Therapeutic Targeting of Hyaluronan in the Tumor Stroma

Abstract
:Abbreviation
| BRCA1 | breast cancer 1 |
| BSA | bovine serum albumin |
| CD44 | cluster of differentiation 44 |
| Col1α1 | collagen, type 1, alpha 1 |
| Col5α1 | collagen, type 5, alpha 1 |
| CS | chondroitin sulfate |
| DS | dermatan sulfate |
| ECM | extracellular matrix |
| EGF | epidermal growth factor |
| ERK1/2 | extracellular signal-regulated kinase 1/2 |
| ERM | ezrin-radixin-moesin |
| FAK | focal adhesion kinase |
| GFP | green fluorescent protein |
| HABP | hyaluronan binding protein |
| GPI | glycosylphosphatidylinositol |
| GTPase | guanosine triphosphatase |
| HA | hyaluronan |
| HAS | hyaluronan synthase |
| hdf | heart defect |
| HER | human epidermal growth factor receptor |
| HS | heparan sulfate |
| HYAL | hyaluronidase |
| HYALP-1 | hyaluronidase pseudogene 1 |
| IαI | inter-α-inhibitor |
| KPC | LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre |
| MAPK | mitogen-activated protein kinase |
| NSCLC | non-small cell lung cancer |
| PDA | pancreatic ductal adenocarcinoma |
| PDGFR | platelet-derived growth factor receptor |
| PEG | polyethylene glycol |
| PEGPH20 | pegylated human recombinant PH20 hyaluronidase enzyme |
| PLN | para-aortic lymph node |
| PI3K | phosphoinositide 3-kinase |
| RHAMM | receptor for hyaluronan-mediated motility |
| rHuPH20 | recombinant human PH20 hyaluronidase |
| RTK | receptor tyrosine kinase |
| SEM | standard error of the mean |
| SHAP | serum-derived hyaluronan-associated protein |
| SPAM-1 | sperm adhesion molecule-1 |
| Stab2 | stabilin-2 |
| tIFP | tumor interstitial fluid pressure |
| TGFβR | transforming growth factor beta receptor |
| TGI | tumor growth inhibition |
| TLR | toll-like receptor |
| TNC | tenascin-C |
| TNF-α | tumor necrosis factor-alpha |
| TSG-6 | tumor necrosis factor-inducible gene 6 protein |
| UDP | uridine 5' diphosphate |
| VEGF-A | vascular endothelial growth factor A |
1. Introduction—The Tumor Stroma
2. Hyaluronan (HA)
3. The Role of Hyaluronan in the Tumor Stroma
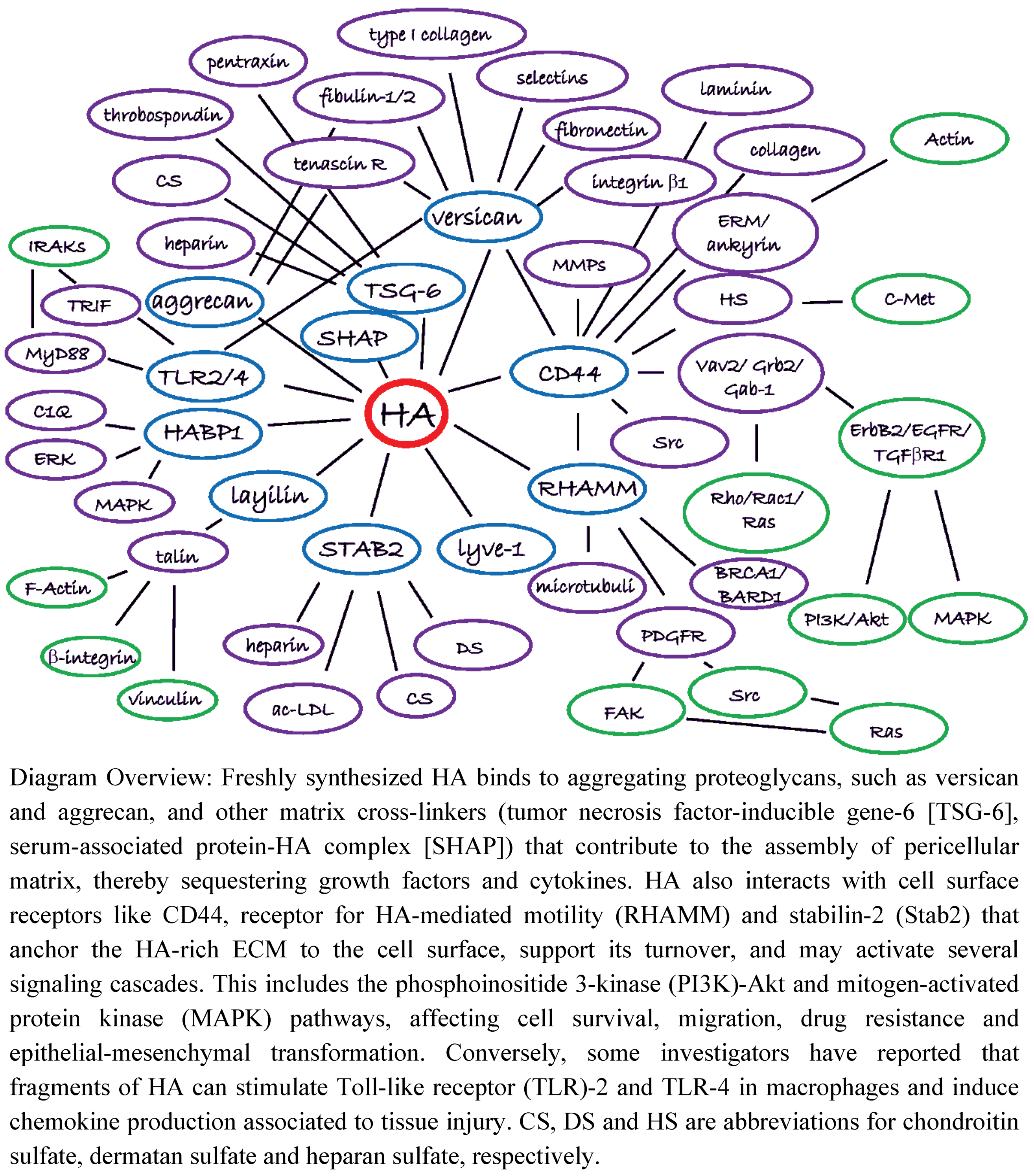
4. The HA Interactome in Cancer

4.1. HA Interactions with ECM Components
4.2. HA Interactions with Cell Surface Receptors
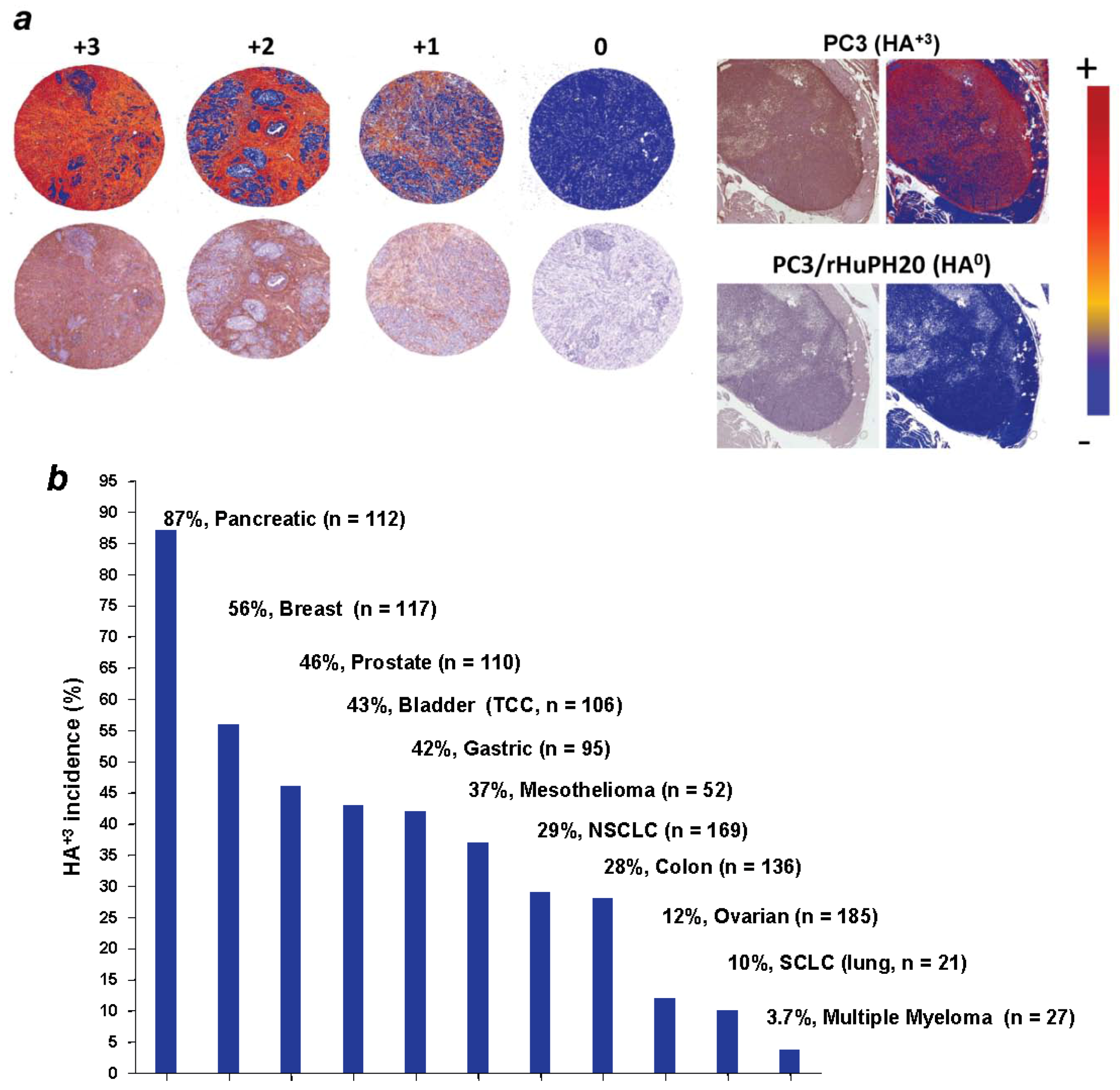
5. Significance of HA Accumulation in Cancer and the Early History of Hyaluronidase Therapies
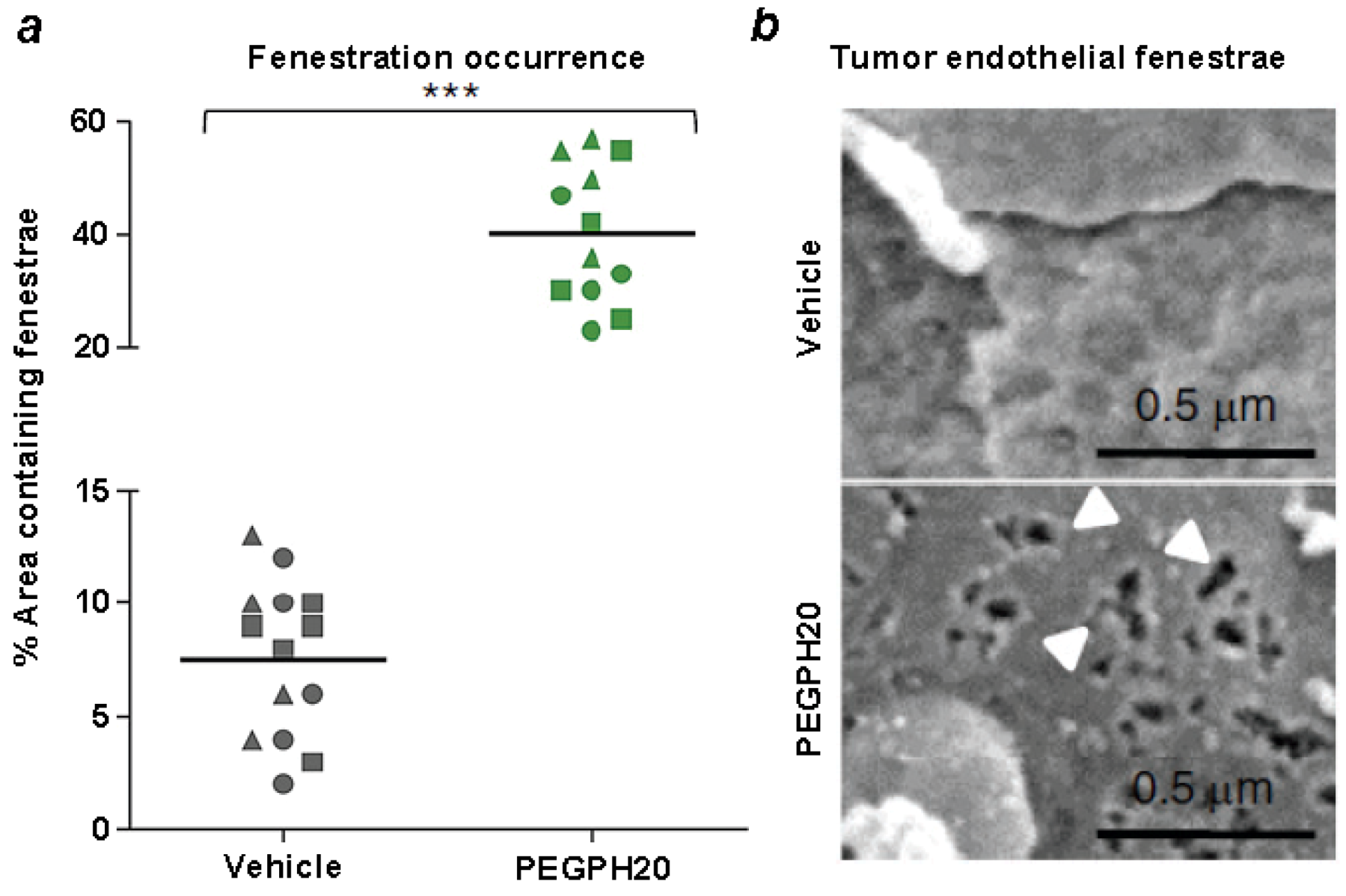
6. Preclinical Proof of Concept for Therapeutic Targeting of HA in the Tumor Stroma
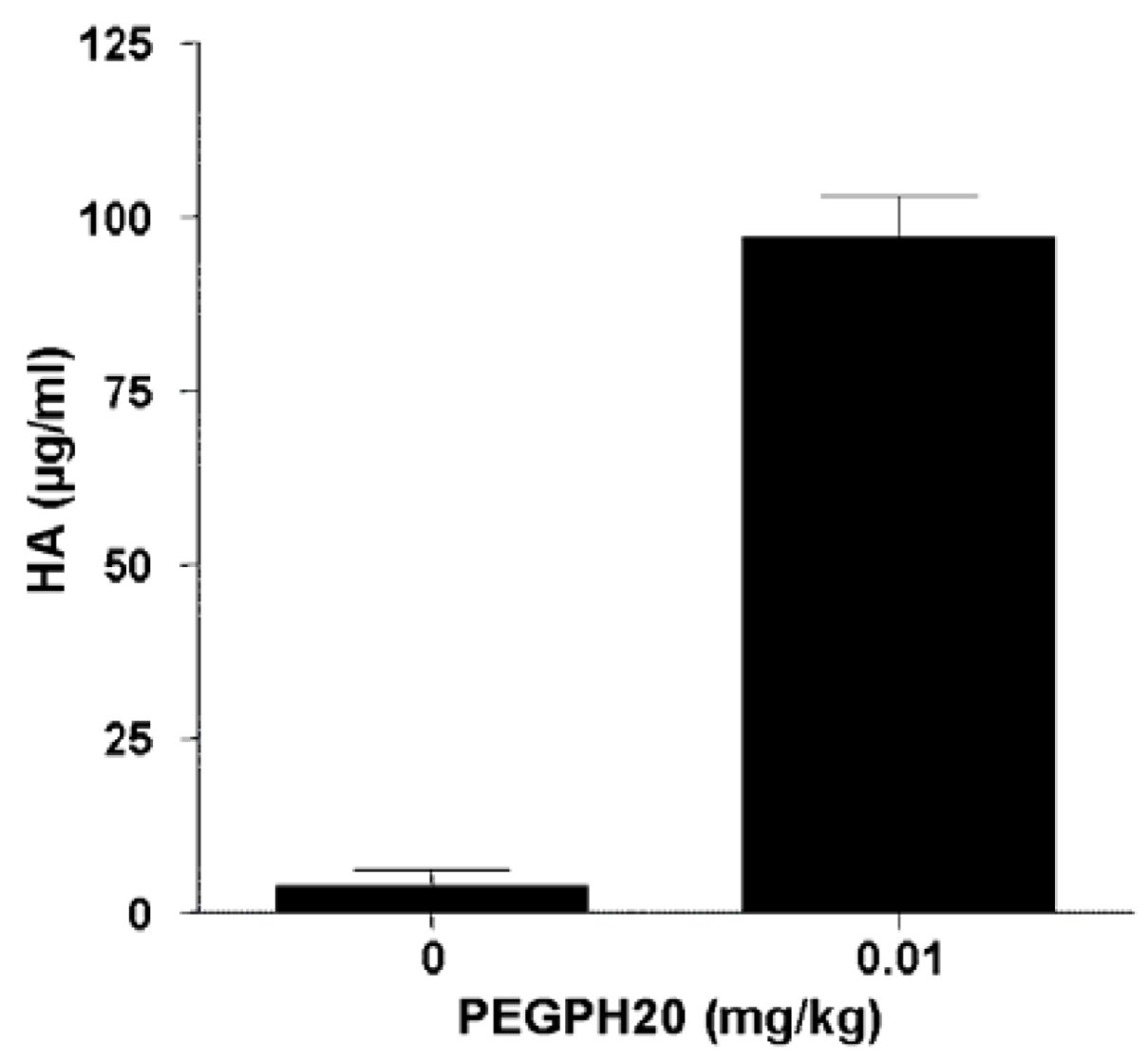
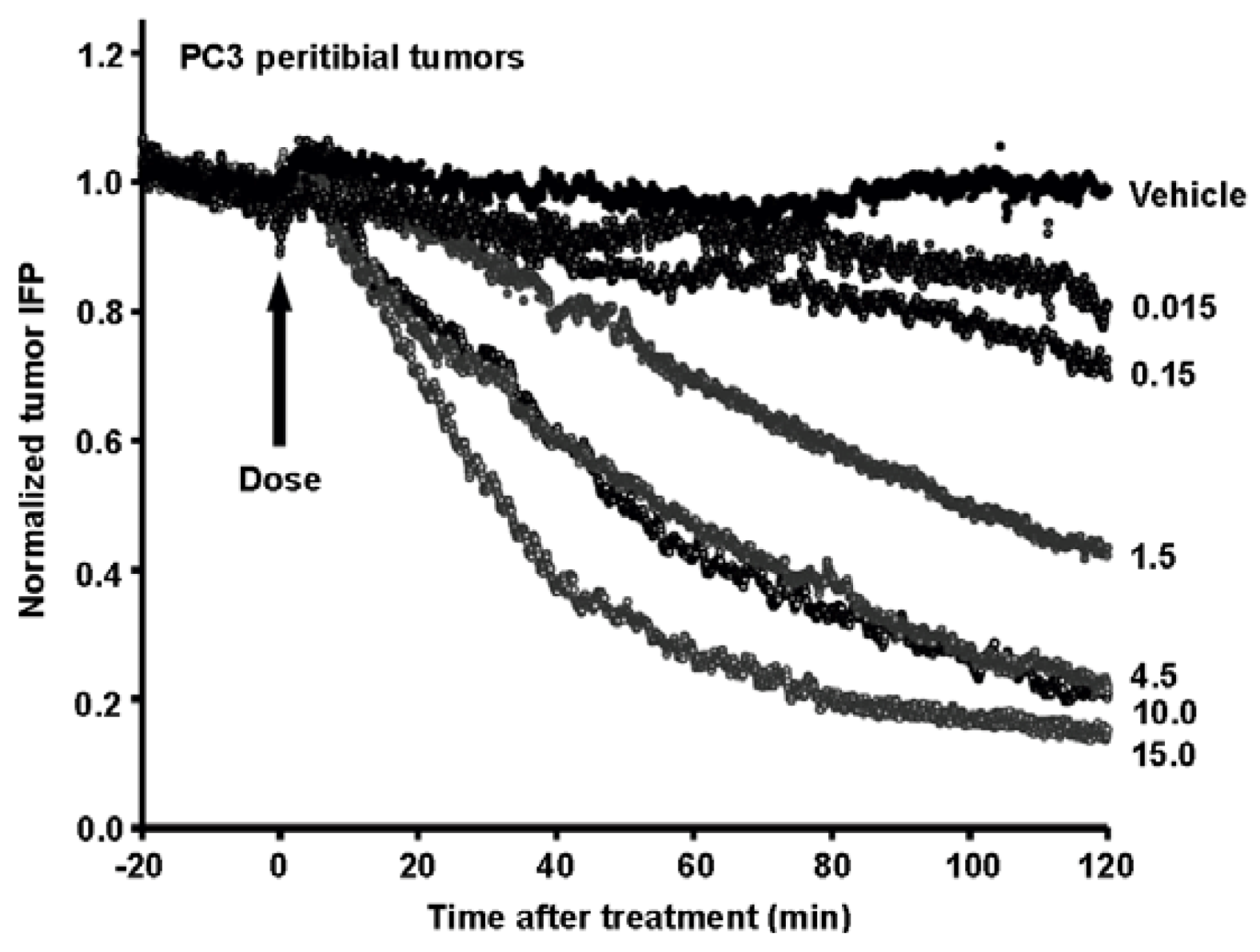
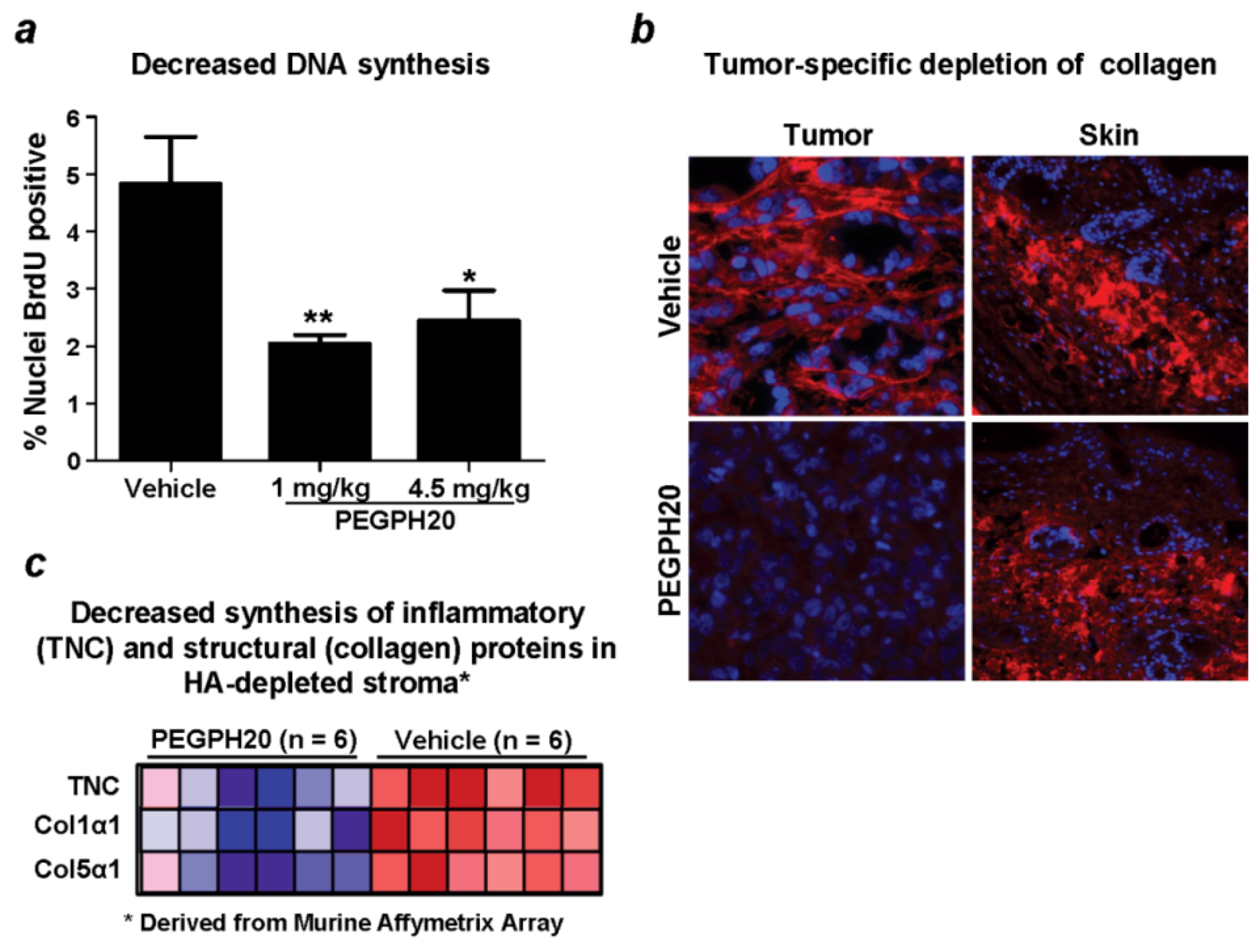
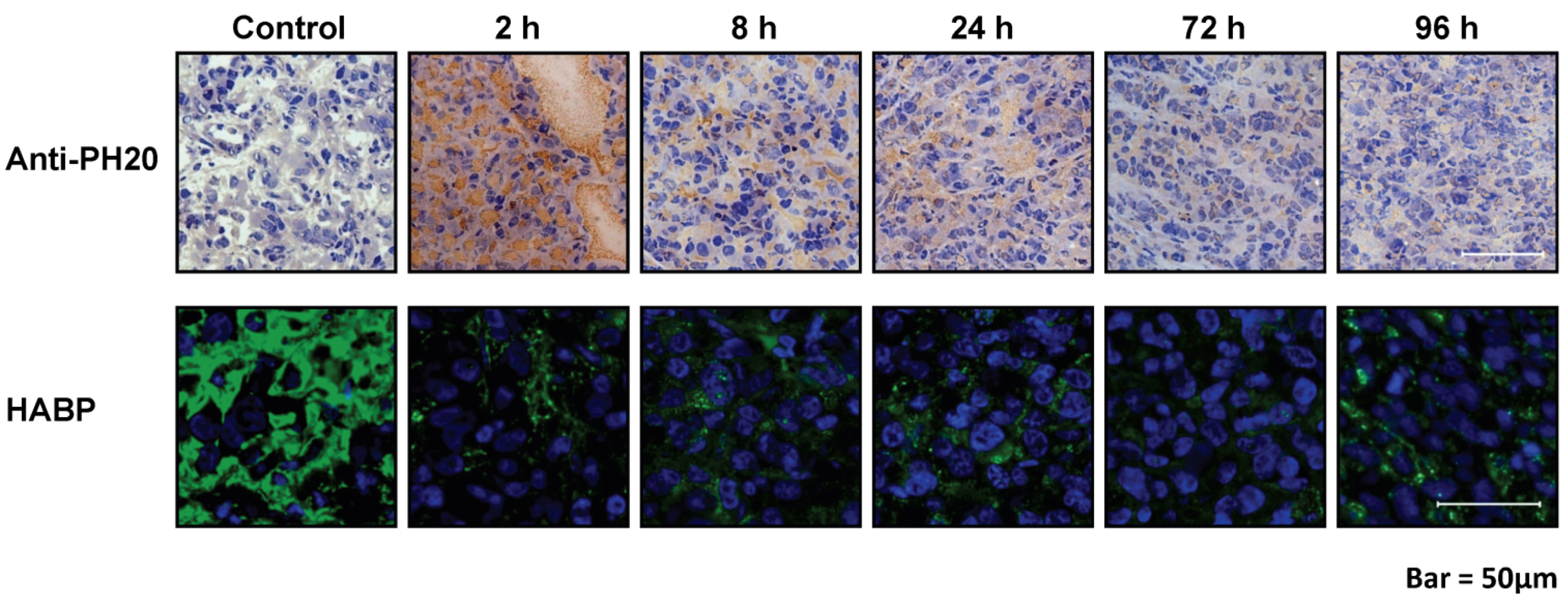
7. Enzymatic Targeting of Tumor HA


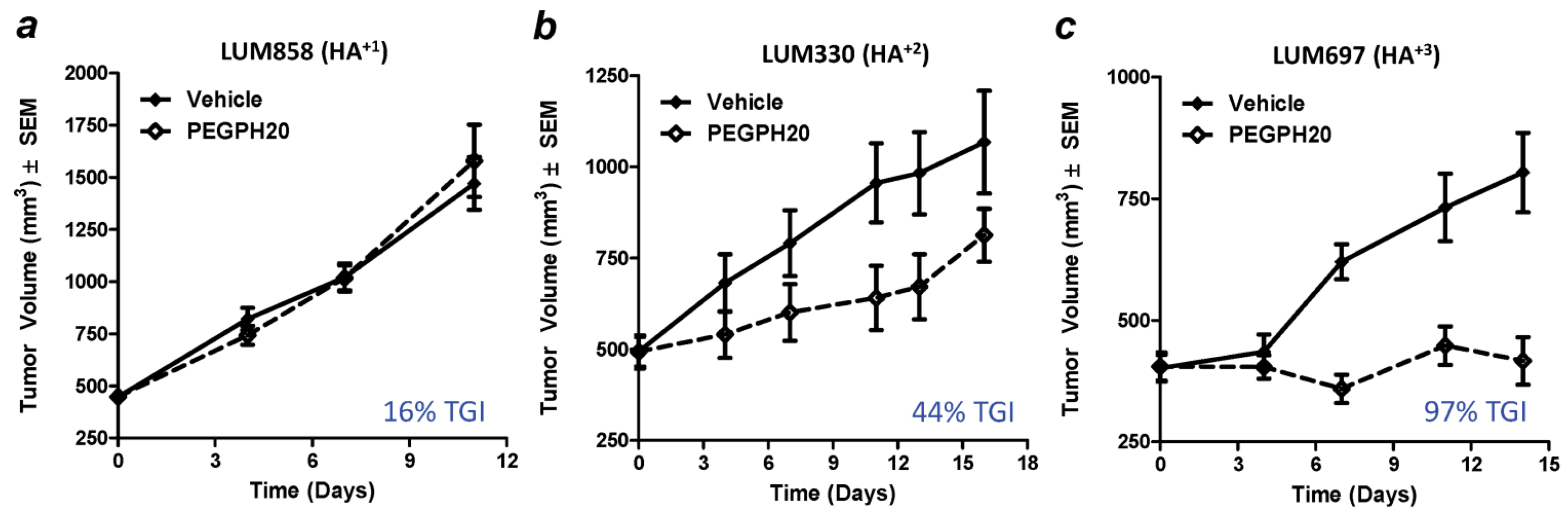
8. Combining Therapeutic Targeting of Tumor HA with Chemotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
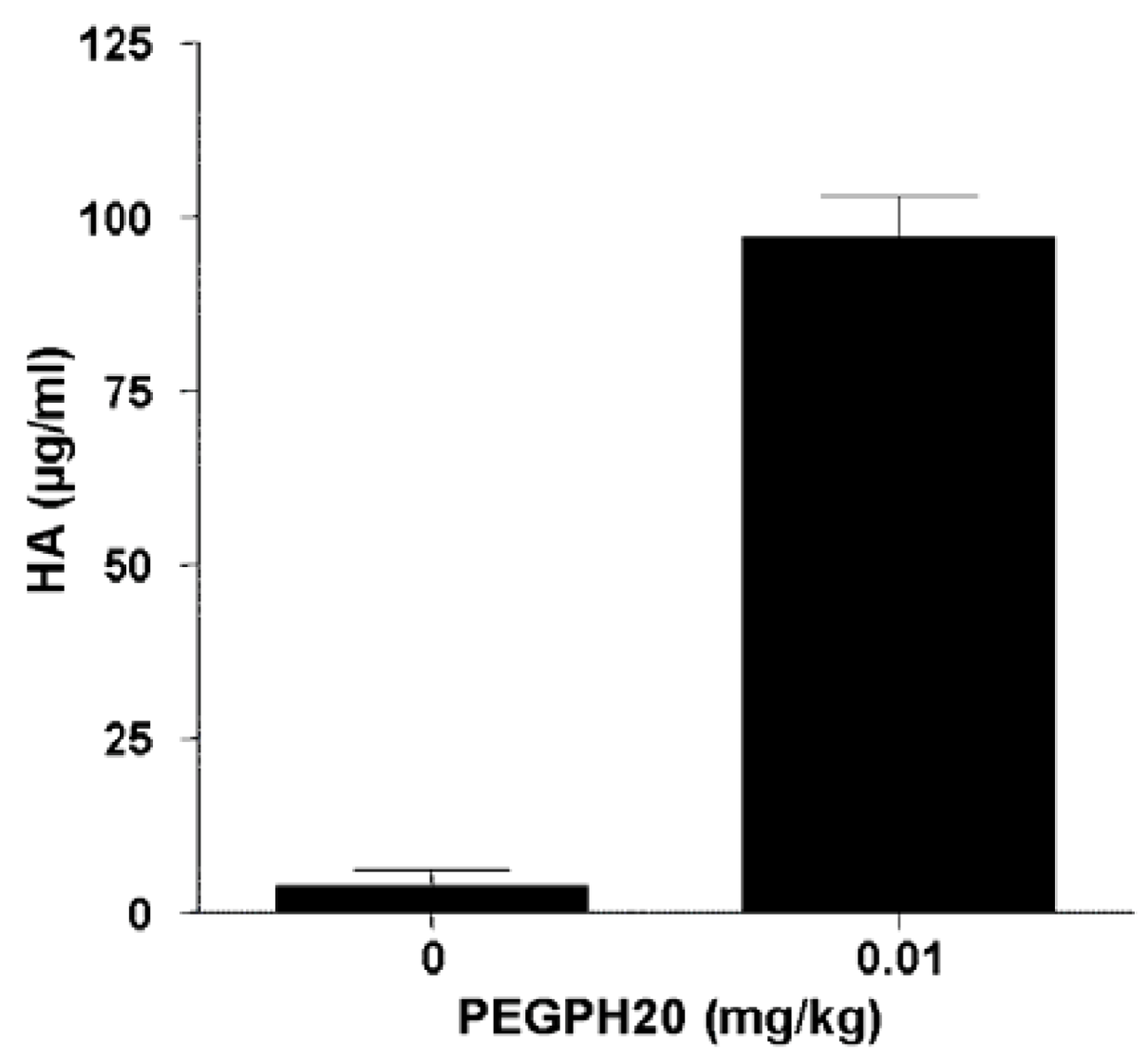{kind=link}
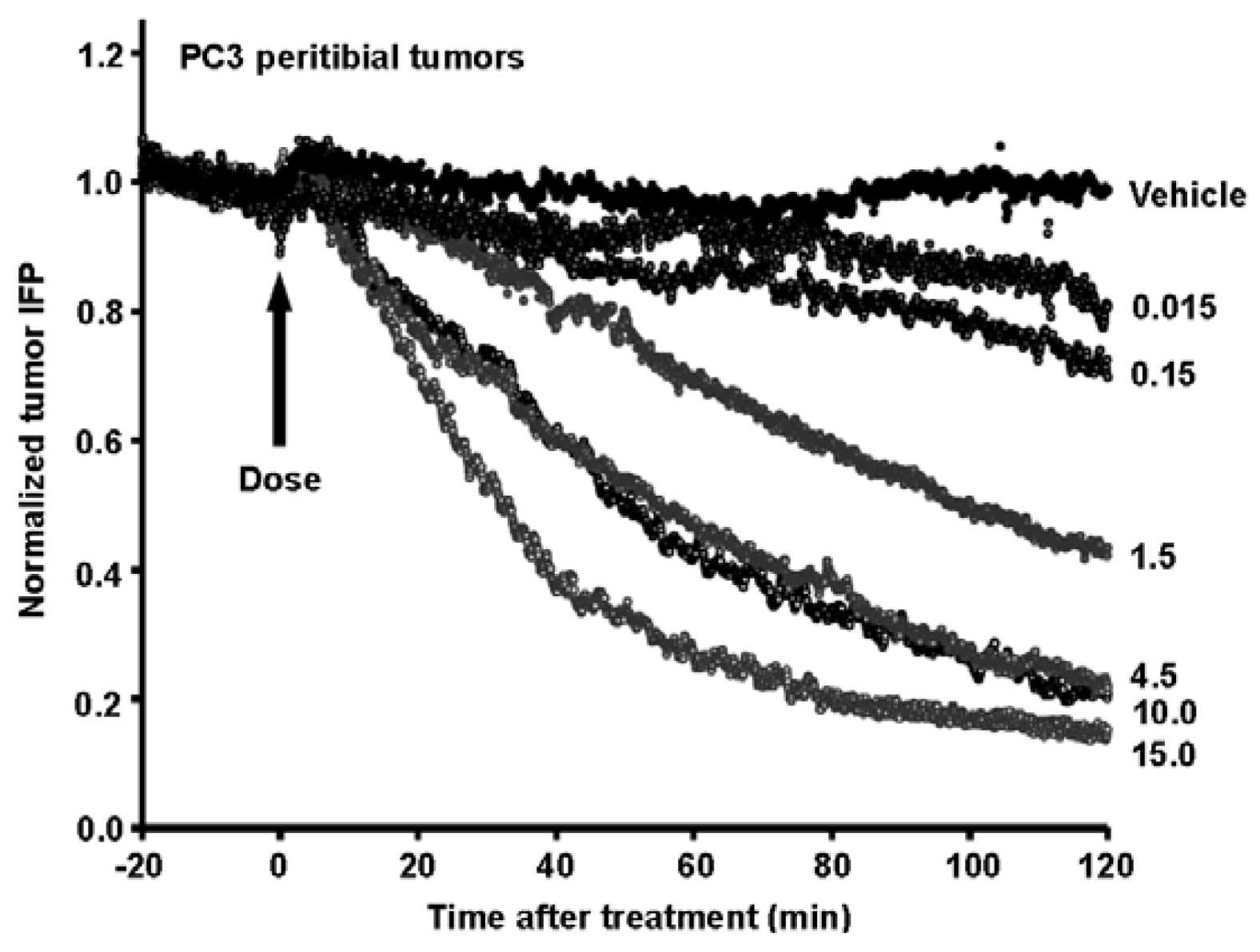{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment 1 | n | Median Survival (days) | Increase in Survival 2 (%) |
|---|---|---|---|
| Vehicle | 7 | 10.5 | 0 |
| Gemcitabine | 11 | 15.0 | 43 |
| PEGPH20 | 10 | 9.0 | 0 |
| Gemcitabine + PEGPH20 | 11 | 28.5 | 271 |
9. Endogenous Hyaluronidases and HA Oligosaccharides as Tumor Promotors or Suppressors
9.1. Endogenous Hyaluronidase Activity
9.2. HA Oligosaccharides in Cancer
10. Therapeutic Targeting of Tumor HA and Metastasis
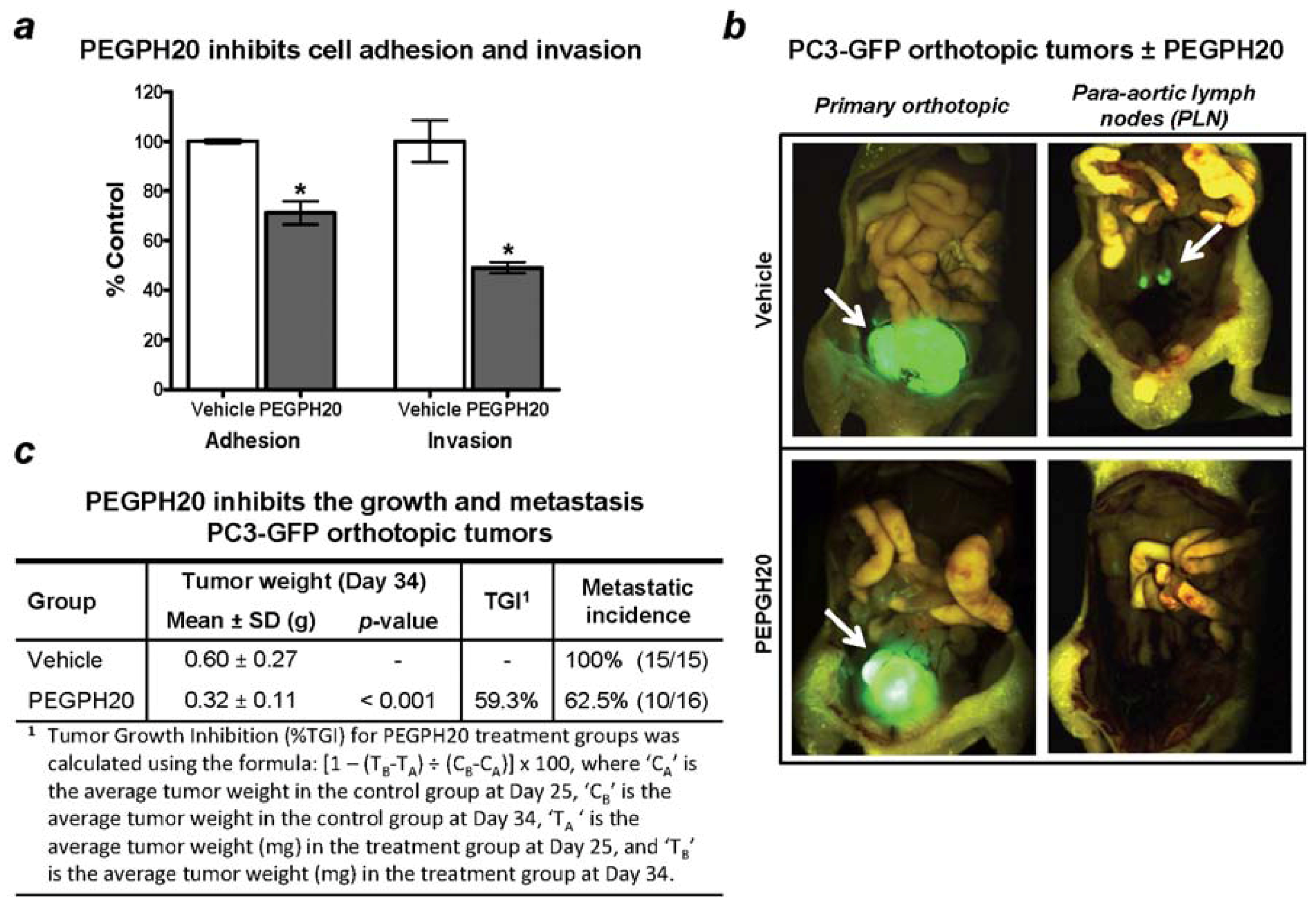
11. Conclusions
Acknowledgments
References
- Sund, M.; Kalluri, R. Tumor stroma derived biomarkers in cancer. Cancer Metastasis Rev. 2009, 28, 177–183. [Google Scholar] [CrossRef]
- Weinberg, R.A. Coevolution in the tumor microenvironment. Nat. Genet. 2008, 40, 494–495. [Google Scholar] [CrossRef]
- Shimoda, M.; Mellody, K.T.; Orimo, A. Carcinoma-associated fibroblasts are a rate-limiting determinant for tumour progression. Semin. Cell Dev. Biol. 2010, 21, 19–25. [Google Scholar]
- Johansson, A.; Ganss, R. Remodeling of tumor stroma and response to therapy. Cancers 2012, 4, 340–353. [Google Scholar] [CrossRef]
- Bissell, M.J.; Hines, W.C. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011, 17, 320–329. [Google Scholar] [CrossRef]
- Shepard, H.M.; Brdlik, C.M.; Schreiber, H. Signal integration: A framework for understanding the efficacy of therapeutics targeting the human EGFR family. J. Clin. Invest. 2008, 118, 3574–3581. [Google Scholar] [CrossRef]
- Ferrara, N. VEGF as a therapeutic target in cancer. Oncology 2005, 69, 11–16. [Google Scholar] [CrossRef]
- Kelleher, F.C. Hedgehog signaling and therapeutics in pancreatic cancer. Carcinogenesis 2011, 32, 445–451. [Google Scholar] [CrossRef]
- Liu, Q.; Sun, J.D.; Wang, J.; Ahluwalia, D.; Baker, A.F.; Cranmer, L.D.; Ferraro, D.; Wang, Y.; Duan, J.X.; Ammons, W.S.; et al. TH-302, a hypoxia-activated prodrug with broad in vivo preclinical combination therapy efficacy: Optimization of dosing regimens and schedules. Cancer Chemother. Pharmacol. 2012, 69, 1487–1498. [Google Scholar]
- Weiss, G.J.; Infante, J.R.; Chiorean, E.G.; Borad, M.J.; Bendell, J.C.; Molina, J.R.; Tibes, R.; Ramanathan, R.K.; Lewandowski, K.; Jones, S.F.; et al. Phase 1 study of the safety, tolerability, and pharmacokinetics of TH-302, a hypoxia-activated prodrug, in patients with advanced solid malignancies. Clin. Cancer Res. 2011, 17, 2997–3004. [Google Scholar] [CrossRef]
- Jacobetz, M.A.; Chan, D.S.; Neesse, A.; Bapiro, T.E.; Cook, N.; Frese, K.K.; Feig, C.; Nakagawa, T.; Caldwell, M.E.; Zecchini, H.I.; et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut 2012. [Google Scholar] [CrossRef]
- Provenzano, P.P.; Cuevas, C.; Chang, A.E.; Goel, V.K.; von Hoff, D.D.; Hingorani, S.R. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell 2012, 21, 418–429. [Google Scholar] [CrossRef]
- Jiang, P.; Li, X.; Thompson, C.B.; Huang, Z.; Araiza, F.; Osgood, R.; Wei, G.; Feldmann, M.; Frost, G.I.; Shepard, H.M. Effective targeting of the tumor microenvironment for cancer therapy. Anticancer Res. 2012, 32, 1203–1212. [Google Scholar]
- Thompson, C.B.; Shepard, H.M.; O’Connor, P.M.; Kadhim, S.; Jiang, P.; Osgood, R.J.; Bookbinder, L.H.; Li, X.; Sugarman, B.J.; Connor, R.J.; et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Mol. Cancer Ther. 2010, 9, 3052–3064. [Google Scholar] [CrossRef]
- Phase 1 Study of PEGPH20 With Initial Dexamethasone Premedication Given Intravenously to Patients With Advanced Solid Tumors (Sponsor: Halozyme Therapeutics). Available online: http://clinicaltrials.gov/ct2/show/NCT01170897?term=NCT01170897&rank=1/ (accessed on 7 August 2012).
- Phase 1B/2 Study of Gemcitabine + PEGPH20 versus Gemcitabine Alone in Stage IV Previously Untreated Pancreatic Cancer (Sponsor: Halozyme Therapeutics). Available online: http://clinicaltrials.gov/ct2/show/NCT01453153?term=NCT01453153&rank=1/ (accessed on 7 August 2012).
- Weigel, P.H.; DeAngelis, P.L. Hyaluronan synthases: A decade-plus of novel glycosyltransferases. J. Biol. Chem. 2007, 282, 36777–36781. [Google Scholar]
- Jokela, T.A.; Makkonen, K.M.; Oikari, S.; Kärnä, R.; Koli, E.; Hart, G.W.; Tammi, R.H.; Carlberg, C.; Tammi, M.I. Cellular content of UDP-N-acetylhexosamines controls hyaluronan synthase 2 expression and correlates with O-linked N-acetylglucosamine modification of transcription factors YY1 and SP1. J. Biol. Chem. 2011, 286, 33632–33640. [Google Scholar]
- Itano, N.; Sawai, T.; Yoshida, M.; Lenas, P.; Yamada, Y.; Imagawa, M.; Shinomura, T.; Hamaguchi, M.; Yoshida, Y.; Ohnuki, Y.; et al. Three isoforms of mammalian hyaluronan synthases have distinct enzymatic properties. J. Biol. Chem. 1999, 274, 25085–25092. [Google Scholar]
- Viola, M.; Vigetti, D.; Genasetti, A.; Rizzi, M.; Karousou, E.; Moretto, P.; Clerici, M.; Bartolini, B.; Pallotti, F.; de Luca, G.; et al. Molecular control of the hyaluronan biosynthesis. Connect. Tissue Res. 2008, 49, 111–114. [Google Scholar] [CrossRef]
- Li, L.; Asteriou, T.; Bernert, B.; Heldin, C.H.; Heldin, P. Growth factor regulation of hyaluronan synthesis and degradation in human dermal fibroblasts: Importance of hyaluronan for the mitogenic response of PDGF-BB. Biochem. J. 2007, 404, 327–336. [Google Scholar]
- Wilkinson, T.S.; Bressler, S.L.; Evanko, S.P.; Braun, K.R.; Wight, T.N. Overexpression of hyaluronan synthases alters vascular smooth muscle cell phenotype and promotes monocyte adhesion. J. Cell. Physiol. 2006, 206, 378–385. [Google Scholar] [CrossRef]
- Camenisch, T.D.; Spicer, A.P.; Brehm-Gibson, T.; Biesterfeldt, J.; Augustine, M.L.; Calabro, A., Jr.; Kubalak, S.; Klewer, S.E.; McDonald, J.A. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Invest. 2000, 106, 349–360. [Google Scholar]
- Tien, J.Y.; Spicer, A.P. Three vertebrate hyaluronan synthases are expressed during mouse development in distinct spatial and temporal patterns. Dev. Dyn. 2005, 233, 130–141. [Google Scholar] [CrossRef]
- Jokela, T. Regulation of hyaluronan synthesis by UDP-sugars. Ph.D. Thesis, University of Eastern Finland, Kuopio, Finland, 2011. [Google Scholar]
- Toole, B.P. Hyaluronan: From extracellular glue to pericellular cue. Nat. Rev. Cancer 2004, 4, 528–539. [Google Scholar] [CrossRef]
- Spicer, A.P.; Tien, J.Y. Hyaluronan and morphogenesis. Birth Defects Res. C Embryo Today 2004, 72, 89–108. [Google Scholar] [CrossRef]
- Spicer, A.P.; Tien, J.L.; Joo, A.; Bowling, R.A., Jr. Investigation of hyaluronan function in the mouse through targeted mutagenesis. Glycoconj. J. 2002, 19, 341–345. [Google Scholar] [CrossRef]
- Koyama, H.; Hibi, T.; Isogai, Z.; Yoneda, M.; Fujimori, M.; Amano, J.; Kawakubo, M.; Kannagi, R.; Kimata, K.; Taniguchi, S.; et al. Hyperproduction of hyaluronan in neu-induced mammary tumor accelerates angiogenesis through stromal cell recruitment: Possible involvement of versican/PG-M. Am. J. Pathol. 2007, 170, 1086–1099. [Google Scholar]
- Csoka, A.B.; Frost, G.I.; Stern, R. The six hyaluronidase-like genes in the human and mouse genomes. Matrix Biol. 2001, 20, 499–508. [Google Scholar] [CrossRef]
- Jadin, L.; Wu, X.; Ding, H.; Frost, G.I.; Onclinx, C.; Triggs-Raine, B.; Flamion, B. Skeletal and hematological anomalies in HYAL2-deficient mice: A second type of mucopolysaccharidosis IX? FASEB J. 2008, 22, 4316–4326. [Google Scholar] [CrossRef]
- Atmuri, V.; Martin, D.C.; Hemming, R.; Gutsol, A.; Byers, S.; Sahebjam, S.; Thliveris, J.A.; Mort, J.S.; Carmona, E.; Anderson, J.E.; et al. Hyaluronidase 3 (HYAL3) knockout mice do not display evidence of hyaluronan accumulation. Matrix Biol. 2008, 27, 653–660. [Google Scholar]
- Martin, D.C.; Atmuri, V.; Hemming, R.J.; Farley, J.; Mort, J.S.; Byers, S.; Hombach-Klonisch, S.; Csoka, A.B.; Stern, R.; Triggs-Raine, B.L. A mouse model of human mucopolysaccharidosis IX exhibits osteoarthritis. Hum. Mol. Genet. 2008, 17, 1904–1915. [Google Scholar] [CrossRef]
- Kimura, M.; Kim, E.; Kang, W.; Yamashita, M.; Saigo, M.; Yamazaki, T.; Nakanishi, T.; Kashiwabara, S.; Baba, T. Functional roles of mouse sperm hyaluronidases, HYAL5 and SPAM1, in fertilization. Biol. Reprod. 2009, 81, 939–947. [Google Scholar]
- Triggs-Raine, B.; Salo, T.J.; Zhang, H.; Wicklow, B.A.; Natowicz, M.R. Mutations in HYAL1, a member of a tandemly distributed multigene family encoding disparate hyaluronidase activities, cause a newly described lysosomal disorder, mucopolysaccharidosis IX. Proc. Natl. Acad. Sci. USA 1999, 96, 6296–6300. [Google Scholar]
- Baba, D.; Kashiwabara, S.; Honda, A.; Yamagata, K.; Wu, Q.; Ikawa, M.; Okabe, M.; Baba, T. Mouse sperm lacking cell surface hyaluronidase PH-20 can pass through the layer of cumulus cells and fertilize the egg. J. Biol. Chem. 2002, 277, 30310–30314. [Google Scholar]
- Wynne, C.; Harvey, V.; Schwabe, C.; Waaka, D.; McIntyre, C.; Bittner, B. Comparison of subcutaneous and intravenous administration of trastuzumab: A phase I/Ib trial in healthy male volunteers and patients With HER2-positive breast cancer. J. Clin. Pharmacol. 2012. [Google Scholar] [CrossRef]
- Frost, G.I. Recombinant human hyaluronidase (rHuPH20): An enabling platform for subcutaneous drug and fluid administration. Expert Opin. Drug Deliv. 2007, 4, 427–440. [Google Scholar] [CrossRef]
- Bookbinder, L.H.; Hofer, A.; Haller, M.F.; Zepeda, M.L.; Keller, G.A.; Lim, J.E.; Edgington, T.S.; Shepard, H.M.; Patton, J.S.; Frost, G.I. A recombinant human enzyme for enhanced interstitial transport of therapeutics. J. Control. Release 2006, 114, 230–241. [Google Scholar] [CrossRef]
- Dea, I.C.; Moorhouse, R.; Rees, D.A.; Arnott, S.; Guss, J.M.; Balazs, E.A. Hyaluronic acid: A novel, double helical molecule. Science 1973, 179, 560–562. [Google Scholar]
- Tammi, R.H.; Kultti, A.; Kosma, V.M.; Pirinen, R.; Auvinen, P.; Tammi, M.I. Hyaluronan in human tumors: Pathobiological and prognostic messages from cell-associated and stromal hyaluronan. Semin. Cancer Biol. 2008, 18, 288–295. [Google Scholar] [CrossRef]
- Simpson, M.A.; Lokeshwar, V.B. Hyaluronan and hyaluronidase in genitourinary tumors. Front. Biosci. 2008, 13, 5664–5680. [Google Scholar]
- Itano, N.; Kimata, K. Altered hyaluronan biosynthesis in cancer progression. Semin. Cancer Biol. 2008, 18, 268–274. [Google Scholar] [CrossRef]
- Erickson, M.; Stern, R. Chain gangs: New aspects of hyaluronan metabolism. Biochem. Res. Int. 2012, 2012, 893947:1–893947:9. [Google Scholar]
- Meyer, F.A. Macromolecular basis of globular protein exclusion and of swelling pressure in loose connective tissue (umbilical cord). Biochim. Biophys. Acta 1983, 755, 388–399. [Google Scholar] [CrossRef]
- Jain, R.K. Delivery of molecular medicine to solid tumors: Lessons from in vivo imaging of gene expression and function. J. Control. Release 2001, 74, 7–25. [Google Scholar] [CrossRef]
- Jacobson, A.; Salnikov, A.; Lammerts, E.; Roswall, P.; Sundberg, C.; Heldin, P.; Rubin, K.; Heldin, N.E. Hyaluronan content in experimental carcinoma is not correlated to interstitial fluid pressure. Biochem. Biophys. Res. Commun. 2003, 305, 1017–1023. [Google Scholar]
- Tse, J.M.; Cheng, G.; Tyrrell, J.A.; Wilcox-Adelman, S.A.; Boucher, Y.; Jain, R.K.; Munn, L.L. Mechanical compression drives cancer cells toward invasive phenotype. Proc. Natl. Acad. Sci. USA 2012, 109, 911–916. [Google Scholar]
- Wang, S.; Basson, M.D. Akt directly regulates focal adhesion kinase through association and serine phosphorylation: Implication for pressure-induced colon cancer metastasis. Am. J. Physiol. Cell Physiol. 2011, 300, C657–C670. [Google Scholar] [CrossRef]
- Toole, B.P. Hyaluronan and its binding proteins, the hyaladherins. Curr. Opin. Cell Biol. 1990, 2, 839–844. [Google Scholar] [CrossRef]
- Day, A.J. The structure and regulation of hyaluronan-binding proteins. Biochem. Soc. Trans. 1999, 27, 115–121. [Google Scholar]
- Toole, B.P. Hyaluronan is not just a goo! J. Clin. Invest. 2000, 106, 335–336. [Google Scholar] [CrossRef]
- Varga, I.; Hutóczki, G.; Szemcsák, C.D.; Zahuczky, G.; Tóth, J.; Adamecz, Z.; Kenyeres, A.; Bognár, L.; Hanzély, Z.; Klekner, A. Brevican, neurocan, tenascin-C and versican are mainly responsible for the invasiveness of low-grade astrocytoma. Pathol. Oncol. Res. 2012, 18, 413–420. [Google Scholar] [CrossRef]
- Kodama, J.; Hasengaowa; Kusumoto, T.; Seki, N.; Matsuo, T.; Nakamura, K.; Hongo, A.; Hiramatsu, Y. Versican expression in human cervical cancer. Eur. J. Cancer 2007, 43, 1460–1466. [Google Scholar]
- Pukkila, M.; Kosunen, A.; Ropponen, K.; Virtaniemi, J.; Kellokoski, J.; Kumpulainen, E.; Pirinen, R.; Nuutinen, J.; Johansson, R.; Kosma, V.M. High stromal versican expression predicts unfavourable outcome in oral squamous cell carcinoma. J. Clin. Pathol. 2007, 60, 267–272. [Google Scholar]
- Said, N.; Sanchez-Carbayo, M.; Smith, S.C.; Theodorescu, D. RhoGDI2 suppresses lung metastasis in mice by reducing tumor versican expression and macrophage infiltration. J. Clin. Invest. 2012, 122, 1503–1518. [Google Scholar] [CrossRef]
- Wu, Y.J.; La Pierre, D.P.; Wu, J.; Yee, A.J.; Yang, B.B. The interaction of versican with its binding partners. Cell Res. 2005, 15, 483–494. [Google Scholar] [CrossRef]
- Aspberg, A.; Miura, R.; Bourdoulous, S.; Shimonaka, M.; Heinegârd, D.; Schachner, M.; Ruoslahti, E.; Yamaguchi, Y. The C-type lectin domains of lecticans, a family of aggregating chondroitin sulfate proteoglycans, bind tenascin-R by protein-protein interactions independent of carbohydrate moiety. Proc. Natl. Acad. Sci. USA 1997, 94, 10116–10121. [Google Scholar]
- Aspberg, A.; Adam, S.; Kostka, G.; Timpl, R.; Heinegård, D. Fibulin-1 is a ligand for the C-type lectin domains of aggrecan and versican. J. Biol. Chem. 1999, 274, 20444–20449. [Google Scholar]
- Olin, A.I.; Mörgelin, M.; Sasaki, T.; Timpl, R.; Heinegård, D.; Aspberg, A. The proteoglycans aggrecan and Versican form networks with fibulin-2 through their lectin domain binding. J. Biol. Chem. 2001, 276, 1253–1261. [Google Scholar]
- Yamagata, M.; Yamada, K.M.; Yoneda, M.; Suzuki, S.; Kimata, K. Chondroitin sulfate proteoglycan (PG-M-like proteoglycan) is involved in the binding of hyaluronic acid to cellular fibronectin. J. Biol. Chem. 1986, 261, 13526–13535. [Google Scholar]
- Isogai, Z.; Aspberg, A.; Keene, D.R.; Ono, R.N.; Reinhardt, D.P.; Sakai, L.Y. Versican interacts with fibrillin-1 and links extracellular microfibrils to other connective tissue networks. J. Biol. Chem. 2002, 277, 4565–4572. [Google Scholar]
- Mjaatvedt, C.H.; Yamamura, H.; Capehart, A.A.; Turner, D.; Markwald, R.R. The Cspg2 gene, disrupted in the hdf mutant, is required for right cardiac chamber and endocardial cushion formation. Dev. Biol. 1998, 202, 56–66. [Google Scholar] [CrossRef]
- Huang, L.; Yoneda, M.; Kimata, K. A serum-derived hyaluronan-associated protein (SHAP) is the heavy chain of the inter alpha-trypsin inhibitor. J. Biol. Chem. 1993, 268, 26725–26730. [Google Scholar]
- Milner, C.M.; Tongsoongnoen, W.; Rugg, M.S.; Day, A.J. The molecular basis of inter-alpha-inhibitor heavy chain transfer on to hyaluronan. Biochem. Soc. Trans. 2007, 35, 672–676. [Google Scholar]
- Yabushita, H.; Iwasaki, K.; Kanyama, K.; Obayashi, Y.; Zhuo, L.; Itano, N.; Kimata, K.; Wakatsuki, A. Clinicopathological role of serum-derived hyaluronan-associated protein (SHAP)-hyaluronan complex in endometrial cancer. Obstet. Gynecol. Int. 2011, 2011, 739150:1–739150:10. [Google Scholar]
- Salustri, A.; Garlanda, C.; Hirsch, E.; de Acetis, M.; Maccagno, A.; Bottazzi, B.; Doni, A.; Bastone, A.; Mantovani, G.; Beck Peccoz, P.; et al. PTX3 plays a key role in the organization of the cumulus oophorus extracellular matrix and in in vivo fertilization. Development 2004, 131, 1577–1586. [Google Scholar]
- Kuznetsova, S.A.; Day, A.J.; Mahoney, D.J.; Rugg, M.S.; Mosher, D.F.; Roberts, D.D. The N-terminal module of thrombospondin-1 interacts with the link domain of TSG-6 and enhances its covalent association with the heavy chains of inter-alpha-trypsin inhibitor. J. Biol. Chem. 2005, 280, 30899–30908. [Google Scholar]
- Kultti, A.; Rilla, K.; Tiihonen, R.; Spicer, A.P.; Tammi, R.H.; Tammi, M.I. Hyaluronan synthesis induces microvillus-like cell surface protrusions. J. Biol. Chem. 2006, 281, 15821–15828. [Google Scholar]
- Rilla, K.; Tiihonen, R.; Kultti, A.; Tammi, M.; Tammi, R. Pericellular hyaluronan coat visualized in live cells with a fluorescent probe is scaffolded by plasma membrane protrusions. J. Histochem. Cytochem. 2008, 56, 901–910. [Google Scholar] [CrossRef]
- Misra, S.; Toole, B.P.; Ghatak, S. Hyaluronan constitutively regulates activation of multiple receptor tyrosine kinases in epithelial and carcinoma cells. J. Biol. Chem. 2006, 281, 34936–34941. [Google Scholar]
- Toole, B.P. Hyaluronan-CD44 interactions in cancer: Paradoxes and possibilities. Clin. Cancer Res. 2009, 15, 7462–7468. [Google Scholar] [CrossRef]
- Bennett, K.L.; Modrell, B.; Greenfield, B.; Bartolazzi, A.; Stamenkovic, I.; Peach, R.; Jackson, D.G.; Spring, F.; Aruffo, A. Regulation of CD44 binding to hyaluronan by glycosylation of variably spliced exons. J. Cell Biol. 1995, 131, 1623–1633. [Google Scholar] [CrossRef]
- Thankamony, S.P.; Knudson, W. Acylation of CD44 and its association with lipid rafts are required for receptor and hyaluronan endocytosis. J. Biol. Chem. 2006, 281, 34601–34609. [Google Scholar] [CrossRef]
- Bourguignon, L.Y. Hyaluronan-mediated CD44 activation of RhoGTPase signaling and cytoskeleton function promotes tumor progression. Semin. Cancer Biol. 2008, 18, 251–259. [Google Scholar] [CrossRef]
- Bourguignon, L.Y. Hyaluronan-Mediated CD44 Interaction with Receptor and Non-Receptor Kinases Promotes Oncogenic Signaling, Cytoskeleton Activation and Tumor Progression. In Hyaluronan in Cancer Biology, 1st; Stern, R., Ed.; Academic Press/Elsevier: San Diego, CA, USA, 2009; pp. 89–107. [Google Scholar]
- Toole, B.P.; Slomiany, M.G. Hyaluronan, CD44 and Emmprin: Partners in cancer cell chemoresistance. Drug Resist. Updat. 2008, 11, 110–121. [Google Scholar] [CrossRef]
- Bourguignon, L.Y.; Lokeshwar, V.B.; He, J.; Chen, X.; Bourguignon, G.J. A CD44-like endothelial cell transmembrane glycoprotein (GP116) interacts with extracellular matrix and ankyrin. Mol. Cell. Biol. 1992, 12, 4464–4471. [Google Scholar]
- Tsukita, S.; Oishi, K.; Sato, N.; Sagara, J.; Kawai, A.; Tsukita, S. ERM family members as molecular linkers between the cell surface glycoprotein CD44 and actin-based cytoskeletons. J. Cell Biol. 1994, 126, 391–401. [Google Scholar] [CrossRef]
- Maxwell, C.A.; McCarthy, J.; Turley, E. Cell-surface and mitotic-spindle RHAMM: moonlighting or dual oncogenic functions? J. Cell Sci. 2008, 121, 925–932. [Google Scholar] [CrossRef]
- Hall, C.L.; Wang, C.; Lange, L.A.; Turley, E.A. Hyaluronan and the hyaluronan receptor RHAMM promote focal adhesion turnover and transient tyrosine kinase activity. J. Cell Biol. 1994, 126, 575–588. [Google Scholar]
- Hall, C.L.; Wang, F.S.; Turley, E. Src−/− fibroblasts are defective in their ability to disassemble focal adhesions in response to phorbol ester/hyaluronan treatment. Cell Commun. Adhes. 2002, 9, 273–283. [Google Scholar]
- Itano, N.; Sawai, T.; Atsumi, F.; Miyaishi, O.; Taniguchi, S.; Kannagi, R.; Hamaguchi, M.; Kimata, K. Selective expression and functional characteristics of three mammalian hyaluronan synthases in oncogenic malignant transformation. J. Biol. Chem. 2004, 279, 18679–18687. [Google Scholar]
- Maxwell, C.A.; Keats, J.J.; Crainie, M.; Sun, X.; Yen, T.; Shibuya, E.; Hendzel, M.; Chan, G.; Pilarski, L.M. RHAMM is a centrosomal protein that interacts with dynein and maintains spindle pole stability. Mol. Biol. Cell 2003, 14, 2262–2276. [Google Scholar]
- Tolg, C.; Hamilton, S.R.; Morningstar, L.; Zhang, J.; Zhang, S.; Esguerra, K.V.; Telmer, P.G.; Luyt, L.G.; Harrison, R.; McCarthy, J.B.; et al. RHAMM promotes interphase microtubule instability and mitotic spindle integrity through MEK1/ERK1/2 activity. J. Biol. Chem. 2010, 285, 26461–26474. [Google Scholar]
- Joukov, V.; Groen, A.C.; Prokhorova, T.; Gerson, R.; White, E.; Rodriguez, A.; Walter, J.C.; Livingston, D.M. The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell 2006, 127, 539–552. [Google Scholar] [CrossRef]
- Maxwell, C.A.; Benítez, J.; Gómez-Baldó, L.; Osorio, A.; Bonifaci, N.; Fernández-Ramires, R.; Costes, S.V.; Guinó, E.; Chen, H.; Evans, G.J.; et al. Interplay between BRCA1 and RHAMM regulates epithelial apicobasal polarization and may influence risk of breast cancer. PLoS Biol. 2011, 9, e1001199. [Google Scholar]
- Pujana, M.A.; Han, J.D.; Starita, L.M.; Stevens, K.N.; Tewari, M.; Ahn, J.S.; Rennert, G.; Moreno, V.; Kirchhoff, T.; Gold, B.; et al. Network modeling links breast cancer susceptibility and centrosome dysfunction. Nat. Genet. 2007, 39, 1338–1349. [Google Scholar]
- Hamilton, S.R.; Fard, S.F.; Paiwand, F.F.; Tolg, C.; Veiseh, M.; Wang, C.; McCarthy, J.B.; Bissell, M.J.; Koropatnick, J.; Turley, E.A. The hyaluronan receptors CD44 and Rhamm (CD168) form complexes with ERK1,2 that sustain high basal motility in breast cancer cells. J. Biol. Chem. 2007, 282, 16667–16680. [Google Scholar]
- Jiang, D.; Liang, J.; Fan, J.; Yu, S.; Chen, S.; Luo, Y.; Prestwich, G.D.; Mascarenhas, M.M.; Garg, H.G.; Quinn, D.A.; et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nat. Med. 2005, 11, 1173–1179. [Google Scholar]
- Jiang, D.; Liang, J.; Noble, P.W. Hyaluronan as an immune regulator in human diseases. Physiol. Rev. 2011, 91, 221–264. [Google Scholar] [CrossRef]
- Termeer, C.; Benedix, F.; Sleeman, J.; Fieber, C.; Voith, U.; Ahrens, T.; Miyake, K.; Freudenberg, M.; Galanos, C.; Simon, J.C. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J. Exp. Med. 2002, 195, 99–111. [Google Scholar] [CrossRef]
- Kouvidi, K.; Berdiaki, A.; Nikitovic, D.; Katonis, P.; Afratis, N.; Hascall, V.C.; Karamanos, N.K.; Tzanakakis, G.N. Role of receptor for hyaluronic acid-mediated motility (RHAMM) in low molecular weight hyaluronan (LMWHA)-mediated fibrosarcoma cell adhesion. J. Biol. Chem. 2011, 286, 38509–38520. [Google Scholar]
- Gao, F.; Yang, C.X.; Mo, W.; Liu, Y.W.; He, Y.Q. Hyaluronan oligosaccharides are potential stimulators to angiogenesis via RHAMM mediated signal pathway in wound healing. Clin. Invest. Med. 2008, 31, E106–E116. [Google Scholar]
- Lokeshwar, V.B.; Selzer, M.G. Differences in hyaluronic acid-mediated functions and signaling in arterial, microvessel, and vein-derived human endothelial cells. J. Biol. Chem. 2000, 275, 27641–27649. [Google Scholar]
- Wang, W.; Xu, G.L.; Jia, W.D.; Ma, J.L.; Li, J.S.; Ge, Y.S.; Ren, W.H.; Yu, J.H.; Liu, W.B. Ligation of TLR2 by versican: A link between inflammation and metastasis. Arch. Med. Res. 2009, 40, 321–323. [Google Scholar] [CrossRef]
- Critchley, D.R. Cytoskeletal proteins talin and vinculin in integrin-mediated adhesion. Biochem. Soc. Trans. 2004, 32, 831–836. [Google Scholar] [CrossRef]
- Hirose, Y.; Saijou, E.; Sugano, Y.; Takeshita, F.; Nishimura, S.; Nonaka, H.; Chen, Y.R.; Sekine, K.; Kido, T.; Nakamura, T.; et al. Inhibition of Stabilin-2 elevates circulating hyaluronic acid levels and prevents tumor metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 4263–4268. [Google Scholar]
- Chen, Y.B.; Jiang, C.T.; Zhang, G.Q.; Wang, J.S.; Pang, D. Increased expression of hyaluronic acid binding protein 1 is correlated with poor prognosis in patients with breast cancer. J. Surg. Oncol. 2009, 100, 382–386. [Google Scholar] [CrossRef]
- Kim, K.B.; Yi, J.S.; Nguyen, N.; Lee, J.H.; Kwon, Y.C.; Ahn, B.Y.; Cho, H.; Kim, Y.K.; Yoo, H.J.; Lee, J.S.; et al. Cell-surface receptor for complement component C1q (gC1qR) is a key regulator for lamellipodia formation and cancer metastasis. J. Biol. Chem. 2011, 286, 23093–23101. [Google Scholar]
- Truedsson, E. A case of mesothelioma of the pleura and peritoneum producing hyaluronic acid. Acta Soc. Med. Ups. 1951, 56, 39–45. [Google Scholar]
- Jensen, C.E. Hyaluronic acid. IV. Isolation of hyaluronic acid from pseudomucinous ovarian cysts. Acta Pharmacol. Toxicol. (Copenh.) 1954, 10, 83–88. [Google Scholar]
- Pillwein, K.; Fuiko, R.; Slavc, I.; Czech, T.; Hawliczek, G.; Bernhardt, G.; Nirnberger, G.; Köller, U. Hyaluronidase additional to standard chemotherapy improves outcome for children with malignant brain tumors. Cancer Lett. 1998, 131, 101–108. [Google Scholar] [CrossRef]
- Baumgartner, G.; Gomar-Höss, C.; Sakr, L.; Ulsperger, E.; Wogritsch, C. The impact of extracellular matrix on the chemoresistance of solid tumors—Experimental and clinical results of hyaluronidase as additive to cytostatic chemotherapy. Cancer Lett. 1998, 131, 85–99. [Google Scholar] [CrossRef]
- Klocker, J.; Sabitzer, H.; Raunik, W.; Wieser, S.; Schumer, J. Hyaluronidase as additive to induction chemotherapy in advanced squamous cell carcinoma of the head and neck. Cancer Lett. 1998, 131, 113–115. [Google Scholar] [CrossRef]
- Spruss, T.; Bernhardt, G.; Schönenberger, H.; Schiess, W. Hyaluronidase significantly enhances the efficacy of regional vinblastine chemotherapy of malignant melanoma. J. Cancer Res. Clin. Oncol. 1995, 121, 193–202. [Google Scholar] [CrossRef]
- Brekken, C.; de Lange Davies, C. Hyaluronidase reduces the interstitial fluid pressure in solid tumours in a non-linear concentration-dependent manner. Cancer Lett. 1998, 131, 65–70. [Google Scholar] [CrossRef]
- Eikenes, L.; Tari, M.; Tufto, I.; Bruland, O.S.; de Lange Davies, C. Hyaluronidase induces a transcapillary pressure gradient and improves the distribution and uptake of liposomal doxorubicin (Caelyx) in human osteosarcoma xenografts. Br. J. Cancer 2005, 93, 81–88. [Google Scholar] [CrossRef]
- Kosaki, R.; Watanabe, K.; Yamaguchi, Y. Overproduction of hyaluronan by expression of the hyaluronan synthase Has2 enhances anchorage-independent growth and tumorigenicity. Cancer Res. 1999, 59, 1141–1145. [Google Scholar]
- Koyama, H.; Kobayashi, N.; Harada, M.; Takeoka, M.; Kawai, Y.; Sano, K.; Fujimori, M.; Amano, J.; Ohhashi, T.; Kannagi, R.; et al. Significance of tumor-associated stroma in promotion of intratumoral lymphangiogenesis: Pivotal role of a hyaluronan-rich tumor microenvironment. Am. J. Pathol. 2008, 172, 179–193. [Google Scholar] [CrossRef]
- Shuster, S.; Frost, G.I.; Csoka, A.B.; Formby, B.; Stern, R. Hyaluronidase reduces human breast cancer xenografts in SCID mice. Int. J. Cancer 2002, 102, 192–197. [Google Scholar] [CrossRef]
- Simpson, M.A.; Wilson, C.M.; McCarthy, J.B. Inhibition of prostate tumor cell hyaluronan synthesis impairs subcutaneous growth and vascularization in immunocompromised mice. Am. J. Pathol. 2002, 161, 849–857. [Google Scholar]
- Kim, H.R.; Wheeler, M.A.; Wilson, C.M.; Iida, J.; Eng, D.; Simpson, M.A.; McCarthy, J.B.; Bullard, K.M. Hyaluronan facilitates invasion of colon carcinoma cells in vitro via interaction with CD44. Cancer Res. 2004, 64, 4569–4576. [Google Scholar]
- Nishida, Y.; Knudson, W.; Knudson, C.B.; Ishiguro, N. Antisense inhibition of hyaluronan synthase-2 in human osteosarcoma cells inhibits hyaluronan retention and tumorigenicity. Exp. Cell Res. 2005, 307, 194–203. [Google Scholar] [CrossRef]
- Udabage, L.; Brownlee, G.R.; Nilsson, S.K.; Brown, T.J. The over-expression of HAS2, Hyal-2 and CD44 is implicated in the invasiveness of breast cancer. Exp. Cell Res. 2005, 310, 205–217. [Google Scholar] [CrossRef]
- Kudo, D.; Kon, A.; Yoshihara, S.; Kakizaki, I.; Sasaki, M.; Endo, M.; Takagaki, K. Effect of a hyaluronan synthase suppressor, 4-methylumbelliferone, on B16F-10 melanoma cell adhesion and locomotion. Biochem. Biophys. Res. Commun. 2004, 321, 783–787. [Google Scholar]
- Urakawa, H.; Nishida, Y.; Wasa, J.; Arai, E.; Zhuo, L.; Kimata, K.; Kozawa, E.; Futamura, N.; Ishiguro, N. Inhibition of hyaluronan synthesis in breast cancer cells by 4-methylumbelliferone suppresses tumorigenicity in vitro and metastatic lesions of bone in vivo. Int. J. Cancer 2012, 130, 454–466. [Google Scholar] [CrossRef]
- Kultti, A.; Pasonen-Seppänen, S.; Jauhiainen, M.; Rilla, K.J.; Kärnä, R.; Pyöriä, E.; Tammi, R.H.; Tammi, M.I. 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Exp. Cell Res. 2009, 315, 1914–1923. [Google Scholar] [CrossRef]
- Yoshihara, S.; Kon, A.; Kudo, D.; Nakazawa, H.; Kakizaki, I.; Sasaki, M.; Endo, M.; Takagaki, K. A hyaluronan synthase suppressor, 4-methylumbelliferone, inhibits liver metastasis of melanoma cells. FEBS Lett. 2005, 579, 2722–2726. [Google Scholar]
- Auvinen, P.; Tammi, R.; Parkkinen, J.; Tammi, M.; Agren, U.; Johansson, R.; Hirvikoski, P.; Eskelinen, M.; Kosma, V.M. Hyaluronan in peritumoral stroma and malignant cells associates with breast cancer spreading and predicts survival. Am. J. Pathol. 2000, 156, 529–536. [Google Scholar] [CrossRef]
- Setälä, L.P.; Tammi, M.I.; Tammi, R.H.; Eskelinen, M.J.; Lipponen, P.K.; Agren, U.M.; Parkkinen, J.; Alhava, E.M.; Kosma, V.M. Hyaluronan expression in gastric cancer cells is associated with local and nodal spread and reduced survival rate. Br. J. Cancer 1999, 79, 1133–1138. [Google Scholar]
- Ropponen, K.; Tammi, M.; Parkkinen, J.; Eskelinen, M.; Tammi, R.; Lipponen, P.; Agren, U.; Alhava, E.; Kosma, V.M. Tumor cell-associated hyaluronan as an unfavorable prognostic factor in colorectal cancer. Cancer Res. 1998, 58, 342–347. [Google Scholar]
- Anttila, M.A.; Tammi, R.H.; Tammi, M.I.; Syrjänen, K.J.; Saarikoski, S.V.; Kosma, V.M. High levels of stromal hyaluronan predict poor disease outcome in epithelial ovarian cancer. Cancer Res. 2000, 60, 150–155. [Google Scholar]
- Josefsson, A.; Adamo, H.; Hammarsten, P.; Granfors, T.; Stattin, P.; Egevad, L.; Laurent, A.E.; Wikström, P.; Bergh, A. Prostate cancer increases hyaluronan in surrounding nonmalignant stroma, and this response is associated with tumor growth and an unfavorable outcome. Am. J. Pathol. 2011, 179, 1961–1968. [Google Scholar]
- Kramer, M.W.; Escudero, D.O.; Lokeshwar, S.D.; Golshani, R.; Ekwenna, O.O.; Acosta, K.; Merseburger, A.S.; Soloway, M.; Lokeshwar, V.B. Association of hyaluronic acid family members (HAS1, HAS2, and HYAL-1) with bladder cancer diagnosis and prognosis. Cancer 2011, 117, 1197–1209. [Google Scholar]
- Pirinen, R.; Tammi, R.; Tammi, M.; Hirvikoski, P.; Parkkinen, J.J.; Johansson, R.; Böhm, J.; Hollmén, S.; Kosma, V.M. Prognostic value of hyaluronan expression in non-small-cell lung cancer: Increased stromal expression indicates unfavorable outcome in patients with adenocarcinoma. Int. J. Cancer 2001, 95, 12–17. [Google Scholar] [CrossRef]
- Hertweck, M.K.; Erdfelder, F.; Kreuzer, K.A. CD44 in hematological neoplasias. Ann. Hematol. 2011, 90, 493–508. [Google Scholar] [CrossRef]
- Tzankov, A.; Strasser, U.; Dirnhofer, S.; Menter, T.; Arber, C.; Jotterand, M.; Rovo, A.; Tichelli, A.; Stauder, R.; Günthert, U. In situ RHAMM protein expression in acute myeloid leukemia blasts suggests poor overall survival. Ann. Hematol. 2011, 90, 901–909. [Google Scholar] [CrossRef]
- Lugli, A.; Zlobec, I.; Günthert, U.; Minoo, P.; Baker, K.; Tornillo, L.; Terracciano, L.; Jass, J.R. Overexpression of the receptor for hyaluronic acid mediated motility is an independent adverse prognostic factor in colorectal cancer. Mod. Pathol. 2006, 19, 1302–1309. [Google Scholar] [CrossRef]
- Godar, S.; Ince, T.A.; Bell, G.W.; Feldser, D.; Donaher, J.L.; Bergh, J.; Liu, A.; Miu, K.; Watnick, R.S.; Reinhardt, F.; et al. Growth-inhibitory and tumor- suppressive functions of p53 depend on its repression of CD44 expression. Cell 2008, 134, 62–73. [Google Scholar] [CrossRef]
- Sohr, S.; Engeland, K. RHAMM is differentially expressed in the cell cycle and downregulated by the tumor suppressor p53. Cell Cycle 2008, 7, 3448–3460. [Google Scholar] [CrossRef]
- Willenberg, A.; Saalbach, A.; Simon, J.C.; Anderegg, U. Melanoma cells control HA synthesis in peritumoral fibroblasts via PDGF-AA and PDGF-CC: Impact on melanoma cell proliferation. J. Invest. Dermatol. 2012, 132, 385–393. [Google Scholar] [CrossRef]
- Hingorani, S.R.; Wang, L.; Multani, A.S.; Combs, C.; Deramaudt, T.B.; Hruban, R.H.; Rustgi, A.K.; Chang, S.; Tuveson, D.A. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 2005, 7, 469–483. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar]
- Beckenlehner, K.; Bannke, S.; Spruss, T.; Bernhardt, G.; Schönenberg, H.; Schiess, W. Hyaluronidase enhances the activity of adriamycin in breast cancer models in vitro and in vivo. J. Cancer Res. Clin. Oncol. 1992, 118, 591–596. [Google Scholar]
- Smith, K.J.; Skelton, H.G.; Turiansky, G.; Wagner, K.F. Hyaluronidase enhances the therapeutic effect of vinblastine in intralesional treatment of Kaposi’s sarcoma. Military Medical Consortium for the Advancement of Retroviral Research (MMCARR). J. Am. Acad. Dermatol. 1997, 36, 239–242. [Google Scholar]
- Novak, U.; Stylli, S.S.; Kaye, A.H.; Lepperdinger, G. Hyaluronidase-2 overexpression accelerates intracerebral but not subcutaneous tumor formation of murine astrocytoma cells. Cancer Res. 1999, 59, 6246–6250. [Google Scholar]
- Tan, J.X.; Wang, X.Y.; Li, H.Y.; Su, X.L.; Wang, L.; Ran, L.; Zheng, K.; Ren, G.S. HYAL1 overexpression is correlated with the malignant behavior of human breast cancer. Int. J. Cancer 2011, 128, 1303–1315. [Google Scholar] [CrossRef]
- Lokeshwar, V.B.; Young, M.J.; Goudarzi, G.; Iida, N.; Yudin, A.I.; Cherr, G.N.; Selzer, M.G. Identification of bladder tumor-derived hyaluronidase: Its similarity to HYAL1. Cancer Res. 1999, 59, 4464–4470. [Google Scholar]
- Lokeshwar, V.B.; Rubinowicz, D.; Schroeder, G.L.; Forgacs, E.; Minna, J.D.; Block, N.L.; Nadji, M.; Lokeshwar, B.L. Stromal and epithelial expression of tumor markers hyaluronic acid and HYAL1 hyaluronidase in prostate cancer. J. Biol. Chem. 2001, 276, 11922–11932. [Google Scholar]
- Benitez, A.; Yates, T.J.; Lopez, L.E.; Cerwinka, W.H.; Bakkar, A.; Lokeshwar, V.B. Targeting hyaluronidase for cancer therapy: Antitumor activity of sulfated hyaluronic acid in prostate cancer cells. Cancer Res. 2011, 71, 4085–4095. [Google Scholar]
- Lokeshwar, V.B.; Lokeshwar, B.L.; Pham, H.T.; Block, N.L. Association of elevated levels of hyaluronidase, a matrix-degrading enzyme, with prostate cancer progression. Cancer Res. 1996, 56, 651–657. [Google Scholar]
- Droller, M.J. Tumor-derived hyaluronidase: A diagnostic urine marker for high-grade bladder cancer. J. Urol. 1998, 160, 619–620. [Google Scholar]
- Franzmann, E.J.; Schroeder, G.L.; Goodwin, W.J.; Weed, D.T.; Fisher, P.; Lokeshwar, V.B. Expression of tumor markers hyaluronic acid and hyaluronidase (HYAL1) in head and neck tumors. Int. J. Cancer 2003, 106, 438–445. [Google Scholar] [CrossRef]
- Bertrand, P.; Girard, N.; Duval, C.; d’Anjou, J.; Chauzy, C.; Ménard, J.F.; Delpech, B. Increased hyaluronidase levels in breast tumor metastases. Int. J. Cancer 1997, 73, 327–331. [Google Scholar] [CrossRef]
- Delpech, B.; Laquerriere, A.; Maingonnat, C.; Bertrand, P.; Freger, P. Hyaluronidase is more elevated in human brain metastases than in primary brain tumours. Anticancer Res. 2002, 22, 2423–2427. [Google Scholar]
- Pham, H.T.; Block, N.L.; Lokeshwar, V.B. Tumor-derived hyaluronidase: A diagnostic urine marker for high-grade bladder cancer. Cancer Res. 1997, 57, 778–783. [Google Scholar]
- Stern, M.; Longaker, M.T.; Adzick, N.S.; Harrison, M.R.; Stern, R. Hyaluronidase levels in urine from Wilms’ tumor patients. J. Natl. Cancer Inst. 1991, 83, 1569–1574. [Google Scholar] [CrossRef]
- Nykopp, T.K.; Rilla, K.; Sironen, R.; Tammi, M.I.; Tammi, R.H.; Hämäläinen, K.; Heikkinen, A.M.; Komulainen, M.; Kosma, V.M.; Anttila, M. Expression of hyaluronan synthases (HAS1-3) and hyaluronidases (HYAL1-2) in serous ovarian carcinomas: Inverse correlation between HYAL1 and hyaluronan content. BMC Cancer 2009, 9, 143. [Google Scholar]
- Lokeshwar, V.B.; Selzer, M.G. Hyaluronidase: Both a tumor promoter and suppressor. Semin. Cancer Biol. 2008, 18, 281–287. [Google Scholar] [CrossRef]
- Jacobson, A.; Rahmanian, M.; Rubin, K.; Heldin, P. Expression of hyaluronan synthase 2 or hyaluronidase 1 differentially affect the growth rate of transplantable colon carcinoma cell tumors. Int. J. Cancer 2002, 102, 212–219. [Google Scholar] [CrossRef]
- Lokeshwar, V.B.; Cerwinka, W.H.; Isoyama, T.; Lokeshwar, B.L. HYAL1 hyaluronidase in prostate cancer: A tumor promoter and suppressor. Cancer Res. 2005, 65, 7782–7789. [Google Scholar]
- Camenisch, T.D.; McDonald, J.A. Hyaluronan: Is bigger better? Am. J. Respir. Cell Mol. Biol. 2000, 23, 431–433. [Google Scholar]
- Deed, R.; Rooney, P.; Kumar, P.; Norton, J.D.; Smith, J.; Freemont, A.J.; Kumar, S. Early-response gene signalling is induced by angiogenic oligosaccharides of hyaluronan in endothelial cells. Inhibition by non-angiogenic, high-molecular-weight hyaluronan. Int. J. Cancer 1997, 71, 251–256. [Google Scholar]
- Rooney, P.; Kumar, S.; Ponting, J.; Wang, M. The role of hyaluronan in tumour neovascularization (review). Int. J. Cancer 1995, 60, 632–636. [Google Scholar] [CrossRef]
- Matou-Nasri, S.; Gaffney, J.; Kumar, S.; Slevin, M. Oligosaccharides of hyaluronan induce angiogenesis through distinct CD44 and RHAMM-mediated signalling pathways involving Cdc2 and gamma-adducin. Int. J. Oncol. 2009, 35, 761–773. [Google Scholar]
- Rahmanian, M.; Pertoft, H.; Kanda, S.; Christofferson, R.; Claesson-Welsh, L.; Heldin, P. Hyaluronan oligosaccharides induce tube formation of a brain endothelial cell line in vitro. Exp. Cell Res. 1997, 237, 223–230. [Google Scholar] [CrossRef]
- Rooney, P.; Wang, M.; Kumar, P.; Kumar, S. Angiogenic oligosaccharides of hyaluronan enhance the production of collagens by endothelial cells. J. Cell Sci. 1993, 105, 213–218. [Google Scholar]
- Lokeshwar, V.B.; Obek, C.; Soloway, M.S.; Block, N.L. Tumor-associated hyaluronic acid: A new sensitive and specific urine marker for bladder cancer. Cancer Res. 1997, 57, 773–777. [Google Scholar]
- Urakawa, H.; Nishida, Y.; Knudson, W.; Knudson, C.B.; Arai, E.; Kozawa, E.; Futamura, N.; Wasa, J.; Ishiguro, N. Therapeutic potential of hyaluronan oligosaccharides for bone metastasis of breast cancer. J. Orthop. Res. 2012, 30, 662–672. [Google Scholar]
- Zeng, C.; Toole, B.P.; Kinney, S.D.; Kuo, J.W.; Stamenkovic, I. Inhibition of tumor growth in vivo by hyaluronan oligomers. Int. J. Cancer 1998, 77, 396–401. [Google Scholar]
- Slomiany, M.G.; Dai, L.; Tolliver, L.B.; Grass, G.D.; Zeng, Y.; Toole, B.P. Inhibition of functional hyaluronan-CD44 interactions in CD133-positive primary human ovarian carcinoma cells by small hyaluronan oligosaccharides. Clin. Cancer Res. 2009, 15, 7593–7601. [Google Scholar] [CrossRef]
- Hosono, K.; Nishida, Y.; Knudson, W.; Knudson, C.B.; Naruse, T.; Suzuki, Y.; Ishiguro, N. Hyaluronan oligosaccharides inhibit tumorigenicity of osteosarcoma cell lines MG-63 and LM-8 in vitro and in vivo via perturbation of hyaluronan-rich pericellular matrix of the cells. Am. J. Pathol. 2007, 171, 274–286. [Google Scholar] [CrossRef]
- Slomiany, M.G.; Dai, L.; Bomar, P.A.; Knackstedt, T.J.; Kranc, D.A.; Tolliver, L.; Maria, B.L.; Toole, B.P. Abrogating drug resistance in malignant peripheral nerve sheath tumors by disrupting hyaluronan-CD44 interactions with small hyaluronan oligosaccharides. Cancer Res. 2009, 69, 4992–4998. [Google Scholar] [CrossRef]
- Ghatak, S.; Misra, S.; Toole, B.P. Hyaluronan oligosaccharides inhibit anchorage-independent growth of tumor cells by suppressing the phosphoinositide 3-kinase/Akt cell survival pathway. J. Biol. Chem. 2002, 277, 38013–38020. [Google Scholar]
- Misra, S.; Ghatak, S.; Zoltan-Jones, A.; Toole, B.P. Regulation of multidrug resistance in cancer cells by hyaluronan. J. Biol. Chem. 2003, 278, 25285–25288. [Google Scholar]
- Stern, R.; Asari, A.A.; Sugahara, K.N. Hyaluronan fragments: An information-rich system. Eur. J. Cell Biol. 2006, 85, 699–715. [Google Scholar] [CrossRef]
- Rembrink, K.; Romijn, J.C.; van der Kwast, T.H.; Rübben, H.; Schröder, F.H. Orthotopic implantation of human prostate cancer cell lines: A clinically relevant animal model for metastatic prostate cancer. Prostate 1997, 31, 168–174. [Google Scholar] [CrossRef]
- Cos, S.; Fernández, R.; Güézmes, A.; Sánchez-Barceló, E.J. Influence of melatonin on invasive and metastatic properties of MCF-7 human breast cancer cells. Cancer Res. 1998, 58, 4383–4390. [Google Scholar]
- Richter, U.; Wicklein, D.; Geleff, S.; Schumacher, U. The interaction between CD44 on tumour cells and hyaluronan under physiologic flow conditions: Implications for metastasis formation. Histochem. Cell Biol. 2012, 137, 687–695. [Google Scholar] [CrossRef]
- Stephenson, R.A.; Dinney, C.P.; Gohji, K.; Ordóñez, N.G.; Killion, J.J.; Fidler, I.J. Metastatic model for human prostate cancer using orthotopic implantation in nude mice. J. Natl. Cancer Inst. 1992, 17, 951–957. [Google Scholar]
- Veiseh, M.; Turley, E.A. Hyaluronan metabolism in remodeling extracellular matrix: Probes for imaging and therapy of breast cancer. Integr. Biol. (Camb.) 2011, 4, 304–315. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kultti, A.; Li, X.; Jiang, P.; Thompson, C.B.; Frost, G.I.; Shepard, H.M. Therapeutic Targeting of Hyaluronan in the Tumor Stroma. Cancers 2012, 4, 873-903. https://doi.org/10.3390/cancers4030873
Kultti A, Li X, Jiang P, Thompson CB, Frost GI, Shepard HM. Therapeutic Targeting of Hyaluronan in the Tumor Stroma. Cancers. 2012; 4(3):873-903. https://doi.org/10.3390/cancers4030873
Chicago/Turabian StyleKultti, Anne, Xiaoming Li, Ping Jiang, Curtis B. Thompson, Gregory I. Frost, and H. Michael Shepard. 2012. "Therapeutic Targeting of Hyaluronan in the Tumor Stroma" Cancers 4, no. 3: 873-903. https://doi.org/10.3390/cancers4030873