Tumor-Promoting Circuits That Regulate a Cancer-Related Chemokine Cluster: Dominance of Inflammatory Mediators Over Oncogenic Alterations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
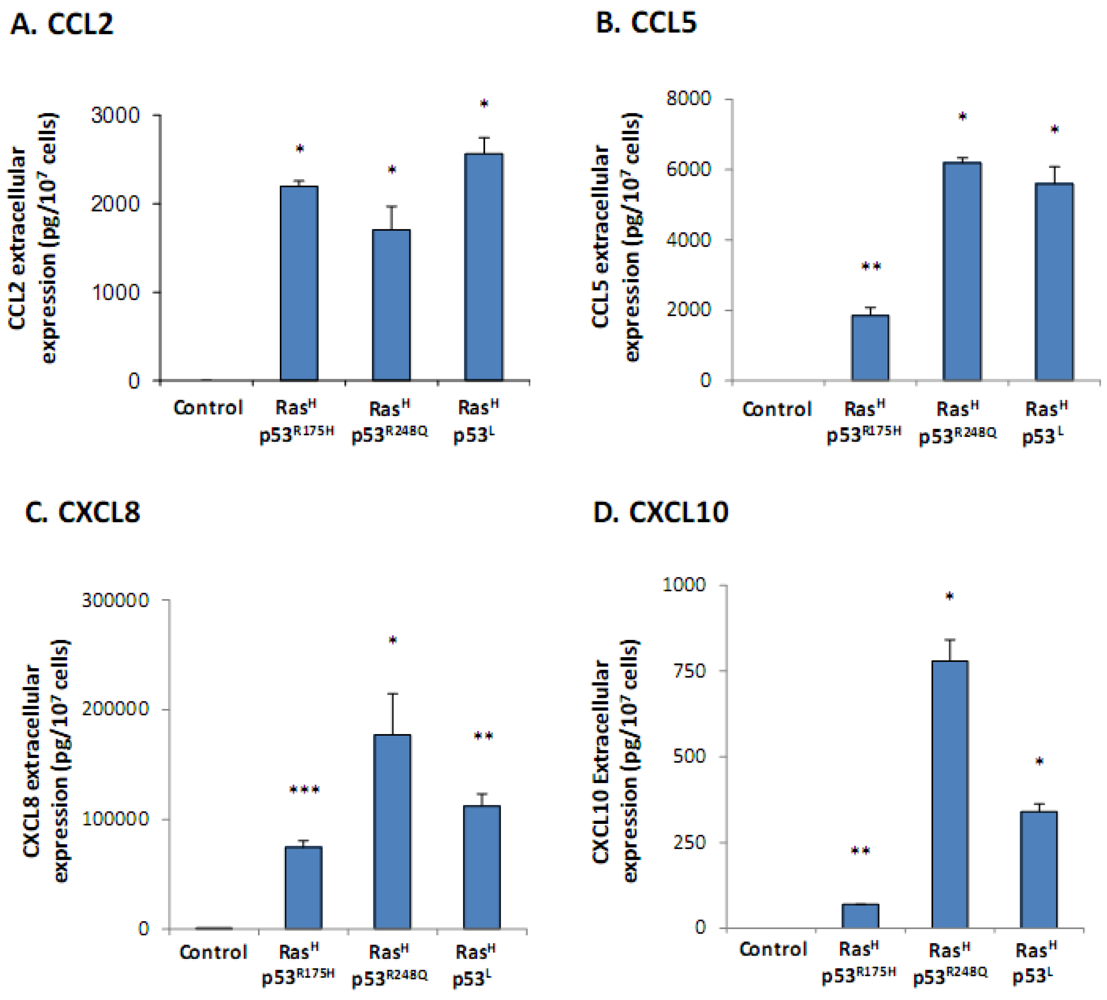
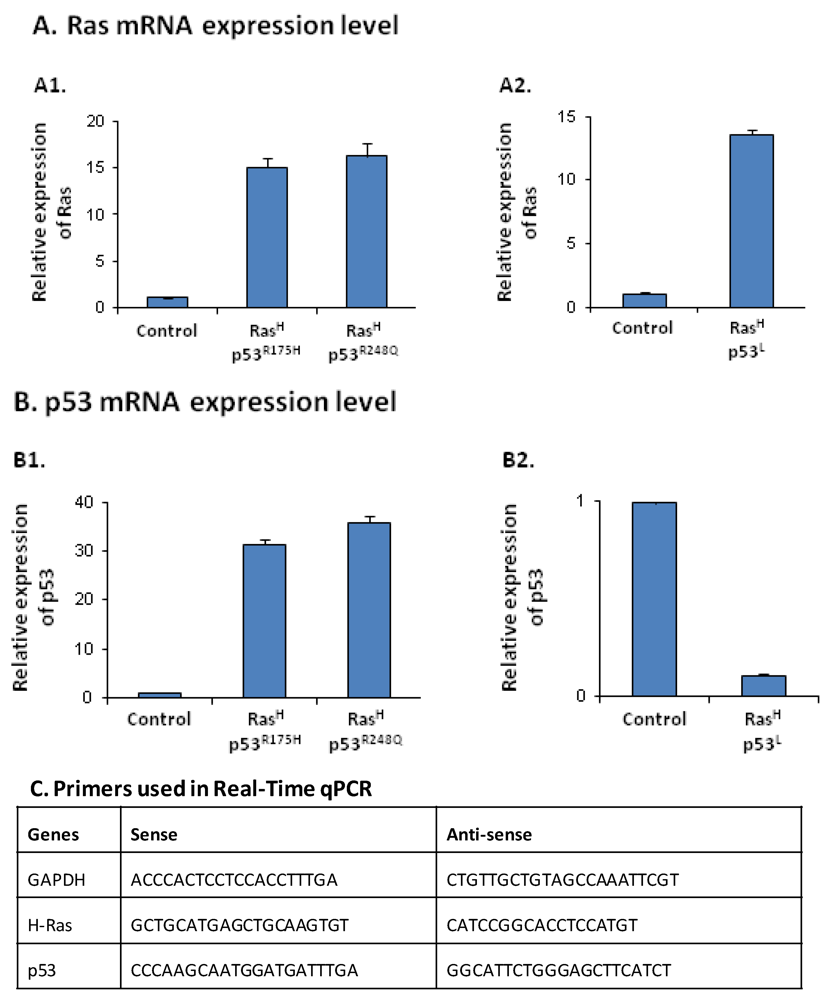
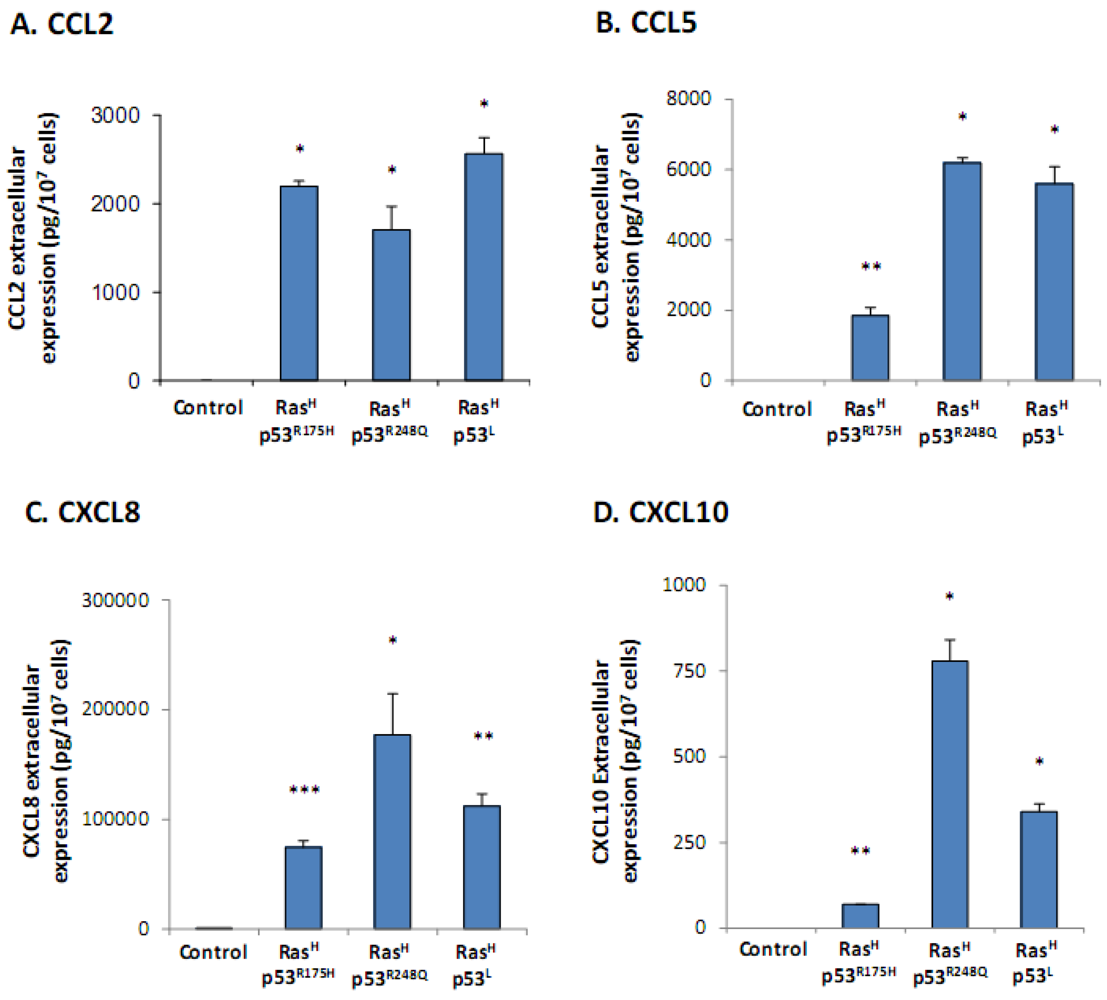
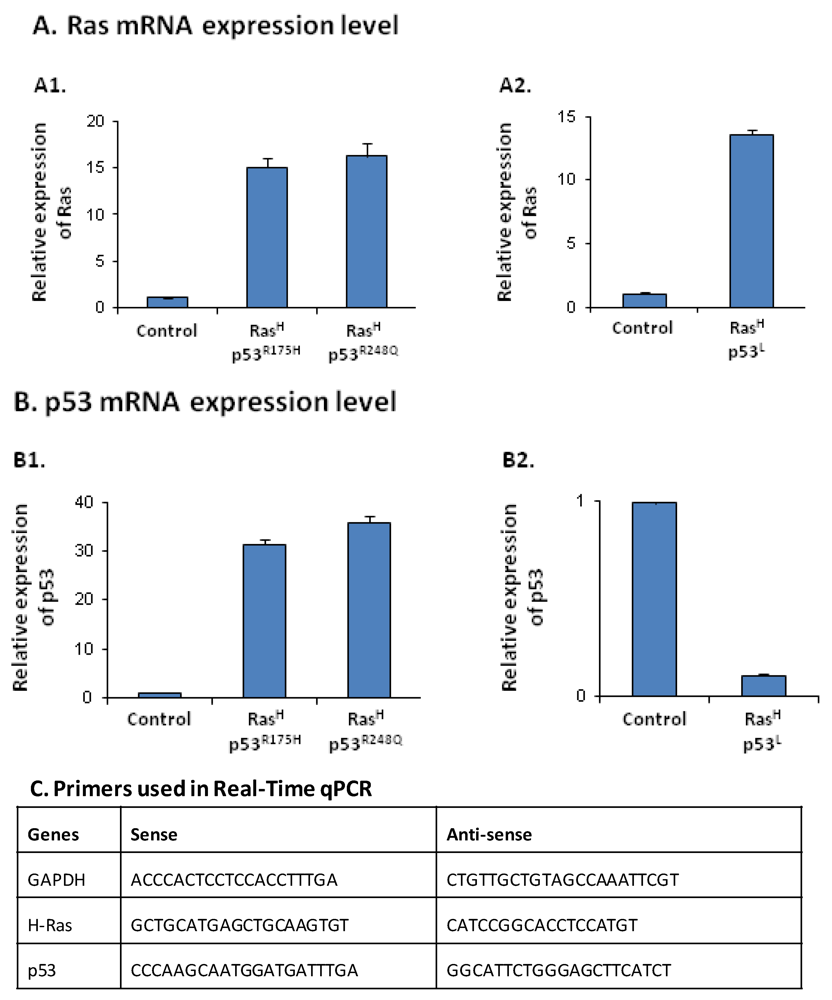
2.1. Co-Expression of Hyper-Activated Ras and Dysfunctional p53 Together, in Non-Transformed Cells, Leads to Excessive Release of the Cancer-Related Chemokine Cluster
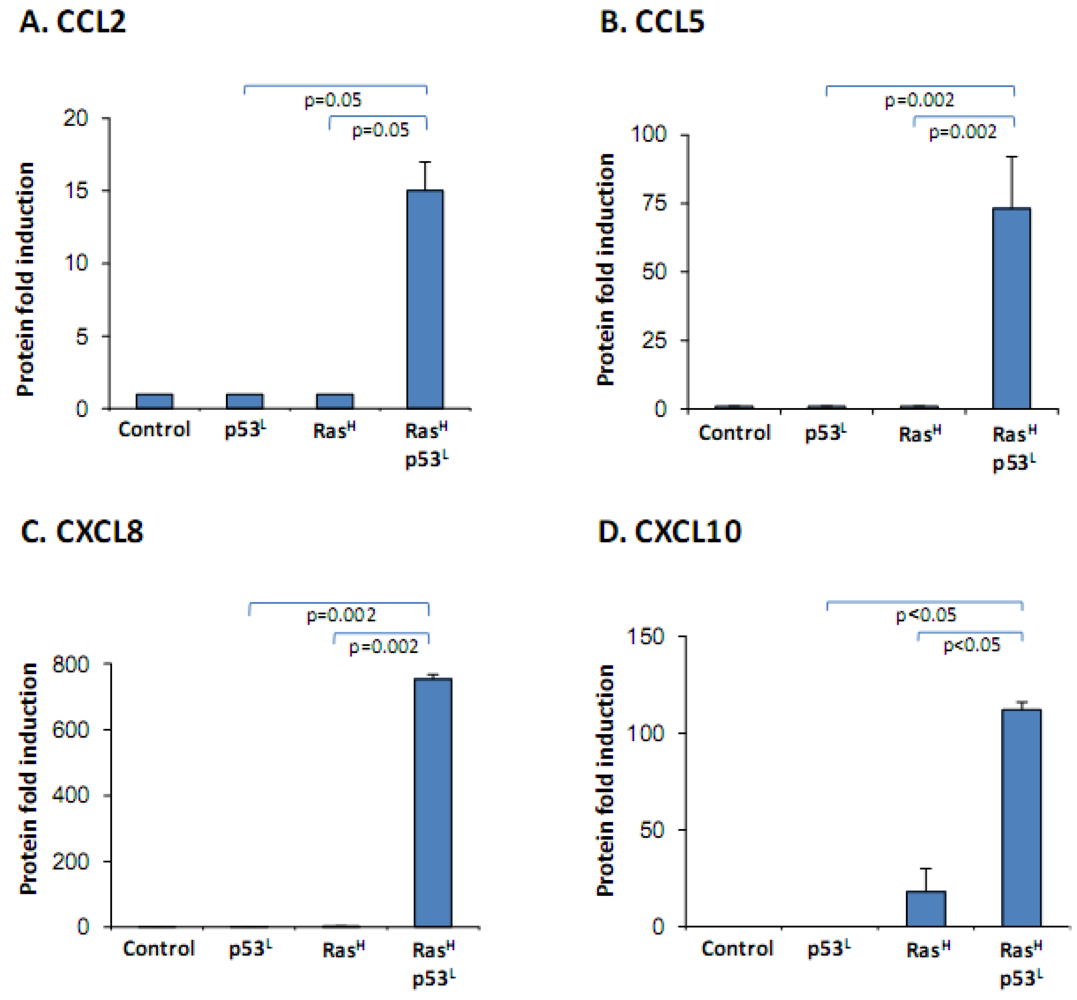
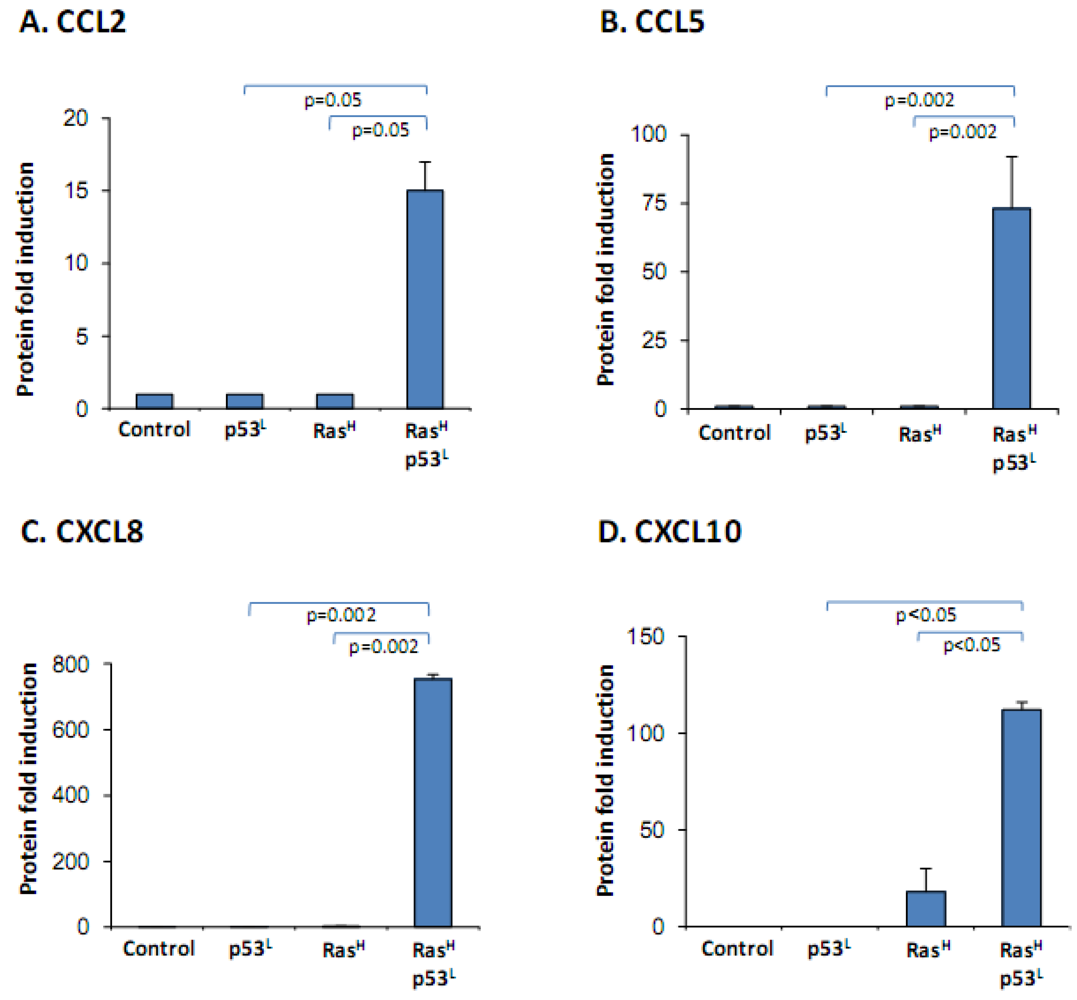
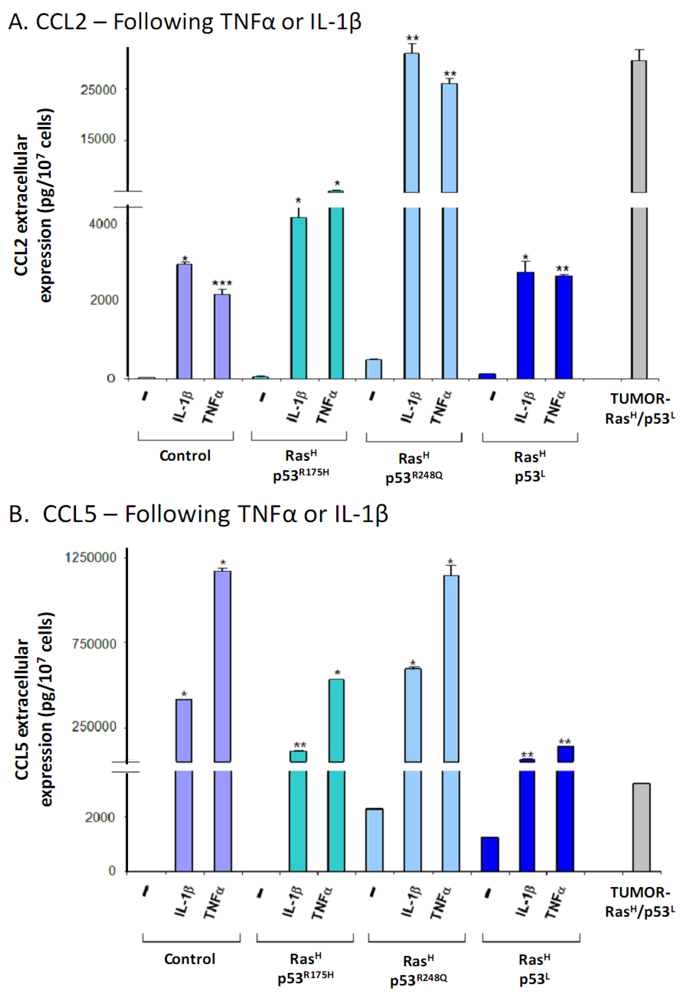
2.2. The Release of the Cancer-Related Chemokine Cluster Requires Cooperation between Ras Hyper-Activation and p53 Down-Regulation
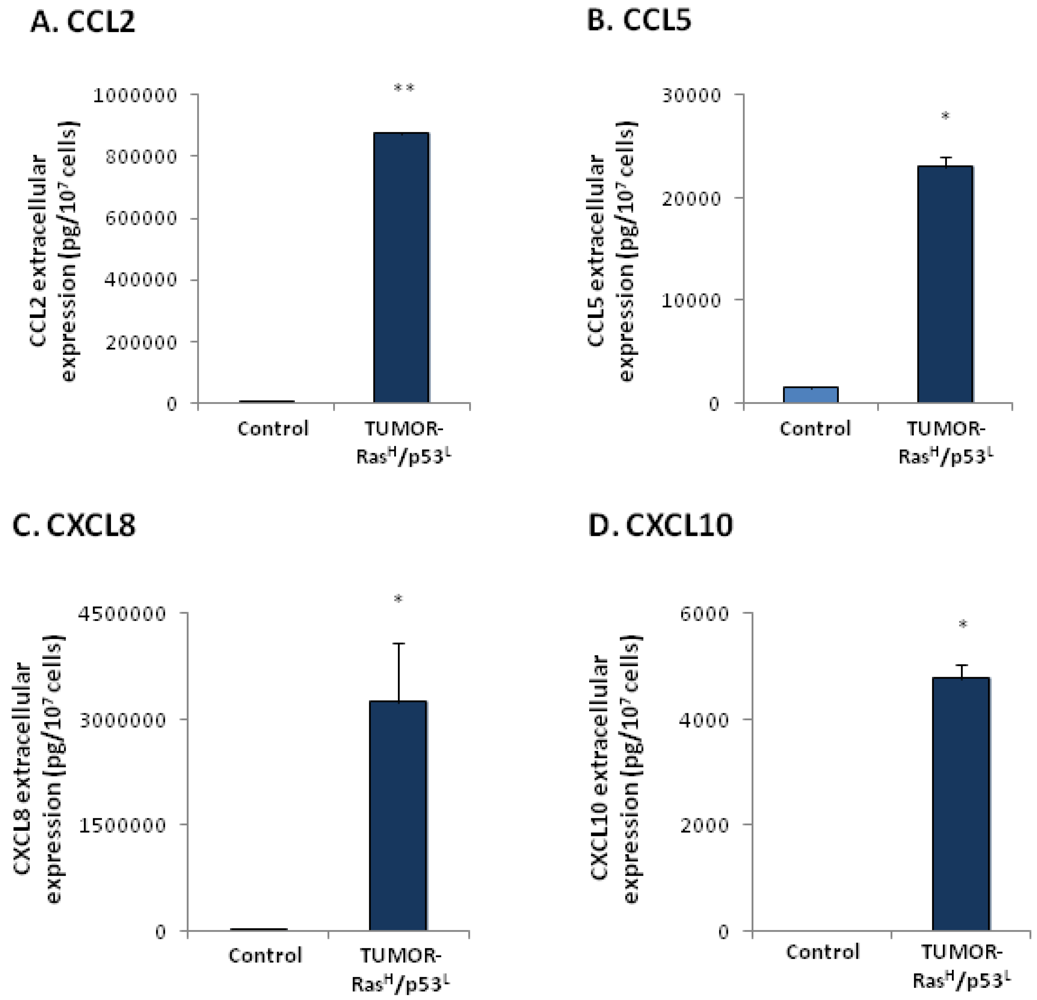
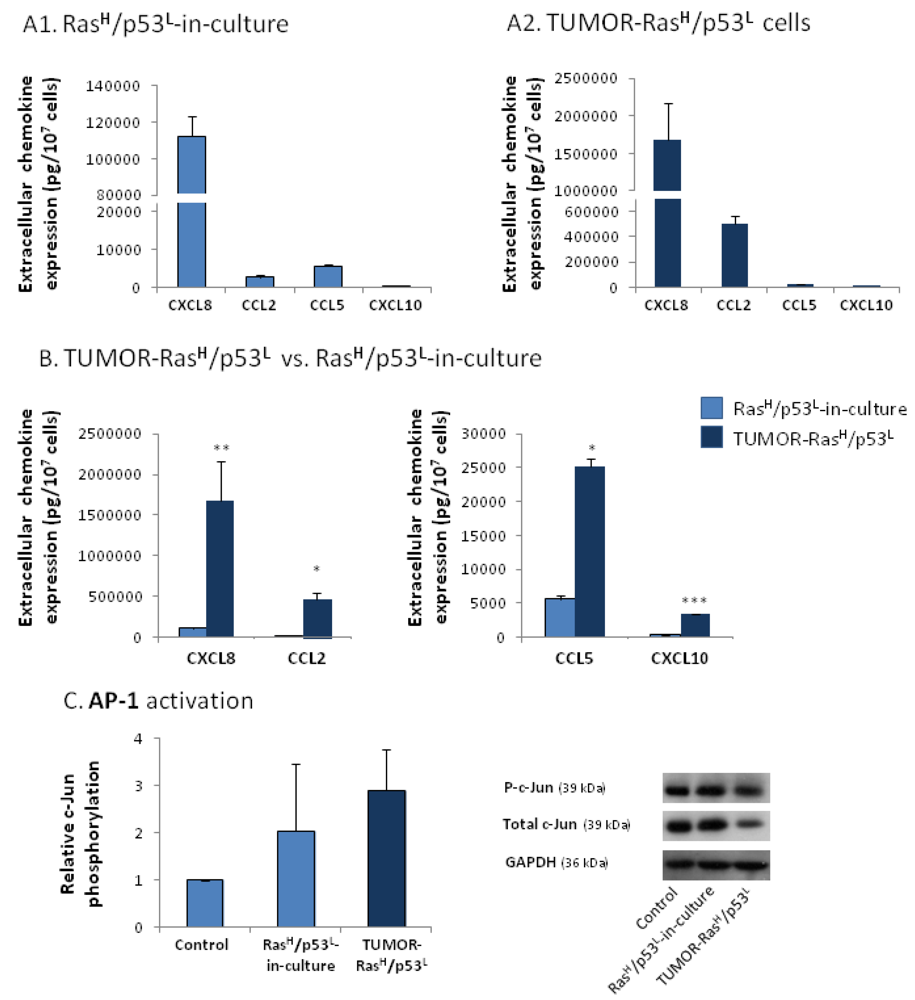
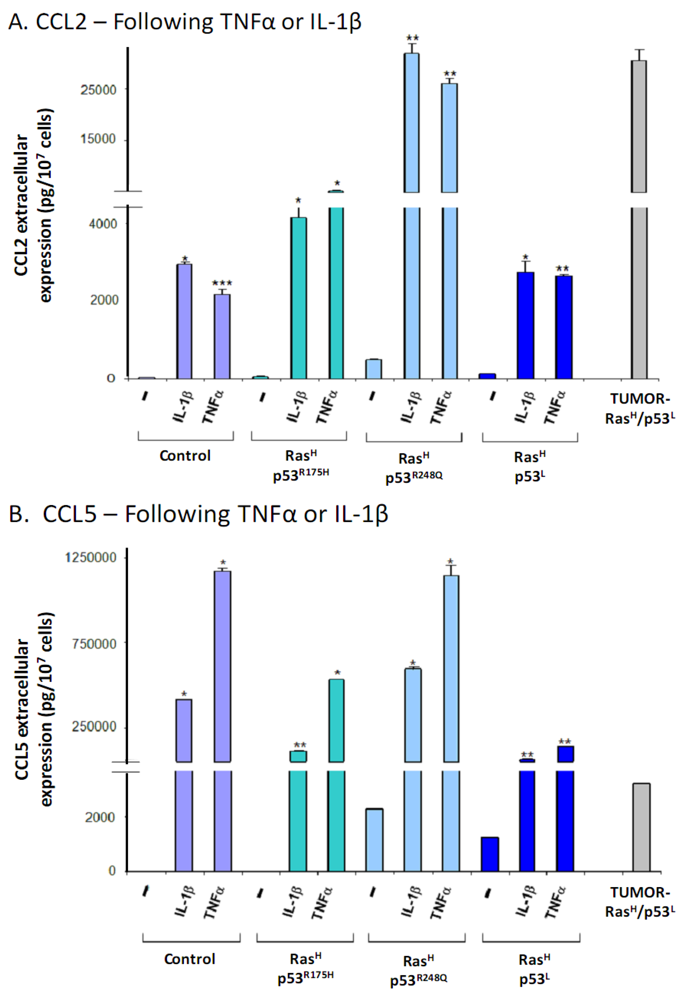
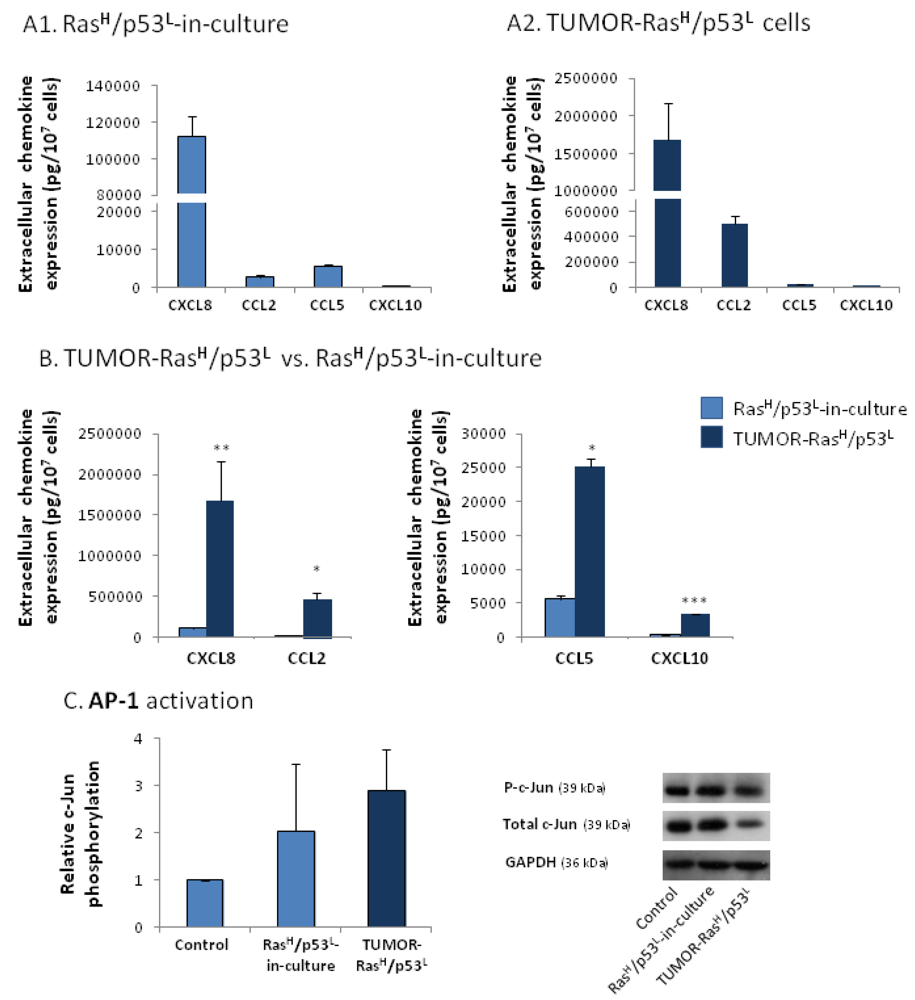
2.3. Exposure to the Host Microenvironment Leads to Further Increase in the Expression of the Cancer-Related Chemokine Cluster

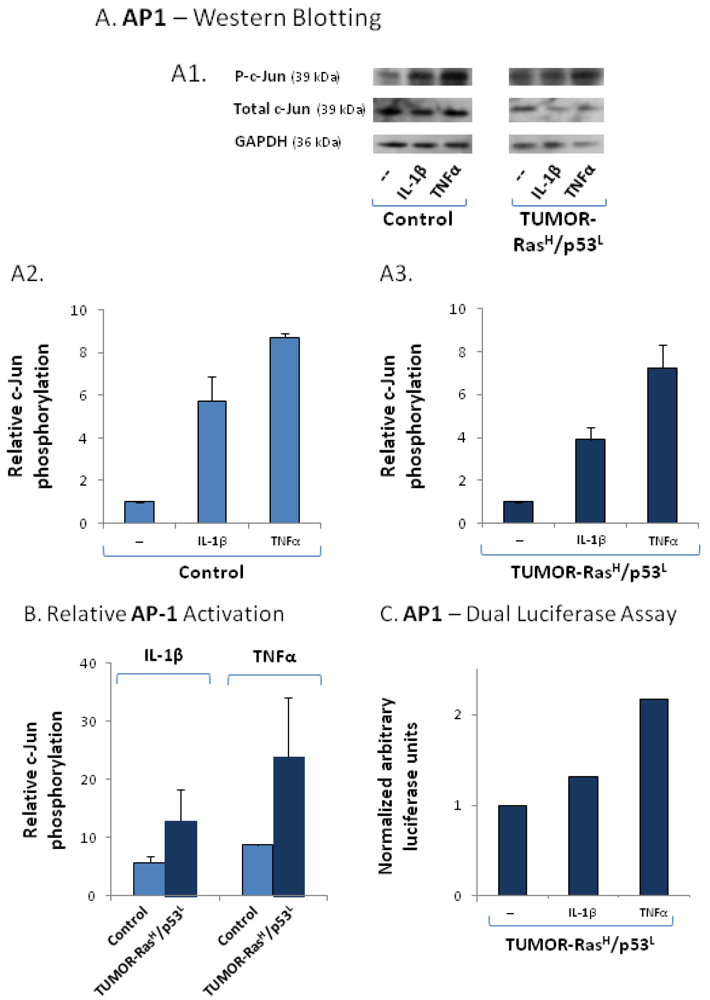
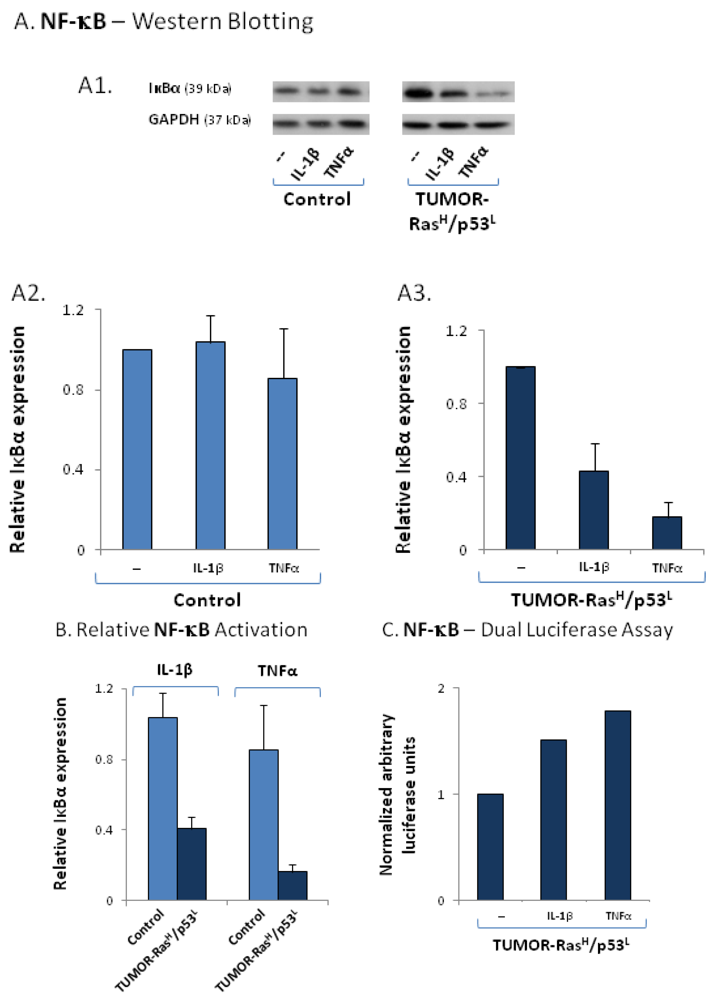
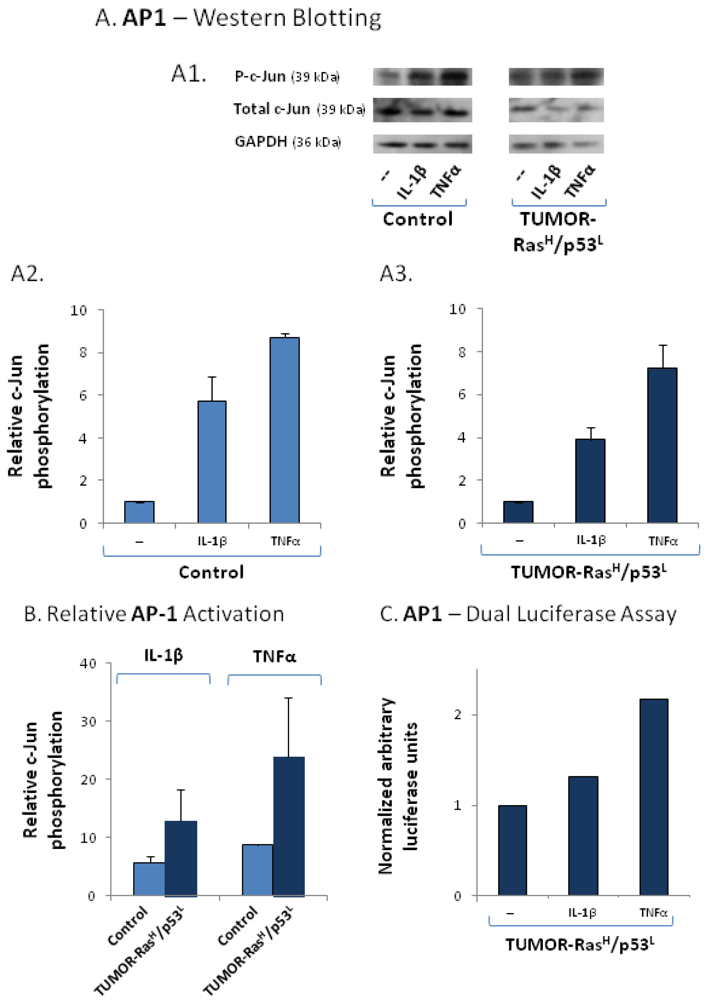
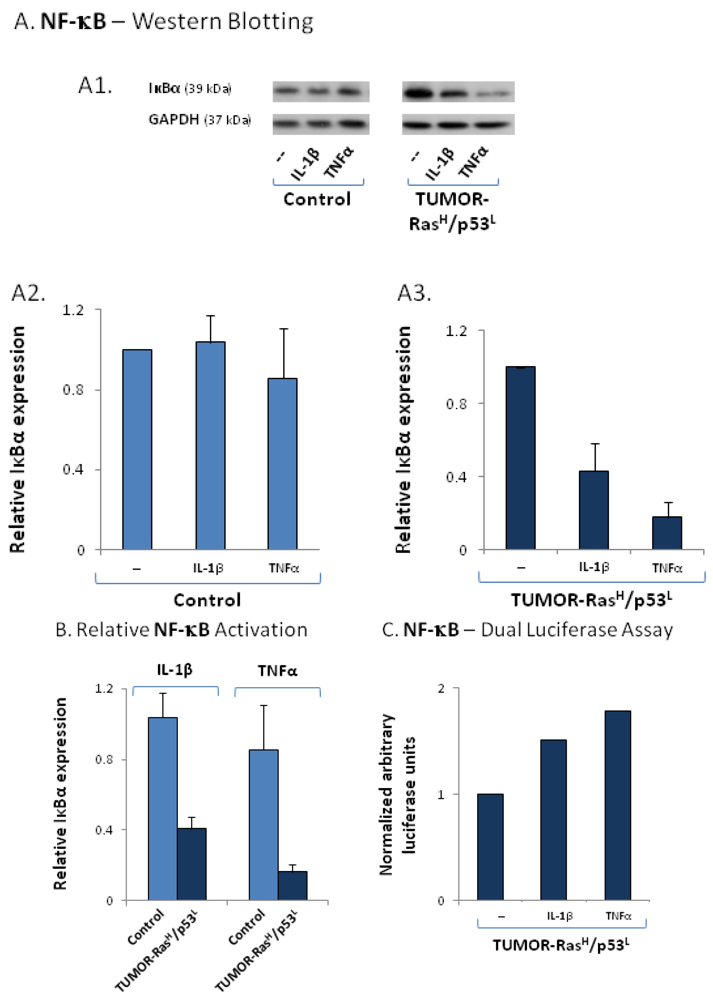
2.4. The Exacerbated Release of the Chemokine Cluster Following Exposure to the Host Can be Recapitulated by Stimulation with Inflammatory Cytokines
3. Experimental Section
3.1. Cells
3.2. Determination of Chemokine Release by ELISA
3.3. Western Blot Analyses
3.4. Dual Luciferase Assays
4. Conclusions
Acknowledgements
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Joyce, J.A.; Pollard, J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer 2009, 9, 239–252. [Google Scholar] [CrossRef]
- Milyavsky, M.; Tabach, Y.; Shats, I.; Erez, N.; Cohen, Y.; Tang, X.; Kalis, M.; Kogan, I.; Buganim, Y.; Goldfinger, N.; et al. Transcriptional programs following genetic alterations in p53, INK4A, and H-Ras genes along defined stages of malignant transformation. Cancer Res. 2005, 65, 4530–4543. [Google Scholar]
- Buganim, Y.; Solomon, H.; Rais, Y.; Kistner, D.; Nachmany, I.; Brait, M.; Madar, S.; Goldstein, I.; Kalo, E.; Adam, N.; et al. 53 Regulates the Ras circuit to inhibit the expression of a cancer-related gene signature by various molecular pathways. Cancer Res. 2010, 70, 2274–2284. [Google Scholar]
- Bonecchi, R.; Locati, M.; Mantovani, A. Chemokines and cancer: A fatal attraction. Cancer Cell 2011, 19, 434–435. [Google Scholar] [CrossRef]
- Lazennec, G.; Richmond, A. Chemokines and chemokine receptors: New insights into cancer-related inflammation. Trends Mol. Med. 2010, 16, 133–144. [Google Scholar] [CrossRef]
- Yadav, A.; Saini, V.; Arora, S. MCP-1: Chemoattractant with a role beyond immunity: A review. Clin. Chim. Acta 2010, 411, 1570–1579. [Google Scholar] [CrossRef]
- Soria, G.; Ben-Baruch, A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. 2008, 267, 271–285. [Google Scholar] [CrossRef]
- Conti, I.; Rollins, B.J. CCL2 (monocyte chemoattractant protein-1) and cancer. Semin. Cancer Biol. 2004, 14, 149–154. [Google Scholar] [CrossRef]
- Waugh, D.J.; Wilson, C. The interleukin-8 pathway in cancer. Clin. Cancer Res. 2008, 14, 6735–6741. [Google Scholar] [CrossRef]
- Richmond, A.; Yang, J.; Su, Y. The good and the bad of chemokines/chemokine receptors in melanoma. Pigment Cell Melanoma Res. 2009, 22, 175–186. [Google Scholar] [CrossRef]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. CXC chemokines in cancer angiogenesis and metastases. Adv. Cancer Res. 2010, 106, 91–111. [Google Scholar] [CrossRef]
- Keeley, E.C.; Mehrad, B.; Strieter, R.M. Chemokines as mediators of neovascularization. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1928–1936. [Google Scholar] [CrossRef]
- Fulton, A.M. The chemokine receptors CXCR4 and CXCR3 in cancer. Curr. Oncol. Rep. 2009, 11, 125–131. [Google Scholar] [CrossRef]
- Ben-Baruch, A. Expert commentary: The chemokine receptor CXCR3 and its ligands in malignancy: Do they act as double-edged swords? In Chemokine Research Trends; Grinwald, L.R., Ed.; Nova Science Publishers: Hauppauge, NY, USA, 2007. [Google Scholar]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: Split personalities. Nat. Rev. Mol. Cell Biol. 2008, 9, 517–531. [Google Scholar] [CrossRef]
- Schubbert, S.; Shannon, K.; Bollag, G. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 2007, 7, 295–308. [Google Scholar] [CrossRef]
- Blum, R.; Cox, A.D.; Kloog, Y. Inhibitors of chronically active ras: Potential for treatment of human malignancies. Recent Pat. Anticancer Drug Discov. 2008, 3, 31–47. [Google Scholar] [CrossRef]
- Olivier, M.; Hollstein, M.; Hainaut, P. TP53 mutations in human cancers: Origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010, 2, a001008. [Google Scholar] [CrossRef]
- Goldstein, I.; Marcel, V.; Olivier, M.; Oren, M.; Rotter, V.; Hainaut, P. Understanding wild-type and mutant p53 activities in human cancer: New landmarks on the way to targeted therapies. Cancer Gene Ther. 2011, 18, 2–11. [Google Scholar] [CrossRef]
- Oren, M.; Rotter, V. Mutant p53 gain-of-function in cancer. Cold Spring Harb. Perspect. Biol. 2010, 2, a001107. [Google Scholar] [CrossRef]
- Toledo, F.; Bardot, B. Cancer: Three birds with one stone. Nature 2009, 460, 466–467. [Google Scholar] [CrossRef]
- Brosh, R.; Rotter, V. When mutants gain new powers: News from the mutant p53 field. Nat. Rev. Cancer 2009, 9, 701–713. [Google Scholar]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef]
- Balkwill, F. TNF-alpha in promotion and progression of cancer. Cancer Metastasis Rev. 2006, 25, 409–416. [Google Scholar] [CrossRef]
- ten Hagen, T.L.; Seynhaeve, A.L.; Eggermont, A.M. Tumor necrosis factor-mediated interactions between inflammatory response and tumor vascular bed. Immunol. Rev. 2008, 222, 299–315. [Google Scholar] [CrossRef]
- Mocellin, S.; Rossi, C.R.; Pilati, P.; Nitti, D. Tumor necrosis factor, cancer and anticancer therapy. Cytokine Growth Factor Rev. 2005, 16, 35–53. [Google Scholar] [CrossRef]
- Dinarello, C.A. Why not treat human cancer with interleukin-1 blockade? Cancer Metastasis Rev. 2010, 29, 317–329. [Google Scholar] [CrossRef]
- Apte, R.N.; Voronov, E. Is interleukin-1 a good or bad ‘guy’ in tumor immunobiology and immunotherapy? Immunol. Rev. 2008, 222, 222–241. [Google Scholar] [CrossRef]
- Meylan, E.; Dooley, A.L.; Feldser, D.M.; Shen, L.; Turk, E.; Ouyang, C.; Jacks, T. Requirement for NF-kappaB signalling in a mouse model of lung adenocarcinoma. Nature 2009, 462, 104–107. [Google Scholar]
- Motohara, T.; Masuko, S.; Ishimoto, T.; Yae, T.; Onishi, N.; Muraguchi, T.; Hirao, A.; Matsuzaki, Y.; Tashiro, H.; Katabuchi, H.; et al. Transient depletion of p53 followed by transduction of c-Myc and K-Ras converts ovarian stem-like cells into tumor-initiating cells. Carcinogenesis 2011, 32, 1597–1606. [Google Scholar] [CrossRef]
- Parada, L.F.; Land, H.; Weinberg, R.A.; Wolf, D.; Rotter, V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature 1984, 312, 649–651. [Google Scholar] [CrossRef]
- Xia, M.; Land, H. Tumor suppressor p53 restricts Ras stimulation of RhoA and cancer cell motility. Nat. Struct. Mol. Biol. 2007, 14, 215–223. [Google Scholar] [CrossRef]
- Hahn, W.C.; Counter, C.M.; Lundberg, A.S.; Beijersbergen, R.L.; Brooks, M.W.; Weinberg, R.A. Creation of human tumour cells with defined genetic elements. Nature 1999, 400, 464–468. [Google Scholar] [CrossRef]
- Drayton, S.; Rowe, J.; Jones, R.; Vatcheva, R.; Cuthbert-Heavens, D.; Marshall, J.; Fried, M.; Peters, G. Tumor suppressor p16INK4a determines sensitivity of human cells to transformation by cooperating cellular oncogenes. Cancer Cell 2003, 4, 301–310. [Google Scholar] [CrossRef]
- Rangarajan, A.; Hong, S.J.; Gifford, A.; Weinberg, R.A. Species- and cell type-specific requirements for cellular transformation. Cancer Cell 2004, 6, 171–183. [Google Scholar] [CrossRef]
- Milyavsky, M.; Shats, I.; Erez, N.; Tang, X.; Senderovich, S.; Meerson, A.; Tabach, Y.; Goldfinger, N.; Ginsberg, D.; Harris, C.C.; et al. Prolonged culture of telomerase-immortalized human fibroblasts leads to a premalignant phenotype. Cancer Res. 2003, 63, 7147–7157. [Google Scholar]
- Solomon, H.; Brosh, R.; Buganim, Y.; Rotter, V. Inactivation of the p53 tumor suppressor gene and activation of the Ras oncogene: Cooperative events in tumorigenesis. Discov. Med. 2010, 9, 448–454. [Google Scholar]
- Zlotnik, A. Involvement of chemokine receptors in organ-specific metastasis. Contrib. Microbiol. 2006, 13, 191–199. [Google Scholar] [CrossRef]
- Zlotnik, A. New insights on the role of CXCR4 in cancer metastasis. J. Pathol. 2008, 215, 211–213. [Google Scholar] [CrossRef]
- Su, H.; Sobrino Najul, E.J.; Toth, T.A.; Mei Ng, C.; Lelievre, S.A.; Fred, M.; Tang, C.K. Chemokine receptor CXCR4-mediated transformation of mammary epithelial cells by enhancing multiple RTKs expression and deregulation of the p53/MDM2 axis. Cancer Lett. 2011, 307, 132–140. [Google Scholar] [CrossRef]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef]
- Creighton, C.J.; Bromberg-White, J.L.; Misek, D.E.; Monsma, D.J.; Brichory, F.; Kuick, R.; Giordano, T.J.; Gao, W.; Omenn, G.S.; Webb, C.P.; et al. Analysis of tumor-host interactions by gene expression profiling of lung adenocarcinoma xenografts identifies genes involved in tumor formation. Mol. Cancer Res. 2005, 3, 119–129. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Fidler, I.J. Enhanced metastatic potential of tumor cells harvested from spontaneous metastases of heterogeneous murine tumors. J. Natl. Cancer Inst. 1982, 69, 975–980. [Google Scholar]
- Halachmi, E.; Witz, I.P. Differential tumorigenicity of 3T3 cells transformed in vitro with polyoma virus and in vivo selection for high tumorigenicity. Cancer Res. 1989, 49, 2383–2389. [Google Scholar]
- Roebuck, K.A.; Carpenter, L.R.; Lakshminarayanan, V.; Page, S.M.; Moy, J.N.; Thomas, L.L. Stimulus-specific regulation of chemokine expression involves differential activation of the redox-responsive transcription factors AP-1 and NF-kappaB. J. Leukoc. Biol. 1999, 65, 291–298. [Google Scholar]
- Murugan, V.; Peck, M.J. Signal transduction pathways linking the activation of alveolar macrophages with the recruitment of neutrophils to lungs in chronic obstructive pulmonary disease. Exp. Lung Res. 2009, 35, 439–485. [Google Scholar] [CrossRef]
- Adcock, I.M. Transcription factors as activators of gene transcription: AP-1 and NF-kappa B. Monaldi Arch. Chest Dis. 1997, 52, 178–186. [Google Scholar]
- Baeuerle, P.A.; Henkel, T. Function and activation of NF-kappa B in the immune system. Annu. Rev. Immunol. 1994, 12, 141–179. [Google Scholar] [CrossRef]
- Ueda, A.; Ishigatsubo, Y.; Okubo, T.; Yoshimura, T. Transcriptional regulation of the human monocyte chemoattractant protein-1 gene. Cooperation of two NF-kappaB sites and NF-kappaB/Rel subunit specificity. J. Biol. Chem. 1997, 272, 31092–31099. [Google Scholar]
- Martin, T.; Cardarelli, P.M.; Parry, G.C.; Felts, K.A.; Cobb, R.R. Cytokine induction of monocyte chemoattractant protein-1 gene expression in human endothelial cells depends on the cooperative action of NF-kappa B and AP-1. Eur. J. Immunol. 1997, 27, 1091–1097. [Google Scholar] [CrossRef]
- Mantovani, A.; Bonecchi, R.; Locati, M. Tuning inflammation and immunity by chemokine sequestration: Decoys and more. Nat. Rev. Immunol. 2006, 6, 907–918. [Google Scholar] [CrossRef]
- Rot, A.; von Andrian, U.H. Chemokines in innate and adaptive host defense: Basic chemokinese grammar for immune cells. Annu. Rev. Immunol. 2004, 22, 891–928. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, O.; Nomiyama, H. The chemokine and chemokine receptor superfamilies and their molecular evolution. Genome Biol. 2006, 7, 243. [Google Scholar] [CrossRef]
- Sedgwick, J.D.; Riminton, D.S.; Cyster, J.G.; Korner, H. Tumor necrosis factor: A master-regulator of leukocyte movement. Immunol. Today 2000, 21, 110–113. [Google Scholar] [CrossRef]
- Furuichi, K.; Wada, T.; Iwata, Y.; Kokubo, S.; Hara, A.; Yamahana, J.; Sugaya, T.; Iwakura, Y.; Matsushima, K.; Asano, M.; et al. Interleukin-1-dependent sequential chemokine expression and inflammatory cell infiltration in ischemia-reperfusion injury. Crit. Care Med. 2006, 34, 2447–2455. [Google Scholar]
- Soria, G.; Ofri-Shahak, M.; Haas, I.; Yaal-Hahoshen, N.; Leider-Trejo, L.; Leibovich-Rivkin, T.; Weitzenfeld, P.; Meshel, T.; Shabtai, E.; Gutman, M.; et al. Inflammatory mediators in breast cancer: Coordinated expression of TNFa & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer 2011, 11, 130–149. [Google Scholar]
- Madhusudan, S.; Foster, M.; Muthuramalingam, S.R.; Braybrooke, J.P.; Wilner, S.; Kaur, K.; Han, C.; Hoare, S.; Balkwill, F.; Talbot, D.C.; et al. A phase II study of etanercept (Enbrel), a tumor necrosis factor alpha inhibitor in patients with metastatic breast cancer. Clin. Cancer Res. 2004, 10, 6528–6534. [Google Scholar]
- Brown, E.R.; Charles, K.A.; Hoare, S.A.; Rye, R.L.; Jodrell, D.I.; Aird, R.E.; Vora, R.; Prabhakar, U.; Nakada, M.; Corringham, R.E.; et al. A clinical study assessing the tolerability and biological effects of infliximab, a TNF-alpha inhibitor, in patients with advanced cancer. Ann. Oncol. 2008, 19, 1340–1346. [Google Scholar]
- Harrison, M.L.; Obermueller, E.; Maisey, N.R.; Hoare, S.; Edmonds, K.; Li, N.F.; Chao, D.; Hall, K.; Lee, C.; Timotheadou, E.; et al. Tumor necrosis factor alpha as a new target for renal cell carcinoma: Two sequential phase II trials of infliximab at standard and high dose. J. Clin. Oncol. 2007, 25, 4542–4549. [Google Scholar]
Supplementary Material
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Leibovich-Rivkin, T.; Buganim, Y.; Solomon, H.; Meshel, T.; Rotter, V.; Ben-Baruch, A. Tumor-Promoting Circuits That Regulate a Cancer-Related Chemokine Cluster: Dominance of Inflammatory Mediators Over Oncogenic Alterations. Cancers 2012, 4, 55-76. https://doi.org/10.3390/cancers4010055
Leibovich-Rivkin T, Buganim Y, Solomon H, Meshel T, Rotter V, Ben-Baruch A. Tumor-Promoting Circuits That Regulate a Cancer-Related Chemokine Cluster: Dominance of Inflammatory Mediators Over Oncogenic Alterations. Cancers. 2012; 4(1):55-76. https://doi.org/10.3390/cancers4010055
Chicago/Turabian StyleLeibovich-Rivkin, Tal, Yosef Buganim, Hilla Solomon, Tsipi Meshel, Varda Rotter, and Adit Ben-Baruch. 2012. "Tumor-Promoting Circuits That Regulate a Cancer-Related Chemokine Cluster: Dominance of Inflammatory Mediators Over Oncogenic Alterations" Cancers 4, no. 1: 55-76. https://doi.org/10.3390/cancers4010055