTOR pathway involved in carcinoma metastasis. The figure focuses on the biological process of peritoneal metastasis, which is one of the most common metastatic types of gastric carcinoma, although these principles apply to most patterns of metastases. The PI3K/Akt/mTOR pathway is involved in each step of metastasis; thus, numerous inhibitors and agents can affect these processes by modulation of PI3K signaling. Stimulatory events are indicated by upward arrows and inhibitory events are indicated by downward arrows.
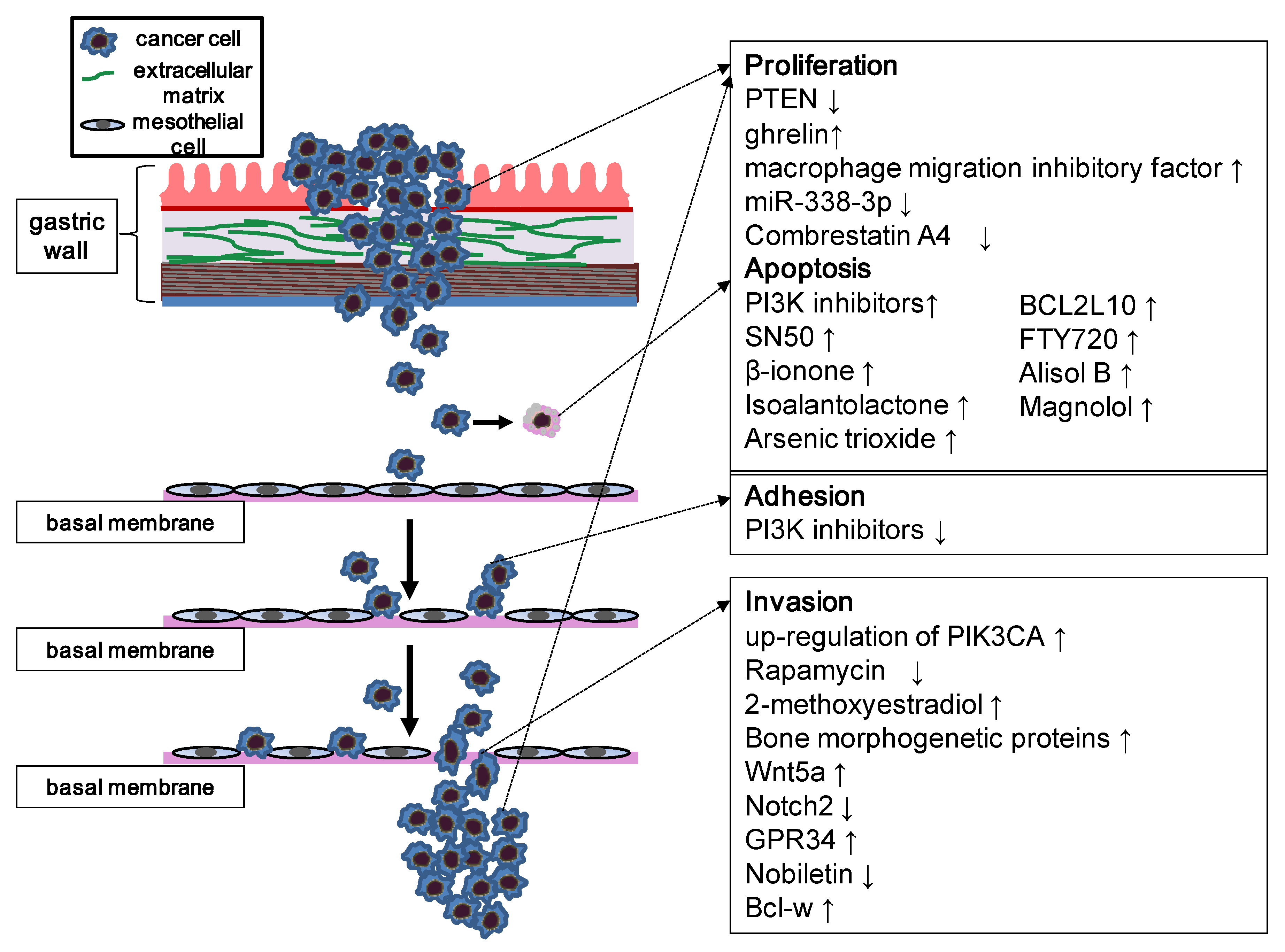
Upregulation of PIK3CA expression is likely related to lymph node metastasis in gastric cancer [51]. Upregulated PIK3CA enhances the activity of PI3K/Akt signaling through increased activity of PI3K p110α, thereby promoting the invasion and metastasis of gastric carcinoma cells [51]. Downregulation of PIK3CA in these cells reduces their ability to proliferate, migrate, and invade. Robust knockdown of PIK3CA by siRNA results in decreased catalytic activity of PI3K and subsequent dephosphorylation of Akt in these cells [57]. PI3K signaling regulates Rac1 activity, resulting in the induction of hypoxia-inducible factor 1α and VEGF [58].
PTEN-mediated dephosphorylation of FAK can influence the growth and invasion of gastric carcinoma [59]. Rapamycin has been reported to inhibit the migration of gastric cancer cells. Chemokine (CXC motif) ligand 12 (CXCL12) could be an independent prognostic factor in gastric carcinoma. The mTOR pathway was also found to play an important role in CXCL12/CXCR4-mediated cell migration [60].
Several factors mediate the metastatic capacity of gastric carcinoma through the PI3K/Akt/mTOR pathway [61,62,63,64,65]. Administration of 2-methoxyestradiol, a metabolite of estradiol-17b, decreased cell invasion and metastasis of gastric carcinoma through attenuation of Akt activity [66]. The bone morphogenetic protein signaling pathway enhances tumor invasiveness and metastasis in gastric carcinoma by sequential activation of PI3K/Akt followed by induction of NF-κB and MMP-9 activity [67]. Wnt5a dose-dependently stimulates the migration of human gastric carcinoma cells by enhancing phosphorylation of PI3K/Akt and GSK3β and by activating RhoA [68]. Notch2, a transmembrane receptor that mediates local cell-cell interaction in mammals, negatively regulates cell invasion and expression of MMP-9 by inhibiting the PI3K/Akt signaling pathway [69]. Micro-RNA (miRNA) miR-338-3p inhibits cell proliferation and clonogenicity and induces G1/S arrest and apoptosis in gastric carcinoma cells. Hyperactivation of miR-338-3p and silencing of P-Rex2a reduces the expression of P-Rex2a, resulting in the activation of PTEN and a decline in the phosphorylation of Akt [70]. miR-214 could also post-transcriptionally mediate PTEN expression by binding to the 3'-UTR of its mRNA. The expression level of miR-214 increases in gastric carcinoma and downregulates miR-214, leading to attenuated proliferation and tumor invasion [50].
5. The Role of the PI3K/Akt/mTOR Pathway in Resistance to Chemotherapy in Gastric Carcinoma
Anticancer agents and radiation therapy attack target cells by inducing apoptosis. In contrast, failure to activate apoptotic signaling represents an important mode of drug resistance in tumor cells [71]. Thus, resistance to chemotherapy is a major clinical problem in the treatment of all malignancies, including gastric carcinoma. The PI3K pathway is commonly activated in advanced gastric carcinoma [72,73] and survival signals induced by several receptors are mediated mainly by the PI3K/Akt/mTOR pathway [8]. A previous report has shown that LY294002 enhances the sensitivity of Fas-mediated apoptosis of gastric cancer cells by inducing p27/Kip accumulation and by dowregulating Mcl-1 expression [74]. LY294002 and vincristine were shown to synergistically promote growth inhibition, which was associated with decreased expression of MDR1/P-gp, Bcl-2, and XIAP, as well as with upregulation of Bax and caspase-3 expression [75]. BEZ235, a novel inhibitor of PI3K and mTOR, exerts antitumor effects against gastric carcinoma and enhances the effects of nab-paclitaxel through inhibition of cell proliferation and modulation of the PI3K/mTOR pathway [76]. Therefore, inhibition of PI3K/Akt activation may overcome the resistance of cancer cells to anticancer therapy.
The use of etoposide and doxorubicin in gastric carcinoma enhanced Akt activity in a manner that was dependent on PI3K [77]. Akt activity was correlated with increased resistance to multiple chemotherapeutic agents such as 5-fluorouracil (5-FU), doxorubicin, and cisplatin [73]. NF-κB is another factor mediating chemoresistance induced by Akt. Chemotherapy facilitates the activation of NF-κB, thereby inhibiting the PI3K/Akt pathway [78]. PrPC is a glycosylphosphatidylinositol-anchored membrane protein. Inhibition of PI3K/Akt signaling results in reduction of multidrug resistance of gastric carcinoma cells by downregulation of P-glycoprotein induced by PrPC [79]. Upregulation of p-Akt confers resistance of gastric carcinoma cells to anticancer agents and suppresses adriamycin-induced apoptosis [80]. Inhibition of p-Akt expression reverses Akt-mediated multidrug resistance, upregulates p53 expression, and downregulates the expression of P-glycoprotein and transcription of multidrug resistance gene 1. The chemosensitivity of gastric carcinoma cells to cisplatin increased significantly after RNAi knockdown of Akt1 expression. This increase may be associated with the inactivation of the PI3K/Akt1 signaling pathway followed by the induction of Bax expression and a decrease in Bcl-2 expression [81].
Several studies have shown that PTEN is involved in drug resistance in various types of malignancies, including gastric carcinoma [82,83]. Downregulation of PTEN can lead to chemotherapeutic drug resistance, including DDP resistance, in gastric cancer patients [35,73]. Interestingly, the mTOR inhibitor everolimus specifically enhanced fluorouracil-induced apoptosis in two cell lines with HER2 amplification. This result suggests that mTOR inhibitors may be alternative drugs in gastric cancers with HER2 amplification [84].
Several other factors, including transcription factors, have been confirmed as mediators of chemoresistance through the PI3K/Akt/mTOR pathway in gastric carcinomas. Recent studies have suggested that miRNAs such as those in the miR-17 family, are novel candidate targets in the advancement of resistance to chemotherapy [85]. miR-106a, which belongs to the miR-17 family, is upregulated in DDP-resistant cells. Its overexpression regulates apoptosis and DDP resistance in gastric carcinoma cells. Enhanced expression of miR-106a also leads to downregulation of PTEN and more active signaling through the PI3K/Akt pathway. miR-21 also confers cisplatin resistance in gastric carcinoma cells by regulating PTEN [85]. Expression of miR-34a has been shown to be downregulated in cisplatin-resistant gastric carcinoma cell lines. miR-34a overexpression could improve the sensitivity of cells against cisplatin-based chemotherapies via the PI3K/Akt/survivin signaling pathway. miRNA-19a/b affects multidrug resistance in gastric carcinoma cells by targeting PTEN through regulation of Bcl-2 and Bax [86]. p21-activated kinase 4 (PAK4) induces CDDP resistance in gastric carcinoma cells by activation of the MEK/Erk and PI3K/Akt pathways. There exists mutual activation between the PAK4 and PI3K/Akt pathways in gastric carcinoma cells [87]. The forkhead transcription factor FOXO1 mediates cisplatin resistance in gastric carcinoma cells by activating the PI3K/Akt pathway. CDDP-induced FOXO1 activation is accompanied by an increase in p110α and p-Akt expression [88]. Interestingly, Tamoxifen reverses drug efflux transporters in P-gp-mediated multidrug resistance of human estrogen-receptor-negative gastric carcinoma cells by inhibiting the PI3K/Akt signaling pathway in these cells [89]. Activation of PI3K/Akt and NF-κB by TRAIL (TNF-related apoptosis-inducing ligand) confers resistance of human gastric cancer cells to TRAIL. Interference of survival signals significantly enhances apoptosis induced by TRAIL [90].
6. Targeting PI3K/Akt/mTOR as a Therapy for Gastric Carcinoma
Inhibitors and antibodies targeting specific molecules, including PI3K pathways, are becoming more advanced; clinical trials have demonstrated some of these to be effective. The significant role of the PI3K/Akt/mTOR pathway in the initiation and development of gastric carcinoma suggests that this pathway may be an appropriate target for cancer therapy (Figure 3). Such therapy may involve inhibiting cell proliferation, enhancing apoptosis, and restoring the sensitivity of cancer cells to chemotherapy. Although these strategies are promising, clinical development of PI3K/Akt/mTOR pathway inhibitors still faces many problems [91]. PI3K inhibitors may be classified into several functional categories: PI3K inhibitors, dual mTOR1/mTOR2 inhibitors, Akt inhibitors, mTOR1 inhibitors, and dual PI3K/mTOR inhibitors. These inhibitors may exhibit differences in their affinity for different isoforms of these proteins. Derivatives of these inhibitors are more selective and are highly effective in targeting the PI3K/Akt/mTOR pathway, either alone or in combination.
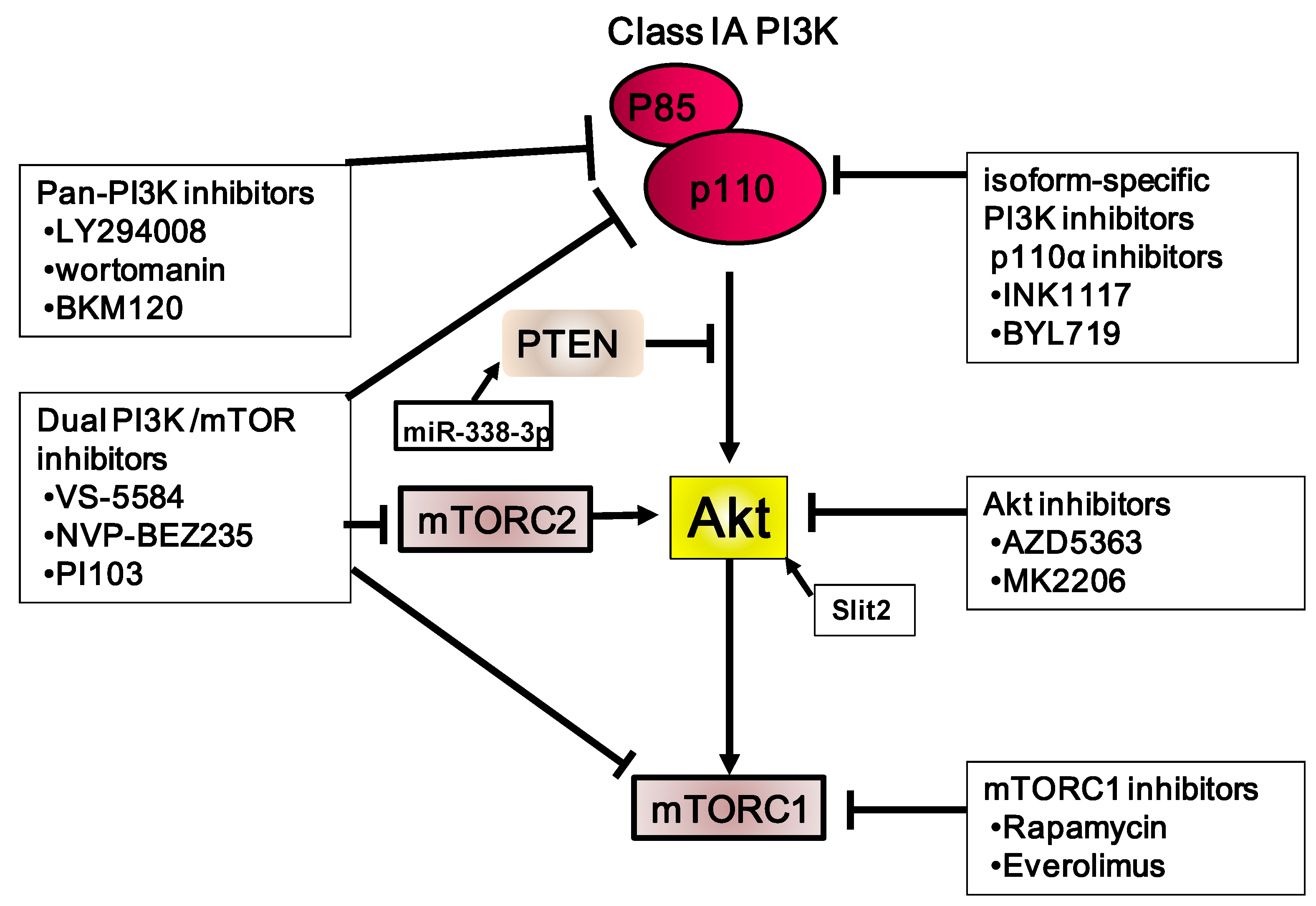
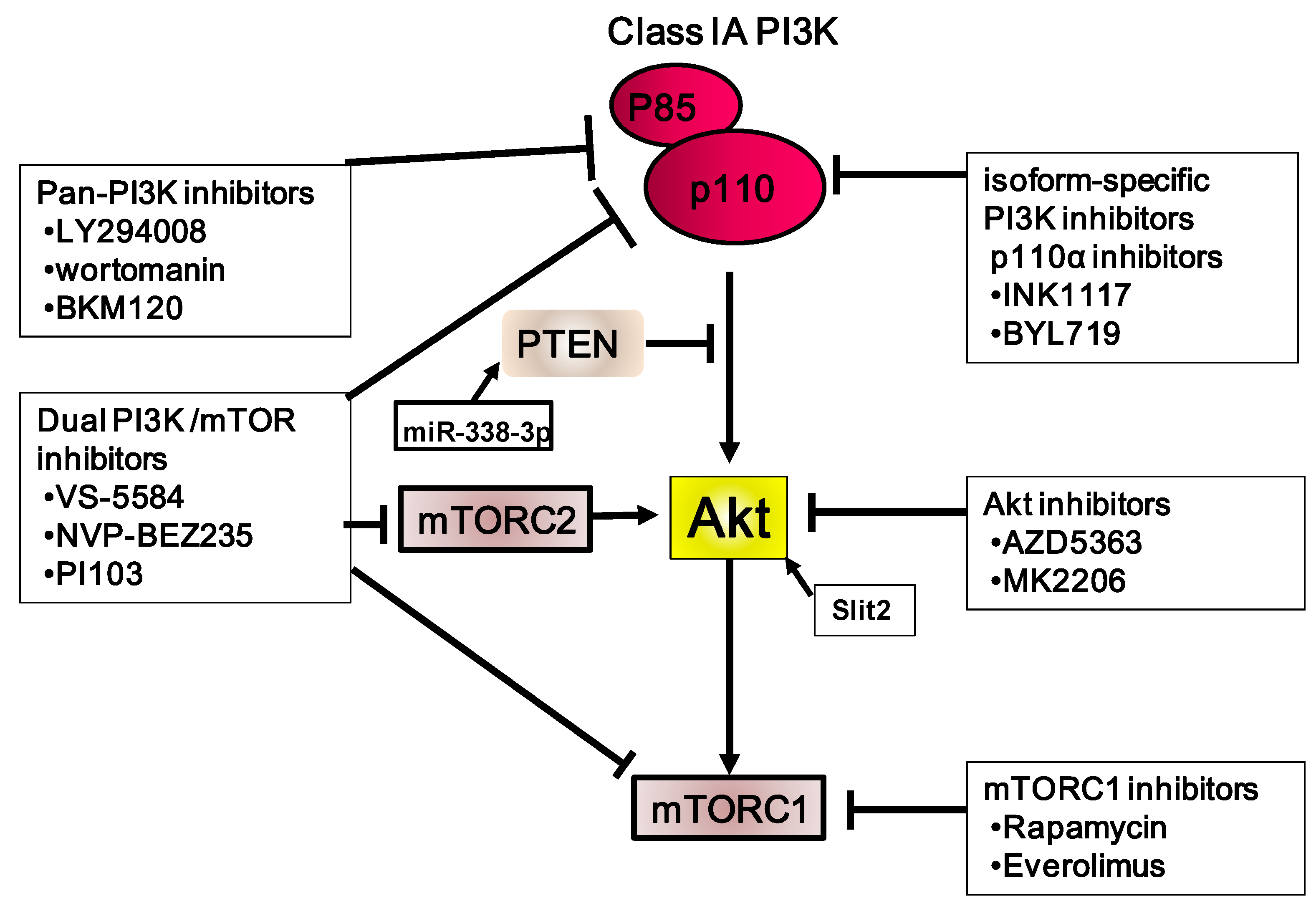
Figure 3.
Summary of the PI3K/Akt/mTOR inhibitors used in gastric carcinoma, including those used in clinical trials. Stimulatory events are indicated by arrows and inhibitory events are indicated by lines ending in flat lines.
Figure 3.
Summary of the PI3K/Akt/mTOR inhibitors used in gastric carcinoma, including those used in clinical trials. Stimulatory events are indicated by arrows and inhibitory events are indicated by lines ending in flat lines.
6.1. PI3K Inhibitors
Several clinical trials targeting the PI3K/Akt/mTOR pathway are ongoing (Table 2). The first PI3K inhibitors identified were LY294002 [92] and wortmannin, a natural product from Penicillium wortmannii [93]. Our previous study showed that LY294002 reduces the total volume of metastatic nodules per mouse of OCUM-2MD3 in the peritoneal cavity in an experimental metastatic model [56]. However, these two inhibitors have been found to be toxic in animals. Several different types of PI3K inhibitors have also been tested against various tumor types in clinical trials. The combination of NVP-BKM120, a pan-class I PI3K inhibitor, and AG490, a STAT3 inhibitor, shows synergistic induction of apoptosis; however, this effect was observed only in cells harboring mutant KRAS. This result suggests that dual inhibition of signals may be an effective therapeutic strategy for KRAS-mutant gastric cancer patients [94]. An HER3-neutralizing antibody, LJM716, and an ATP-competitive p110α-specific inhibitor, BYL719, synergistically inhibit the growth of tumor xenografts. Gastric tumors in mice treated with a combination of these two inhibitors exhibited reduced p-Akt expression [95]. A phase I study on the safety of BYL719 and an HSP90 inhibitor, AUY922, in patients with advanced gastric cancer is ongoing.
| Therapeutic Agent | Target | Clinical Trial | Efficacy | Year | Ref |
|---|---|---|---|---|---|
| Everolimus vs. placebo | mTORC1 | Phase III (GRANITE-1) | PFS 1.68 vs. 1.41, p < 0.0001
OS 1.68 vs. 1.41, p = 0.1244 | 2013 | [97] |
| Everolimus ± paclitaxel | mTORC1 | Phase III (AIO-STO-0111) | Enrolling | ||
| MK-2206 + Trastuzumab | Akt | Phase I | 1 of 4 patient archive SD | 2013 | [97] |
| BYL719 | p110α | Phase I | Enrolling | ||
| BKM120 | PI3K | Phase I | Enrolling |
PFS, progression free survival; OS, overall survival.
6.2. Akt Inhibitors
Several classes of Akt inhibitors are currently being developed. The effect of the Akt inhibitor, AZD5363, was determined according to the mutation of PIK3CA in gastric carcinoma cells. AZD5363 and taxotere administered in combination showed significant antitumor activity in PTEN loss in an in vivo model [98]. Targeting all Akt isozymes is better than inhibition of a single enzyme, although toxicity may be an issue. GSK690693 is an ATP-competitive, low-nanomolar, pan-Akt kinase inhibitor [99]. A potent, highly selective pan-Akt inhibitor, MK-2206, is now in early clinical trials [97]. Preliminary evidence shows that simultaneous administration of MK-2206 and trastuzumab has therapeutic efficacy in patients with HER2+ breast or gastroesophageal carcinoma, with a clinical benefit response rate of ~24% and a median time of progression of 72 days. The synergism of the two agents is as tolerable as the same dose of MK-2206 alone.
6.3. mTOR Inhibitors
Proof-of-principle that the PI3K pathway in cancer can be successfully targeted has been demonstrated by the development of rapamycin analogs that inhibit mTORC1 kinase (e.g., temsirolimus and everolimus) [100]. However, recent results have been disappointing when these analogs have been administered as single agents in gastric carcinoma.
Everolimus, a derivative of rapamycin, is an oral inhibitor of mTOR. Everolimus has been shown to be effective in several solid tumors, including gastric carcinomas. Several clinical trials have evaluated the efficacy and safety of this agent for gastric carcinoma. In a multicenter phase II study, everolimus monotherapy resulted in a promising DCR in patients with previously treated advanced gastric carcinoma. In general, this treatment is well-tolerated and has manageable side effects [101]. Based on these results, a randomized, double-blind phase III study (GRANITE-1) was performed to compare everolimus with BSC against placebo and against BSC in patients with gastric carcinoma [96]. Compared with BSC, everolimus did not significantly improve overall survival for patients with advanced gastric carcinoma that progressed after one or two sessions of previous systemic chemotherapy. Nevertheless, everolimus reduced the risk of progression by 34%, and the PFS was 1.7 versus 1.4 months, respectively, suggesting promise of combination therapy using everolimus with other molecular targeted or antitumor agents. Interestingly, MK-2206 and everolimus may be combined to synergistically inhibit PI3K/Akt/mTOR signaling and growth of gastric carcinoma cells. This enhancement is due to MAPK-dependent autophagy, and not to the apoptotic pathway [102].
6.4. Dual mTORC1/2 Inhibitors
Limiting mTORC1 inhibition correlates with the negative feedback loops that regulate the PI3K pathway controlled by mTORC1. Inhibition of mTORC1 alone can lead to a paradoxical increase in mTORC2 activity and thus continuation of Akt signaling [103]. A recent study revealed that rapamycin activates p-Akt and mTORC2 in gastric carcinoma cells. Downregulation of RICROR, which is regulated by mTORC2-induced Akt activation, reduces phosphorylation of GSK3 and cancer cell motility, suggesting that mTORC2 inhibition may abrogate the unfavorable signaling effects of mTOR inhibitors [104]. Therefore, mTORC1 and mTORC2 inhibition may be more effective in blocking the mTOR pathway Development of small-molecule inhibitors for mTORC1/2 has made progress. AZD2014, AZD8055, and OSI-027 are currently in clinical trials in patients with solid tumors. However, there is still no information about their activity in gastric carcinoma.
6.5. PI3K and mTOR Inhibitors
Inhibition of PI3K and mTOR1/2 results in inhibition of growth of gastric carcinoma cell lines greater than that attained with mono-inhibition alone. VS-5584 is a novel, low-molecular-weight compound with high and equivalent potency against mTOR and against PI3K class I isoforms. This compound has no activity against many lipid and protein kinases; therefore, it has a profile different from that of compounds in the clinical stages at present. Interestingly, VS-5584 was compared with 5-FU in a HER2-overexpressing gastric xenograft model. 5-FU and VS-5584 inhibited gastric carcinoma tumor growth by 32% and 121%, respectively, with a statistically significant difference for VS-5584 only [105]. Monotreatment of gastric-tumor-bearing mice with VS-5584 or with gefitinib resulted in tumor growth inhibition of 88% and 17%, respectively, with statistically significance for VS-5584 only. Combination therapy at the same dose levels resulted in tumor growth inhibition of 121%, with good tolerance for the therapy and no significant loss of body weight. The PI3K and mTOR inhibitor, PI103, acted in synergy with 5-FU in PIK3CA-mutated gastric carcinoma cells. This effect was associated with reductions in E2F1 and thymidylate synthase, and with increased DNA damage [106]. BEZ235 and BKM120 induces pro-apoptotic effects in all cell lines, with a particularly high response in PI3KCA-mutated gastric carcinoma cell lines [107]. On the other hand, cancers harboring KRAS mutations are likely to be insensitive to single-agent PI3K inhibitors and this type of malignancies show synergism in combination treatment with MEK inhibitors [108]. Thus, it is believed that combined inhibition is required to suppress the activation of other pathways and feedback-loop-induced activation of other signaling pathways and thereby to potentially lead to greater induction of apoptosis. Furthermore, BEZ235 has been shown to have antitumor effects against gastric carcinoma. It enhances the inhibitory effects of paclitaxel and reduces cell proliferation [76].
6.6. Other Therapeutic Approaches
Suppression of Akt1 and PIK3R1 expression using Akt1 and PIK3R1 small hairpin RNA RNAi can inhibit gastric carcinoma cell growth in vitro and in vivo [109]. Adenovirus PI3K(I) RNAi GFP induces mitochondrial dysfunction and activates apoptosis in gastric carcinoma cells, leading to increased formation of autophagosomes. Additionally, cells treated with adenovirus PI3K(I) RNAi GFP exhibit typical signs of autophagy [110].
Recent studies have shown that expression of HGF and its receptor c-Met is enhanced in gastric carcinoma. Thus, several therapeutic approaches targeting c-Met have been explored [111]. The small-molecule inhibitor, PHA-665752, which targets the catalytic activity of c-Met kinase, decreases PI3K signaling and correlates with potent cytoreductive activity in a gastric carcinoma xenograft model [112]. Moreover, knockdown of c-Met by RNA interference in vitro leads to reduction of Akt and to apoptotic cell death in vitro [113]. The c-Met inhibitor, KRC-408, exerts antitumor effects by directly affecting tumor cell growth and survival through attenuation of Akt and Erk phosphorylation as well CD34 expression in gastric tumors [114].
7. Conclusions and Future Perspectives
Deregulation of the PI3K/Akt/mTOR pathway is frequently encountered in gastric carcinoma. This crucial pathway plays important roles in tumor initiation and progression, including those in proliferative activity and in apoptosis. PI3K signaling is also commonly associated with the metastatic cascade in gastric carcinoma, which includes proteolytic activity, cytoskeletal remodeling, and resistance to chemotherapy. Although several aspects of tumor inhibition are not fully understood, numerous small molecule inhibitors targeting the PI3K/Akt/mTOR pathway are currently being studied in clinical trials for gastric carcinoma. Notably, gastric carcinoma is not a homogeneous disease and thus requires more personalized treatment. The heterogeneity of gastric carcinomas often causes delays in clinical trials for treatments against this cancer. One important approach to overcome this problem may be elucidation of feedback loops within the PI3k/Akt/mTOR pathway and of crosstalk with other signaling pathways [115]. Combination therapy with other cytotoxic or targeted agents and development of multitargeting agents may eliminate resistance and may result in the synergistic improvement of clinical benefits [116].
Trastuzumab has already been shown to have therapeutic utility in gastric carcinoma. The PI3K pathway is one of the main downstream signals of HER2. In gastric carcinoma, high expression levels of HER2 result in enhanced activity of Akt, which is closely related to poor prognosis. Consequently, PI3K pathway alterations are may be biomarkers for predicting the effectiveness of trastuzumab therapy. Understanding the specific association between HER2 expression and PI3K/Akt/mTOR pathway alterations in gastric carcinoma may lead to the development of new therapeutic strategies using trastuzumab.
Considering that activation of PI3K/Akt/mTOR pathway is necessary for each event in the metastatic cascade, this pathway is likely to be a key underlying process for metastasis; therefore, focusing on this pathway may be worthwhile. Evaluation of the signaling networks and molecules that are involved in the metastasis of gastric carcinoma, for example, PI3K, may lead to novel therapeutic strategies. Management of metastasis is critical for good prognosis in patients. Rational use of biological therapies that have different molecular targets, such as integrin signaling and proteolytic factors, may have synergistic efficacy.
Comprehensive knowledge of the PI3K/Akt/mTOR pathway would be useful in understanding the mechanism of tumor development and in identifying the ideal targeted anticancer therapy for gastric carcinoma. Appropriate clinical trials that incorporate predictive biomarkers need to be developed, as these may lead to personalized therapy.
Acknowledgments
This study is partially founded by KAKENHI (Grant-in-Aid for Scientific Research, Nos. 20591573, 22390262, and 23390329), by the National Cancer Center Research and Development Fund (23-A-9), and by Priority Research Fund of Osaka City University.
Conflicts of Interest
There are not any financial or other interests with regard to the submitted manuscript that might be construed as a conflict of interest.
References
- Guggenheim, D.E.; Shah, M.A. Gastric cancer epidemiology and risk factors. J. Surg. Oncol. 2013, 107, 230–236. [Google Scholar] [CrossRef]
- Bang, Y.J.; van Cutsem, E.; Feyereislova, A.; Chung, H.C.; Shen, L.; Sawaki, A.; Lordick, F.; Ohtsu, A.; Omuro, Y.; Satoh, T.; et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): A phase 3, open-label, randomised controlled trial. Lancet 2010, 376, 687–697. [Google Scholar] [CrossRef]
- Yashiro, M.; Shinto, O.; Nakamura, K.; Tendo, M.; Matsuoka, T.; Matsuzaki, T.; Kaizaki, R.; Miwa, A.; Hirakawa, K. Synergistic anti-tumor effects of FGFR2 inhibitor with 5-fluorouracil on scirrhous gastric carcinoma. Int. J. Cancer 2010, 126, 1004–1016. [Google Scholar]
- Nakamura, K.; Yashiro, M.; Matsuoka, T.; Tendo, M.; Shimizu, T.; Miwa, A.; Hirakawa, K. A novel molecular targeting compound as K-samII/FGF-R2 phosphorylation inhibitor, Ki23057, for Scirrhous gastric cancer. Gastroenterology 2006, 131, 1530–1541. [Google Scholar] [CrossRef]
- Deng, N.; Goh, L.K.; Wang, H.; Das, K.; Tao, J.; Tan, I.B.; Zhang, S.; Lee, M.; Wu, J.; Lim, K.H.; et al. A comprehensive survey of genomic alterations in gastric cancer reveals systematic patterns of molecular exclusivity and co-occurrence among distinct therapeutic targets. Gut 2012, 61, 673–684. [Google Scholar] [CrossRef]
- Wadhwa, R.; Song, S.; Lee, J.S.; Yao, Y.; Wei, Q.; Ajani, J.A. Gastric cancer-molecular and clinical dimensions. Nat. Rev. Clin. Oncol. 2013, 10, 643–655. [Google Scholar] [CrossRef]
- Willems, L.; Tamburini, J.; Chapuis, N.; Lacombe, C.; Mayeux, P.; Bouscary, D. PI3K and mTOR signaling pathways in cancer: New data on targeted therapies. Curr. Oncol. Rep. 2012, 14, 129–138. [Google Scholar] [CrossRef]
- Fresno Vara, J.A.; Casado, E.; de Castro, J.; Cejas, P.; Belda-Iniesta, C.; Gonzalez-Baron, M. PI3K/Akt signalling pathway and cancer. Cancer Treat. Rev. 2004, 30, 193–204. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, G.B.; Tang, Y.C.; Sinha, R.A.; Wu, Y.; Yap, C.S.; Wang, G.; Hu, J.; Xia, X.; Tan, P.; et al. Genetic and bioinformatic analyses of the expression and function of PI3K regulatory subunit PIK3R3 in an Asian patient gastric cancer library. BMC Med. Genomics 2012, 5. [Google Scholar] [CrossRef]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High frequency of mutations of the PIK3CA gene in human cancers. Science 2004, 304. [Google Scholar] [CrossRef]
- Siegfried, Z.; Bonomi, S.; Ghigna, C.; Karni, R. Regulation of the Ras-MAPK and PI3K-mTOR signalling pathways by alternative splicing in cancer. Int. J. Cell. Biol. 2013. [Google Scholar] [CrossRef]
- Staal, S.P.; Hartley, J.W.; Rowe, W.P. Isolation of transforming murine leukemia viruses from mice with a high incidence of spontaneous lymphoma. Proc. Natl. Acad. Sci. USA 1977, 74, 3065–3067. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Almhanna, K.; Strosberg, J.; Malafa, M. Targeting AKT protein kinase in gastric cancer. Anticancer Res. 2011, 31, 4387–4392. [Google Scholar]
- Ortega-Molina, A.; Serrano, M. PTEN in cancer, metabolism, and aging. Trends Endocrinol. Metab. 2013, 24, 184–189. [Google Scholar] [CrossRef]
- Dowling, R.J.; Topisirovic, I.; Fonseca, B.D.; Sonenberg, N. Dissecting the role of mTOR: Lessons from mTOR inhibitors. Biochim. Biophys. Acta 2010, 1804, 433–439. [Google Scholar] [CrossRef]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef]
- Brader, S.; Eccles, S.A. Phosphoinositide 3-kinase signalling pathways in tumor progression, invasion and angiogenesis. Tumori 2004, 90, 2–8. [Google Scholar]
- Zhou, H.; Huang, S. mTOR signaling in cancer cell motility and tumor metastasis. Crit. Rev. Eukaryot. Gene Expr. 2010, 20, 1–16. [Google Scholar] [CrossRef]
- Velho, S.; Oliveira, C.; Ferreira, A.; Ferreira, A.C.; Suriano, G.; Schwartz, S., Jr.; Duval, A.; Carneiro, F.; Machado, J.C.; Hamelin, R.; et al. The prevalence of PIK3CA mutations in gastric and colon cancer. Eur. J. Cancer 2005, 41, 1649–1654. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Costa, A.M.; Pereira-Castro, I.; Salvatore, G.; Hernandez, R.; Hermsem, M.J.; Herrero, A.; Fusco, A.; Cameselle-Teijeiro, J.; Santoro, M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005, 65, 10199–10207. [Google Scholar] [CrossRef]
- Barbi, S.; Cataldo, I.; de Manzoni, G.; Bersani, S.; Lamba, S.; Mattuzzi, S.; Bardelli, A.; Scarpa, A. The analysis of PIK3CA mutations in gastric carcinoma and metanalysis of literature suggest that exon-selectivity is a signature of cancer type. J. Exp. Clin. Cancer Res. 2010. [Google Scholar] [CrossRef]
- Sukawa, Y.; Yamamoto, H.; Nosho, K.; Ito, M.; Igarashi, H.; Naito, T.; Mitsuhashi, K.; Matsunaga, Y.; Takahashi, T.; Mikami, M.; et al. HER2 expression and PI3K-Akt pathway alterations in gastric cancer. Digestion 2014, 89, 12–17. [Google Scholar] [CrossRef]
- Corso, G.; Velho, S.; Paredes, J.; Pedrazzani, C.; Martins, D.; Milanezi, F.; Pascale, V.; Vindigni, C.; Pinheiro, H.; Leite, M.; et al. Oncogenic mutations in gastric cancer with microsatellite instability. Eur. J. Cancer 2011, 47, 443–451. [Google Scholar] [CrossRef]
- Shi, J.; Yao, D.; Liu, W.; Wang, N.; Lv, H.; Zhang, G.; Ji, M.; Xu, L.; He, N.; Shi, B.; et al. Highly frequent PIK3CA amplification is associated with poor prognosis in gastric cancer. BMC Cancer 2012, 12. [Google Scholar] [CrossRef]
- Nam, S.Y.; Lee, H.S.; Jung, G.A.; Choi, J.; Cho, S.J.; Kim, M.K.; Kim, W.H.; Lee, B.L. Akt/PKB activation in gastric carcinomas correlates with clinicopathologic variables and prognosis. Apmis 2003, 111, 1105–1113. [Google Scholar] [CrossRef]
- Cinti, C.; Vindigni, C.; Zamparelli, A.; La Sala, D.; Epistolato, M.C.; Marrelli, D.; Cevenini, G.; Tosi, P. Activated Akt as an indicator of prognosis in gastric cancer. Virchows Arch. 2008, 453, 449–455. [Google Scholar] [CrossRef]
- Kobayashi, I.; Semba, S.; Matsuda, Y.; Kuroda, Y.; Yokozaki, H. Significance of Akt phosphorylation on tumor growth and vascular endothelial growth factor expression in human gastric carcinoma. Pathobiology 2006, 73, 8–17. [Google Scholar] [CrossRef]
- Sukawa, Y.; Yamamoto, H.; Nosho, K.; Kunimoto, H.; Suzuki, H.; Adachi, Y.; Nakazawa, M.; Nobuoka, T.; Kawayama, M.; Mikami, M.; et al. Alterations in the human epidermal growth factor receptor 2-phosphatidylinositol 3-kinase-v-Akt pathway in gastric cancer. World J. Gastroenterol. 2012, 18, 6577–6586. [Google Scholar] [CrossRef]
- Staal, S.P. Molecular cloning of the akt oncogene and its human homologues AKT1 and AKT2: Amplification of AKT1 in a primary human gastric adenocarcinoma. Proc. Natl. Acad. Sci. USA 1987, 84, 5034–5037. [Google Scholar] [CrossRef]
- Cho, Y.J.; Kim, J.H.; Yoon, J.; Cho, S.J.; Ko, Y.S.; Park, J.W.; Lee, H.S.; Lee, H.E.; Kim, W.H.; Lee, B.L. Constitutive activation of glycogen synthase kinase-3beta correlates with better prognosis and cyclin-dependent kinase inhibitors in human gastric cancer. BMC Gastroenterol. 2010, 10. [Google Scholar] [CrossRef]
- Wen, Y.G.; Wang, Q.; Zhou, C.Z.; Qiu, G.Q.; Peng, Z.H.; Tang, H.M. Mutation analysis of tumor suppressor gene PTEN in patients with gastric carcinomas and its impact on PI3K/AKT pathway. Oncol. Rep. 2010, 24, 89–95. [Google Scholar]
- Mina, S.; Bohn, B.A.; Simon, R.; Krohn, A.; Reeh, M.; Arnold, D.; Bokemeyer, C.; Sauter, G.; Izbicki, J.R.; Marx, A.; et al. PTEN deletion is rare but often homogeneous in gastric cancer. J. Clin. Pathol. 2011, 65, 693–698. [Google Scholar]
- Byun, D.S.; Cho, K.; Ryu, B.K.; Lee, M.G.; Park, J.I.; Chae, K.S.; Kim, H.J.; Chi, S.G. Frequent monoallelic deletion of PTEN and its reciprocal associatioin with PIK3CA amplification in gastric carcinoma. Int. J. Cancer 2003, 104, 318–327. [Google Scholar] [CrossRef]
- Kang, Y.H.; Lee, H.S.; Kim, W.H. Promoter methylation and silencing of PTEN in gastric carcinoma. Lab. Investig. 2002, 82, 285–291. [Google Scholar] [CrossRef]
- Li, M.; Sun, H.; Song, L.; Gao, X.; Chang, W.; Qin, X. Immunohistochemical expression of mTOR negatively correlates with PTEN expression in gastric carcinoma. Oncol. Lett. 2012, 4, 1213–1218. [Google Scholar]
- Vasudevan, K.M.; Barbie, D.A.; Davies, M.A.; Rabinovsky, R.; McNear, C.J.; Kim, J.J.; Hennessy, B.T.; Tseng, H.; Pochanard, P.; Kim, S.Y.; et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell 2009, 16, 21–32. [Google Scholar] [CrossRef]
- Marone, R.; Cmiljanovic, V.; Giese, B.; Wymann, M.P. Targeting phosphoinositide 3-kinase: Moving towards therapy. Biochim. Biophys. Acta 2008, 1784, 159–185. [Google Scholar] [CrossRef]
- Xing, C.G.; Zhu, B.S.; Liu, H.H.; Lin, F.; Yao, H.H.; Liang, Z.Q.; Qin, Z.H. LY294002 induces p53-dependent apoptosis of SGC7901 gastric cancer cells. Acta Pharmacol. Sin. 2008, 29, 489–498. [Google Scholar] [CrossRef]
- Xing, C.G.; Zhu, B.S.; Fan, X.Q.; Liu, H.H.; Hou, X.; Zhao, K.; Qin, Z.H. Effects of LY294002 on the invasiveness of human gastric cancer in vivo in nude mice. World J. Gastroenterol. 2009, 15, 5044–5052. [Google Scholar] [CrossRef]
- Chao, X.; Zao, J.; Xiao-Yi, G.; Li-Jun, M.; Tao, S. Blocking of PI3K/AKT induces apoptosis by its effect on NF-kappaB activity in gastric carcinoma cell line SGC7901. Biomed. Pharmacother. 2010, 64, 600–604. [Google Scholar] [CrossRef]
- Zhao, K.; Zhu, B.S.; Gong, W.; Zhu, M.L.; Gao, Z.T.; Wu, Y.Y.; Chen, Q.; Yang, X.D.; Xing, C.G. SN50 enhances the effects of LY294002 on cell death induction in gastric cancer cell line SGC7901. Arch. Med. Sci. 2011, 9, 990–998. [Google Scholar]
- Liu, Q.; Dong, H.W.; Sun, W.G.; Liu, M.; Ibla, J.C.; Liu, L.X.; Parry, J.W.; Han, X.H.; Li, M.S.; Liu, J.R. Apoptosis initiation of beta-ionone in SGC-7901 gastric carcinoma cancer cells via a PI3K-AKT pathway. Arch. Toxicol. 2013, 87, 481–490. [Google Scholar] [CrossRef]
- Gao, Y.H.; Zhang, H.P.; Yang, S.M.; Yang, Y.; Ma, Y.Y.; Zhang, X.Y.; Yang, Y.M. Inactivation of Akt by arsenic trioxide induces cell death via mitochondrial-mediated apoptotic signaling in SGC-7901 human gastric cancer cells. Oncol. Rep. 2014, 31, 1645–1652. [Google Scholar]
- Xu, J.D.; Cao, X.X.; Long, Z.W.; Liu, X.P.; Furuya, T.; Xu, J.W.; Liu, X.L.; de Xu, Z.; Sasaki, K.; Li, Q.Q. BCL2L10 protein regulates apoptosis/proliferation through differential pathways in gastric cancer cells. J. Pathol. 2011, 223, 400–409. [Google Scholar] [CrossRef]
- Zheng, T.; Meng, X.; Wang, J.; Chen, X.; Yin, D.; Liang, Y.; Song, X.; Pan, S.; Jiang, H.; Liu, L. PTEN- and p53-mediated apoptosis and cell cycle arrest by FTY720 in gastric cancer cells and nude mice. J. Cell. Biochem. 2010, 111, 218–228. [Google Scholar] [CrossRef]
- Xu, Y.H.; Zhao, L.J.; Li, Y. Alisol B acetate induces apoptosis of SGC7901 cells via mitochondrial and phosphatidylinositol 3-kinases/Akt signaling pathways. World J. Gastroenterol. 2009, 15, 2870–2877. [Google Scholar] [CrossRef]
- Rasul, A.; Khan, M.; Yu, B.; Ali, M.; Bo, Y.J.; Yang, H.; Ma, T. Isoalantolactone, a sesquiterpene lactone, induces apoptosis in SGC-7901 cells via mitochondrial and phosphatidylinositol 3-kinase/Akt signaling pathways. Arch. Pharm. Res. 2013, 36, 1262–1269. [Google Scholar]
- Xu, W.T.; Yang, Z.; Lu, N.H. Roles of PTEN (phosphatase and tensin homolog) in gastric cancer development and progression. Asian Pac. J. Cancer Prev. 2014, 15, 17–24. [Google Scholar] [CrossRef]
- Liu, J.F.; Zhou, X.K.; Chen, J.H.; Yi, G.; Chen, H.G.; Ba, M.C.; Lin, S.Q.; Qi, Y.C. Up-regulation of PIK3CA promotes metastasis in gastric carcinoma. World J. Gastroenterol. 2010, 16, 4986–4991. [Google Scholar] [CrossRef]
- Zhou, X.D.; Chen, H.X.; Guan, R.N.; Lei, Y.P.; Shu, X.; Zhu, Y.; Lv, N.H. Protein kinase B phosphorylation correlates with vascular endothelial growth factor A and microvessel density in gastric adenocarcinoma. J. Int. Med. Res. 2012, 40, 2124–2134. [Google Scholar] [CrossRef]
- Han, Z.; Wu, K.; Shen, H.; Li, C.; Han, S.; Hong, L.; Shi, Y.; Liu, N.; Guo, C.; Xue, Y.; et al. Akt1/protein kinase B alpha is involved in gastric cancer progression and cell proliferation. Dig. Dis. Sci. 2008, 53, 1801–1810. [Google Scholar] [CrossRef]
- Ye, B.; Jiang, L.L.; Xu, H.T.; Zhou, D.W.; Li, Z.S. Expression of PI3K/AKT pathway in gastric cancer and its blockade suppresses tumor growth and metastasis. Int. J. Immunopathol. Pharmacol. 2012, 25, 627–636. [Google Scholar]
- Lin, H.L.; Chiou, S.H.; Wu, C.W.; Lin, W.B.; Chen, L.H.; Yang, Y.P.; Tsai, M.L.; Uen, Y.H.; Liou, J.P.; Chi, C.W. Combretastatin A4-induced differential cytotoxicity and reduced metastatic ability by inhibition of AKT function in human gastric cancer cells. J. Pharmacol. Exp. Ther. 2007, 323, 365–373. [Google Scholar] [CrossRef]
- Matsuoka, T.; Yashiro, M.; Nishioka, N.; Hirakawa, K.; Olden, K.; Roberts, J.D. PI3K/Akt signalling is required for the attachment and spreading, and growth in vivo of metastatic scirrhous gastric carcinoma. Br. J. Cancer 2012, 106, 1535–1542. [Google Scholar] [CrossRef]
- Zhou, X.K.; Tang, S.S.; Yi, G.; Hou, M.; Chen, J.H.; Yang, B.; Liu, J.F.; He, Z.M. RNAi knockdown of PIK3CA preferentially inhibits invasion of mutant PIK3CA cells. World J. Gastroenterol. 2011, 17, 3700–3708. [Google Scholar] [CrossRef]
- Xue, Y.; Li, N.L.; Yang, J.Y.; Chen, Y.; Yang, L.L.; Liu, W.C. Phosphatidylinositol 3'-kinase signaling pathway is essential for Rac1-induced hypoxia-inducible factor-1(alpha) and vascular endothelial growth factor expression. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H2169–H2176. [Google Scholar] [CrossRef]
- Zhang, L.L.; Liu, J.; Lei, S.; Zhang, J.; Zhou, W.; Yu, H.G. PTEN inhibits the invasion and metastasis of gastric cancer via downregulation of FAK expression. Cell. Signal. 2014, 26, 1011–1020. [Google Scholar] [CrossRef]
- Chen, G.; Chen, S.M.; Wang, X.; Ding, X.F.; Ding, J.; Meng, L.H. Inhibition of chemokine (CXC motif) ligand 12/chemokine (CXC motif) receptor 4 axis (CXCL12/CXCR4)-mediated cell migration by targeting mammalian target of rapamycin (mTOR) pathway in human gastric carcinoma cells. J. Biol. Chem. 2012, 287, 12132–12141. [Google Scholar]
- Yu, W.; Ma, S.; Wang, L.; Zuo, B.; Li, M.; Qiao, Z.; Pan, X.; Liu, Y.; Wang, J. Upregulation of GPR34 expression affects the progression and prognosis of human gastric adenocarcinoma by PI3K/PDK1/AKT pathway. Histol. Histopathol. 2013, 28, 1629–1638. [Google Scholar]
- Tian, P.Y.; Fan, X.M. The proliferative effects of ghrelin on human gastric cancer AGS cells. J. Dig. Dis. 2012, 13, 453–458. [Google Scholar] [CrossRef]
- Lee, Y.C.; Cheng, T.H.; Lee, J.S.; Chen, J.H.; Liao, Y.C.; Fong, Y.; Wu, C.H.; Shih, Y.W. Nobiletin, a citrus flavonoid, suppresses invasion and migration involving FAK/PI3K/Akt and small GTPase signals in human gastric adenocarcinoma AGS cells. Mol. Cell. Biochem. 2011, 347, 103–115. [Google Scholar] [CrossRef]
- Li, G.Q.; Xie, J.; Lei, X.Y.; Zhang, L. Macrophage migration inhibitory factor regulates proliferation of gastric cancer cells via the PI3K/Akt pathway. World J. Gastroenterol. 2009, 15, 5541–5548. [Google Scholar] [CrossRef]
- Bae, I.H.; Park, M.J.; Yoon, S.H.; Kang, S.W.; Lee, S.S.; Choi, K.M.; Um, H.D. Bcl-w promotes gastric cancer cell invasion by inducing matrix metalloproteinase-2 expression via phosphoinositide 3-kinase, Akt, and Sp1. Cancer Res. 2006, 66, 4991–4995. [Google Scholar] [CrossRef]
- Lin, H.L.; Yang, M.H.; Wu, C.W.; Chen, P.M.; Yang, Y.P.; Chu, Y.R.; Kao, C.L.; Ku, H.H.; Lo, J.F.; Liou, J.P.; et al. 2-Methoxyestradiol attenuates phosphatidylinositol 3-kinase/Akt pathway-mediated metastasis of gastric cancer. Int. J. Cancer 2007, 121, 2547–2555. [Google Scholar] [CrossRef]
- Kang, M.H.; Oh, S.C.; Lee, H.J.; Kang, H.N.; Kim, J.L.; Kim, J.S.; Yoo, Y.A. Metastatic function of BMP-2 in gastric cancer cells: The role of PI3K/AKT, MAPK, the NF-kappaB pathway, and MMP-9 expression. Exp. Cell Res. 2011, 317, 1746–1762. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, Y.; Xu, R.; Du, J.; Hu, Z.; Yang, L.; Chen, Y.; Zhu, Y.; Gu, L. PI3K/Akt-dependent phosphorylation of GSK3beta and activation of RhoA regulate Wnt5a-induced gastric cancer cell migration. Cell. Signal. 2013, 25, 447–456. [Google Scholar] [CrossRef]
- Guo, L.Y.; Li, Y.M.; Qiao, L.; Liu, T.; Du, Y.Y.; Zhang, J.Q.; He, W.T.; Zhao, Y.X.; He, D.Q. Notch2 regulates matrix metallopeptidase 9 via PI3K/AKT signaling in human gastric carcinoma cell MKN-45. World J. Gastroenterol. 2012, 18, 7262–7270. [Google Scholar] [CrossRef]
- Guo, B.; Liu, L.; Yao, J.; Ma, R.; Chang, D.; Li, Z.; Song, T.; Huang, C. miR-338–3p suppresses progression of gastric cancer through PTEN-AKT signaling pathways by targeting P-REX2a. Mol. Cancer Res. 2014, 12, 313–321. [Google Scholar] [CrossRef]
- Fodale, V.; Pierobon, M.; Liotta, L.; Petricoin, E. Mechanism of cell adaptation: When and how do cancer cells develop chemoresistance? Cancer J. 2011, 17, 89–95. [Google Scholar] [CrossRef]
- Ang, K.L.; Shi, D.L.; Keong, W.W.; Epstein, R.J. Upregulated Akt signaling adjacent to gastric cancers: Implications for screening and chemoprevention. Cancer Lett. 2005, 225, 53–59. [Google Scholar] [CrossRef]
- Oki, E.; Baba, H.; Tokunaga, E.; Nakamura, T.; Ueda, N.; Futatsugi, M.; Mashino, K.; Yamamoto, M.; Ikebe, M.; Kakeji, Y.; et al. Akt phosphorylation associates with LOH of PTEN and leads to chemoresistance for gastric cancer. Int. J. Cancer 2005, 117, 376–380. [Google Scholar] [CrossRef]
- Osaki, M.; Kase, S.; Adachi, K.; Takeda, A.; Hashimoto, K.; Ito, H. Inhibition of the PI3K-Akt signaling pathway enhances the sensitivity of Fas-mediated apoptosis in human gastric carcinoma cell line, MKN-45. J. Cancer Res. Clin. Oncol. 2004, 130, 8–14. [Google Scholar] [CrossRef]
- Xie, X.; Tang, B.; Zhou, J.; Gao, Q.; Zhang, P. Inhibition of the PI3K/Akt pathway increases the chemosensitivity of gastric cancer to vincristine. Oncol. Rep. 2013, 30, 773–782. [Google Scholar]
- Zhang, C.; Awasthi, N.; Schwarz, M.A.; Schwarz, R.E. The dual PI3K/mTOR inhibitor NVP-BEZ235 enhances nab-paclitaxel antitumor response in experimental gastric cancer. Int. J. Oncol. 2013, 43, 1627–1635. [Google Scholar]
- Yu, H.G.; Ai, Y.W.; Yu, L.L.; Zhou, X.D.; Liu, J.; Li, J.H.; Xu, X.M.; Liu, S.; Chen, J.; Liu, F.; et al. Phosphoinositide 3-kinase/Akt pathway plays an important role in chemoresistance of gastric cancer cells against etoposide and doxorubicin induced cell death. Int. J. Cancer 2008, 122, 433–443. [Google Scholar] [CrossRef]
- Yu, L.L.; Dai, N.; Yu, H.G.; Sun, L.M.; Si, J.M. Akt associates with nuclear factor kappaB and plays an important role in chemoresistance of gastric cancer cells. Oncol. Rep. 2012, 24, 113–119. [Google Scholar]
- Liang, J.; Ge, F.; Guo, C.; Luo, G.; Wang, X.; Han, G.; Zhang, D.; Wang, J.; Li, K.; Pan, Y.; et al. Inhibition of PI3K/Akt partially leads to the inhibition of PrP(C)-induced drug resistance in gastric cancer cells. FEBS J. 2009, 276, 685–694. [Google Scholar] [CrossRef]
- Han, Z.; Hong, L.; Han, Y.; Wu, K.; Han, S.; Shen, H.; Li, C.; Yao, L.; Qiao, T.; Fan, D. Phospho Akt mediates multidrug resistance of gastric cancer cells through regulation of P-gp, Bcl-2 and Bax. J. Exp. Clin. Cancer Res. 2007, 26, 261–268. [Google Scholar]
- Zhou, W.; Fu, X.Q.; Liu, J.; Yu, H.G. RNAi knockdown of the Akt1 gene increases the chemosensitivity of gastric cancer cells to cisplatin both in vitro and in vivo. Regul. Pept. 2012, 176, 13–21. [Google Scholar] [CrossRef]
- Steelman, L.S.; Navolanic, P.M.; Sokolosky, M.L.; Taylor, J.R.; Lehmann, B.D.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.; Stadelman, K.M.; Terrian, D.M.; et al. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene 2008, 27, 4086–4095. [Google Scholar] [CrossRef]
- McCubrey, J.A.; Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Montalto, G.; Cervello, M.; Nicoletti, F.; Fagone, P.; Malaponte, G.; Mazzarino, M.C.; et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012, 3, 954–987. [Google Scholar]
- Tomioka, H.; Mukohara, T.; Kataoka, Y.; Ekyalongo, R.C.; Funakoshi, Y.; Imai, Y.; Kiyota, N.; Fujiwara, Y.; Minami, H. Inhibition of the mTOR/S6K signal is necessary to enhance fluorouracil-induced apoptosis in gastric cancer cells with HER2 amplification. Int. J. Oncol. 2012, 41, 551–558. [Google Scholar]
- Yang, S.M.; Huang, C.; Li, X.F.; Yu, M.Z.; He, Y.; Li, J. miR-21 confers cisplatin resistance in gastric cancer cells by regulating PTEN. Toxicology 2013, 306, 162–168. [Google Scholar] [CrossRef]
- Wang, F.; Li, T.; Zhang, B.; Li, H.; Wu, Q.; Yang, L.; Nie, Y.; Wu, K.; Shi, Y.; Fan, D. MicroRNA-19a/b regulates multidrug resistance in human gastric cancer cells by targeting PTEN. Biochem. Biophys. Res. Commun. 2013, 434, 688–694. [Google Scholar]
- Fu, X.; Feng, J.; Zeng, D.; Ding, Y.; Yu, C.; Yang, B. PAK4 confers cisplatin resistance in gastric cancer cells via PI3K/Akt- and MEK/Erk-dependent pathways. Biosci. Rep. 2014. [Google Scholar] [CrossRef]
- Park, J.; Ko, Y.S.; Yoon, J.; Kim, M.A.; Park, J.W.; Kim, W.H.; Choi, Y.; Kim, J.H.; Cheon, Y.; Lee, B.L. The forkhead transcription factor FOXO1 mediates cisplatin resistance in gastric cancer cells by activating phosphoinositide 3-kinase/Akt pathway. Gastric Cancer 2013. [Google Scholar] [CrossRef]
- Mao, Z.; Zhou, J.; Luan, J.; Sheng, W.; Shen, X.; Dong, X. Tamoxifen reduces P-gp-mediated multidrug resistance via inhibiting the PI3K/Akt signaling pathway in ER-negative human gastric cancer cells. Biomed. Pharmacother. 2014, 68, 179–183. [Google Scholar]
- Liu, J.; Qu, X.; Xu, L.; Zhang, Y.; Qu, J.; Hou, K.; Liu, Y. Phosphoinositide 3-kinase/Akt and nuclear factor kappaB pathways are involved in tumor necrosis factor-related apoptosis-inducing ligand resistance in human gastric cancer cells. Mol. Med. Rep. 2010, 3, 491–496. [Google Scholar]
- Pal, I.; Mandal, M. PI3K and Akt as molecular targets for cancer therapy: Current clinical outcomes. Acta Pharmacol. Sin. 2012, 33, 1441–1458. [Google Scholar] [CrossRef]
- Vlahos, C.J.; Matter, W.F.; Hui, K.Y.; Brown, R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J. Biol. Chem. 1994, 269, 5241–5248. [Google Scholar]
- Powis, G.; Bonjouklian, R.; Berggren, M.M.; Gallegos, A.; Abraham, R.; Ashendel, C.; Zalkow, L.; Matter, W.F.; Dodge, J.; Grindey, G.; et al. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994, 54, 2419–2423. [Google Scholar]
- Park, E.; Park, J.; Han, S.W.; Im, S.A.; Kim, T.Y.; Oh, D.Y.; Bang, Y.J. NVP-BKM120, a novel PI3K inhibitor, shows synergism with a STAT3 inhibitor in human gastric cancer cells harboring KRAS mutations. Int. J. Oncol. 2012, 40, 1259–1266. [Google Scholar]
- Garrett, J.T.; Sutton, C.R.; Kurupi, R.; Bialucha, C.U.; Ettenberg, S.A.; Collins, S.D.; Sheng, Q.; Wallweber, J.; Defazio-Eli, L.; Arteaga, C.L. Combination of antibody that inhibits ligand-independent HER3 dimerization and a p110alpha inhibitor potently blocks PI3K signaling and growth of HER2+ breast cancers. Cancer Res. 2013, 73, 6013–6023. [Google Scholar] [CrossRef]
- Ohtsu, A.; Ajani, J.A.; Bai, Y.X.; Bang, Y.J.; Chung, H.C.; Pan, H.M.; Sahmoud, T.; Shen, L.; Yeh, K.H.; Chin, K.; et al. Everolimus for previously treated advanced gastric cancer: Results of the randomized, double-blind, phase III GRANITE-1 study. J. Clin. Oncol. 2013, 31, 3935–3943. [Google Scholar]
- Hudis, C.; Swanton, C.; Janjigian, Y.Y.; Lee, R.; Sutherland, S.; Lehman, R.; Chandarlapaty, S.; Hamilton, N.; Gajria, D.; Knowles, J.; et al. A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res. 2013, 15. [Google Scholar] [CrossRef]
- Li, J.; Davies, B.R.; Han, S.; Zhou, M.; Bai, Y.; Zhang, J.; Xu, Y.; Tang, L.; Wang, H.; Liu, Y.J.; et al. The AKT inhibitor AZD5363 is selectively active in PI3KCA mutant gastric cancer, and sensitizes a patient-derived gastric cancer xenograft model with PTEN loss to Taxotere. J. Transl. Med. 2013, 11. [Google Scholar] [CrossRef]
- 99 Rhodes, N.; Heerding, D.A.; Duckett, D.R.; Eberwein, D.J.; Knick, V.B.; Lansing, T.J.; McConnell, R.T.; Gilmer, T.M.; Zhang, S.Y.; Robell, K.; et al. Characterization of an Akt kinase inhibitor with potent pharmacodynamic and antitumor activity. Cancer Res. 2008, 68, 2366–2374. [Google Scholar] [CrossRef]
- Matsuzaki, T.; Yashiro, M.; Kaizaki, R.; Yasuda, K.; Doi, Y.; Sawada, T.; Ohira, M.; Hirakawa, K. Synergistic antiproliferative effect of mTOR inhibitors in combination with 5-fluorouracil in scirrhous gastric cancer. Cancer Sci. 2009, 100, 2402–2410. [Google Scholar] [CrossRef]
- Doi, T.; Muro, K.; Boku, N.; Yamada, Y.; Nishina, T.; Takiuchi, H.; Komatsu, Y.; Hamamoto, Y.; Ohno, N.; Fujita, Y.; et al. Multicenter phase II study of everolimus in patients with previously treated metastatic gastric cancer. J. Clin. Oncol. 2010, 28, 1904–1910. [Google Scholar] [CrossRef]
- Ji, D.; Zhang, Z.; Cheng, L.; Chang, J.; Wang, S.; Zheng, B.; Zheng, R.; Sun, Z.; Wang, C.; Zhang, Z.; et al. The combination of RAD001 and MK-2206 exerts synergistic cytotoxic effects against PTEN mutant gastric cancer cells: Involvement of MAPK-dependent autophagic, but not apoptotic cell death pathway. PLoS One 2014, 9, e85116. [Google Scholar]
- O’Reilly, K.E.; Rojo, F.; She, Q.B.; Solit, D.; Mills, G.B.; Smith, D.; Lane, H.; Hofmann, F.; Hicklin, D.J.; Ludwig, D.L.; et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006, 66, 1500–1508. [Google Scholar] [CrossRef]
- Lang, S.A.; Hackl, C.; Moser, C.; Fichtner-Feigl, S.; Koehl, G.E.; Schlitt, H.J.; Geissler, E.K.; Stoeltzing, O. Implication of RICTOR in the mTOR inhibitor-mediated induction of insulin-like growth factor-I receptor (IGF-IR) and human epidermal growth factor receptor-2 (Her2) expression in gastrointestinal cancer cells. Biochim. Biophys. Acta 2010, 1803, 435–442. [Google Scholar] [CrossRef]
- Hart, S.; Novotny-Diermayr, V.; Goh, K.C.; Williams, M.; Tan, Y.C.; Ong, L.C.; Cheong, A.; Ng, B.K.; Amalini, C.; Madan, B.; et al. VS-5584, a novel and highly selective PI3K/mTOR kinase inhibitor for the treatment of cancer. Mol. Cancer Ther. 2013, 12, 151–161. [Google Scholar]
- Bhattacharya, B.; Akram, M.; Balasubramanian, I.; Tam, K.K.; Koh, K.X.; Yee, M.Q.; Soong, R. Pharmacologic synergy between dual phosphoinositide-3-kinase and mammalian target of rapamycin inhibition and 5-fluorouracil in PIK3CA mutant gastric cancer cells. Cancer Biol. Ther. 2012, 13, 34–42. [Google Scholar] [CrossRef]
- Mueller, A.; Bachmann, E.; Linnig, M.; Khillimberger, K.; Schimanski, C.C.; Galle, P.R.; Moehler, M. Selective PI3K inhibition by BKM120 and BEZ235 alone or in combination with chemotherapy in wild-type and mutated human gastrointestinal cancer cell lines. Cancer Chemother. Pharmacol. 2012, 69, 1601–1615. [Google Scholar] [CrossRef]
- Engelman, J.A.; Chen, L.; Tan, X.; Crosby, K.; Guimaraes, A.R.; Upadhyay, R.; Maira, M.; McNamara, K.; Perera, S.A.; Song, Y.; et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 2008, 14, 1351–1356. [Google Scholar] [CrossRef]
- Fu, Y.; Zhang, Q.; Kang, C.; Zhang, J.; Zhang, K.; Pu, P.; Wang, G.; Wang, T. Inhibitory effects of adenovirus mediated Akt1 and PIK3R1 shRNA on the growth of malignant tumor cells in vitro and in vivo. Cancer Biol. Ther. 2009, 8, 1002–1009. [Google Scholar] [CrossRef]
- Zhu, B.S.; Yu, L.Y.; Zhao, K.; Wu, Y.Y.; Cheng, X.L.; Wu, Y.; Zhong, F.Y.; Gong, W.; Chen, Q.; Xing, C.G. Effects of small interfering RNA inhibit Class I phosphoinositide 3-kinase on human gastric cancer cells. World J. Gastroenterol. 2013, 19, 1760–1769. [Google Scholar] [CrossRef]
- Yashiro, M.; Nishii, T.; Hasegawa, T.; Matsuzaki, T.; Morisaki, T.; Fukuoka, T.; Hirakawa, K. A c-Met inhibitor increases the chemosensitivity of cancer stem cells to the irinotecan in gastric carcinoma. Br. J. Cancer 2013, 109, 2619–2628. [Google Scholar] [CrossRef]
- Christensen, J.G.; Schreck, R.; Burrows, J.; Kuruganti, P.; Chan, E.; Le, P.; Chen, J.; Wang, X.; Ruslim, L.; Blake, R.; et al. A selective small molecule inhibitor of c-Met kinase inhibits c-Met-dependent phenotypes in vitro and exhibits cytoreductive antitumor activity in vivo. Cancer Res. 2003, 63, 7345–7355. [Google Scholar]
- Shinomiya, N.; Gao, C.F.; Xie, Q.; Gustafson, M.; Waters, D.J.; Zhang, Y.W.; Vande Woude, G.F. RNA interference reveals that ligand-independent met activity is required for tumor cell signaling and survival. Cancer Res. 2004, 64, 7962–7970. [Google Scholar] [CrossRef]
- Hong, S.W.; Jung, K.H.; Park, B.H.; Zheng, H.M.; Lee, H.S.; Choi, M.J.; Yun, J.I.; Kang, N.S.; Lee, J.; Hong, S.S. KRC-408, a novel c-Met inhibitor, suppresses cell proliferation and angiogenesis of gastric cancer. Cancer Lett. 2013, 332, 74–82. [Google Scholar] [CrossRef]
- Lu, Y.; Muller, M.; Smith, D.; Dutta, B.; Komurov, K.; Iadevaia, S.; Ruths, D.; Tseng, J.T.; Yu, S.; Yu, Q.; et al. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene 2011, 30, 4567–4577. [Google Scholar] [CrossRef]
- Wong, H.; Yau, T. Targeted therapy in the management osf advanced gastric cancer: Are we making progress in the era of personalized medicine? Oncologist 2012, 17, 346–358. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Matsuoka, T.; Yashiro, M. The Role of PI3K/Akt/mTOR Signaling in Gastric Carcinoma. Cancers 2014, 6, 1441-1463. https://doi.org/10.3390/cancers6031441
AMA Style
Matsuoka T, Yashiro M. The Role of PI3K/Akt/mTOR Signaling in Gastric Carcinoma. Cancers. 2014; 6(3):1441-1463. https://doi.org/10.3390/cancers6031441
Chicago/Turabian StyleMatsuoka, Tasuku, and Masakazu Yashiro. 2014. "The Role of PI3K/Akt/mTOR Signaling in Gastric Carcinoma" Cancers 6, no. 3: 1441-1463. https://doi.org/10.3390/cancers6031441