Cancer Stem Cells in Head and Neck Cancer

Abstract
: Head and neck cancer (HNC) is the sixth most common malignancy world-wide, however the survival rate has not improved for the past 20 years. In recent years, the cancer stem cell (CSC) hypothesis has gained ground in several malignancies and there is mounting evidence suggesting CSCs mediate tumor resistance to chemotherapy and radiation therapy. However, the CSC theory is also challenged at least in certain types of cancer. Here we review the progress of CSC studies in HNC, which suggest that HNC conforms to the CSC model. The identified CSC markers and their tumor initiation properties provide a framework for the development of novel therapeutic strategies for HNC.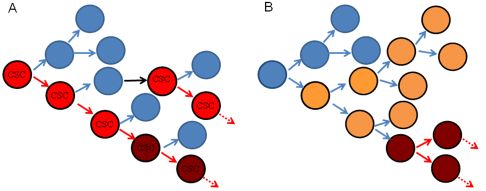
{kind=link}
{kind=link}
{kind=link}
1. Introduction
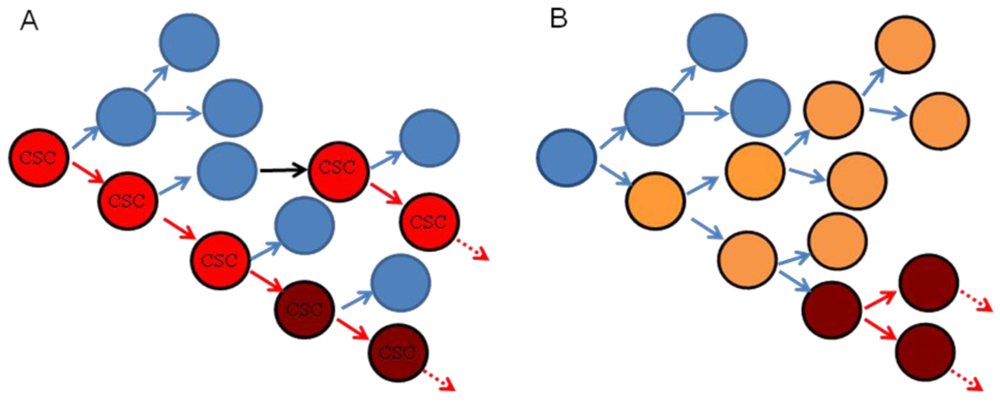
Head and neck cancer (HNC) is the sixth most common malignancy worldwide [1]. The head and neck is an anatomically complex region that gives rise to a wide variety of cancers varying in phenotype, histology, differentiation, tumorigenicity, and invasiveness [2]. Cancer heterogeneity has been explained by both clonal evolution and cancer stem cell models (Figure 1). In the clonal evolution model, all tumor cells are tumorigenic and heterogeneity arises from genetic or epigenetic changes occurring stochastically within individual clones [3]. The cancer stem cell (CSC) model proposes a hierarchical organization whereby tumor growth is dependent on CSCs that have the capacity for self renewal and to give rise to more differentiated tumor cells, analogous to the role of stem cells in normal tissue [3]. In this setting, small numbers of CSCs can produce a tumor recapitulating the heterogeneity of the original tumor when transplanted into immunocompromised mice [3]. In this model, differentiated daughter cells should not be tumorigenic and cannot recapitulate original tumor heterogeneity. It should be noted that the CSC model does not exclude the possibility that CSCs undergo clonal evolution, and thus these models are not mutually exclusive [3]. Furthermore, it is possible that different tumors follow either the clonal evolution or CSC hypothesis or some combination thereof.
Another important feature of CSCs is resistance to cytotoxic chemotherapy and ionizing radiation [4]. This has profound clinical implications as the clonal evolution model requires treatment directed at all tumor cells, while the CSC model suggests specifically targeting CSCs could affect the entire tumor. In fact, most conventional chemotherapeutic regimens do not kill CSCs and instead enrich the CSC population [4], presumably because CSCs are slowly cycling and over-express efflux pumps capable of extruding these toxic agents [5]. Indeed, the ability to efflux Hoechst 33342 dye has been used to identify CSC populations [6]. Accordingly, it is clinically relevant to understand whether given cancers follow a CSC hierarchy or a clonal evolution hypothesis. For example, Quintana et al. recently found that the majority of melanoma cells were tumorgenic, regardless of disease stage [7]. Furthermore, despite using an array of 22 different markers, they were unable to identify a substantial sub-population of non-tumorigenic cells, suggesting that melanoma is a striking example of a non-hierarchically organized cancer [7].
There has been controversy regarding the term ‘cancer stem cell’, as it seems to imply that CSCs have originated through mutation of normal stem cells, however, this is not necessarily true since CSCs might also arise from de-differentiation of tumor cells that acquire alterations imparting “stemness” [8]. Moreover, because ‘CSC’ terminology might imply pluripotency (like embryonic stem cells), some authors prefer the term ‘tumor initiating cells.’ However Gupta et al. have argued that since the identification and characterization of CSCs does not rely on their origin and CSCs are oligo-potent, if not pluripotent, these arguments do not contradict a CSC model [8].
A CSC model was first suggested in Acute Myeloid Leukemia (AML), where only a rare population of CD34+ CD38- cells in the peripheral blood collected from AML patients could engraft in immunodeficient mice and recapitulate morphological features of the original malignancy [9]. This landmark discovery, involving isolation of a tumor initiating population by flow cytometry using cell surface markers, paved the way for subsequent experiments in solid tumors, which pose additional challenges requiring dissociation of cells from the tumor. Subsequently, Al-Hajj et al. identified the tumor initiating (TI) population from human breast tumors and showed that as few as 100 CD44(+)CD24(-/low)Lineage(-) cells could form tumors while 10,000 cells of the opposite phenotype could not [10]. Work in other solid cancers has since provided evidence of CSCs in ovary, colon, prostate, pancreas and brain tumors [11-18].
In addition to cell surface markers, assays using the ability of CSCs to efflux dyes and aldehyde dehydrogenase 1 (ALDH) activity have been used to identify these cells [4,19]. Efflux pumps of the ATP-binding cassette transporter (ABC transporter) superfamily are overexpressed in CSCs, a property that also enables their isolation by flow cytometry [4]. ALDH is an enzyme responsible for detoxification of intracellular aldehydes [19]. Similar to the high ALDH activity possessed by neural and hematopoietic stem cells, CSCs in AML were found to share this phenotype [19].
Several in vitro assays have been used to assess CSC potential, including colony formation in soft agar and sphere formation in serum free media supplemented with growth factors [20]. The sphere formation assay is based on the neurosphere formation assay in which neural stem cells form clonally derived spheres under appropriate culture conditions [20]. The long-term proliferation potential of CSCs is assessed by dissociating tumor spheres into single cells, plating to limiting dilution, then serial passaging [21]. In HNC, CD44+ cells from the Gun-1 cell line express stem cell markers such as CD44 and ABCG2, show increased resistance to chemotherapeutic drugs, and can form spheres in vitro, however in vivo tumorigenicity of these cells has not been demonstrated [22]. CD133, a marker for both normal and CSC, has also been used in HNC to characterize a tumor initiating cell (TIC) population [21,23-26]. CD133+ cells from the oral SCC cell line UPCI: SCC-016 were characterized to possess cancer stem-like properties of increased tumorigenicity, chemoresistance to paclitaxel, and greater sphere-forming ability than the CD133- population. Moreover these spheres express the stem cell genes Oct-4, and hTERT [27]. The side populations (SP, capable of extruding Hoechst dye) of two highly metastatic HNSCC cell lines M3a2 and M4e had greater sphere formation ability than the non-SP cells and these spheres could be serially passaged [28].
One must bear in mind, however, that while in vitro assays can identify populations with stem cell characteristics they cannot replace the most stringent CSC test, the limiting dilution transplantation assay (LDA) in immunocompromised mice that determines the minimum number of CSCs for tumor formation. In fact, the heterogeneity in the fraction of CSCs in a given tumor is likely dependent on cancer-specific characteristics (stage, grade, tissue of origin), contribution of surrounding stromal elements (e.g. fibroblasts) and the immune-competence of the murine host used to assess tumorigenicity [8]. Consistent with previous observations in other cancers, the frequency of tumor initiating cells from HNC increases when transplanted into NOD/SCID mice lacking the interleukin 2 receptor γ chain (IL2Rγnull) compared to NOD/SCID mice [3,29]. NOD/SCID mice lacking the interleukin 2 receptor γ chain (IL2Rγnull), do not have natural killer cells and are more immunocompromised than NOD/SCID mice, which are only devoid of T and B cells [30].
The study of CSC in HNC has started fairly recently and has been reviewed previously, we have presented here an update to highlight some of the exciting new developments in the area of chemoresistance and metastatic ability of HN-TIC [31-33].
2. HNCSC Tumorigenicity
Prince and co-workers published the first report with evidence of CSCs in HNC when they identified a CD44+ population from primary human head and neck cancers that was tumorigenic in nude mice, could be serially passaged, and recapitulated the heterogeneity of the original human tumor [34]. These CD44+ tumor cells expressed basal cell markers, cytokeratin 5 and 14, and the stem cell gene BMI1, consistent with a progenitor function. In contrast, not only could CD44-cells not form tumors in nude mice, but these cells also exhibited features of more differentiated squamous epithelium such as involucrin expression. The fraction of CD44+ cells in the tumor was as high as 10%, and ∼5000 cells was required for tumor formation, suggesting that CD44+ cells are not a pure HNC stem cell population. There are several contradictory reports on the utility of CD44 as a marker for HN-TIC. CD44 is a transmembrane glycoprotein, with several isoforms that differ in their extracellular domains, and serve as a receptor for hyaluronan [35]. Since CD44 variants are known to play a causal role in metastasis in other cancers, therefore the expression of CD44 variants CD44v3, CD44v6 and CD44v10 has been studied in primary tumor and lymph node samples from HNC patients [36,37]. CD44v10 expression correlated with distant metastasis, whereas v3 and v6 were associated with regional and peri-neural metastasis, respectively [37]. Significant association of CD44v6 and v10 with shorter disease-free survival further highlights their importance in HNC [37]. However, other groups have shown that CD44 is expressed by a majority of tumor cells in HNSCC and also by normal HN epithelium as well as benign lesions of the head and neck [38]. A possible explanation for such contradictory results could be the specificity or lack thereof of the CD44 antibodies.
These observations highlight the need for more specific markers or combinations of markers and activity assays for identifying HNCSCs. In breast cancer, for example, the use of a combination of cell surface markers and activity assays, namely CD44+CD24-Lin-ALDH+, has enabled the enrichment of the CSC population such that as few as 20 CSCs form tumors in NOD/SCID mice [39]. Using a similar approach, 1000 CD44+CD24-ALDH+ cells from tumors of oral cancer patients were found to form tumors in SCID mice [40]. In this study, the lowest number of tumor cells implanted was 1000, however, in a separate study as few as 500 ALDH high cells from human HNSCC tumors exhibited tumorigenicity in NOD/SCID mice [41]. It should be noted that in the former study, the lowest number of tumor cells implanted was 1000 so it is not possible to directly compare the two studies in terms of tumorigenicity of the TIC populations.
The combined use of the ALDH and CD44 has recently enabled isolation of HNCSC from primary human tumors wherein 1000 (ALDH+CD44+Lin-) cells were shown to be tumorigenic in SCID mice [42]. These xenografted tumors could be serially passaged in mice, while a 10-fold higher number of ALDH-CD44-Lin-cells could not. Interestingly, these HNCSC were located in close proximity (within 100 μm, the radius of diffusion of oxygen and nutrients from blood vessels) to blood vessels in human tumors, reminiscent of neural and brain tumor stem cells that reside in perivascular niches [42-44]. Analogous to normal stem cells which reside in a protective microenvironment, termed ‘niche’, consisting of a number of cell types such as endothelial cells, and fibroblasts that ensure survival and quiescence, CSCs are also proposed to be dependent on their microenvironment [45,46]. Preliminary experiments suggest that endothelial cells in the niche promote the survival and self renewal of these HNCSC, presumably through secreted growth factors and signaling molecules [42]. These findings, if validated through longer-term in vivo experiments to assess the effect of anti-angiogenic therapeutics, may lead to better therapies for HNC patients.
Recently, glucose regulated protein 78 (GRP78) was used to identify HN-TIC from the HNSCC cell line, SAS [47]. GRP78 is an endoplasmic reticulum chaperone protein that is also expressed on the plasma membrane and is essential for survival of embryonic stem cells, presumably by acting in the ER stress response pathway [48]. GRP78 is overexpressed in several cancers including HNSCC, and co-expression of the stem cell marker Nanog with GRP78 is associated with reduced survival of HNSCC patients [47,49,50]. GRP78 is required for tumorigenicty, invasion, and metastasis of HNSCC and as few as 100 plasma membrane GRP78mem+ cells from a HNSCC cell line can form tumors in nude mice [47,51]. While the tumorigenicity of GRP78mem+ from primary human HNSCC remains to be demonstrated, GRP78mem+ cells from SAS, a HNSCC cell line, have stem-cell properties of self-renewal, differentiation and radioresistance. Importantly, knockdown of GRP78 reduces self-renewal and tumorigenicity in nude mice suggesting GRP78 is not merely a marker for HN-TIC but seems to be also involved in their stemness [47]. The molecular mechanism by which GRP78 mediates stemness of HNCSC is an exciting question that needs to be addressed.
3. CSC Chemoresistance and Radioresistance
The resistance of CSCs to chemotherapeutic agents has driven much research and CSCs typically overexpress ABC transporters that efflux these agents and are also associated with increased DNA repair and relative quiescence [4]. ABC transporter expression has facilitated CSC isolation by flow cytometry on the basis of their ability to efflux fluorescent dyes like Hoechst [52]. This population of Hoechst dye effluxing cells are referred to as ‘side population’ (SP) cells. SP cells isolated from laryngeal cancer cell lines are radioresistant, have increased in vivo tumorigenicity, and can give rise to both SP and non-SP progeny [53]. However, not all SP cells were tumorigenic, and some non-SP cells gave rise to SP cells, suggesting that the ability to efflux Hoechst dye, while characteristic of CSCs, is not limited exclusively to this cell population. This underscores the need to use a combination of cell surface markers in conjunction with dye exclusion or other activity assays to select and define CSC populations. SP cells isolated from aerodigestive squamous cell carcinoma cell lines are more tumorigenic than non-SP cells when transplanted into NOD/SCID mice and overexpress ABC transporters, ABCG2 and ABCC1 [54]. Importantly, the SP population is resistant to the cytotoxic drug mitoxantrone and this resistance can be reversed by chemical inhibition of ABC transporters [54]. SP populations have been characterized from several oral cancer cell lines and primary tumors from patients and were found to be more tumorigenic in mice, express the stem cell gene Bmi1, and ABC transporters in comparison to non-SP cells from the same source [54]. In accordance with the CSC model, the SP cells from an oral cancer cell line could give rise to both SP and non-SP cells under in vivo and in vitro conditions, whereas the non-SP cells could not generate SP cells [55]. In sum, these observations suggest that inhibition of ABC transporters may be a viable strategy for CSC targeting, and ABCB1 transporter inhibitors are in clinical trials for breast cancer, multiple myeloma and leukemia [56-58].
Because radiation therapy is commonly used to treat HNC, the radioresistance of HNCSCs is of immense interest. Mechanisms of radioresistance have been investigated in brain and breast tumors. Irradiation of tumor bearing mice enriches the rare population of CD133+ glioma stem cells in the brain [59]. Radiation preferentially activates the DNA damage checkpoint and DNA repair pathways in CD133+ cells isolated from human glioblastoma samples compared to CD133- cells, and CD133+ cell radioresistance requires the checkpoint kinases Chk1 and Chk2 [59]. Another mechanism of CSC radioresistance is increased free radical scavenging and reduced levels of reactive oxygen species (ROS); accordingly, pharmacological depletion of ROS scavengers radiosensitizes breast CSCs [60]. Whether HNCSC radioresistance is mediated through similar molecular mechanisms is unknown but holds promise for novel therapeutic strategies for HNC.
4. CSCs in Metastasis
Mortality in HNC often results from metastasis to regional lymph nodes and distant organs and CSCs may be critical purveyors of metastasis [61]. Since CSCs have been isolated from primary tumors as well as metastatic sites, an important question is if there are distinct subsets of CSCs that are either stationary or migratory [62]. If these are two distinct pools it will be necessary to therapeutically target both. In a pancreatic cancer cell line, two distinct populations, namely CD133+CXCR4+ and CD133+CXCR4-, are both tumorigenic but only the former migratory cells cause metastasis [14]. Significant progress has been made in colorectal cancer on this subject; screening of several cell surface markers led to the identification of CD26 as a marker for metastatic CSC. The cell surface glycoprotein CD26, is a multifunctional molecule with dipeptidyl peptidase activity and also interacts with extracellular matrix proteins, it plays an important role in tumor progression [63]. Transplantation of only CD26+ cells in the cecal wall of SCID mice gave rise to both cecal tumors and liver metastasis whereas CD26- cells could only form primary tumors and no metastasis occurred [64]. CD26 appears to be directly involved in metastases and CD26 knockdown decreases the expression of proteins related to epithelial to mesenchymal transition (EMT) and in vitro invasiveness [64]. This work has profound clinical implications for patient prognosis because the presence of CD26+ cells in primary tumors of patients predicted liver metastasis [64]. Preliminary work in HNSCC cell lines shows the highly metastatic M3a2 and M4e lines have almost 20-times more SP cells compared to their weakly metastatic parental cell line 686LN [28]. However additional work is required to assess the in vivo metastatic ability of these populations and answer the questions mentioned above. These SP populations did show stem-like characteristics of chemoresistance, increased in vitro invasiveness, and activation of Wnt/β-catenin signaling that is involved in stem cell self-renewal, as compared to non-SP cells [28].
EMT is a developmental program that, when activated in cancer cells, increases invasiveness and motility [65]. Given the importance of EMT in facilitating dissemination of cancer cells, a pertinent question is the molecular mechanism by which these cells in distant tissues self-renew in order to give rise to micrometastasis, and the contribution of EMT to the process of self renewal of metastatic CSCs. The answer to this important question came from the work of Mani and co-workers, who showed that induction of the EMT program in immortalized mammary epithelial cells causes them to acquire stem-like properties in addition to the mesenchymal phenotype [66]. Subsequent work cemented this connection by showing the EMT promoter ZEB1 also promotes stemness by repressing miRNAs that target stemness factors [67].
The connection between EMT and self-renewal has also been explored in HNC. ALDH+ cells isolated from tumors of head and neck patients exhibit stem-like properties such as greater tumorigenicity, sphere formation, and increased radioresistance compared to ALDH- cells [40]. These ALDH+ cells also have features consistent with EMT, namely loss of expression of epithelial markers like E-cadherin and acquisition of mesenchymal markers such as Vimentin and Snail [40]. Snail is a master regulator of EMT and controls invasiveness and metastasis in many cancers [68,69]. The stem-like properties of these ALDH+ HNCs was dependent on Snail expression, suggesting a causal link between EMT and stemness. [40].
Bmi1 is a transcriptional repressor regulating gene expression by changing chromatin structure; it regulates self-renewal of neural, hematopoietic and intestinal stem cells [70-73]. Bmi1 is also involved in HNSC carcinogenesis [55,74]. The ALDH+ HNSCC cells, while exhibiting stemness, also overexpress Bmi1 and Snail in comparison to the ALDH-cells [75]. The tumorigenicity, radioresistance and lung metastasis of these HNSCC ALDH+ cells was dependent on Bmi1 [75]. It is pertinent to point out that this is the first report of lung metastasis of HNC stem-like cells [75]. Clinically, co-expression of ALDH, Bmi1 and Snail predicted the worst prognosis in HNC patients [75]. This study further strengthened the link between EMT and stemness in HNC.
Seminal work by Yang and co-workers has provided the mechanistic link between EMT and self renewal in HNSCC [76]. The stemness factor Bmi1 is directly regulated by the EMT regulator, Twist. Using HNSCC cell lines they show that the transcription factors Bmi1 and Twist act cooperatively to regulate p16INK4a and E-cadherin, mediating self renewal and EMT, respectively. These observations provide the vital explanation underlying the worse prognosis of HNSCC patients overexpressing both Bmi1 and Twist as compared to those expressing either protein alone.
5. Conclusion and Future Perspectives
Research on HNCSC started fairly recently in 2007, with the use of CD44 as a marker for isolation of stem cells from human HN tumors [34]. Subsequent work has expanded the repertoire of cell surface markers and activity assays used for HNCSC isolation to include CD133, ALDH, Hoechst dye exclusion (SP), and GRP78 [27,40,47,54]. Proteomic analysis of the SAS HNSCC cell line grown in regular media and sphere cells (presumably a TIC population) led to identification of GRP78 as a putative HNCSC marker [47]. This study highlights the use of proteomics to identify novel markers. Additional markers are needed to isolate pure populations of HNCSC and combinations of makers and activity assays may provide further enrichment of the CSC pool.
Molecular characterization of gene expression by microarray analysis of the HNCSC populations has provided evidence of activation of the EMT program and of Wnt/β-catenin signaling in these cells [28,40]. The Wnt/β-catenin signaling pathway is involved in diverse aspects of normal stem cell biology such as maintenance of pluripotency, differentiation and proliferation [77-79]. This signaling pathway has also been implicated in CSCs, and inhibitors of this pathway are in clinical trials in several cancers [80]. These observations need to be extended to HNCSCs isolated from primary human tumor samples to identify additional signaling pathways that are preferentially activated in HNCSC, and may constitute novel therapeutic targets. In summary, HNC seems to follow the CSC model of tumorigenesis, though additional work is necessary for refinement of molecular mechanisms. Observations made in HNSCC cell lines need to be confirmed in patient samples.
References
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA Cancer J. Clin. 2005, 55, 74–108. [Google Scholar]
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. Lancet 2008, 371, 1695–1709. [Google Scholar]
- Shackleton, M.; Quintana, E.; Fearon, E.R.; Morrison, S.J. Heterogeneity in cancer: Cancer stem cells versus clonal evolution. Cell 2009, 138, 822–829. [Google Scholar]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar]
- Donnenberg, V.S.; Donnenberg, A.D. Multiple drug resistance in cancer revisited: The cancer stem cell hypothesis. J. Clin. Pharmaco. l 2005, 45, 872–877. [Google Scholar]
- Wu, C.; Alman, B.A. Side population cells in human cancers. Cancer Lett. 2008, 268, 1–9. [Google Scholar]
- Quintana, E.; Shackleton, M.; Foster, H.R.; Fullen, D.R.; Sabel, M.S.; Johnson, T.M.; Morrison, S.J. Phenotypic heterogeneity among tumorigenic melanoma cells from patients that is reversible and not hierarchically organized. Cancer Cell 2010, 18, 510–523. [Google Scholar]
- Gupta, P.B.; Chaffer, C.L.; Weinberg, R.A. Cancer stem cells: Mirage or reality? Nat. Med. 2009, 15, 1010–1012. [Google Scholar]
- Lapidot, T.; Sirard, C.; Vormoor, J.; Murdoch, B.; Hoang, T.; Caceres-Cortes, J.; Minden, M.; Paterson, B.; Caligiuri, M.A.; Dick, J.E. A cell initiating human acute myeloid leukaemia after transplantation into scid mice. Nature 1994, 367, 645–648. [Google Scholar]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar]
- Curley, M.D.; Therrien, V.A.; Cummings, C.L.; Sergent, P.A.; Koulouris, C.R.; Friel, A.M.; Roberts, D.J.; Seiden, M.V.; Scadden, D.T.; Rueda, B.R.; Foster, R. Cd133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells 2009, 27, 2875–2883. [Google Scholar]
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; Shelton, A.A.; Parmiani, G.; Castelli, C.; Clarke, M.F. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar]
- Hermann, P.C.; Huber, S.L.; Herrler, T.; Aicher, A.; Ellwart, J.W.; Guba, M.; Bruns, C.J.; Heeschen, C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell 2007, 1, 313–323. [Google Scholar]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar]
- O'Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar]
- Ma, I.; Allan, A.L. The role of human aldehyde dehydrogenase in normal and cancer stem cells. Stem Cell Rev. 2010. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P.B. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003, 63, 5821–5828. [Google Scholar]
- Okamoto, A.; Chikamatsu, K.; Sakakura, K.; Hatsushika, K.; Takahashi, G.; Masuyama, K. Expansion and characterization of cancer stem-like cells in squamous cell carcinoma of the head and neck. Oral Oncol. 2009, 45, 633–639. [Google Scholar]
- Ito, Y.; Hamazaki, T.S.; Ohnuma, K.; Tamaki, K.; Asashima, M.; Okochi, H. Isolation of murine hair-inducing cells using the cell surface marker prominin-1/cd133. J. Invest. Dermatol. 2007, 127, 1052–1060. [Google Scholar]
- Miraglia, S.; Godfrey, W.; Yin, A.H.; Atkins, K.; Warnke, R.; Holden, J.T.; Bray, R.A.; Waller, E.K.; Buck, D.W. A novel five-transmembrane hematopoietic stem cell antigen: Isolation, characterization, and molecular cloning. Blood 1997, 90, 5013–5021. [Google Scholar]
- Uchida, N.; Buck, D.W.; He, D.; Reitsma, M.J.; Masek, M.; Phan, T.V.; Tsukamoto, A.S.; Gage, F.H.; Weissman, I.L. Direct isolation of human central nervous system stem cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14720–14725. [Google Scholar]
- Yin, A.H.; Miraglia, S.; Zanjani, E.D.; Almeida-Porada, G.; Ogawa, M.; Leary, A.G.; Olweus, J.; Kearney, J.; Buck, D.W. Ac133, a novel marker for human hematopoietic stem and progenitor cells. Blood 1997, 90, 5002–5012. [Google Scholar]
- Zhang, Q.; Shi, S.; Yen, Y.; Brown, J.; Ta, J.Q.; Le, A.D. A subpopulation of cd133(+) cancer stem-like cells characterized in human oral squamous cell carcinoma confer resistance to chemotherapy. Cancer Lett. 2010, 289, 151–160. [Google Scholar]
- Song, J.; Chang, I.; Chen, Z.; Kang, M.; Wang, C.Y. Characterization of side populations in hnscc: Highly invasive, chemoresistant and abnormal wnt signaling. PLoS One 2010, 5, e11456. [Google Scholar]
- Ishizawa, K.; Rasheed, Z.A.; Karisch, R.; Wang, Q.; Kowalski, J.; Susky, E.; Pereira, K.; Karamboulas, C.; Moghal, N.; Rajeshkumar, N.V.; Hidalgo, M.; Tsao, M.; Ailles, L.; Waddell, T.K.; Maitra, A.; Neel, B.G.; Matsui, W. Tumor-initiating cells are rare in many human tumors. Cell Stem Cell 2010, 7, 279–282. [Google Scholar]
- McKenzie, J.L.; Gan, O.I.; Doedens, M.; Dick, J.E. Human short-term repopulating stem cells are efficiently detected following intrafemoral transplantation into nod/scid recipients depleted of cd122+ cells. Blood 2005, 106, 1259–1261. [Google Scholar]
- Monroe, M.M.; Anderson, E.C.; Clayburgh, D.R.; Wong, M.H. Cancer stem cells in head and neck squamous cell carcinoma. J. Oncol. 2011. Article ID 762780. [Google Scholar] [CrossRef]
- Prince, M.E.; Ailles, L.E. Cancer stem cells in head and neck squamous cell cancer. J. Clin. Oncol. 2008, 26, 2871–2875. [Google Scholar]
- Chen, Z.G. The cancer stem cell concept in progression of head and neck cancer. J. Oncol. 2009. Article ID 894064. [Google Scholar] [CrossRef]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar]
- Ponta, H.; Sherman, L.; Herrlich, P.A. Cd44: From adhesion molecules to signalling regulators. Nat. Rev. Mol. Cell. Biol. 2003, 4, 33–45. [Google Scholar]
- Gunthert, U.; Hofmann, M.; Rudy, W.; Reber, S.; Zoller, M.; Haussmann, I.; Matzku, S.; Wenzel, A.; Ponta, H.; Herrlich, P. A new variant of glycoprotein cd44 confers metastatic potential to rat carcinoma cells. Cell 1991, 65, 13–24. [Google Scholar]
- Wang, S.J.; Wong, G.; de Heer, A.M.; Xia, W.; Bourguignon, L.Y. Cd44 variant isoforms in head and neck squamous cell carcinoma progression. Laryngoscope 2009, 119, 1518–1530. [Google Scholar]
- Mack, B.; Gires, O. Cd44s and cd44v6 expression in head and neck epithelia. PLoS One 2008, 3, e3360. [Google Scholar]
- Ginestier, C.; Hur, M.H.; Charafe-Jauffret, E.; Monville, F.; Dutcher, J.; Brown, M.; Jacquemier, J.; Viens, P.; Kleer, C.G.; Liu, S.; Schott, A.; Hayes, D.; Birnbaum, D.; Wicha, M.S.; Dontu, G. Aldh1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007, 1, 555–567. [Google Scholar]
- Chen, Y.C.; Chen, Y.W.; Hsu, H.S.; Tseng, L.M.; Huang, P.I.; Lu, K.H.; Chen, D.T.; Tai, L.K.; Yung, M.C.; Chang, S.C.; Ku, H.H.; Chiou, S.H.; Lo, W.L. Aldehyde dehydrogenase 1 is a putative marker for cancer stem cells in head and neck squamous cancer. Biochem. Biophys. Res. Commun. 2009, 385, 307–313. [Google Scholar]
- Clay, M.R.; Tabor, M.; Owen, J.H.; Carey, T.E.; Bradford, C.R.; Wolf, G.T.; Wicha, M.S.; Prince, M.E. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck 2010, 32, 1195–1201. [Google Scholar]
- Krishnamurthy, S.; Dong, Z.; Vodopyanov, D.; Imai, A.; Helman, J.I.; Prince, M.E.; Wicha, M.S.; Nor, J.E. Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer Res. 2010, 70, 9969–9978. [Google Scholar]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; Frank, A.; Bayazitov, I.T.; Zakharenko, S.S.; Gajjar, A.; Davidoff, A.; Gilbertson, R.J. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar]
- Shen, Q.; Goderie, S.K.; Jin, L.; Karanth, N.; Sun, Y.; Abramova, N.; Vincent, P.; Pumiglia, K.; Temple, S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 2004, 304, 1338–1340. [Google Scholar]
- LaBarge, M.A. The difficulty of targeting cancer stem cell niches. Clin. Cancer. Res. 2010, 16, 3121–3129. [Google Scholar]
- Moore, K.A.; Lemischka, I.R. Stem cells and their niches. Science 2006, 311, 1880–1885. [Google Scholar]
- Wu, M.J.; Jan, C.I.; Tsay, Y.G.; Yu, Y.H.; Huang, C.Y.; Lin, S.C.; Liu, C.J.; Chen, Y.S.; Lo, J.F.; Yu, C.C. Elimination of head and neck cancer initiating cells through targeting glucose regulated protein78 signaling. Mol. Cancer 2010, 9, 283. [Google Scholar]
- Luo, S.; Mao, C.; Lee, B.; Lee, A.S. Grp78/bip is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell. Biol. 2006, 26, 5688–5697. [Google Scholar]
- Lee, E.; Nichols, P.; Spicer, D.; Groshen, S.; Yu, M.C.; Lee, A.S. Grp78 as a novel predictor of responsiveness to chemotherapy in breast cancer. Cancer Res. 2006, 66, 7849–7853. [Google Scholar]
- Uramoto, H.; Sugio, K.; Oyama, T.; Nakata, S.; Ono, K.; Yoshimastu, T.; Morita, M.; Yasumoto, K. Expression of endoplasmic reticulum molecular chaperone grp78 in human lung cancer and its clinical significance. Lung Cancer 2005, 49, 55–62. [Google Scholar]
- Chiu, C.C.; Lin, C.Y.; Lee, L.Y.; Chen, Y.J.; Kuo, T.F.; Chang, J.T.; Liao, C.T.; Wang, H.M.; Yen, T.C.; Shen, C.R.; Liao, S.K.; Cheng, A.J. Glucose-regulated protein 78 regulates multiple malignant phenotypes in head and neck cancer and may serve as a molecular target of therapeutic intervention. Mol. Cancer Ther. 2008, 7, 2788–2797. [Google Scholar]
- Goodell, M.A.; Brose, K.; Paradis, G.; Conner, A.S.; Mulligan, R.C. Isolation and functional properties of murine hematopoietic stem cells that are replicating in vivo. J. Exp. Med. 1996, 183, 1797–1806. [Google Scholar]
- Wan, G.; Zhou, L.; Xie, M.; Chen, H.; Tian, J. Characterization of side population cells from laryngeal cancer cell lines. Head Neck 2010, 32, 1302–1309. [Google Scholar]
- Loebinger, M.R.; Giangreco, A.; Groot, K.R.; Prichard, L.; Allen, K.; Simpson, C.; Bazley, L.; Navani, N.; Tibrewal, S.; Davies, D.; Janes, S.M. Squamous cell cancers contain a side population of stem-like cells that are made chemosensitive by abc transporter blockade. Br. J. Cancer 2008, 98, 380–387. [Google Scholar]
- Zhang, P.; Zhang, Y.; Mao, L.; Zhang, Z.; Chen, W. Side population in oral squamous cell carcinoma possesses tumor stem cell phenotypes. Cancer Lett. 2009, 277, 227–234. [Google Scholar]
- Belpomme, D.; Gauthier, S.; Pujade-Lauraine, E.; Facchini, T.; Goudier, M.J.; Krakowski, I.; Netter-Pinon, G.; Frenay, M.; Gousset, C.; Marie, F.N.; Benmiloud, M.; Sturtz, F. Verapamil increases the survival of patients with anthracycline-resistant metastatic breast carcinoma. Ann. Oncol. 2000, 11, 1471–1476. [Google Scholar]
- List, A.F.; Kopecky, K.J.; Willman, C.L.; Head, D.R.; Persons, D.L.; Slovak, M.L.; Dorr, R.; Karanes, C.; Hynes, H.E.; Doroshow, J.H.; Shurafa, M.; Appelbaum, F.R. Benefit of cyclosporine modulation of drug resistance in patients with poor-risk acute myeloid leukemia: A southwest oncology group study. Blood 2001, 98, 3212–3220. [Google Scholar]
- Sonneveld, P.; Suciu, S.; Weijermans, P.; Beksac, M.; Neuwirtova, R.; Solbu, G.; Lokhorst, H.; van der Lelie, J.; Dohner, H.; Gerhartz, H.; Segeren, C.M.; Willemze, R.; Lowenberg, B. Cyclosporin a combined with vincristine, doxorubicin and dexamethasone (vad) compared with vad alone in patients with advanced refractory multiple myeloma: An eortc-hovon randomized phase iii study (06914). Br. J. Haematol. 2001, 115, 895–902. [Google Scholar]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; Joshua, B.; Kaplan, M.J.; Wapnir, I.; Dirbas, F.M.; Somlo, G.; Garberoglio, C.; Paz, B.; Shen, J.; Lau, S.K.; Quake, S.R.; Brown, J.M.; Weissman, I.L.; Clarke, M.F. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar]
- Lo, W.L.; Kao, S.Y.; Chi, L.Y.; Wong, Y.K.; Chang, R.C. Outcomes of oral squamous cell carcinoma in taiwan after surgical therapy: Factors affecting survival. J. Oral. Maxillofac. Surg. 2003, 61, 751–758. [Google Scholar]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Opinion: Migrating cancer stem cells - an integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar]
- Pro, B.; Dang, N.H. Cd26/dipeptidyl peptidase iv and its role in cancer. Histol. Histopathol. 2004, 19, 1345–1351. [Google Scholar]
- Pang, R.; Law, W.L.; Chu, A.C.; Poon, J.T.; Lam, C.S.; Chow, A.K.; Ng, L.; Cheung, L.W.; Lan, X.R.; Lan, H.Y.; Tan, V.P.; Yau, T.C.; Poon, R.T.; Wong, B.C. A subpopulation of cd26+ cancer stem cells with metastatic capacity in human colorectal cancer. Cell Stem Cell 6, 603–615.
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; Campbell, L.L.; Polyak, K.; Brisken, C.; Yang, J.; Weinberg, R.A. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; Brunton, V.G.; Morton, J.; Sansom, O.; Schuler, J.; Stemmler, M.P.; Herzberger, C.; Hopt, U.; Keck, T.; Brabletz, S.; Brabletz, T. The emt-activator zeb1 promotes tumorigenicity by repressing stemness-inhibiting micrornas. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia De Herreros, A. The transcription factor snail is a repressor of e-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar]
- Zhou, B.P.; Deng, J.; Xia, W.; Xu, J.; Li, Y.M.; Gunduz, M.; Hung, M.C. Dual regulation of snail by gsk-3beta-mediated phosphorylation in control of epithelial-mesenchymal transition. Nat. Cell Biol. 2004, 6, 931–940. [Google Scholar]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.K.; Clarke, M.F.; Morrison, S.J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003, 425, 962–967. [Google Scholar]
- Park, I.K.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003, 423, 302–305. [Google Scholar]
- Sangiorgi, E.; Capecchi, M.R. Bmi1 is expressed in vivo in intestinal stem cells. Nat. Genet. 2008, 40, 915–920. [Google Scholar]
- Widschwendter, M.; Fiegl, H.; Egle, D.; Mueller-Holzner, E.; Spizzo, G.; Marth, C.; Weisenberger, D.J.; Campan, M.; Young, J.; Jacobs, I.; Laird, P.W. Epigenetic stem cell signature in cancer. Nat. Genet. 2007, 39, 157–158. [Google Scholar]
- Brunner, M.; Thurnher, D.; Pammer, J.; Geleff, S.; Heiduschka, G.; Reinisch, C.M.; Petzelbauer, P.; Erovic, B.M. Expression of vegf-a/c, vegf-r2, pdgf-alpha/beta, c-kit, egfr, her-2/neu, mcl-1 and bmi-1 in merkel cell carcinoma. Mod. Pathol. 2008, 21, 876–884. [Google Scholar]
- Yu, C.C.; Lo, W.L.; Chen, Y.W.; Huang, P.I.; Hsu, H.S.; Tseng, L.M.; Hung, S.C.; Kao, S.Y.; Chang, C.J.; Chiou, S.H. Bmi-1 regulates snail expression and promotes metastasis ability in head and neck squamous cancer-derived aldh1 positive cells. J. Oncol. 2011. Article ID 609259. [Google Scholar] [CrossRef]
- Yang, M.H.; Hsu, D.S.; Wang, H.W.; Wang, H.J.; Lan, H.Y.; Yang, W.H.; Huang, C.H.; Kao, S.Y.; Tzeng, C.H.; Tai, S.K.; Chang, S.Y.; Lee, O.K.; Wu, K.J. Bmi1 is essential in twist1-induced epithelial-mesenchymal transition. Nat. Cell Biol. 2010, 12, 982–992. [Google Scholar]
- Chenn, A.; Walsh, C.A. Regulation of cerebral cortical size by control of cell cycle exit in neural precursors. Science 2002, 297, 365–369. [Google Scholar]
- Sato, N.; Meijer, L.; Skaltsounis, L.; Greengard, P.; Brivanlou, A.H. Maintenance of pluripotency in human and mouse embryonic stem cells through activation of wnt signaling by a pharmacological gsk-3-specific inhibitor. Nat. Med. 2004, 10, 55–63. [Google Scholar]
- Zechner, D.; Fujita, Y.; Hulsken, J.; Muller, T.; Walther, I.; Taketo, M.M.; Crenshaw, E.B., 3rd.; Birchmeier, W.; Birchmeier, C. Beta-catenin signals regulate cell growth and the balance between progenitor cell expansion and differentiation in the nervous system. Dev. Biol. 2003, 258, 406–418. [Google Scholar]
- Takahashi-Yanaga, F.; Kahn, M. Targeting wnt signaling: Can we safely eradicate cancer stem cells? Clin. Cancer Res. 2010, 16, 3153–3162. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mitra, D.; Malkoski, S.P.; Wang, X.-J. Cancer Stem Cells in Head and Neck Cancer. Cancers 2011, 3, 415-427. https://doi.org/10.3390/cancers3010415
Mitra D, Malkoski SP, Wang X-J. Cancer Stem Cells in Head and Neck Cancer. Cancers. 2011; 3(1):415-427. https://doi.org/10.3390/cancers3010415
Chicago/Turabian StyleMitra, Doyel, Stephen P. Malkoski, and Xiao-Jing Wang. 2011. "Cancer Stem Cells in Head and Neck Cancer" Cancers 3, no. 1: 415-427. https://doi.org/10.3390/cancers3010415