Cancer Stem Cells, EMT, and Developmental Pathway Activation in Pancreatic Tumors


Abstract
:1. Introduction
1.1. Incidence and Mortality of Pancreatic Cancer
1.2. Onset of Metastasis Formation
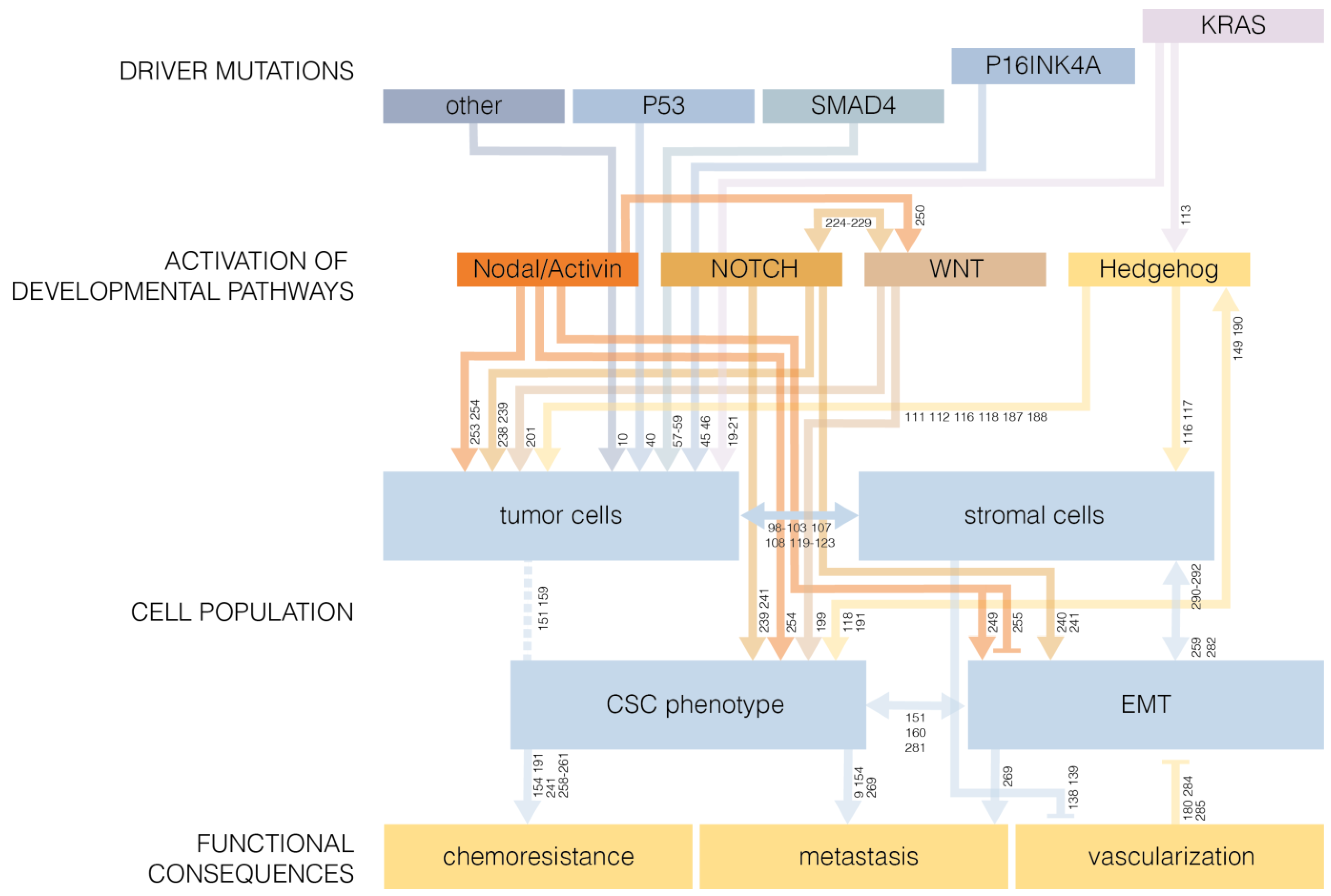
2. Mutations that Drive Pancreatic Cancer
2.1. KRAS
2.2. P53
2.3. P16INK4A
2.4. SMAD4
3. Treatment of Pancreatic Cancer
3.1. Tumor Resection
3.2. Chemotherapy
4. Tumor Micro-Environment
4.1. Pancreatitis
4.2. Pancreatic Stellate Cells and Fibroblasts
4.3. Stromal Factors Can Mediate Chemoresistance
4.4. Tumor Vascularization
5. Cancer Stem Cells in Pancreatic Cancer
5.1. The Cancer Stem Cell Concept
5.2. Identification of Cancer Stem Cells

{kind=link}
| CD44/CD24/ESA | CD133 | ALDH | c-MET | dye exclusion | miRNA | functional assay | |
|---|---|---|---|---|---|---|---|
| CD44/CD24/ESA | n/a | 154 | 159 160 | 161 | – | 169 | 149 150 160 |
| CD133 | 154 | n/a | 159 | 161 | 162 163 | 169 | 154 |
| ALDH | 159 160 | 159 | n/a | 161 | – | – | 159 160 |
| c-MET | 161 | 161 | 161 | n/a | – | – | 161 |
| dye exclusion | – | 162 163 | – | – | n/a | – | 162 163 |
| miRNA | 169 | 169 | – | – | – | n/a | 168 169 |
| functional assay | 149 150 160 | 154 | 159 160 | 161 | 162 163 | 168 169 | n/a |
5.3. CD44, CD24, and ESA
5.4. CD133
5.5. Aldehyde Dehydrogenase
5.6. c-MET
5.7. Dye Exclusion
5.8. MicroRNAs
6. Developmental Pathways in Cancer Stem Cell Biology
6.1. The Epithelial-to-Mesenchymal Transition
6.2. Hedgehog Signaling
6.3. WNT Signaling
6.4. NOTCH Signaling
6.5. TGF-β Superfamily: Nodal/Activin Signaling
6.6. Developmental Pathways as Drivers of Oncogenesis
7. Pancreatic Cancer Stem Cells and EMT
7.1. Cancer Stem Cells Are Less Sensitive to Chemotherapy
7.2. EMT and Metastasis in Pancreatic Cancer Stem Cells
7.3. Cancer Stem Cells Generated via EMT
7.4. Tumor Micro-Environment Promotes EMT in Pancreatic Cancer
7.5. Contribution of EMT to Give Rise to Stromal Cells
8. Conclusions
8.1. Re-evaluating the Cancer Stem Cell Concept
8.2. Inhibition of Stemness as a Treatment Regimen

Acknowledgments
References
- Siegel, R.; Ward, E.; Brawly, O.; Jemal, A. Cancer Statistics, 2011: The impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin. 2011, 61, 212–236. [Google Scholar] [CrossRef]
- Parkin, D.M.; Bray, F.I.; Devesa, S.S. Cancer burden in the year 2000. The global picture. Eur. J. Cancer 2001, 37, S4–S66. [Google Scholar]
- Fitzgerald, T.L.; Hickner, Z.J.; Schmitz, M.; Kort, E.J. Changing incidence of pancreatic neoplasms: A 16-year review of statewide tumor registry. Pancreas 2008, 37, 134–138. [Google Scholar] [CrossRef]
- Andea, A.; Sarkar, F.; Adsay, V.N. Clinicopathological correlates of pancreatic intraepithelial neoplasia: A comparative analysis of 82 cases with and 152 cases without pancreatic ductal adenocarcinoma. Mod. Pathol. 2003, 16, 996–1006. [Google Scholar] [CrossRef]
- Niederhuber, J.E.; Brennan, M.F.; Menck, H.R. The national cancer data base report on pancreatic cancer. Cancer 1995, 9, 1671–1677. [Google Scholar]
- Haeno, H.; Gonen, M.; Davis, M.B.; Herman, J.M.; Iacobuzio-Donahue, C.A.; Michor, F. Computational modeling of pancreatic cancer reveals kinetics of metastasis suggesting optimum treatment strategies. Cell 2012, 148, 362–375. [Google Scholar] [CrossRef]
- Yachida, S.; Jones, S.; Bozic, I.; Antal, T.; Leary, R.; Fu, B.; Kamiyama, M.; Hruban, R.H.; Eshleman, J.R.; Nowak, M.A.; et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 2010, 467, 1114–1147. [Google Scholar] [CrossRef]
- Campbell, P.J.; Yachida, S.; Mudie, L.J.; Stephens, P.J.; Pleasance, E.D.; Stebbings, L.A.; Morsberger, L.A.; Latimer, C.; McLaren, S.; et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 2010, 467, 1109–1113. [Google Scholar] [CrossRef]
- Rhim, A.D.; Mirek, E.T.; Aiello, N.M.; Maitra, A.; Bailey, J.M.; McAllister, F.; Reichert, M.; Beatty, G.L.; Rustgi, A.K.; Vonderheide, R.H.; et al. EMT and dissemination precede pancreatic tumor formation. Cell 2012, 148, 349–361. [Google Scholar] [CrossRef]
- Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Kamiyama, H.; Jimeno, A.; et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008, 321, 1801–1806. [Google Scholar] [CrossRef]
- Hruban, R.H.; Mansfeld, A.D.M.V.; Offerhaus, G.J.A.; Weering, D.H.J.V.; Allison, D.C.; Goodman, S.N.; Kensler, T.W.; Bose, K.K.; Cameron, J.L.; Bost, J.L. K-ras oncogene activation in adenocarcinoma of the human pancreas: A study of 82 carcinomas using a combination of mutant-enriched polymerase chain reaction analysis and allele specific oligonucleotide hybridization. Am. J. Pathol. 1993, 143, 545–554. [Google Scholar]
- Rozenblum, E.; Schutte, M.; Goggins, M.; Hahn, S.A.; Panzer, S.; Zahurak, M.; Goodman, S.N.; Sohn, T.A.; Hruban, R.H.; Yeo, C.J.; et al. Tumor-suppresive pathways in pancreatic carcinoma. Cancer Res. 1997, 57, 1731–1734. [Google Scholar]
- Sweet, R.W.; Yokoyama, S.; Kamata, T.; Feramisco, J.R.; Rosenberg, M.; Gross, M. The product of ras is a GTPase and the T24 oncogenic mutant is deficient in this activity. Nature 1984, 311, 273–275. [Google Scholar] [CrossRef]
- Field, J.; Broek, D.; Kataoka, T.; Wigler, M. Guanine nucleotide activation of, and competition between, RAS proteins from Saccharomyces cerevisiae. Mol. Cell. Biol. 1987, 7, 2128–2133. [Google Scholar]
- Field, J.; Nikawa, J.; Broek, D.; MacDonald, B.; Rodgers, L.; Wilson, I.A.; Lerner, R.A.; Wigler, M. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol. Cell. Biol. 1988, 8, 2159–2165. [Google Scholar]
- De Vendittis, E.; Zahn, R.; Fasano, O. Regeneration of the GTP-bound from the GDP-bound form of human and yeast ras proteins by nucleotide exchange. Stimulatory effect of organic and inorganic polyphosphates. J. Biochem. 1986, 161, 473–478. [Google Scholar]
- Lowenstein, E.J.; Daly, R.J.; Batzer, A.G.; Li, W.; Margolis, B.; Lammers, R.; Ullrich, A.; Skolnik, E.Y.; Bar-Sagi, D.; Schlessinger, J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992, 70, 431–442. [Google Scholar] [CrossRef]
- McGrath, J.P.; Capon, D.J.; Goeddel, D.V.; Levinson, A.D. Comparative biochemical properties of normal and activated human ras p21 protein. Nature 1984, 310, 644–649. [Google Scholar] [CrossRef]
- Chin, L.; Tam, A.; Pomerantz, J.; Wong, M.; Holash, J.; Bardeesy, N.; Shen, Q.; O’Hagan, R.; Pantginis, J.; Zhou, H.; et al. Essential role for oncogenic Ras in tumour maintenance. Nature 1999, 400, 468–472. [Google Scholar] [CrossRef]
- Shirasawa, S.; Furuse, M.; Yokoyama, N.; Sasazuki, T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science 1993, 260, 85–88. [Google Scholar]
- Muschel, R.J.; Williams, J.E.; Lowy, D.R.; Liotta, L.A. Harvey ras induction of metastatic potential depends upon oncogene activation and the type of recipient cell. Am. J. Pathol. 1985, 121, 1–8. [Google Scholar]
- Van Cutsem, E.; van de Velde, H.; Karasek, P.; Oettle, H.; Vervenne, W.L.; Szawlowski, A.; Schoffski, P.; Post, S.; Verslype, C.; Neumann, H.; et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. Oncol. 2004, 22, 1430–1438. [Google Scholar] [CrossRef]
- Fleming, J.B.; Shen, G.-L.; Holloway, S.E.; Davis, M.; Brekken, R.A. Molecular consequences of silencing mutant K-ras in pancreatic cancer cells: Justification for K-ras-directed therapy. Mol. Cancer Res. 2005, 3, 413–423. [Google Scholar] [CrossRef]
- Ulivi, P.; Arienti, C.; Amadori, D.; Fabbri, F.; Carloni, S.; Tesei, A.; Vannini, I.; Silvestrini, R.; Zoli, W. Role of RAF/MEK/ERK pathway, p-STAT-3 and Mcl-1 in sorafenib activity in human pancreatic cancer cell lines. J. Cell. Physiol. 2009, 220, 214–221. [Google Scholar] [CrossRef]
- Zitzmann, K.; de Toni, E.N.; Brand, S.; Göke, B.; Meinecke, J.; Spöttl, G.; Meyer, H.H.D.; Auernhammer, C.J. The novel mTOR inhibitor RAD001 (everolimus) induces antiproliferative effects in human pancreatic neuroendocrine tumor cells. Neuroendocrinology 2007, 85, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Ito, D.; Fujimoto, K.; Mori, T.; Kami, K.; Koizumi, M.; Toyoda, E.; Kawaguchi, Y.; Doi, R. In vivo antitumor effect of the mTOR inhibitor CCI-779 and gemcitabine in xenograft models of human pancreatic cancer. Int. J. Cancer 2006, 118, 2337–2343. [Google Scholar] [CrossRef]
- Wolpin, B.M.; Hezel, A.F.; Abrams, T.; Blaszkowsky, L.S.; Meyerhardt, J.A.; Chan, J.A.; Enzinger, P.C.; Allen, B.; Clark, J.W.; Ryan, D.P.; et al. Oral mTOR inhibitor everolimus in patients with gemcitabine-refractory metastatic pancreatic cancer. J. Clin. Oncol. 2009, 27, 193–198. [Google Scholar]
- Javle, M.M.; Shroff, R.T.; Xiong, H.; Varadhachary, G.A.; Fogelman, D.; Reddy, S.A.; Davis, D.; Zhang, Y.; Wolff, R.A.; Abbruzzese, J.L. Inhibition of the mammalian target of rapamycin (mTOR) in advanced pancreatic cancer: Results of two phase II studies. BMC Cancer 2010, 10, 368. [Google Scholar]
- Fjällskog, M.H.; Lejonklou, M.H.; Öberg, K.E.; Fja, M.H. Expression of molecular targets for tyrosine kinase receptor antagonists in malignant endocrine pancreatic tumors. Clin. Cancer Res. 2003, 9, 1469–1473. [Google Scholar]
- Xiong, H.Q.; Rosenberg, A.; LoBuglio, A.; Schmidt, W.; Wolff, R.A.; Deutsch, J.; Needle, M.; Abbruzzese, J.L. Cetuximab, a monoclonal antibody targeting the epidermal growth factor receptor, in combination with gemcitabine for advanced pancreatic cancer: A multicenter phase II trial. J. Clin. Oncol. 2004, 22, 2610–2616. [Google Scholar]
- Philip, P.A.; Benedetti, J.; Corless, C.L.; Wong, R.; O’Reilly, E.M.; Flynn, P.J.; Rowland, K.M.; Atkins, J.N.; Mirtsching, B.C.; Rivkin, S.E.; et al. Phase III study comparing gemcitabine plus cetuximab versus gemcitabine in patients with advanced pancreatic adenocarcinoma: Southwest Oncology Group-directed intergroup trial S0205. J. Clin. Oncol. 2010, 28, 3605–3610. [Google Scholar]
- Moore, M.J.; Goldstein, D.; Hamm, J.; Figer, A.; Hecht, J.R.; Gallinger, S.; Au, H.J.; Murawa, P.; Walde, D.; Wolff, R.A.; et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2007, 25, 1960–1966. [Google Scholar] [CrossRef]
- Loewer, A.; Batchelor, E.; Gaglia, G.; Lahav, G. Basal dynamics of p53 reveal transcriptionally attenuated pulses in cycling cells. Cell 2010, 142, 89–100. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Kubbutat, M.H.G.; Jones, S.N.; Vousden, K.H. Regulation of p53 stability by Mdm2. Nature 1997, 387, 299–303. [Google Scholar] [CrossRef]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar]
- Donehower, L.A.; Harvey, M.; Slagle, B.L.; McArthur, M.J.; Montgomery, C.A.; Butel, J.S.; Bradley, A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992, 356, 215–221. [Google Scholar] [CrossRef]
- Barton, C.M.; Staddon, S.L.; Hughes, C.M.; Hall, P.A.; O’Sullivan, C.; Klöppel, G.; Theis, B.; Russell, R.C.G.; Neoptolemos, J.; Williamson, R.C.N.; et al. Abnormalities of the p53 tumour suppressor gene in human pancreatic cancer. Br. J. Cancer 1991, 64, 1076–1082. [Google Scholar] [CrossRef]
- Scarpa, A.; Capelli, P.; Mukai, K.; Zamboni, G.; Oda, T.; Iacono, C.; Hirohashi, S. Pancreatic adenocarcinomas frequently show p53 gene mutations. Am. J. Pathol. 1993, 142, 1534–1543. [Google Scholar]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar]
- Harbour, J.W.; Luo, R.X.; Santi, A.D.; Postigo, A.A.; Dean, D.C. Intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 1999, 98, 859–869. [Google Scholar] [CrossRef]
- Chicas, A.; Wang, X.; Zhang, C.; McCurrach, M.; Zhao, Z.; Mert, O.; Dickins, R.A.; Narita, M.; Zhang, M.; Lowe, S.W. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 2010, 17, 376–387. [Google Scholar] [CrossRef]
- Russo, A.A.; Tong, L.; Lee, J.; Jeffrey, P.D.; Pavletich, N.P. Structural basis for inhibition of the the tumour suppressor p16INK4A. Nature 1998, 395, 237–243. [Google Scholar] [CrossRef]
- Serrano, M.; Hannon, G.J.; Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 1993, 366, 704–707. [Google Scholar] [CrossRef]
- Schutte, M.; Hruban, R.H.; Geradts, J.; Maynard, R.; Hilgers, W.; Rabindran, S.K.; Moskaluk, C.A.; Hahn, S.A.; Schwarte-Waldhoff, I.; Schmiegel, W.; et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997, 57, 3126–3130. [Google Scholar]
- Wheland, A.J.; Bartsch, D.; Goodfellow, P.J. A familial syndrome of pancreatic cancer and melaanoma with a mutation in the CDKN2 tumor-suppressor gene. N. Engl. J. Med. 1995, 333, 975–977. [Google Scholar] [CrossRef]
- Weber, J.D.; Taylor, L.J.; Roussel, M.F.; Sherr, C.J.; Bar-Sagi, D. Nucleolar Arf sequesters Mdm2 and activates p53. Nat. Cell Biol. 1999, 1, 20–26. [Google Scholar] [CrossRef]
- Sharpless, N.E.; Bardeesy, N.; Lee, K.; Carrasco, D.; Castrillon, D.H.; Aguirre, A.J.; Wu, E.A.; Horner, J.W.; Depinho, R.A. Loss of p16INK4A with retention of p19ARF predisposes mice to tumorigenesis. Nature 2001, 413, 86–91. [Google Scholar] [CrossRef]
- Quelle, D.E.; Cheng, M.; Ashmun, R.A.; Sherr, C.J. Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by p16INK4a but not by the alternative reading frame protein p19ARF. Proc. Natl. Acad. Sci. USA 1997, 94, 669–673. [Google Scholar]
- De Winter, J.P.; Roelen, B.A.; ten Dijke, P.; van der Burg, B.; van den Eijnden-van Raaij, A.J. DPC4 (SMAD4) mediates transforming growth factor-beta1 (TGF-beta1) induced growth inhibition and transcriptional response in breast tumour cells. Oncogene 1997, 14, 1891–1899. [Google Scholar]
- Shimizu, A.; Kato, M.; Nakao, A.; Imamura, T.; ten Dijke, P.; Heldin, C.H.; Kawabata, M.; Shimada, S.; Miyazono, K. Identification of receptors and Smad proteins involved in activin signalling in a human epidermal keratinocyte cell line. Genes Cells 1998, 3, 125–134. [Google Scholar] [CrossRef]
- Liu, F.; Hata, A.; Baker, J.C.; Doody, J.; Cárcamo, J.; Harland, R.M.; Massagué, J. A human Mad protein acting as a BMP-regulated transcriptional activator. Nature 1996, 381, 620–623. [Google Scholar] [CrossRef]
- Hoodless, P.A.; Haerry, T.; Abdollah, S.; Stapleton, M.; O’Connor, M.B.; Attisano, L.; Wrana, J.L. MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell 1996, 85, 489–500. [Google Scholar] [CrossRef]
- Lechleider, R.J.; Caestecker, M.P.D.; Dehejia, A.; Polymeropoulos, M.H.; Roberts, A.B. Serine phosphorylation, chromosomal localization, and Transformong Growth Factor-beta signal transduction by human bsp-1. J. Biol. Chem. 1996, 271, 17617–17620. [Google Scholar]
- Macías-Silva, M.; Abdollah, S.; Hoodless, P.A.; Pirone, R.; Attisano, L.; Wrana, J.L. MADR2 is a substrate of the TGFbeta receptor and its phosphorylation is required for nuclear accumulation and signaling. Cell 1996, 87, 1215–1224. [Google Scholar] [CrossRef]
- Zhang, Y.; Feng, X.; We, R.; Derynck, R. Receptor-associated Mad homologues synergize as effectors of the TGF-beta response. Nature 1996, 383, 168–172. [Google Scholar] [CrossRef]
- Ilio, K.Y.; Sensibar, J.A.; Lee, C. Effect of TGF-beta 1, TGF-alpha, and EGF on cell proliferation and cell death in rat ventral prostatic epithelial cells in culture. J. Androl. 1995, 16, 482–489. [Google Scholar]
- Miettinen, P.J.; Ebner, R.; Lopez, A.R.; Derynck, R. TGF-beta induced transdifferentiation of mammary epithelial cells to mesenchymal cells: Involvement of type I receptors. J. Cell Biol. 1994, 127, 2021–2036. [Google Scholar] [CrossRef]
- Ciacci, C.; Lind, S.E.; Podolsky, D.K. Transforming growth factor beta regulation of migration in wounded rat intestinal epithelial monolayers. Gastroenterology 1993, 105, 93–101. [Google Scholar]
- Schutte, M.; Hruban, R.H.; Hedrick, L.; Cho, K.R.; Nadasdy, G.M.; Weinstein, C.L.; Bova, G.S.; Isaacs, W.B.; Cairns, P.; Nawroz, H.; et al. DPC4 gene in various tumor types. Cancer Res. 1996, 56, 2527–2530. [Google Scholar]
- Bardeesy, N.; Cheng, K.; Berger, J.H.; Chu, G.C.; Pahler, J.; Olson, P.; Hezel, A.F.; Horner, J.; Lauwers, G.Y.; Hanahan, D.; et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006, 20, 3130–3146. [Google Scholar] [CrossRef]
- Blackford, A.; Serrano, O.K.; Wolfgang, C.L.; Parmigiani, G.; Jones, S.; Zhang, X.; Parsons, D.W.; Lin, J.C.-H.; Leary, R.J.; Eshleman, J.R.; et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin. Cancer Res. 2009, 15, 4674–4679. [Google Scholar] [CrossRef]
- Conlon, K.C.; Klimstra, D.S.; Brennan, M.F. Long-term survival after curative resection for pancreatic ductal adenocarcinoma: Clinopathologic analysis of 5-year survivors. Ann. Surg. 1996, 223, 273–279. [Google Scholar] [CrossRef]
- Kausch, W. Das Karzinom der Papilla duodeni und seine radikale Entfernung. Beitr. Klin. Chir. 1912, 78, 439–486. [Google Scholar]
- Whipple, A.O.; Parsons, W.B.; Mullins, C.R. Treatment of carcinoma of the ampulla vater. Ann. Surg. 1935, 102, 763–779. [Google Scholar] [CrossRef]
- Sohn, T.A.; Yeo, C.J.; Cameron, J.L.; Koniaris, L.; Kaushal, S.; Abrams, R.A.; Sauter, P.I.C.; Coleman, J.; Hruban, R.H.; Lillemoe, K.D. Resected adenocarcinoma of the pancreas-616 patients: Results, outcomes, and prognostic indicators. J. Gastrointest. Surg. 2000, 4, 567–579. [Google Scholar] [CrossRef]
- Brennen, D.D.D.; Zamboni, G.A.; Raptopoulos, V.D.; Kruskal, J.B. Comprehensive preoperative assessment of pancreatic adenocarcinoma with 64-section volumetric CT. Radiographics 2007, 27, 1653–1666. [Google Scholar] [CrossRef]
- Lu, D.S.K.; Reber, H.A.; Krasny, R.M.; Kadell, B.M.; Sayre, J. Local staging of pancreatic cancer: Criteria for unresectability of major vessels as revealed by pancreatic-phase, thin-section helical CT. AJR Am. J. Roentgenol. 1997, 168, 1439–1443. [Google Scholar]
- Neoptolemos, J.P.; Stocken, D.D.; Dunn, J.A.; Almond, J.; Beger, H.G.; Pederzoli, P.; Bassi, C.; Dervenis, C.; Fernandez-Cruz, L.; Lacaine, F.; et al. Influence of resection margins on survival for patients with pancreatic cancer treated by adjuvant chemoradiation and/or chemotherapy in the ESPAC-1 randomized controlled trial. Ann. Surg. 2001, 234, 758–768. [Google Scholar] [CrossRef]
- Ridwelski, K.; Meyer, F.; Schmidt, U.; Lippert, H. Ergebnisse der chirurgischen Therapie beim Papillen- und Pankreaskarzinom sowie Prognoseparameter nach R0-Resektion Chirurgie und Prognose des Pankreaskarzinoms. Zentralbl. Chir. 2005, 130, 353–361. [Google Scholar] [CrossRef]
- Wagner, M.; Redaelli, C.; Lietz, M.; Seiler, C.A.; Friess, H.; Büchler, M.W. Curative resection is the single most important factor determining outcome in patients with pancreatic adenocarcinoma. Br. J. Surg. 2004, 91, 586–594. [Google Scholar] [CrossRef]
- Sasson, A.R.; Hoffman, J.P.; Ross, E.A.; Kagan, S.A.; Pingpank, J.F.; Eisenberg, B.L. En bloc resection for locally advanced cancer of the pancreas: Is it worthwhile? J. Gastrointest. Surg. 2002, 6, 147–158. [Google Scholar] [CrossRef]
- Shrikhande, S.V.; Kleeff, J.; Reiser, C.; Weitz, J.; Hinz, U.; Esposito, I.; Schmidt, J.; Friess, H.; Büchler, M.W. Pancreatic resection for M1 pancreatic ductal adenocarcinoma. Ann. Surg. Oncol. 2007, 14, 118–127. [Google Scholar]
- Kayahara, M.; Nagakawa, T.; Ueno, K.; Ohta, T.; Takeda, T.; Miyazaki, I. An evaluation of radical resection for pancreatic cancer based on the mode of recurrence as determined by autopsy and diagnostic imaging. Cancer 1993, 72, 2118–2123. [Google Scholar] [CrossRef]
- Van den Broeck, A.; Sergeant, G.; Ectors, N.; van Steenbergen, W.; Aerts, R.; Topal, B. Patterns of recurrence after curative resection of pancreatic ductal adenocarcinoma. Eur. J. Surg. Oncol. 2009, 35, 600–604. [Google Scholar] [CrossRef]
- Sener, S.F.; Fremgen, A.; Menck, H.R.; Winchester, D.P. Pancreatic cancer: A report of treatment and survival trends for 100,313 patients diagnosed from 1985-1995, using the National Cancer Database. J. Am. Coll. Surg. 1999, 189, 1–7. [Google Scholar] [CrossRef]
- Stojadinovic, A.; Hoos, A.; Brennan, M.F.; Conlon, K.C. Randomized clinical trials in pancreatic cancer. Surg. Oncol. Clin. N. Am. 2002, 11, 207–229. [Google Scholar] [CrossRef]
- Geer, R.J.; Brennan, M.F. Prognostic indicators for survival after resection of pancreatic adenocarcinoma. Am. J. Surg. 1993, 165, 68–73. [Google Scholar] [CrossRef]
- Gillen, S.; Schuster, T.; Meyer Zum Büschenfelde, C.; Friess, H.; Kleeff, J. Preoperative/neoadjuvant therapy in pancreatic cancer: A systematic review and meta-analysis of response and resection percentages. PLoS Med. 2010, 7, e1000267. [Google Scholar] [CrossRef]
- Danneberg, P.B.; Montag, B.J.; Heidelberger, C. Studies on fluorinated pyrimidines IV. Effects on nucleic acid metabolism in vivo. Cancer Res. 1958, 18, 329–334. [Google Scholar]
- Guchelaar, H.J.; Vermes, I.; Koopmans, R.P.; Reutelingsperger, C.P.; Haanen, C. Apoptosis- and necrosis-inducing potential of cladribine, cytarabine, cisplatin, and 5-fluorouracil in vitro: A quantitative pharmacodynamic model. Cancer Chemother. Pharmacol. 1999, 44, 439–443. [Google Scholar] [CrossRef]
- Huang, P.; Chubb, S.; Hertel, L.W.; Grindey, G.B.; Plunkett, W. Action of 2',2'-difluorodeoxycytidine on DNA synthesis. Cancer Res. 1991, 51, 6110–6117. [Google Scholar]
- Robertson, B.L.E.; Chubb, S.; Meyn, R.E.; Story, M.; Ford, R.; Hittelman, W.N.; Plunkett, W. Induction of apoptotic cell death in chronic lymphocytic leukemia by 2-chloro-2'-deoxyadenosine and 9-beta-D-arabinosyl-2-fluoroadenine. Blood 1993, 81, 143–150. [Google Scholar]
- Dushinsky, R.; Pleven, E.; Heidelberger, C. The synthesis of 5-fluorpyrimidines. J. Am. Chem. Soc. 1957, 79, 4559–4560. [Google Scholar]
- Heidelberger, C.; Chaudhuri, N.K.; Danneberg, P.; Mooren, D.; Griesbach, L.; Dushinsky, R.; Schnitzer, J.; Pleven, E.; Scheiner, J. Fluorinated pyrimidines, a new class of tumour-inhibitory compunds. Nature 1957, 179, 663–666. [Google Scholar]
- Tajiri, H.; Yoshimori, M.; Okazaki, N.; Miyaji, M. Phase II study of continuous venous infusion of 5-fluorouracil in advanced pancreatic cancer. Oncology 1991, 48, 18–21. [Google Scholar] [CrossRef]
- DeCaprio, J.A.; Mayer, R.J.; Gonin, R.; Arbruck, S.G. Fluorouracil and high-dose leucovorin in previously untreated patients with advanced adenocarcinoma of the pancreas: Results of a phase II trial. J. Clin. Oncol. 1991, 9, 2128–2133. [Google Scholar]
- Burris, H.A.; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar]
- Lawrence, T.S.; Chang, E.Y.; Hahn, T.M.; Hertel, L.W.; Shewach, D.S. Radiosensitization of pancreatic cancer cells by 2',2'-difluoro-2'-deoxycytidine. Int. J. Radiat. Oncol. Biol. Phys. 1996, 34, 867–872. [Google Scholar] [CrossRef]
- Colucci, G.; Giuliani, F.; Gebbia, V.; Biglietto, M.; Rabitti, P.; Uomo, G.; Cigolari, S.; Testa, A.; Maiello, E.; Lopez, M. Gemcitabine alone or with cisplatin for the treatment of patients with locally advanced and/or metastatic pancreatic cancinoma: A prospective, randomized phase III study of the Gruppo Oncologia dell’Italia Meridionale. Cancer 2002, 94, 902–910. [Google Scholar] [CrossRef]
- Berlin, J.D.; Catalano, P.; Thomas, J.P.; Kugler, J.W.; Haller, D.G.; Benson, A.B.I. Phase III study of gemcitabine in combination with fluorouracil versus gemcitabine alone in patients with advanced pancreatic carcinoma: Eastern Cooperative Oncology Group trial E2297. J. Clin. Oncol. 2002, 20, 3270–3275. [Google Scholar] [CrossRef]
- Cunningham, D.; Chau, I.; Stocken, D.D.; Valle, J.W.; Smith, D.; Steward, W.; Harper, P.G.; Dunn, J.; Tudur-Smith, C.; West, J.; et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. 2009, 27, 5513–5518. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.-L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1818–1825. [Google Scholar]
- Raimondi, S.; Lowenfels, A.B.; Morselli-Labate, A.M.; Maisonneuve, P.; Pezzilli, R. Pancreatic cancer in chronic pancreatitis; aetiology, incidence, and early detection. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 349–358. [Google Scholar] [CrossRef]
- Farrow, B.; Sugiyama, Y.; Chen, A.; Uffort, E.; Nealon, W.; Mark Evers, B. Inflammatory mechanisms contributing to pancreatic pancer development. Ann. Surg. 2004, 239, 763–771. [Google Scholar] [CrossRef]
- Guerra, C.; Schuhmacher, A.J.; Cañamero, M.; Grippo, P.J.; Verdaguer, L.; Pérez-Gallego, L.; Dubus, P.; Sandgren, E.P.; Barbacid, M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell 2007, 11, 291–302. [Google Scholar] [CrossRef]
- Guerra, C.; Collado, M.; Navas, C.; Schuhmacher, A.J.; Hernández-Porras, I.; Cañamero, M.; Rodriguez-Justo, M.; Serrano, M.; Barbacid, M. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell 2011, 19, 728–739. [Google Scholar] [CrossRef]
- Roland, C.L.; Dineen, S.P.; Toombs, J.E.; Carbon, J.G.; Smith, C.W.; Brekken, R.A.; Barnett, C.C. Tumor-derived intercellular adhesion molecule-1 mediates tumor-associated leukocyte infiltration in orthotopic pancreatic xenografts. Exp. Biol. Med. 2010, 235, 263–270. [Google Scholar]
- Dineen, S.P.; Lynn, K.D.; Holloway, S.E.; Miller, A.F.; Sullivan, J.P.; Shames, D.S.; Beck, A.W.; Barnett, C.C.; Fleming, J.B.; Brekken, R.A. Vascular endothelial growth factor receptor 2 mediates macrophage infiltration into orthotopic pancreatic tumors in mice. Cancer Res. 2008, 68, 4340–4346. [Google Scholar]
- Kurahara, H.; Shinchi, H.; Mataki, Y.; Maemura, K.; Noma, H.; Kubo, F.; Sakoda, M.; Ueno, S.; Natsugoe, S.; Takao, S. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J. Surg. Res. 2011, 167, e211–e219. [Google Scholar] [CrossRef]
- Campbell, A.S.; Albo, D.; Kimsey, T.F.; White, S.L.; Wang, T.N. Macrophage inflammatory protein-3α promotes pancreatic cancer cell invasion. J. Surg. Res. 2005, 123, 96–101. [Google Scholar] [CrossRef]
- Esposito, I.; Menicagli, M.; Funel, N.; Bergmann, F.; Boggi, U.; Mosca, F.; Bevilacqua, G.; Campani, D. Inflammatory cells contribute to the generation of an angiogenic phenotype in pancreatic ductal adenocarcinoma. J. Clin. Pathol. 2004, 57, 630–636. [Google Scholar] [CrossRef]
- Strouch, M.J.; Cheon, E.C.; Salabat, M.R.; Krantz, S.B.; Gounaris, E.; Melstrom, L.G.; Dangi-garimella, S.; Wang, E.; Munshi, H.G.; Khazaie, K.; et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin. Cancer Res. 2011, 16, 2257–2265. [Google Scholar]
- Bachem, M.G.; Schünemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef]
- Aoki, H.; Ohnishi, H.; Hama, K.; Ishijima, T.; Yukihiro, S.; Hanatsuka, K.; Ohashi, A.; Wada, S.; Miyata, T.; Kita, H.; et al. Autocrine loop between TGF-beta1 and IL-1beta through Smad3- and ERK-dependent pathways in rat pancreatic stellate cells. Am. J. Physiol. Cell Physiol. 2005, 290, C1100–C1108. [Google Scholar] [CrossRef]
- Mews, P.; Phillips, P.; Fahmy, R.; Korsten, M.; Pirola, R.; Wilson, J.; Apte, M. Pancreatic stellate cells respond to inflammatory cytokines: Potential role in chronic pancreatitis. Gut 2002, 50, 535–541. [Google Scholar] [CrossRef]
- Phillips, P.A.; McCarroll, J.A.; Park, S.; Wu, M.J.; Pirola, R.; Korsten, M.; Wilson, J.S.; Apte, M.V. Rat pancreatic stellate cells secrete matrix metalloproteinases: Implications for extracellular matrix turnover. Gut 2003, 52, 275–282. [Google Scholar] [CrossRef]
- Pryczynicz, A.; Uziñska-ustymowicz, K.; Dymicka-piekarska, V.; Czy, J.; Kemona, A. Expression of matrix metalloproteinase 9 in pancreatic ductal carcinoma is associated with tumor metastasis formation. Folia Histochem. Cytobiol. 2007, 45, 37–40. [Google Scholar]
- Aoki, H.; Ohnishi, H.; Hama, K.; Shinozaki, S.; Kita, H.; Yamamoto, H.; Osawa, H.; Sato, K.; Tamada, K.; Sugano, K. Existence of autocrine loop between interleukin-6 and tranforming growth factor-β1 in activated rat pancreatic stellate cells. J. Cell. Biochem. 2006, 99, 221–228. [Google Scholar] [CrossRef]
- Ohnishi, H.; Miyata, T.; Yasuda, H.; Yukihiro, S.; Hanatsuka, K.; Kita, H.; Ohashi, A.; Tamada, K.; Makita, N.; Iiri, T.; et al. Distinct roles of Smad2-, Smad3-, and ERK-dependent pathways in Transforming Growth Factor-β1 regulation of pancreatic stellate cellular functions. J. Biol. Chem. 2004, 279, 8873–8878. [Google Scholar]
- Nakashima, H.; Nakamura, M.; Yamaguchi, H.; Yamanaka, N.; Akiyoshi, T.; Koga, K.; Yamaguchi, K.; Tsuneyoshi, M.; Tanaka, M.; Katano, M. Nuclear factor-kappaB contributes to Hedgehog signaling pathway activation through sonic hedgehog induction in pancreatic cancer. Cancer Res. 2006, 66, 7041–7049. [Google Scholar] [CrossRef]
- Yamasaki, A.; Kameda, C.; Xu, R.; Tanaka, H.; Tasaka, T.; Chikazawa, N.; Suzuki, H.; Morisaki, T.; Kubo, M.; Onishi, H.; et al. Nuclear factor kappaB-activated monocytes contribute to pancreatic cancer progression through the production of Shh. Cancer Immunol. Immunother. 2010, 59, 675–686. [Google Scholar] [CrossRef]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates Hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.-J.; Verrecchia, F.; Mauviel, A. Induction of Sonic Hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, A.; Shimokawa, T.; Tostar, U.; Jin, Q.; Fendrich, V.; Guerra, C.; Barbacid, M.; Toftgård, R. DYRK1B-dependent autocrine-to-paracrine shift of Hedgehog signaling by mutant RAS. Nat. Struct. Mol. Biol. 2010, 17, 718–725. [Google Scholar]
- Bailey, J.M.; Swanson, B.J.; Hamada, T.; Eggers, J.P.; Ouellette, M.M.; Hollingsworth, M.A. Sonic Hedgehog promotes desmoplasia in pancreatic cancer. Clin. Cancer Res. 2008, 14, 5995–6004. [Google Scholar] [CrossRef]
- Jung, I.H.; Jung, D.E.; Park, Y.N.; Song, S.Y.; Park, S.W. Aberrant Hedgehog ligands induce progressive pancreatic fibrosis by paracrine activation of myofibroblasts and ductular cells in transgenic zebrafish. PLoS One 2011, 6, e27941. [Google Scholar]
- Feldmann, G.; Dhara, S.; Fendrich, V.; Bedja, D.; Beaty, R.; Mullendore, M.; Karikari, C.; Alvarez, H.; Iacobuzio-Donahue, C.; Jimeno, A.; et al. Blockade of Hedgehog signaling inhibits pancreatic cancer invasion and metastases: A new paradigm for combination therapy in solid cancers. Cancer Res. 2007, 67, 2187–2196. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Gorbachevski, A.; Reiser, C.; Mitkus, T.; Esposito, I.; Giese, T.; Büchler, M.W.; Giese, N.A.; Friess, H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 2007, 132, 1447–1464. [Google Scholar] [CrossRef]
- Baril, P.; Gangeswaran, R.; Mahon, P.C.; Caulee, K.; Kocher, H.M.; Harada, T.; Zhu, M.; Kalthoff, H.; Crnogorac-Jurcevic, T.; Lemoine, N.R. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: Role of the beta4 integrin and the PI3k pathway. Oncogene 2007, 26, 2082–2094. [Google Scholar]
- Hwang, R.F.; Moore, T.; Arumugam, T.; Ramachandran, V.; Amos, K.D.; Rivera, A.; Ji, B.; Evans, D.B.; Logsdon, C.D. Cancer-associated stromal fibroblasts promote pancreatic tumor progression. Cancer Res. 2008, 68, 918–926. [Google Scholar]
- Vonlaufen, A.; Joshi, S.; Qu, C.; Phillips, P.A.; Xu, Z.; Parker, N.R.; Toi, C.S.; Pirola, R.C.; Wilson, J.S.; Goldstein, D.; et al. Pancreatic stellate cells: Partners in crime with pancreatic cancer cells. Cancer Res. 2008, 68, 2085–2093. [Google Scholar] [CrossRef]
- Neesse, A.; Wagner, M.; Ellenrieder, V.; Bachem, M.; Gress, T.M.; Buchholz, M. Pancreatic stellate cells potentiate proinvasive effects of SERPINE2 expression in pancreatic cancer xenograft tumors. Pancreatology 2007, 7, 380–385. [Google Scholar] [CrossRef]
- Buchholz, M.; Biebl, A.; Nee, A.; Wagner, M.; Iwamura, T.; Leder, G.; Adler, G.; Gress, T.M. SERPINE2 (protease nexin I) promotes extracellular matrix production and local invasion of pancreatic tumors in vivo. Cancer Res. 2003, 63, 4945–4951. [Google Scholar]
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44. [Google Scholar] [CrossRef]
- Arlt, A.; Vorndamm, J.; Müerköster, S.; Yu, H.; Schmidt, W.E.; Fölsch, U.R.; Schäfer, H. Autocrine production of interleukin 1beta confers constitutive nuclear factor kappaB activity and chemoresistance in pancreatic carcinoma cell lines. Cancer Res. 2002, 62, 910–916. [Google Scholar]
- Müerköster, S.; Wegehenkel, K.; Arlt, A.; Witt, M.; Sipos, B.; Kruse, M.-L.; Sebens, T.; Klöppel, G.; Kalthoff, H.; Fölsch, U.R.; et al. Tumor stroma interactions induce chemoresistance in pancreatic ductal carcinoma cells involving increased secretion and paracrine effects of nitric oxide and interleukin-1β. Cancer Res. 2004, 64, 1331–1337. [Google Scholar] [CrossRef]
- Arlt, A.; Gehrz, A.; Müerköster, S.; Vorndamm, J.; Kruse, M.-L.; Fölsch, U.R.; Schäfer, H. Role of NF-kappaB and Akt/PI3K in the resistance of pancreatic carcinoma cell lines against gemcitabine-induced cell death. Oncogene 2003, 22, 3243–3251. [Google Scholar] [CrossRef]
- Arlt, A.; Vorndamm, J.; Breitenbroich, M.; Fölsch, U.R.; Kalthoff, H.; Schmidt, W.E.; Schäfer, H. Inhibition of NF-kB sensitizes human pancreatic carcinoma cells to apoptosis induced by etoposide (VP16) or doxorubicin. Oncogene 2001, 20, 859–868. [Google Scholar] [CrossRef]
- Schniewind, B.; Christgen, M.; Kurdow, R.; Haye, S.; Kremer, B.; Kalthoff, H.; Ungefroren, H. Resistance of pancreatic cancer to gemcitabine treatment is dependent on mitochondria-mediated apoptosis. Int. J. Cancer 2004, 109, 182–188. [Google Scholar] [CrossRef]
- Shi, X.; Liu, S.; Kleeff, J.; Friess, H.; Büchler, M.W. Acquired resistance of pancreatic cancer cells towards 5-fluorouracil and gemcitabine is associated with altered expression of apoptosis-regulating genes. Oncology 2002, 62, 354–362. [Google Scholar] [CrossRef]
- Nozawa, H.; Chiu, C.; Hanahan, D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 12493–12498. [Google Scholar] [CrossRef]
- Inoue, M.; Hager, J.H.; Ferrara, N.; Gerber, H.-P.; Hanahan, D. VEGF-A has a critical, nonredundant role in angiogenic switching and pancreatic beta cell carcinogenesis. Cancer Cell 2002, 1, 193–202. [Google Scholar] [CrossRef]
- Bergers, G.; Brekken, R.; Mcmahon, G.; Vu, T.H.; Itoh, T.; Tamaki, K.; Tanzawa, K.; Thorpe, P.; Itohara, S.; Hanahan, D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell. Biol. 2000, 2, 737–744. [Google Scholar] [CrossRef]
- Ikeda, N.; Adachi, M.; Taki, T.; Huang, C.; Hashida, H.; Takabayashi, A.; Sho, M.; Nakajima, Y.; Kanehiro, H.; Hisanaga, M.; et al. Prognostic significance of angiogenesis in human pancreatic cancer. Br. J. Cancer 1999, 79, 1553–1563. [Google Scholar] [CrossRef]
- Fujimoto, K.; Hosotani, R.; Wada, M.; Lee, J.; Koshiba, T.; Miyamoto, Y.; Tsuji, S.; Nakajima, S.; Doi, R.; Imamura, M. Expression of two angiogenic factors, vascular endothelial growth factor and platelet-deriver endothelial growth factor in human pancreatic cancer, and its relationship to angiogenesis. Eur. J. Cancer 1998, 34, 1439–1447. [Google Scholar] [CrossRef]
- Fujioka, S.; Yoshida, K.; Yanagisawa, S.; Kawakami, M.; Aoki, T.; Yamazaki, Y. Angiogenesis in pancreatic carcinoma: Thymidine phosphorylase expression in stromal cells and intratumoral microvessel density as independent predictors of overall and relapse-free survival. Cancer 2001, 92, 1788–1797. [Google Scholar] [CrossRef]
- Komar, G.; Kauhanen, S.; Liukko, K.; Seppanen, M.; Kajander, S.; Ovaska, J.; Nuutila, P.; Minn, H. Decreased blood flow with increased metabolic activity: A novel sign of pancreatic tumor aggressiveness. Clin. Cancer Res. 2009, 15, 5511–5517. [Google Scholar] [CrossRef]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2012. [Google Scholar] [PubMed]
- Poste, G.; Greig, R. On the genesis and regulation of cellular heterogeneity in malignant tumors. Invasion Metastasis 1982, 2, 137–176. [Google Scholar]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.M.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells-Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006, 66, 9339–9344. [Google Scholar]
- Rasheed, Z.; Wang, Q.; Matsui, W. Isolation of stem cells from human pancreatic cancer xenografts. J. Vis. Exp. 2010. [Google Scholar] [CrossRef]
- Gou, S.; Liu, T.; Wang, C.; Yin, T.; Li, K.; Yang, M.; Zhou, J. Establishment of clonal colony-forming assay for propagation of pancreatic cancer cells with stem cell properties. Pancreas 2007, 34, 429–435. [Google Scholar] [CrossRef]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 1994, 367, 645–648. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef]
- Huang, P.; Wang, C.-Y.; Gou, S.-M.; Wu, H.-S.; Liu, T.; Xiong, J.-X. Isolation and biological analysis of tumor stem cells from pancreatic adenocarcinoma. World J. Gastroenterol. 2008, 14, 3903–3907. [Google Scholar] [CrossRef]
- Yin, T.; Wei, H.; Gou, S.; Shi, P.; Yang, Z.; Zhao, G.; Wang, C. Cancer stem-like cells enriched in panc-1 spheres possess increased migration ability and resistance to gemcitabine. Int. J. Mol. Sci. 2011, 12, 1595–1604. [Google Scholar] [CrossRef]
- Lu, Y.; Zhu, H.; Wang, Z.; Fan, X.; Zhu, S.; Li, X.; Wang, Y.; Lu, J.; Zhu, M. Altered expressions of embryonic stem-related genes in pancreatic cancer stem cell. Zhonghua Yi Xue Za Zhi 2011, 91, 3107–3110. [Google Scholar]
- Zhu, J.; He, J.; Liu, Y.; Simeone, D.M.; Lubman, D.M. Identification of glycoprotein markers for pancreatic cancer CD24+CD44+ stem-like cells using nano-LC-MS/MS and tissue microarray. J. Proteome Res. 2012, 11, 2272–2281. [Google Scholar] [CrossRef]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem cell 2007, 1, 313–323. [Google Scholar] [CrossRef]
- Immervoll, H.; Hoem, D.; Sakariassen, P.; Steffensen, O.; Molven, A. Expression of the “stem cell marker” CD133 in pancreas and pancreatic ductal adenocarcinomas. BMC Cancer 2008, 8, 48. [Google Scholar] [CrossRef]
- Lardon, J.; Corbeil, D.; Huttner, W.B.; Ling, Z.; Bouwens, L. Stem cell marker prominin-1/AC133 is expressed in duct cells of the adult human pancreas. Pancreas 2008, 36, e1–e6. [Google Scholar]
- Immervoll, H.; Hoem, D.; Steffensen, O.J.; Miletic, H.; Molven, A. Visualization of CD44 and CD133 in normal pancreas and pancreatic ductal adenocarcinomas: Non-overlapping membrane expression in cell populations positive for both markers. J. Histochem. Cytochem. 2011, 59, 441–455. [Google Scholar] [CrossRef]
- Kemper, K.; Sprick, M.R.; de Bree, M.; Scopelliti, A.; Vermeulen, L.; Hoek, M.; Zeilstra, J.; Pals, S.T.; Mehmet, H.; Stassi, G.; et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010, 70, 719–729. [Google Scholar] [CrossRef]
- Kim, M.P.; Fleming, J.B.; Wang, H.; Abbruzzese, J.L.; Choi, W.; Kopetz, S.; McConkey, D.J.; Evans, D.B.; Gallick, G.E. ALDH activity selectively defines an enhanced tumor-initiating cell population relative to CD133 expression in human pancreatic adenocarcinoma. PLoS One 2011, 6, e20636. [Google Scholar]
- Rasheed, Z.A.; Yang, J.; Wang, Q.; Kowalski, J.; Freed, I.; Murter, C.; Hong, S.-M.; Koorstra, J.-B.; Rajeshkumar, N.V.; He, X.; Goggins, M.; et al. Prognostic significance of tumorigenic cells with mesenchymal features in pancreatic adenocarcinoma. J. Natl. Cancer Inst. 2010, 102, 340–351. [Google Scholar] [CrossRef]
- Li, C.; Wu, J.-J.; Hynes, M.; Dosch, J.; Sarkar, B.; Welling, T.H.; Pasca di Magliano, M.; Simeone, D.M. c-Met is a marker of pancreatic cancer stem cells and therapeutic target. Gastroenterology 2011, 141, 2218–2227.e5. [Google Scholar] [CrossRef]
- Zhang, S.-N.; Huang, F.-T.; Huang, Y.-J.; Zhong, W.; Yu, Z. Characterization of a cancer stem cell-like side population derived from human pancreatic adenocarcinoma cells. Tumori 2010, 96, 985–992. [Google Scholar]
- Wang, Y.H.; Li, F.; Wang, X.H.; Sun, H.C.; Liu, S.; Cui, Y.Q.; Xu, X.X. A side population of cells from a human pancreatic carcinoma cell line harbors cancer stem cell characteristics. Neoplasma 2009, 56, 371–378. [Google Scholar] [CrossRef]
- Yekta, S.; Shih, I.-H.; Bartel, D.P. MicroRNA-directed cleavage of HOXB8 mRNA. Science 2004, 304, 594–596. [Google Scholar] [CrossRef]
- Wu, L.; Fan, J.; Belasco, J.G. MicroRNAs direct rapid deadenylation of mRNA. Proc. Natl. Acad. Sci. USA 2006, 103, 4034–4039. [Google Scholar] [CrossRef]
- Mathonnet, G.; Fabian, M.R.; Svitkin, Y.V.; Parsyan, A.; Huck, L.; Murata, T.; Biffo, S.; Merrick, W.C.; Darzynkiewicz, E.; Pillai, R.S.; et al. MicroRNA inhibition of translation initiation in vitro by targeting the cap-binding complex eIF4F. Science 2007, 317, 1764–1767. [Google Scholar]
- Jung, D.E.; Wen, J.; Oh, T.; Song, S.Y. Differentially expressed microRNAs in pancreatic cancer stem cells. Pancreas 2011, 40, 1180–1187. [Google Scholar] [CrossRef]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef]
- Ji, Q.; Hao, X.; Zhang, M.; Tang, W.; Yang, M.; Li, L.; Xiang, D.; Desano, J.T.; Bommer, G.T.; Fan, D.; et al. MicroRNA miR-34 inhibits human pancreatic cancer tumor-initiating cells. PLoS One 2009, 4, e6816. [Google Scholar]
- Lash, J.; Holtzer, S.; Holtzer, H. An experimental analysis of the development of the spinal column: VI. Aspects of cartilage induction. Exp. Cell Res. 1957, 13, 292–303. [Google Scholar] [CrossRef]
- Trelstad, R.L. Mesenchymal cell polarity and morphogenesis of chick cartilage. Dev. Biol. 1977, 59, 153–163. [Google Scholar] [CrossRef]
- Karp, G.C.; Solursh, M. Dynamic activity of the filopodia of sea urchin embryonic cells and their role in directed migration of the primary mesenchyme in vitro. Dev. Biol. 1985, 112, 276–283. [Google Scholar] [CrossRef]
- Trelstad, R.L.; Hay, E.D.; Revel, J.-P. Cell contact during early morphogenesis in the chick embryo. Dev. Biol. 1967, 16, 78–106. [Google Scholar] [CrossRef]
- Behrens, J.; Mareel, M.M.; Roy, F.M.V. Dissecting tumor cell invasion: Epithelial cells acquire invasive properties after the loss of uvomorulin-mediated cell-cell adhesion. J. Cell Biol. 1989, 108, 2435–2447. [Google Scholar] [CrossRef]
- Cano, A.; Pérez-moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor Snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell. Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Eger, A.; Stockinger, A.; Schaffhauser, B.; Beug, H.; Foisner, R. Epithelial mesenchymal transition by c-Fos estrogen receptor activation involves nuclear translocation of beta-catenin and upregulation of beta-catenin/lymphoid enhancer binding factor-1 transcriptional activity. J. Cell Biol. 2000, 148, 173–188. [Google Scholar] [CrossRef]
- Yang, J.; Mani, S.A.; Donaher, J.L.; Ramaswamy, S.; Itzykson, R.A.; Come, C.; Savagner, P.; Gitelman, I.; Richardson, A.; Weinberg, R.A.; et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 2004, 117, 927–939. [Google Scholar] [CrossRef]
- Mazda, M.; Nishi, K.; Naito, Y.; Ui-Tei, K. E-cadherin is transcriptionally activated via suppression of ZEB1 transcriptional repressor by small RNA-mediated gene silencing. PLoS One 2011, 6, e28688. [Google Scholar]
- Mendez, M.G.; Kojima, S.-I.; Goldman, R.D. Vimentin induces changes in cell shape, motility, and adhesion during the epithelial to mesenchymal transition. FASEB J. 2010, 24, 1838–1851. [Google Scholar] [CrossRef]
- Hotz, B.; Arndt, M.; Dullat, S.; Bhargava, S.; Buhr, H.-J.; Hotz, H.G. Epithelial to mesenchymal transition: Expression of the regulators snail, slug, and twist in pancreatic cancer. Clin. Cancer Res. 2007, 13, 4769–4776. [Google Scholar] [CrossRef]
- Nüsslein-Volhard, C.; Wieschaus, E. Mutations affecting segment number and polarity in Drosophila. Nature 1980, 287, 795–801. [Google Scholar] [CrossRef]
- Tabata, T.; Kornberg, T.B. Hedgehog is a signaling protein with a key role in patterning Drosophila imaginal discs. Cell 1994, 76, 89–102. [Google Scholar] [CrossRef]
- Ingham, P.W.; Taylor, A.M.; Nakano, Y. Role of the Drosophila patched gene in positional signalling. Nature 1991, 353, 184–187. [Google Scholar] [CrossRef]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched acts catalytically to suppress the activity of Smoothened. Nature 2002, 418, 892–897. [Google Scholar] [CrossRef]
- Sasaki, H.; Nishizaki, Y.; Hui, C.; Nakafuku, M.; Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: Implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 1999, 126, 3915–3924. [Google Scholar]
- Pan, Y.; Bai, C.B.; Joyner, A.L.; Wang, B. Sonic Hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol. Cell. Biol. 2006, 26, 3915–3924. [Google Scholar]
- Thayer, S.P.; di Magliano, M.P.; Heiser, P.W.; Nielsen, C.M.; Roberts, D.J.; Lauwers, G.Y.; Qi, Y.P.; Gysin, S.; Fernández-del Castillo, C.; Yajnik, V.; et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 2003, 425, 851–856. [Google Scholar] [CrossRef]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Oca, R.M.D.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Bailey, J.M.; Mohr, A.M.; Hollingsworth, M.A. Sonic Hedgehog paracrine signaling regulates metastasis and lymphangiogenesis in pancreatic cancer. Oncogene 2009, 28, 3513–3525. [Google Scholar] [CrossRef]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.-C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO J. 2012, 4, 218–233. [Google Scholar] [Green Version]
- Yao, J.; An, Y.; Wie, J.; Ji, Z.; Lu, Z.; Wu, J.; Jiang, K.; Chen, P.; Xu, Z.; Miao, Y. Cyclopamine reverts acquired chemoresistance and down-regulates cancer stem cell markers in pancreatic cancer cell lines. Swiss Med. Wkly 2011, 141, w13208. [Google Scholar]
- Tang, S.-N.; Fu, J.; Nall, D.; Rodova, M.; Shankar, S.; Srivastava, R.K. Inhibition of Sonic Hedgehog pathway and pluripotency maintaining factors regulate human pancreatic cancer stem cell characteristics. Int. J. Cancer 2011, 40, 30–40. [Google Scholar]
- Bhanot, P.; Brink, M.; Samos, C.; Hsieh, J.; Wang, Y.; Macke, J.; Andrew, D.; Nathans, J.; Nusse, R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 1996, 382, 225–230. [Google Scholar] [CrossRef]
- Cong, F.; Schweizer, L.; Varmus, H. Wnt signals across the plasma membrane to activate the beta-catenin pathway by forming oligomers containing its receptors, Frizzled and LRP. Development 2004, 131, 5103–5115. [Google Scholar] [CrossRef]
- Kishida, S.; Yamamoto, H.; Hino, S.; Ikeda, S.; Kishida, M.; Kikuchi, A. DIX domains of Dvl and Axin are necessary for protein interactions and their ability to regulate β-catenin stability. Mol. Cell. Biol. 1999, 19, 4414–4422. [Google Scholar]
- Rubinfield, B.; Albert, I.; Porfiri, E.; Fiol, C.; Munemitsu, S.; Polakis, P. Binding of GSK3β to the APC-β-catenin complex and regulation of complex assembly. Science 1996, 272, 1023–1026. [Google Scholar]
- Behrens, J.; von Kries, J.P.; Kühl, M.; Bruhn, L.; Wedlich, D.; Grosschedl, R.; Birchmeier, W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature 1996, 382, 638–642. [Google Scholar] [CrossRef]
- Molenaar, M.; van de Wetering, M.; Oosterwegel, M.; Peterson-Maduro, J.; Godsave, S.; Korinek, V.; Roose, J.; Destrée, O.; Clevers, H. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 1996, 86, 391–399. [Google Scholar] [CrossRef]
- Takao, Y.; Yokota, T.; Koide, H. Beta-catenin up-regulates Nanog expression through interaction with Oct-3/4 in embryonic stem cells. Biochem. Biophys. Res. Commun. 2007, 353, 699–705. [Google Scholar] [CrossRef]
- Hochedlinger, K.; Yamada, Y.; Beard, C.; Jaenisch, R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell 2005, 121, 465–477. [Google Scholar] [CrossRef]
- Heiser, P.W.; Cano, D.A.; Landsman, L.; Kim, G.E.; Kench, J.G.; Klimstra, D.S.; Taketo, M.M.; Biankin, A.V.; Hebrok, M. Stabilization of beta-catenin induces pancreas tumor formation. Gastroenterology 2008, 135, 1288–1300. [Google Scholar] [CrossRef]
- Zeng, G.; Germinaro, M.; Micsenyi, A.; Monga, N.K.; Bell, A.; Sood, A.; Malhotra, V.; Sood, N.; Midda, V.; Monga, D.K.; et al. Aberrant Wnt/beta-catenin signaling in pancreatic adenocarcinoma. Neoplasia 2006, 8, 279–289. [Google Scholar] [CrossRef]
- Kim, K.; Lu, Z.; Hay, E.D. Direct evidence for a role of beta-catenin/LEF-1 signaling pathway in induction of EMT. Cell Biol. Int. 2002, 26, 463–476. [Google Scholar] [CrossRef]
- Slusarski, D.; Corces, V.; Moon, R. Interaction of Wnt and a Frizzled homologue triggers G-protein-linked phosphatidylinositol signalling. Nature 1997, 390, 410–413. [Google Scholar] [CrossRef]
- Sheldahl, L.C.; Park, M.; Malbon, C.C.; Moon, R.T. Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in a G-protein-dependent manner. Curr. Biol. 1999, 9, 695–698. [Google Scholar] [CrossRef]
- Kühl, M.; Sheldahl, L.C.; Malbon, C.C.; Moon, R.T. Ca2+/calmodulin-dependent protein kinase II is stimulated by Wnt and Frizzled homologs and promotes ventral cell fates in Xenopus. J. Biol. Chem. 2000, 275, 12701–12711. [Google Scholar] [CrossRef]
- Dissanayake, S.K.; Wade, M.; Johnson, C.E.; O’Connell, M.P.; Leotlela, P.D.; French, A.D.; Shah, K.V.; Hewitt, K.J.; Rosenthal, D.T.; Indig, F.E.; et al. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 17259–17271. [Google Scholar]
- Curtin, J.A.; Quint, E.; Tsipouri, V.; Arkell, R.M.; Cattanach, B.; Copp, A.J.; Henderson, D.J.; Spurr, N.; Stanier, P.; Fisher, E.M.; et al. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr. Biol. 2003, 13, 1129–1133. [Google Scholar] [CrossRef]
- Lu, X.; Borchers, A.G.; Jolicouer, C.; Rayburn, H.; Baker, J.C.; Tessier-Lavigne, M. PTK7/CCK-4 is a novel regulator of planar cell polarity in vertebrates. Nature 2004, 430, 93–98. [Google Scholar] [CrossRef]
- Montcouquiol, M.; Rachel, R.A.; Lanford, P.J.; Copeland, N.G.; Jenkins, N.A.; Kelley, M.W. Identification of Vangl2 and Scrb1 as planar polarity genes in mammals. Nature 2003, 423, 173–177. [Google Scholar] [CrossRef]
- Strutt, D.I.; Weber, U.; Mlodzik, M. The role of RhoA in tissue polarity and Frizzled signalling. Nature 1997, 387, 292–295. [Google Scholar] [CrossRef]
- Fanto, M.; Weber, U.; Strutt, D.I.; Mlodzik, M. Nuclear signaling by Rac and Rho GTPases is required in the establishment of epithelial planar polarity in the Drosophila eye. Curr. Biol. 2000, 10, 979–988. [Google Scholar] [CrossRef]
- Muñoz-Descalzo, S.; Gómez-Cabrero, A.; Mlodzik, M.; Paricio, N. Analysis of the role of the Rac/Cdc42 GTPases during planar cell polarity generation in Drosophila. Int. J. Dev. Biol. 2007, 51, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Winter, C.G.; Wang, B.; Ballew, A.; Royou, A.; Karess, R.; Axelrod, J.D.; Luo, L. Drosophila Rho-associated kinase (Drok) links Frizzled-mediated planar cell polarity signaling to the actin cytoskeleton. Cell 2001, 105, 81–91. [Google Scholar] [CrossRef]
- Vermeulen, L.; de Sousa E Melo, F.; van der Heijden, M.; Cameron, K.; de Jong, J.H.; Borovski, T.; Tuynman, J.B.; Todaro, M.; Merz, C.; Rodermond, H.; et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat. Cell Biol. 2010, 12, 468–476. [Google Scholar] [CrossRef]
- Bettenhausen, B.; Hrabĕ de Angelis, M.; Simon, D.; Guénet, J.L.; Gossler, A. Transient and restricted expression during mouse embryogenesis of Dll1, a murine gene closely related to Drosophila Delta. Development 1995, 121, 2407–2418. [Google Scholar]
- Lindsell, C.E.; Shawber, C.J.; Boulter, J.; Weinmaster, G. Jagged: A mammalian ligand that activates Notch1. Cell 1995, 80, 909–917. [Google Scholar] [CrossRef]
- Rebay, I.; Fleming, R.J.; Fehon, R.G.; Cherbas, L.; Cherbas, P.; Artavanis-Tsakonas, S. Specific EGF repeats of Notch mediate interactions with Delta and Serrate: Implications for Notch as a multifunctional receptor. Cell 1991, 67, 687–699. [Google Scholar] [CrossRef]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998, 393, 382–386. [Google Scholar] [CrossRef]
- Jarriault, S.; Brou, C.; Logeat, F.; Schroeter, E.H.; Kopan, R.; Israel, A. Signalling downstream of activated mammalian Notch. Nature 1995, 377, 355–358. [Google Scholar] [CrossRef]
- Wilkinson, H.A.; Fitzgerald, K.; Greenwald, I. Reciprocal changes in expression of the receptor lin-12 and its ligand lag-2 prior to commitment in a C. elegans cell fate decision. Cell 1994, 79, 1187–1198. [Google Scholar] [CrossRef]
- Panin, V.M.; Papayannopoulos, V.; Wilson, R.; Irvine, K.D. Fringe modulates Notch-ligand interactions. Nature 1997, 387, 908–912. [Google Scholar] [CrossRef]
- Rhyu, M.S.; Jan, L.Y.; Jan, Y.N. Asymmetric distribution of numb protein during division of the sensory organ precursor cell confers distinct fates to daughter cells. Cell 1994, 76, 477–491. [Google Scholar] [CrossRef]
- Axelrod, J.D.; Matsuno, K.; Artavanis-Tsakonas, S.; Perrimon, N. Interaction between Wingless and Notch signaling pathways mediated by Dishevelled. Science 1996, 271, 1826–1832. [Google Scholar]
- Rodilla, V.; Villanueva, A.; Obrador-Hevia, A.; Robert-Moreno, A.; Fernández-Majada, V.; Grilli, A.; López-Bigas, N.; Bellora, N.; Albà, M.M.; Torres, F.; et al. Jagged1 is the pathological link between Wnt and Notch pathways in colorectal cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 6315–6320. [Google Scholar]
- Jayasena, C.S.; Ohyama, T.; Segil, N.; Groves, A.K. Notch signaling augments the canonical Wnt pathway to specify the size of the otic placode. Development 2008, 135, 2251–2261. [Google Scholar] [CrossRef]
- Hayward, P.; Brennan, K.; Sanders, P.; Balayo, T.; DasGupta, R.; Perrimon, N.; Martinez Arias, A. Notch modulates Wnt signalling by associating with Armadillo/beta-catenin and regulating its transcriptional activity. Development 2005, 132, 1819–1830. [Google Scholar] [CrossRef]
- González-Sancho, J.M.; Brennan, K.R.; Leslie, A.; Brown, A.M.C.; Castelo-soccio, L.A. Wnt proteins induce Dishevelled phosphorylation via an LRP5/6-independent mechanism, irrespective of their ability to stabilize β-catenin. Mol. Cell. Biol. 2004, 24, 4757–4768. [Google Scholar] [CrossRef]
- Espinosa, L.; Inglés-Esteve, J.; Aguilera, C.; Bigas, A. Phosphorylation by glycogen synthase kinase-3 beta down-regulates Notch activity, a link for Notch and Wnt pathways. J. Biol. Chem. 2003, 278, 32227–32235. [Google Scholar]
- Poulson, D. Chromosomal deficiencies and the embryonic development of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1937, 23, 133–137. [Google Scholar] [CrossRef]
- Swiatek, P.J.; Lindsell, C.E.; del Amo, F.F.; Weinmaster, G.; Gridley, T. Notch1 is essential for postimplantation development in mice. Genes Dev. 1994, 8, 707–719. [Google Scholar] [CrossRef]
- Parks, A.L.; Huppert, S.S.; Muskavitch, M.A. The dynamics of neurogenic signalling underlying bristle development in Drosophila melanogaster. Mech. Dev. 1997, 63, 61–74. [Google Scholar] [CrossRef]
- Kim, J.; Sebring, A.; Esch, J.J.; Kraus, M.E.; Vorwerk, K.; Magee, J.; Carroll, S.B. Integration of positional signals and regulation of wing formation and identity by Drosophila vestigial gene. Nature 1996, 382, 133–138. [Google Scholar] [CrossRef]
- Li, H.; Sommer, L.; Beatus, P.; Anderson, D.J.; Tasuku, H.; de Angelis, M.H.; Lendahl, U.; Edlund, H. Notch signalling controls pancreatic cell differentiation. Nature 1999, 400, 877–881. [Google Scholar] [CrossRef]
- Hald, J.; Hjorth, J.P.; German, M.S.; Madsen, O.D.; Serup, P.; Jensen, J. Activated Notch1 prevents differentiation of pancreatic acinar cells and attenuate endocrine development. Dev. Biol. 2003, 260, 426–437. [Google Scholar] [CrossRef]
- Giniger, E. A role for Abl in Notch signaling. Neuron 1998, 20, 667–681. [Google Scholar] [CrossRef]
- Hashimoto-Torii, K.; Torii, M.; Sarkisian, M.R.; Bartley, C.M.; Shen, J.; Radtke, F.; Gridley, T.; Sestan, N.; Rakic, P. Interaction between Reelin and Notch signaling regulates neuronal migration in the cerebral cortex. Neuron 2008, 60, 273–284. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Maitra, A.; Ghosh, B.; Zechner, U.; Argani, P.; Iacobuzio-Donahue, C.A.; Sriuranpong, V.; Iso, T.; Meszoely, I.M.; Wolfe, M.S.; et al. Notch mediates TGF alpha-induced changes in epithelial differentiation during pancreatic tumorigenesis. Cancer Cell 2003, 3, 565–576. [Google Scholar] [CrossRef]
- Mullendore, M.E.; Koorstra, J.; Li, Y.; Offerhaus, G.J.; Henderson, C.M.; Bisht, S.; Matsui, W.; Eberhart, C.G.; Feldmann, G. Ligand-dependent Notch signaling is involved in tumor initiation and tumor maintenance in pancreatic cancer. Clin. Cancer Res. 2010, 15, 2291–2301. [Google Scholar]
- Wang, Z.; Li, Y.; Kong, D.; Banerjee, S.; Ahmad, A.; Azmi, A.S.; Ali, S.; Abbruzzese, J.L.; Gallick, G.E.; Sarkar, F.H. Acquisition of epithelial-mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009, 69, 2400–2407. [Google Scholar] [CrossRef]
- Bao, B.; Wang, Z.; Ali, S.; Kong, D.; Li, Y.; Ahmad, A.; Banerjee, S.; Azmi, A.S.; Miele, L.; Sarkar, F.H. Notch-1 induces epithelial-mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett. 2011, 307, 26–36. [Google Scholar] [CrossRef]
- Brabletz, S.; Bajdak, K.; Meidhof, S.; Burk, U.; Niedermann, G.; Firat, E.; Wellner, U.; Dimmler, A.; Faller, G.; Schubert, J.; et al. The ZEB1/miR-200 feedback loop controls Notch signalling in cancer cells. EMBO J. 2011, 30, 770–782. [Google Scholar] [CrossRef]
- James, D.; Levine, A.J.; Besser, D.; Hemmati-Brivanlou, A. TGFβ/activin/nodal signaling is necessary for the maintenance of pluripotency in human embryonic stem cells. Development 2005, 132, 1273–1282. [Google Scholar] [CrossRef]
- Vallier, L.; Reynolds, D.; Pederson, R.A. Nodal inhibits differentiation of human embryonic stem cells along the neuroectodermal default pathway. Dev. Biol. 2004, 275, 403–421. [Google Scholar] [CrossRef]
- Chang-Yeol, Y.; Whitman, M. Nodal signals to Smads through Cripto-dependent and Cripto-independent mechanisms. Mol. Cell 2001, 7, 949–957. [Google Scholar] [CrossRef]
- Reissmann, E.; Jörnvall, H.; Blokzijl, A.; Andersson, O.; Chang, C.; Minchiotti, G.; Persico, M.G.; Ibáñez, C.F.; Brivanlou, A.H. The orphan receptor ALK7 and the Activin receptor ALK4 mediate signaling by Nodal proteins during vertebrate development. Genes Dev. 2001, 15, 2010–2022. [Google Scholar]
- Nomura, M.; Li, E. Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 1998, 393, 786–790. [Google Scholar] [CrossRef]
- Zhang, Y.; Musci, T.; Derynck, R. The tumor suppressor Smad4/DPC 4 as a central mediator of Smad function. Curr. Biol. 1997, 7, 270–276. [Google Scholar] [CrossRef]
- Vallier, L.; Mendjan, S.; Brown, S.; Chng, Z.; Teo, A.; Smithers, L.E.; Trotter, M.W.B.; Cho, C.H.-H.; Martinez, A.; Rugg-Gunn, P.; et al. Activin/Nodal signalling maintains pluripotency by controlling Nanog expression. Development 2009, 136, 1339–1349. [Google Scholar] [CrossRef]
- Xiao, L.; Yuan, X.; Sharkis, S.J. Activin A maintains self-renewal and regulates fibroblast growth factor, Wnt, and bone morphogenic protein pathways in human embryonic stem cells. Stem Cells 2006, 24, 1476–1486. [Google Scholar] [CrossRef]
- Vallier, L.; Touboul, T.; Chng, Z.; Brimpari, M.; Hannan, N.; Millan, E.; Smithers, L.E.; Trotter, M.; Rugg-Gunn, P.; Weber, A.; et al. Early cell fate decisions of human embryonic stem cells and mouse epiblast stem cells are controlled by the same signalling pathways. PLoS One 2009, 4, e6082. [Google Scholar]
- Xu, X.; Browning, V.L.; Odorico, J.S. Activin, BMP and FGF pathways cooperate to promote endoderm and pancreatic lineage cell differentiation from human embryonic stem cells. Mech. Dev. 2011, 128, 412–427. [Google Scholar] [CrossRef]
- Kleeff, J.; Ishiwata, T.; Friess, H.; Büchler, M.; Korc, M. Concomitant over-expression of activin/inhibin beta subunits and their receptors in human pancreatic cancer. Int. J. Cancer 1998, 77, 860–868. [Google Scholar] [CrossRef]
- Lonardo, E.; Hermann, P.C.; Mueller, M.-T.; Huber, S.; Balic, A.; Miranda-Lorenzo, I.; Zagorac, S.; Alcala, S.; Rodriguez-Arabaolaza, I.; Ramirez, J.C.; et al. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 2011, 9, 433–446. [Google Scholar] [CrossRef]
- Gill, J.G.; Langer, E.M.; Lindsley, R.C.; Cai, M.; Murphy, T.L.; Kyba, M.; Murphy, K.M. Snail and the miR-200 family act in opposition to regulate EMT and germ layer fate restriction in differentiating ES cells. Stem Cells 2011, 29, 764–776. [Google Scholar] [CrossRef]
- Verschueren, K.; Remacle, J.E.; Collart, C.; Kraft, H.; Baker, B.S.; Tylzanowski, P.; Nelles, L.; Wuytens, G.; Su, M.T.; Bodmer, R.; et al. SIP1, a novel zinc finger/homeodomain repressor, interacts with Smad proteins and binds to 5'-CACCT sequences in candidate target genes. J. Biol. Chem. 1999, 274, 20489–20498. [Google Scholar]
- Singh, A.; Greninger, P.; Rhodes, D.; Koopman, L.; Violette, S.; Bardeesy, N.; Settleman, J. A gene expression signature associated with “K-Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009, 15, 489–500. [Google Scholar] [CrossRef]
- Hong, S.P.; Wen, J.; Bang, S.; Park, S.; Song, S.Y. CD44-positive cells are responsible for gemcitabine resistance in pancreatic cancer cells. Int. J. Cancer 2009, 125, 2323–2331. [Google Scholar] [CrossRef]
- Shah, A.N.; Summy, J.M.; Zhang, J.; Park, S.I.; Parikh, N.U.; Gallick, G.E. Development and characterization of gemcitabine-resistant pancreatic tumor cells. Ann. Surg. Oncol. 2007, 14, 3629–3637. [Google Scholar] [CrossRef]
- Jimeno, A.; Feldmann, G.; Suárez-Gauthier, A.; Rasheed, Z.; Solomon, A.; Zou, G.-M.; Rubio-Viqueira, B.; García-García, E.; López-Ríos, F.; Matsui, W.; et al. A direct pancreatic cancer xenograft model as a platform for cancer stem cell therapeutic development. Mol. Cancer Ther. 2011, 8, 310–314. [Google Scholar]
- Hu, G.; Ouyang, K.; Xie, F.; Tang, X.; Wang, K.; Han, S.; Jiang, Z.; Zhu, M.; Wen, D.; Qin, X.; et al. Intrinsic gemcitabine resistance in a novel pancreatic cancer cell line is associated with cancer stem cell-like phenotype. Int. J. Oncol. 2012, 40, 798–806. [Google Scholar]
- Zhang, Y.; Wei, J.; Wang, H.; Xue, X.; An, Y.; Tang, D.; Yuan, Z.; Wang, F.; Wu, J.; Zhang, J.; et al. Epithelial mesenchymal transition correlates with CD24+CD44+ and CD133+ cells in pancreatic cancer. Oncol. Rep. 2012, 27, 1599–1605. [Google Scholar]
- Arumugam, T.; Ramachandran, V.; Fournier, K.F.; Wang, H.; Marquis, L.; Abbruzzese, J.L.; Gallick, G.E.; Logsdon, C.D.; McConkey, D.J.; Choi, W. Epithelial to mesenchymal transition contributes to drug resistance in pancreatic cancer. Cancer Res. 2009, 69, 5820–5828. [Google Scholar] [CrossRef]
- Li, Y.; van den Boom, T.G.; Kong, D.; Wang, Z.; Ali, S.; Philip, P.A.; Sarkar, F.H. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009, 69, 6704–6712. [Google Scholar] [CrossRef]
- Singh, S.; Srivastava, S.K.; Bhardwaj, A.; Owen, L.B.; Singh, A.P. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: A novel target for therapy. Br. J. Cancer 2010, 103, 1671–1679. [Google Scholar] [CrossRef]
- Allen, J.D.; Brinkhuis, R.F.; Wijnholds, J.; Schinkel, A.H. The mouse Bcrp1/Mxr/Abcp gene: Amplification and overexpression in cell lines selected for resistance to topotecan, mitoxantrone, or doxorubicin. Cancer Res. 1999, 59, 4237–4241. [Google Scholar]
- Dembinski, J.L.; Krauss, S. Characterization and functional analysis of a slow cycling stem cell-like subpopulation in pancreas adenocarcinoma. Clin. Exp. Metastasis 2009, 26, 611–623. [Google Scholar] [CrossRef]
- Mathews, L.A.; Cabarcas, S.M.; Hurt, E.M.; Zhang, X.; Jaffee, E.M.; Farrar, W.L. Increased expression of DNA repair genes in invasive human pancreatic cancer cells. Pancreas 2012, 40, 730–739. [Google Scholar]
- Ding, Q.; Yoshimitsu, M.; Kuwahata, T.; Maeda, K.; Hayashi, T.; Obara, T.; Miyazaki, Y.; Matsubara, S.; Natsugoe, S.; Takao, S. Establishment of a highly migratory subclone reveals that CD133 contributes to migration and invasion through epithelial-mesenchymal transition in pancreatic cancer. Hum. Cell 2012, 25, 1–8. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Houbracken, I.; Bouwens, L. The quest for tissue stem cells in the pancreas and other organs, and their application in beta-cell replacement. Rev. Diabet. Stud. 2010, 7, 112–123. [Google Scholar] [CrossRef]
- Lagasse, E.; Weissman, I. Bcl-2 inhibits apoptosis of neutrophils but not their engulfment by macrophages. J. Exp. Med. 1994, 179, 1047–1052. [Google Scholar] [CrossRef]
- Turhan, A.G.; Lemoine, F.M.; Debert, C.; Bonnet, M.L.; Baillou, C.; Picard, F.; Macintyre, E.A.; Varet, B. Highly purified primitive hematopoietic stem cells are PML-RARA negative and generate nonclonal progenitors in acute promyelocytic leukemia. Blood 1995, 85, 2154–2161. [Google Scholar]
- Brown, D.; Kogan, S.; Lagasse, E.; Weissman, I.; Alcalay, M.; Pelicci, P.G.; Atwater, S.; Bishop, J.M. A PMLRARα transgene initiates murine acute promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 1997, 94, 2551–2556. [Google Scholar] [CrossRef]
- Bachoo, R.M.; Maher, E.A.; Ligon, K.L.; Sharpless, N.E.; Chan, S.S.; You, M.J.; Tang, Y.; DeFrances, J.; Stover, E.; Weissleder, R.; et al. Epidermal growth factor receptor and Ink4a/Arf: Convergent mechanisms governing terminal differentiation and transformation along the neural stem cell to astrocyte axis. Cancer Cell 2002, 1, 269–277. [Google Scholar] [CrossRef]
- Barnett, S.C.; Robertson, L.; Graham, D.; Allan, D.; Rampling, R. Oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells transformed with c-myc and H-ras form high-grade glioma after stereotactic injection into the rat brain. Carcinogenesis 1998, 19, 1529–1537. [Google Scholar] [CrossRef]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; George, W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef]
- Assou, S.; Lecarrour, T.; Tondeur, S.; Strom, S.; Marty, S.; Nadal, L.; Pantesco, V.; Réme, T.; Hugnot, P.; Gasca, S.; et al. A meta-analysis of human embryonic stem cells transcriptome integrated into a web-based expression atlas. Stem Cells 2007, 25, 961–973. [Google Scholar] [CrossRef]
- Mani, S.A.; Guo, W.; Liao, M.-J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Kabashima, A.; Higuchi, H.; Takaishi, H.; Matsuzaki, Y.; Suzuki, S.; Izumiya, M.; Iizuka, H.; Sakai, G.; Hozawa, S.; Azuma, T.; et al. Side population of pancreatic cancer cells predominates in TGF-beta-mediated epithelial to mesenchymal transition and invasion. Int. J. Cancer 2009, 124, 2771–2779. [Google Scholar] [CrossRef]
- Kikuta, K.; Masamune, A.; Watanabe, T.; Ariga, H.; Itoh, H.; Hamada, S.; Satoh, K.; Egawa, S.; Unno, M.; Shimosegawa, T. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochem. Biophys. Res. Commun. 2010, 403, 380–384. [Google Scholar] [CrossRef]
- Jung, T.; Gross, W.; Zöller, M. CD44v6 coordinates tumor matrix-triggered motility and apoptosis resistance. J. Biochem. 2011, 286, 15862–15874. [Google Scholar]
- Cannito, S.; Novo, E.; Compagnone, A.; Valfrè di Bonzo, L.; Busletta, C.; Zamara, E.; Paternostro, C.; Povero, D.; Bandino, A.; Bozzo, F.; et al. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis 2008, 29, 2267–2278. [Google Scholar] [CrossRef]
- Ide, T.; Kitajima, Y.; Miyoshi, A.; Ohtsuka, T.; Mitsuno, M.; Ohtaka, K.; Miyazaki, K. The hypoxic environment in tumor-stromal cells accelerates pancreatic cancer progression via the activation of paracrine hepatocyte growth factor/c-Met signalling. Ann. Surg. Oncol. 2007, 14, 2600–2607. [Google Scholar] [CrossRef]
- Direkze, N.C.; Hodivala-Dilke, K.; Jeffery, R.; Hunt, T.; Poulsom, R.; Oukrif, D.; Alison, M.R.; Wright, N.A. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004, 64, 8492–8495. [Google Scholar]
- Kurose, K.; Hoshaw-Woodard, S.; Adeyinka, A.; Lemeshow, S.; Watson, P.H.; Eng, C. Genetic model of multi-step breast carcinogenesis involving the epithelium and stroma: Clues to tumour-microenvironment interactions. Hum. Mol. Genet. 2001, 10, 1907–1913. [Google Scholar] [CrossRef]
- Hill, R.; Song, Y.; Cardiff, R.D.; van Dyke, T. Selective evolution of stromal mesenchyme with p53 loss in response to epithelial tumorigenesis. Cell 2005, 123, 1001–1011. [Google Scholar] [CrossRef]
- Kojima, Y.; Acar, A.; Eaton, E.N.; Mellody, K.T.; Scheel, C.; Ben-Porath, I.; Onder, T.T.; Wang, Z.C.; Richardson, A. L.; Weinberg, R.A.; et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc. Natl. Acad. Sci. USA 2010, 107, 20009–20014. [Google Scholar]
- Forino, M.; Torregrossa, R.; Ceol, M.; Murer, L.; Della Vella, M.; Del Prete, D.; D’Angelo, A.; Anglani, F. TGFbeta1 induces epithelial-mesenchymal transition, but not myofibroblast transdifferentiation of human kidney tubular epithelial cells in primary culture. Int. J. Exp. Pathol. 2006, 87, 197–208. [Google Scholar] [CrossRef]
- Iwano, M.; Plieth, D.; Danoff, T.M.; Xue, C.; Okada, H.; Neilson, E.G. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Invest. 2002, 110, 341–350. [Google Scholar]
- Boutet, A.; de Frutos, C.A.; Maxwell, P.H.; Mayol, M.J.; Romero, J.; Nieto, M.A. Snail activation disrupts tissue homeostasis and induces fibrosis in the adult kidney. EMBO J. 2006, 25, 5603–5613. [Google Scholar] [CrossRef]
- Fendrich, V.; Oh, E.; Bang, S.; Karikari, C.; Ottenhof, N.; Bisht, S.; Lauth, M.; Brossart, P.; Katsanis, N.; Maitra, A.; et al. Ectopic overexpression of Sonic Hedgehog (Shh) induces stromal expansion and metaplasia in the adult murine pancreas. Neoplasia 2011, 13, 923–930. [Google Scholar]
- Petersen, O.W.; Nielsen, H.L.; Gudjonsson, T.; Villadsen, R.; Rank, F.; Niebuhr, E.; Bissell, M.J.; Rønnov-Jessen, L. Epithelial to mesenchymal transition in human breast cancer can provide a nonmalignant stroma. Am. J. Pathol. 2003, 162, 391–402. [Google Scholar] [CrossRef]
- Moinfar, F.; Man, Y.G.; Arnould, L.; Bratthauer, G.L.; Ratschek, M.; Tavassoli, F.A. Concurrent and independent genetic alterations in the stromal and epithelial cells of mammary carcinoma: Implications for tumorigenesis. Cancer Res. 2000, 60, 2562–2566. [Google Scholar]
- Kurose, K.; Gilley, K.; Matsumoto, S.; Watson, P.H.; Zhou, X.-P.; Eng, C. Frequent somatic mutations in PTEN and TP53 are mutually exclusive in the stroma of breast carcinomas. Nat. Genet. 2002, 32, 355–357. [Google Scholar] [CrossRef]
- Walter, K.; Omura, N.; Hong, S.; Griffith, M. Pancreatic cancer associated fibroblasts display normal allelotypes. Cancer Biol. Ther. 2009, 7, 882–888. [Google Scholar]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.; Schott, A.; et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar] [CrossRef]
- Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B.M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C.C. Stem cell marker CD133 affects clinical outcome in glioma patients. Clin. Cancer Res. 2008, 14, 123–129. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, Z.-G.; Du, C.-Z.; Wang, H.-W.; Yan, L.; Gu, J. Cancer stem cell marker CD133+ tumour cells and clinical outcome in rectal cancer. Histopathology 2009, 55, 284–293. [Google Scholar] [CrossRef]
- Cioffi, M.; Dorado, J.; Baeuerle, P.A.; Heeschen, C. EpCAM/CD3-Bispecific T-cell engaging antibody MT110 eliminates primary human pancreatic cancer stem cells. Clin. Cancer Res. 2012, 18, 465–474. [Google Scholar] [CrossRef]
- Singh, B.N.; Fu, J.; Srivastava, R.K.; Shankar, S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: Molecular mechanisms. PLoS One 2011, 6, e27306. [Google Scholar]
- LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; Chang, I.; Darbonne, W.C.; et al. Phase I trial of Hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clin. Cancer Res. 2011, 17, 2502–2511. [Google Scholar] [CrossRef]
- Zischek, C.; Niess, H.; Ischenko, I.; Conrad, C.; Huss, R.; Jauch, K.-W.; Nelson, P.J.; Bruns, C. Targeting tumor stroma using engineered mesenchymal stem cells reduces the growth of pancreatic carcinoma. Ann. Surg. 2009, 250, 747–753. [Google Scholar] [CrossRef]
- Kim, H.; Zhai, G.; Samual, S.; Shah, N.; Helman, E.; Knowles, J.; Stockard, C.; Fineberg, N.; Grizzle, W.; Zhou, T.; et al. Extracelluar matrix metalloproteinase as a novel target for pancreatic cancer therapy. Anticancer Drugs 2011, 22, 864–874. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hindriksen, S.; Bijlsma, M.F. Cancer Stem Cells, EMT, and Developmental Pathway Activation in Pancreatic Tumors. Cancers 2012, 4, 989-1035. https://doi.org/10.3390/cancers4040989
Hindriksen S, Bijlsma MF. Cancer Stem Cells, EMT, and Developmental Pathway Activation in Pancreatic Tumors. Cancers. 2012; 4(4):989-1035. https://doi.org/10.3390/cancers4040989
Chicago/Turabian StyleHindriksen, Sanne, and Maarten F. Bijlsma. 2012. "Cancer Stem Cells, EMT, and Developmental Pathway Activation in Pancreatic Tumors" Cancers 4, no. 4: 989-1035. https://doi.org/10.3390/cancers4040989