Targeting of Both the c-Met and EGFR Pathways Results in Additive Inhibition of Lung Tumorigenesis in Transgenic Mice

Abstract
:
1. Introduction
2. Results and Discussion
2.1. Serum Analysis for Circulating c-Met and EGFR Ligands in Lung Cancer Cases and Controls

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Control | Total | p-value* | |||||
|---|---|---|---|---|---|---|---|---|
| N | (%) | N | (%) | N | (%) | |||
| TOTAL Number of subjects | 43 | 28 | 71 | |||||
| SEX | ||||||||
| male | 23 | (53%) | 13 | (46%) | 36 | (51%) | 0.631 | |
| female | 20 | (47%) | 15 | (54%) | 35 | (49%) | ||
| SMOKING STATUS | ||||||||
| current smoker | 20 | (47%) | 14 | (50%) | 34 | (48%) | 1.00 | |
| ex-smoker | 18 | (42%) | 14 | (50%) | 32 | (45%) | ||
| never smoker | 1 | (2%) | 0 | (0%) | 1 | (1%) | ||
| smoker, NOS | 1 | (2%) | 0 | (0%) | 1 | (1%) | ||
| unknown | 3 | (7%) | 0 | (0%) | 3 | (4%) | ||
| COPD/EMPHYSEMA | ||||||||
| yes | 21 | (49%) | 14 | (50%) | 35 | (49%) | 0.456 | |
| no | 14 | (33%) | 14 | (50%) | 28 | (39%) | ||
| unknown | 8 | (19%) | 0 | (0%) | 8 | (11%) | ||
| HISTOLOGY | ||||||||
| squamous | 18 | (42%) | NA | 18 | (25%) | |||
| adenocarcinoma, adeno-squamous | 17 | (40%) | 17 | (24%) | ||||
| NSCLC, small cell, giant cell | 8 | (19%) | 8 | (11%) | ||||
| NA = 28 | (39%) | |||||||
| STAGE | ||||||||
| I | 17 | (40%) | NA | 17 | (24%) | |||
| II | 11 | (26%) | 11 | (15%) | ||||
| III-IV | 14 | (33%) | 14 | (20%) | ||||
| unknown | 1 | (2%) | 1 | (1%) | ||||
| NA = 28 | (39%) | |||||||
| AGE | ||||||||
| mean | 66.81 | 61.75 | 64.82 | 0.036 | ||||
| std.dev | 11.13 | 7.05 | 9.99 | |||||
| median | 67.00 | 65.00 | ||||||
| (min, max) | (38.0, 92.0) | (51.0, 71.0) | (38.0, 92.0) | |||||
| HGF | Group | N | Median (Range) | p-Value |
| Case | 43 | 1657.50 (1020.00–4855.00) | <0.001 | |
| Control | 28 | 1025.72 (592.08–4029.94) | ||
| AREG | Group | N | Median (Range) | p-Value |
| Case | 43 | 25.19 (6.89–37.10) | <0.001 | |
| Control | 28 | 1.33 (0.00–204.30) |
| HGF: Probability of High HGF (HGF ≥ 1510 pg/mL) | |||||||
| Parameter | DF | Wald | Wald 95% | ||||
| Chi-Square | Pr > ChiSq | Odds Ratio | Confidence Limits | ||||
| Intercept | 1 | 2.400 | 0.1219 | ||||
| CASE/CTRL | Case | 1 | 16.000 | <0.0001 | 23.250 | 4.976 | 108.630 |
| AGE | 1 | 1.950 | 0.163 | 1.061 | 0.976 | 1.152 | |
| SEX | Female | 1 | 1.960 | 0.161 | 0.357 | 0.085 | 1.508 |
| SMOKING STATUS | Current Smoker | 1 | 2.770 | 0.096 | 3.825 | 0.788 | 18.558 |
| COPD | COPD | 1 | 0.150 | 0.703 | 1.352 | 2.87 | 6.37 |
| AREG: Probability of High AREG (AREG ≥ 23.8 pg/mL) | |||||||
| Parameter | DF | Wald | Wald 95% | ||||
| Chi-Square | Pr > ChiSq | Odds Ratio | Confidence Limits | ||||
| Intercept | 1 | 0.074 | 0.785 | ||||
| CASE/CTRL | Case | 1 | 20.802 | <0.0001 | 43.048 | 8.547 | 216.83 |
| AGE | 1 | 0.155 | 0.694 | 0.982 | 0.898 | 1.074 | |
| SEX | Female | 1 | 0.192 | 0.662 | 0.722 | 0.168 | 3.102 |
| SMOKING STATUS | Current Smoker | 1 | 0.660 | 0.417 | 1.986 | 0.380 | 10.393 |
| COPD | COPD | 1 | 0.047 | 0.829 | 1.194 | 0.238 | 5.983 |
| TGFα: Probability of High TGFα (TGFα > 0 pg/mL) | |||||||
| Parameter | DF | Wald | Wald 95% | ||||
| Chi-Square | Pr > ChiSq | Odds Ratio | Confidence Limits | ||||
| Intercept | 1 | 4.602 | 0.032 | ||||
| CASE/CTRL | Case | 1 | 1.065 | 0.302 | 0.510 | 0.142 | 1.833 |
| AGE | 1 | 6.001 | 0.014 | 0.889 | 0.809 | 0.977 | |
| SEX | Female | 1 | 2.804 | 0.094 | 0.325 | 0.087 | 1.211 |
| SMOKING STATUS | Current Smoker | 1 | 0.091 | 0.763 | 0.806 | 0.199 | 3.267 |
| COPD | COPD | 1 | 5.803 | 0.016 | 9.193 | 1.512 | 55.891 |
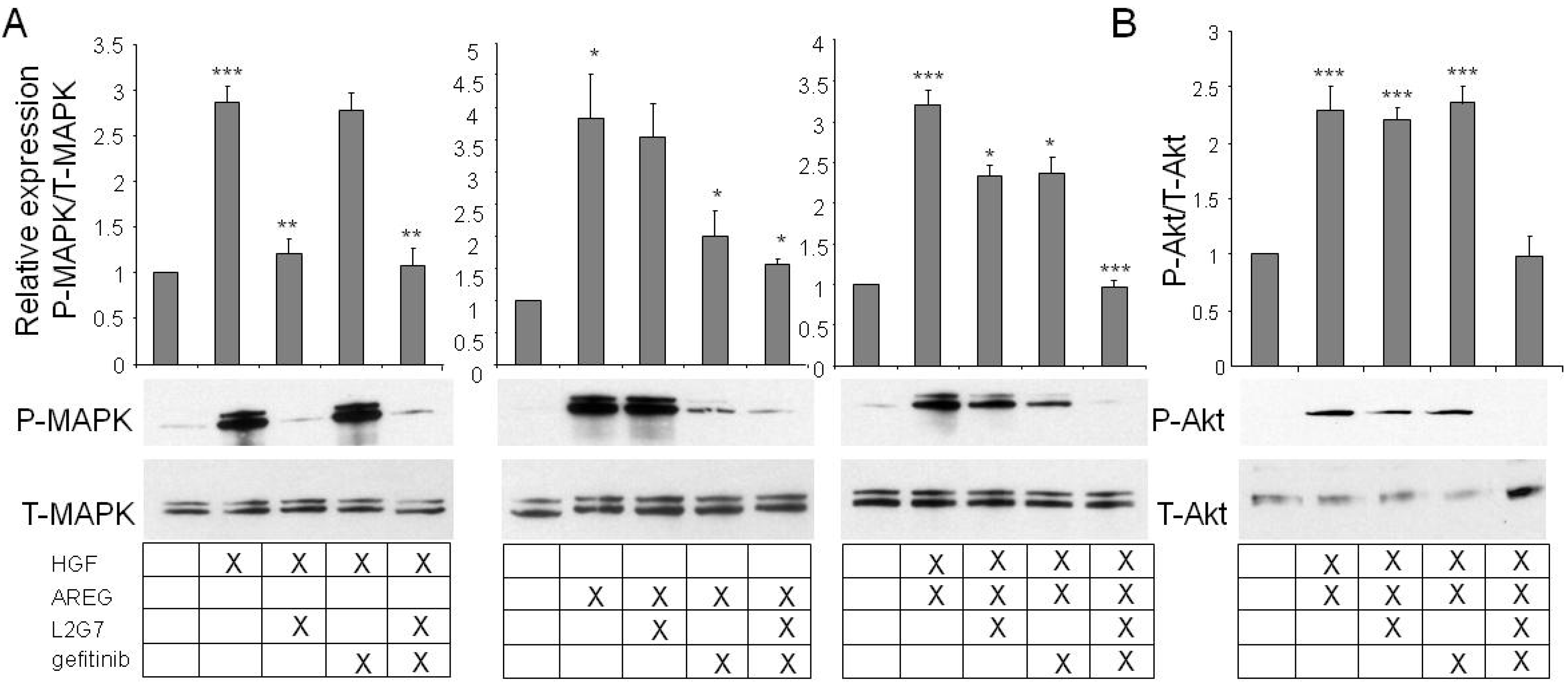
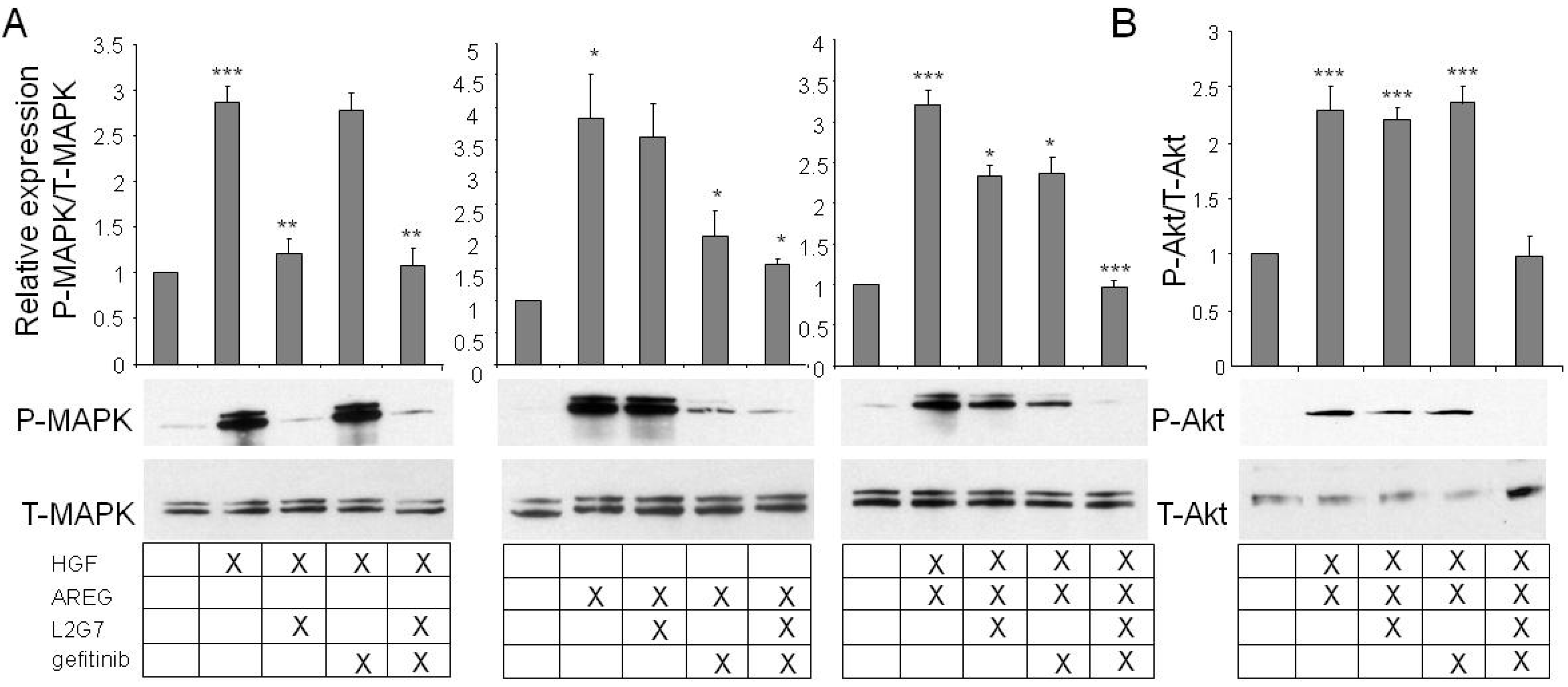
2.2. In the Presence of both c-Met and EGFR Ligands, Inhibitors of Both Pathways Are Necessary for Full Signaling Inhibition
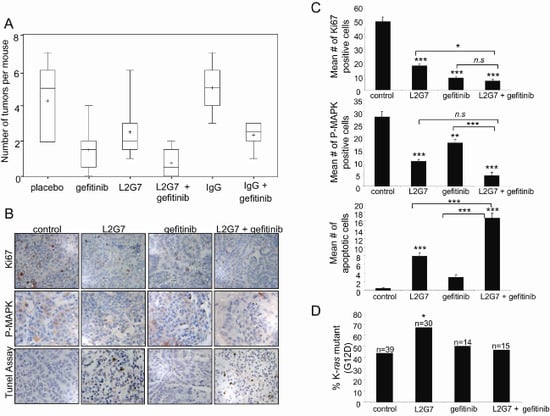
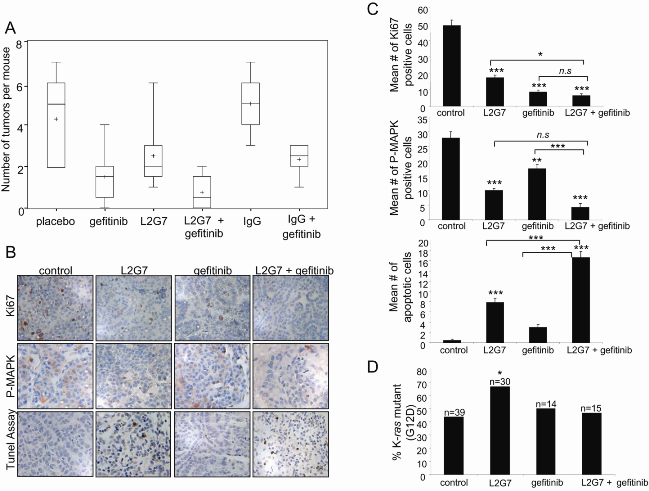
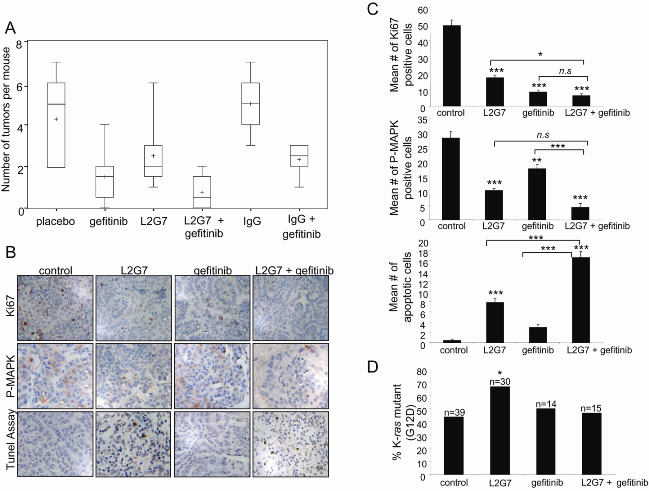
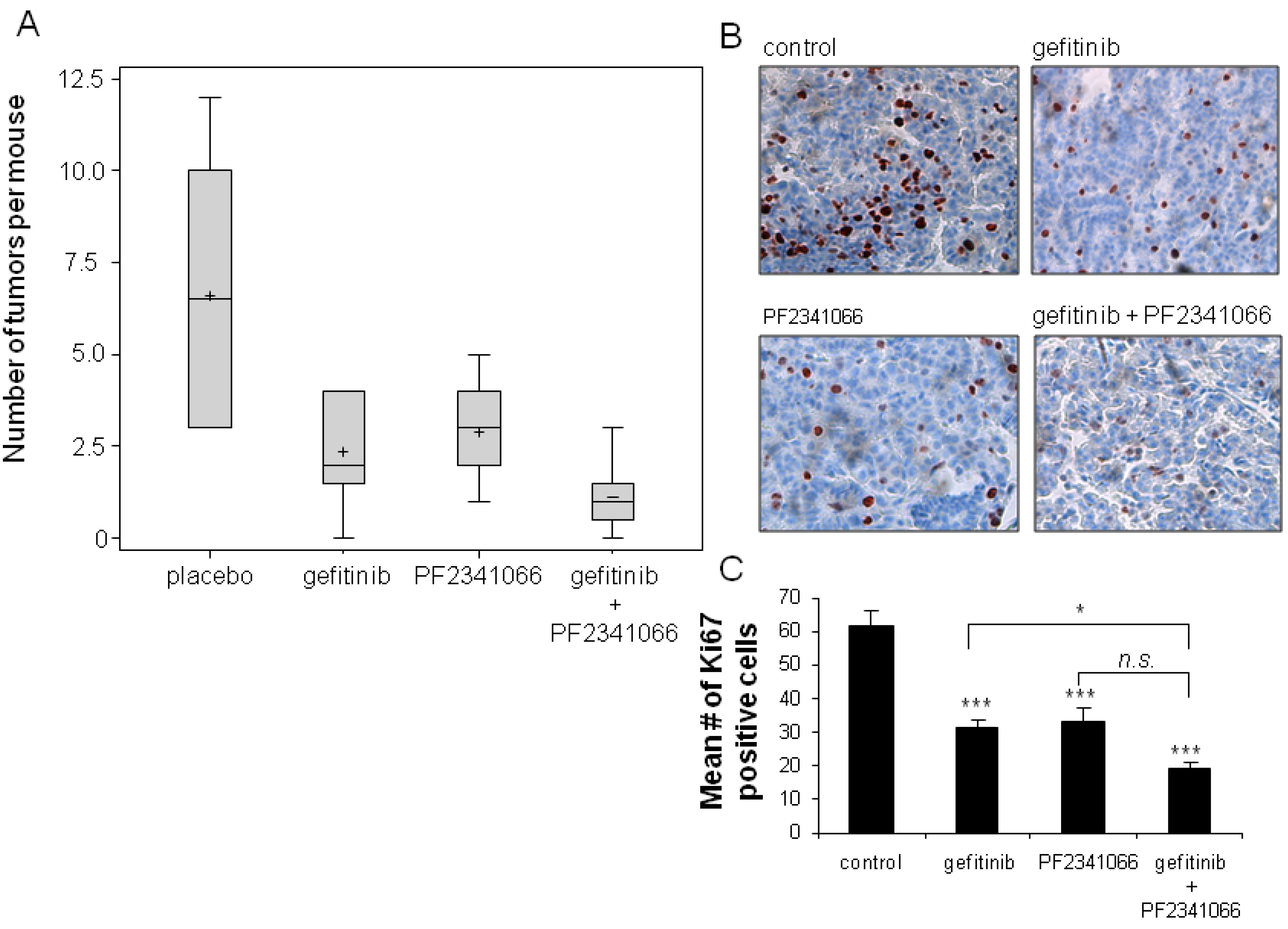
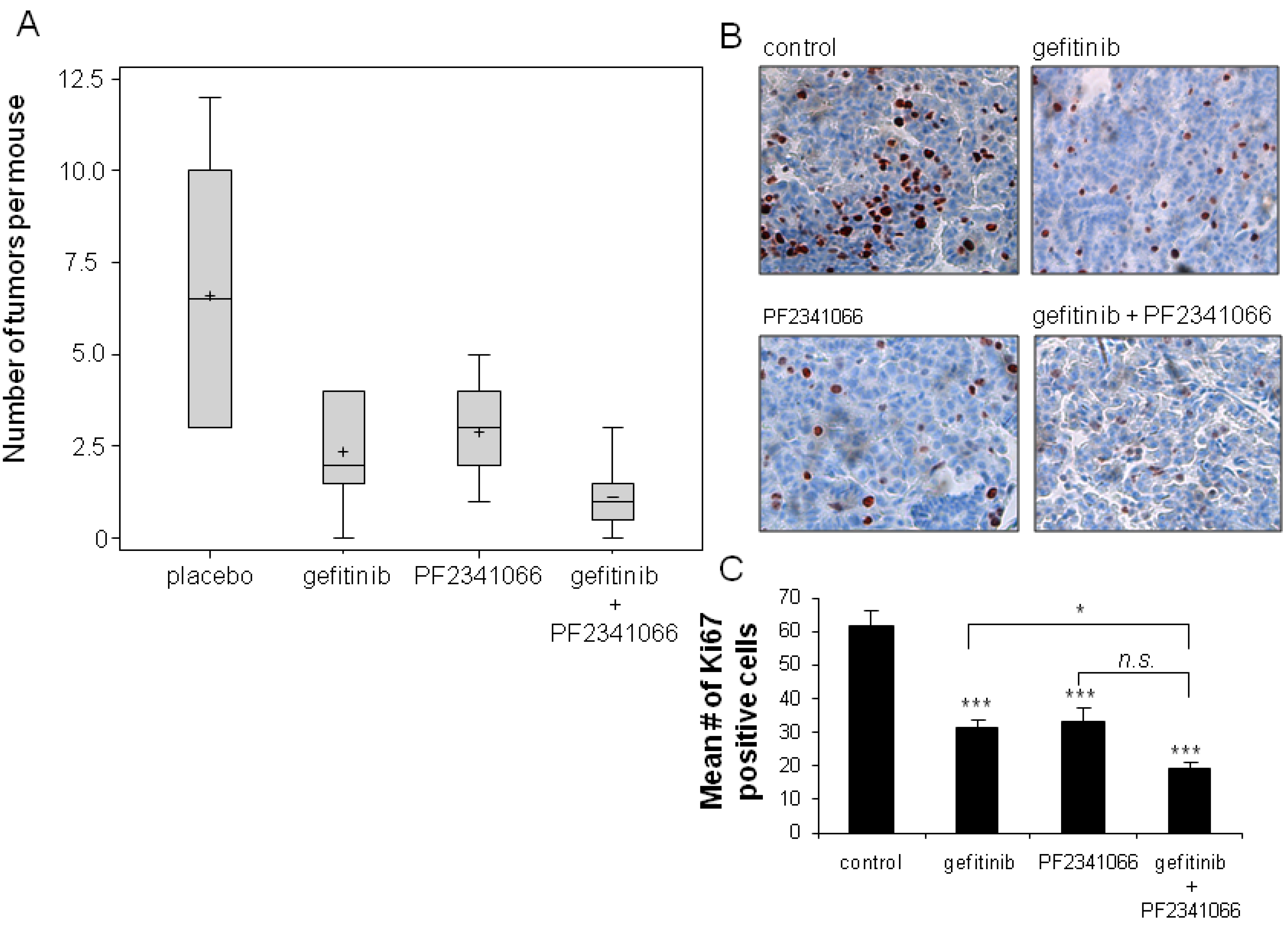
2.3. Inhibition of Tumorigenesis is Maximal with Combination Treatment
2.4. Maximum Inhibition of Indicators of Lung Tumor Growth and Survival Is Observed with Dual Therapy
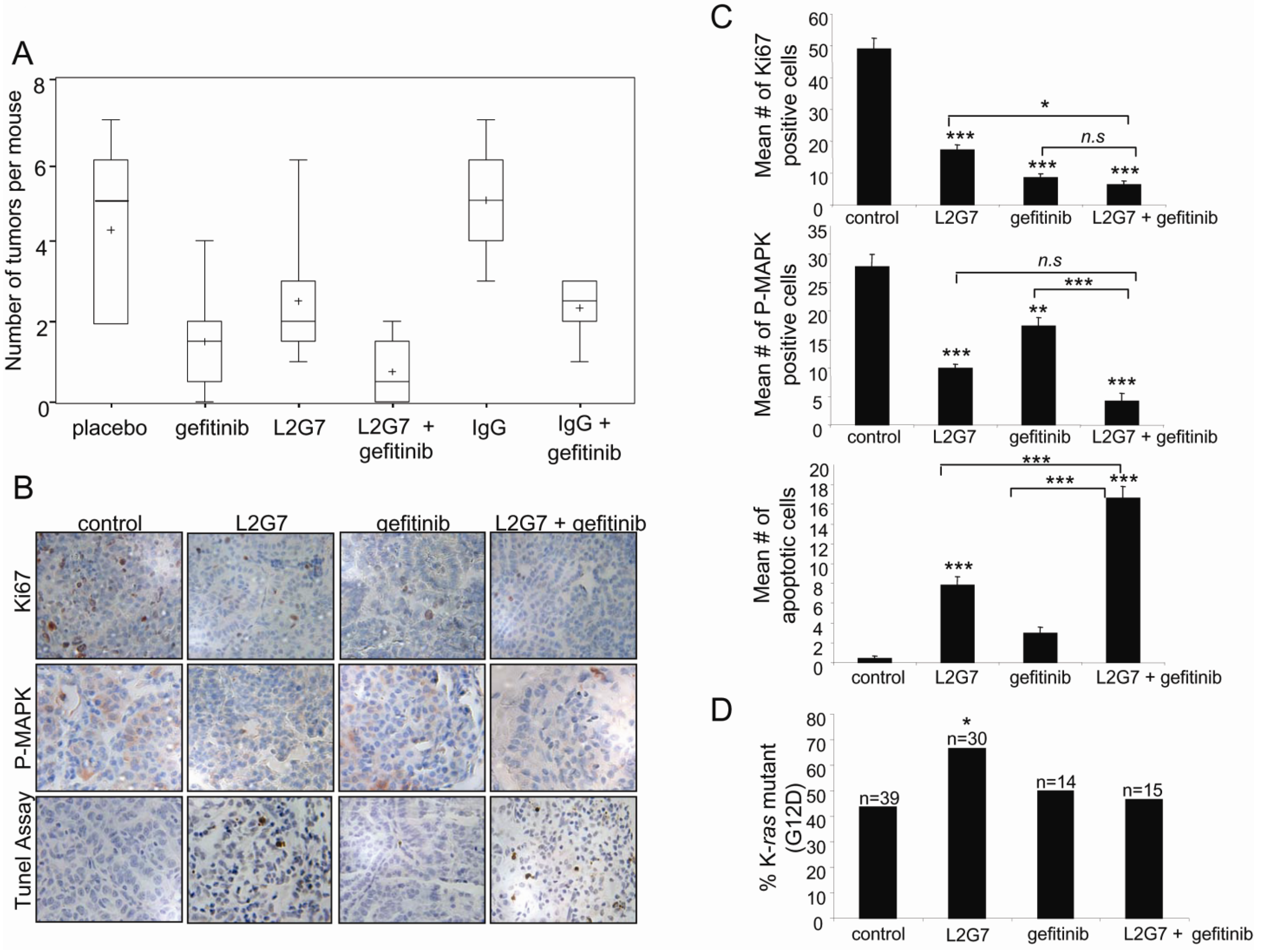
2.5. K-ras Mutant Tumors Are Partially Resistant to HGF Blockade
2.6. Confirmation of Dual Targeting
3. Experimental Section
3.1. Reagents
3.2. Western Analysis
3.3. Mouse Model
3.4. Immunohistochemistry (IHC)
3.5. Laser Capture Microdissection of Tumors and K-ras Mutation Analysis
3.6. Ligand Analysis in Human Serum
3.7. Statistical Analysis
4. Conclusions
Acknowledgements
References
- Tsao, M.S.; Liu, N.; Chen, J.R.; Pappas, J.; Ho, J.; To, C.; Viallet, J.; Park, M.; Zhu, H. Differential expression of Met/hepatocyte growth factor receptor in subtypes of non-small cell lung cancers. Lung Cancer 1998, 20, 1–16. [Google Scholar] [CrossRef]
- Selvaggi, G.; Novello, S.; Torri, V.; Leonardo, E.; De Giuli, P.; Borasio, P.; Mossetti, C.; Ardissone, F.; Lausi, P.; Scagliotti, G.V. Epidermal growth factor receptor overexpression correlates with a poor prognosis in completely resected non-small-cell lung cancer. Ann. Oncol. 2004, 15, 28–32. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haseriat, S.M.; Supko, J.G.; Haluska, F.G.; Louis, D.N.; Christiani, D.C.; Settleman, J.; Haber, D.A. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lineman, N.; Boggon, T.J.; Naoki, K.; Sasaki, H.; Fuji, Y.; Eck, M.J.; Sellers, W.R.; Johnson, B.E.; Meyerson, M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef] [Green Version]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; Kosaka, T.; Holmes, A.J.; Rogers, A.M.; Cappuzzo, F.; Mok, T.; Lee, C.; Johnson, B.E.; Cantley, L.C.; Janne, P.A. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling . Science 2007, 316, 1039–1043. [Google Scholar] [CrossRef]
- Cipriani, N.A.; Abidoye, O.O.; Vokes, E.; Salgia, R. MET as a target for treatment of chest tumors. Lung Cancer 2009, 63, 169–179. [Google Scholar] [CrossRef]
- Stabile, L.P.; Lyker, J.S.; Land, S.R.; Dacic, S.; Zamboni, B.A.; Siegfried, J.M. Transgenic mice overexpressing hepatocyte growth factor in the airways show increased susceptibility to lung cancer. Carcinogenesis 2006, 27, 1547–1555. [Google Scholar]
- Stabile, L.P.; Rothstein, M.E.; Keohavong, P.; Jin, J.; Yin, J.; Land, S.R.; Dacic, S.; Luong, T.M.; Kin, K.J.; Dulak, A.M.; Siegfried, J.M. Therapeutic targeting of human hepatocyte growth factor with a single neutralizing monoclonal antibody reduces lung tumorigenesis. Mol. Cancer Ther. 2008, 7, 1913–1922. [Google Scholar] [CrossRef]
- Siegfried, J.M.; Gubish, C.T.; Rothstein, M.E.; Queiroz de Oliveira, P.E.; Stabile, L.P. Signaling pathways involved in cyclooxygenase-2 induction by hepatocyte growth factor in non small-cell lung cancer. Mol. Pharmacol. 2007, 72, 769–779. [Google Scholar] [CrossRef]
- Dulak, A.M.; Siegfried, J.M. The Src family kinases mediate EGFR-induced prolonged c-Met phosphorylation in non-small cell lung cancer. In American Association for Cancer Research Annual Meeting: Proceedings 2009, Denver, CO, USA, 18–22 April 2009; ACCR: Philadelphia, PA, USA, 2009. [Google Scholar]
- Puri, N.; Salgia, R. Synergism of EGFR and c-Met pathways, cross-talk and inhibition, in non-small cell lung cancer. J. Carcinog. 2008, 7, 9. [Google Scholar] [CrossRef]
- Siegfried, J.M.; Krishnamachary, N.; Gaither Davis, A.; Gubish, C.; Hunt, J.D.; Shriver, S.P. Evidence for autocrine actions of neuromedin B and gastrin-releasing peptide in non-small cell lung cancer. Pulm. Pharmacol. Ther. 1999, 12, 291–302. [Google Scholar] [CrossRef]
- Wilson, D.O.; Weissfeld, J.L.; Fuhrman, C.R.; Fisher, S.N.; Balogh, P.; Landreneau, R.J.; Luketich, J.D.; Siegfried, J.M. The Pittsburgh Lung Screening Study (PLuSS): Outcomes within 3 years of a first computed tomography scan. Am. J. Respir. Crit. Care Med. 2008, 178, 956–961. [Google Scholar] [CrossRef]
- Siegfried, J.M.; Luketich, J.D.; Stabile, L.P.; Christie, N.; Land, S.R. Elevated hepatocyte growth factor level correlates with poor outcome in early-stage and late-stage adenocarcinoma of the lung. Chest 2004, 125, 116–119. [Google Scholar] [CrossRef]
- Stabile, L.P.; Lyker, J.S.; Gubish, C.T.; Zhang, W.; Grandis, J.R.; Siegfried, J.M. Combined targeting of the estrogen receptor and the epidermal growth factor receptor in non-small cell lung cancer shows enhanced antiproliferative effects. Cancer Res. 2005, 65, 1459–1470. [Google Scholar] [CrossRef]
- Guo, A.; Villen, J.; Kornhauser, J.; Lee, K.A.; Stokes, M.P.; Rikova, K.; Possemato, A.; Nardone, J.; Innocenti, G.; Wetzel, R.; Wang, Y.; MacNeil, J.; Mitchell, J.; Gygi, S.P.; Rush, J.; Polakiewicz, R.D.; Comb, M.J. Signaling networks assembled by oncogenic EGFR and c-Met. Proc. Natl. Acad. Sci. USA 2008, 105, 692–697. [Google Scholar] [CrossRef]
- Hammond, D.E.; Hyde, R.; Kratchmarova, I.; Beynon, R.J.; Blagoev, B.; Clague, M.J. Quantitative analysis of HGF and EGF-dependent phosphotyrosine signaling networks. J. Proteome Res. 2010, 9, 2734–2742. [Google Scholar] [CrossRef]
- Kim, C.H.; Lee, J.S.; Kang, S.O.; Bae, J.H.; Hong, S.P.; Kahng, H. Serum hepatocyte growth factor as a marker of tumor activity in head and neck squamous cell carcinoma. Oral Oncol. 2007, 43, 1021–1025. [Google Scholar]
- Kasahara, K.; Arao, T.; Sakai, K.; Matsumoto, K.; Sakai, A,; Kimura, H.; Sone, T.; Horike, A.; Nishio, M.; Ohira, T.; Ikeda, N.; Yamanaka, T.; Saijo, N.; Nishio, K. Impact of serum HGF on treatment response to EGFR tyrosine kinase inhibitors in patients with non-small cell lung adenocarcinoma. Clin. Can. Res. 2010, 16, 4616–4624. [Google Scholar] [CrossRef]
- Ishikawa, N.; Daigo, Y.; Takano, A.; Taniwaki, M.; Kato, T.; Hayama, S.; Murakami, H.; Takeshima, Y.; Inai, K.; Nishimura, H.; Tsuchiya, E.; Kohno, N.; Nakamura, Y. Increases of amphiregulin and transforming growth factor-alpha in serum as predictors of poor response to gefitinib among patients with advanced non-small cell lung cancers. Cancer Res. 2005, 65, 9176–9184. [Google Scholar] [CrossRef]
- Busser, B.; Sancey, L.; Josserand, V.; Niang, C.; Favrot, M.C.; Coll, J.L.; Hurbin, A. Amphiregulin promotes BAX inhibition and resistance to gefitinib in non-small cell lung cancers. Mol. Therapy 2010, 18, 528–535. [Google Scholar] [CrossRef]
- Chang, M.H.; Ahn, H.K.; Lee, J.; Jung, C.K.; Choi, Y.L.; Park, Y.H.; Ahn, J.S.; Park, K.; Ahn, M.J. Clinical impact of amphiregulin expression in patients with epidermal growth factor receptor (EGFR) wild-type non-small cell lung cancer patients treated with EGFR-tyrosine kinase inhibitors. Cancer 2010. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Murakami, H.; Taniguchi, T.; Fujii, M.; Kawata, S.; Fukui, T.; Kondo, Y.; Osada, H.; Usami, N.; Yokoi, K.; Ueda, Y.; Yatabe, Y.; Ito, M.; Hida, T.; Sekido, Y. Combined inhibition of MET and EGFR suppresses proliferation of malignant mesothelioma cells. Carcinogenesis 2009, 30, 1097–1105. [Google Scholar] [CrossRef]
- Seiwert, T.Y.; Jagadeeswaran, R.; Faoro, L.; Janamanchi, V.; Nallasura, V.; El Dinali, M.; Yala, S.; Kanteti, R.; Cohen, E.E.; Lingen, M.W.; Martin, L.; Krishnaswany, S.; Klein-Szanto, A.; Christensen, J.G.; Vokes, E.E.; Salgia, R. The MET receptor tyrosine kinase is a potential novel therapeutic target for head and neck squamous cell carcinoma. Cancer Res. 2009, 69, 3021–3031. [Google Scholar] [CrossRef]
- Lal, B.; Goodwin, C.R.; Sang, Y.; Foss, C.A.; Cornet, K.; Muzamil, S.; Pomper, M.G.; Kim, J.; Laterra, J. EGFRvIII and c-Met pathway inhibitors synergize against PTEN-null/EGFRvIII+ glioblastoma xenografts. Mol. Cancer.Ther. 2009, 8, 1751–1760. [Google Scholar] [CrossRef]
- Zhang, Y.W.; Staal, B.; Essenburg, C.; Su, Y.; Kang, L.; West, R.; Kaufman, D.; Dekoning, T.; Eagleson, B.; Buchanan, S.G.; Vande Woude, G.F. MET kinase inhibitor SGX523 synergizes with epidermal growth factor inhibitor erlotinib in a hepatocyte growth factor-dependent fashion to suppress carcinoma growth. Cancer Res. 2010, 70, 6880–6890. [Google Scholar] [CrossRef]
- Schiller, J.H.; Akerley, W.L.; Brugger, W.; Ferrari, D.; Garmey, E.G.; Gerber, D.E.; Orlov, S.V.; Ramlau, R.; Von Pawel, J.; Sequist, L.V. Results from ARQ 197-209: A global randomized placebo-controlled phase II clinical trial of erlotinib plus ARQ 197 vs. erlotinib plus placebo in previously treated EGFR inhibitor-naive patients with locally advanced or metastatic non-small cell lung cancer (NSCLC). J. Clin. Oncol. 2010, 28, 18–BA7502. [Google Scholar]
- Riely, G.J.; Marks, J.; Pao, W. KRAS mutations in non-small cell lung cancer. Proc Amer. Thorac. Soc. 2009, 6, 201–205. [Google Scholar] [CrossRef]
- Fan, S.; Meng, Q.; Laterra, J.J.; Rosen, E.M. Ras effector pathways modulate scatter factor-stimulated NF-kB signaling and protection against DNA damage. Oncogene 2007, 26, 4774–4796. [Google Scholar] [CrossRef]
Correction
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stabile, L.P.; Rothstein, M.E.; Keohavong, P.; Lenzner, D.; Land, S.R.; Gaither-Davis, A.L.; Kim, K.J.; Kaminski, N.; Siegfried, J.M. Targeting of Both the c-Met and EGFR Pathways Results in Additive Inhibition of Lung Tumorigenesis in Transgenic Mice. Cancers 2010, 2, 2153-2170. https://doi.org/10.3390/cancers2042153
Stabile LP, Rothstein ME, Keohavong P, Lenzner D, Land SR, Gaither-Davis AL, Kim KJ, Kaminski N, Siegfried JM. Targeting of Both the c-Met and EGFR Pathways Results in Additive Inhibition of Lung Tumorigenesis in Transgenic Mice. Cancers. 2010; 2(4):2153-2170. https://doi.org/10.3390/cancers2042153
Chicago/Turabian StyleStabile, Laura P., Mary E. Rothstein, Phouthone Keohavong, Diana Lenzner, Stephanie R. Land, Autumn L. Gaither-Davis, K. Jin Kim, Naftali Kaminski, and Jill M. Siegfried. 2010. "Targeting of Both the c-Met and EGFR Pathways Results in Additive Inhibition of Lung Tumorigenesis in Transgenic Mice" Cancers 2, no. 4: 2153-2170. https://doi.org/10.3390/cancers2042153