Hypoxia Induced Tumor Metabolic Switch Contributes to Pancreatic Cancer Aggressiveness

Abstract
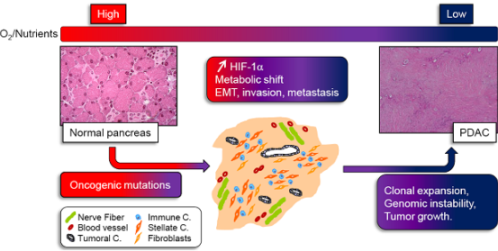
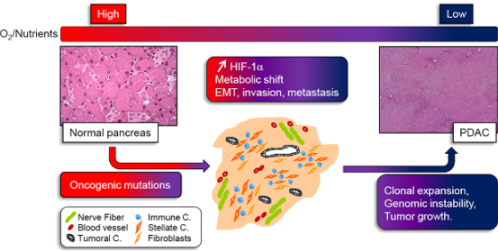
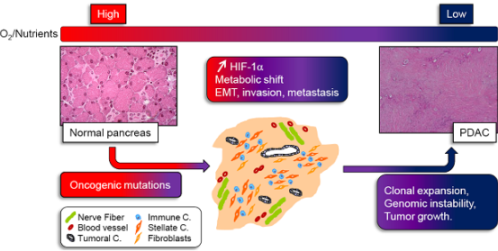
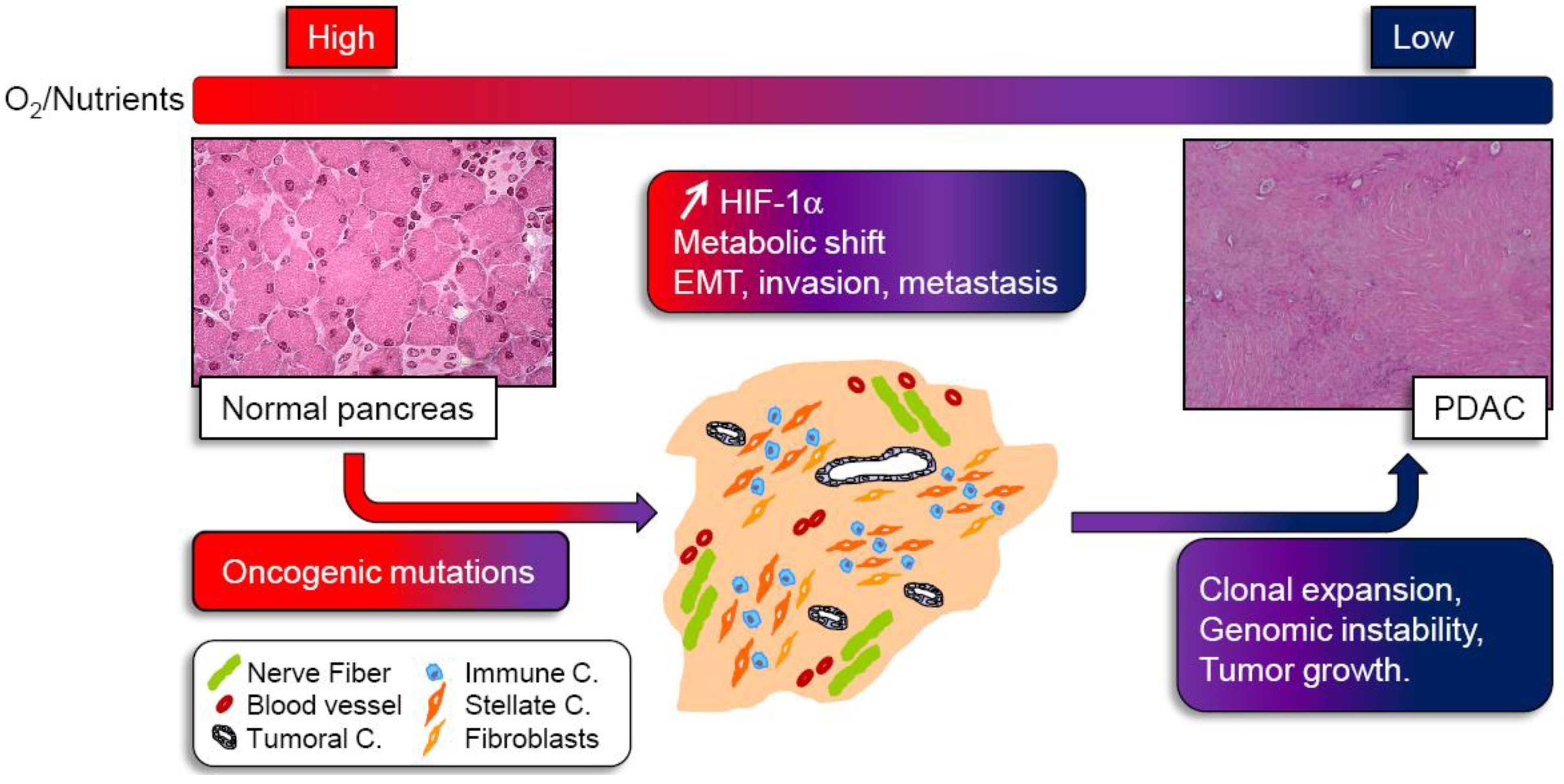
:
{kind=link}
{kind=link}
{kind=link}
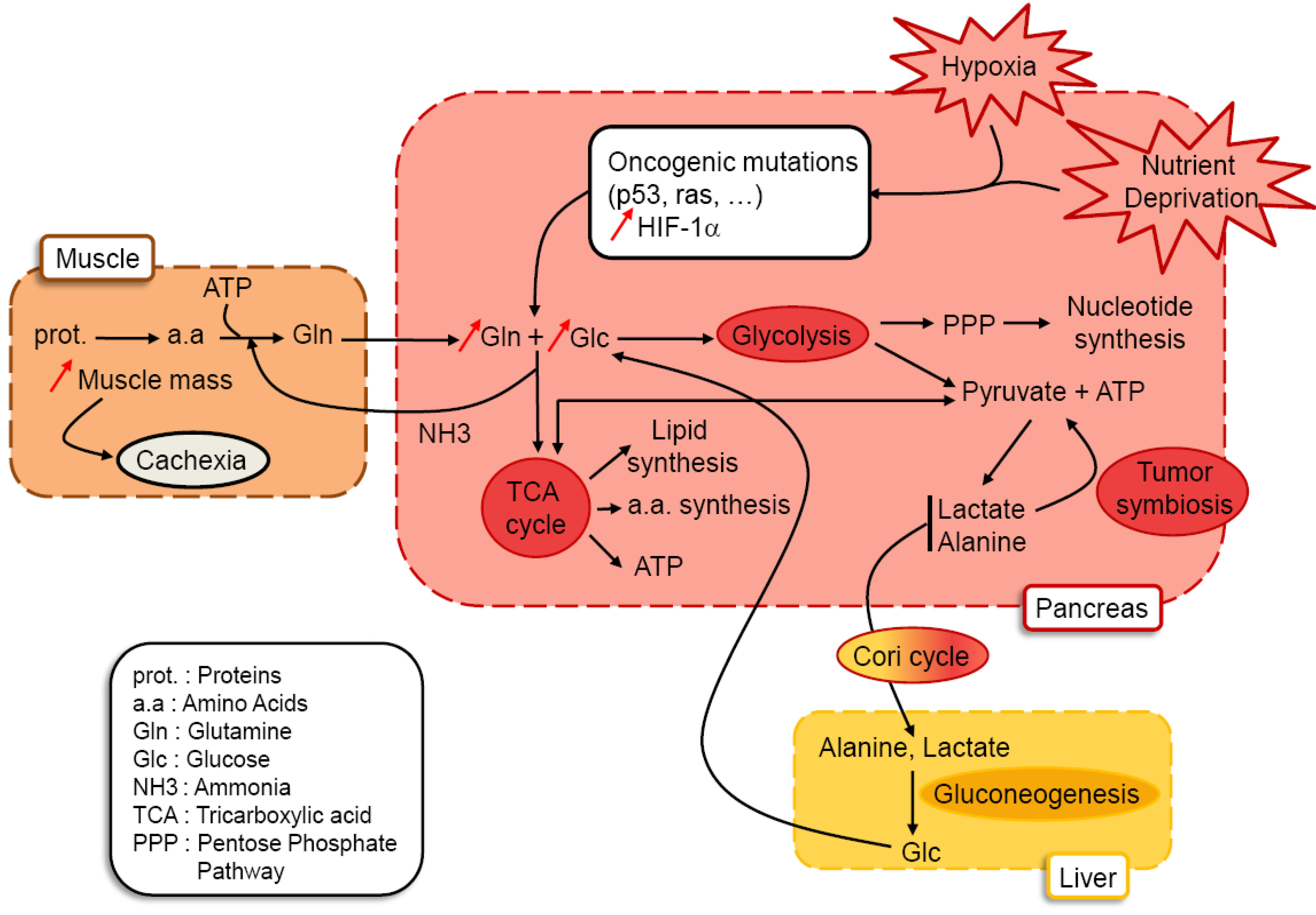
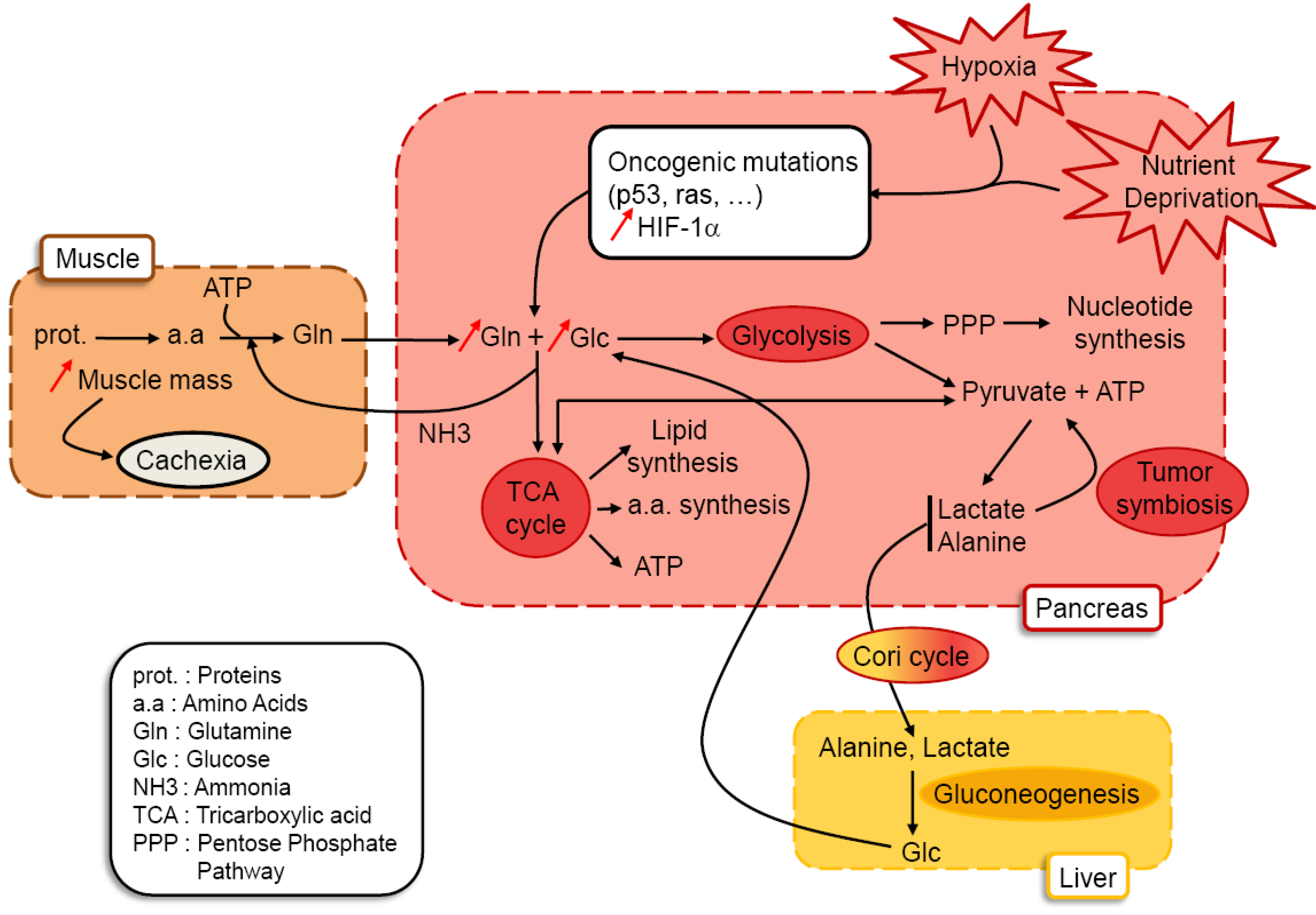{kind=link}
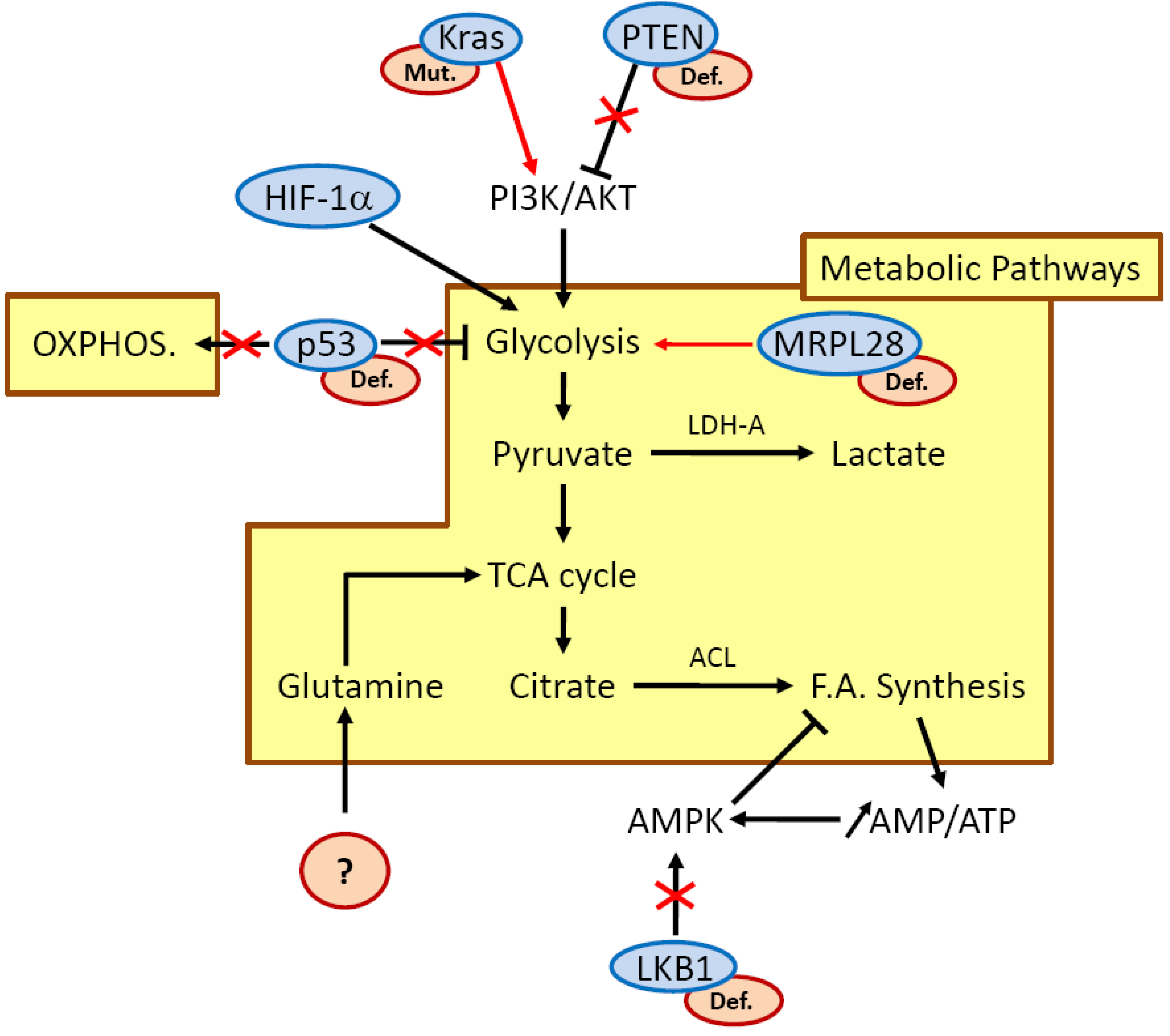{kind=link}
1. Introduction
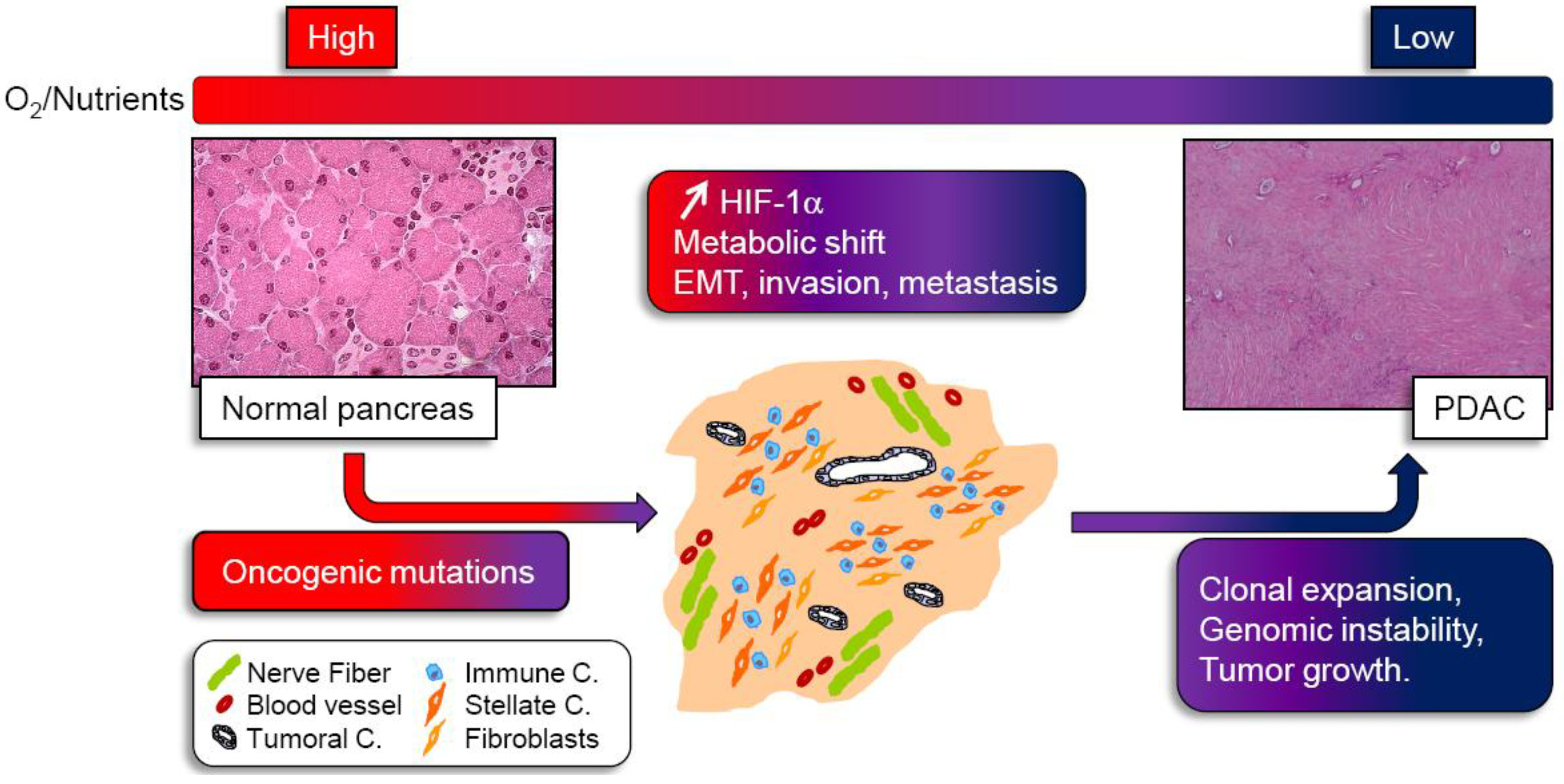
2. Hypoxia: A Master Regulator of Pancreatic Cancer
3. The Metabolic Switch Still Remains Misunderstood
3.1. A tumoral Adaptative Metabolism
3.2. Cachexia as a Consequence of Glutamine and Glucose Metabolism

3.3. LKB1/AMPK Is a Potential Candidate for Treating Pancreatic Cancer Cells by Regulating Their Metabolism
4. Conclusion
References
- Koong, A.C.; Mehta, V.K.; Le, Q.T.; Fisher, G.A.; Terris, D.J.; Brown, J.M.; Bastidas, A.J.; Vierra, M. Pancreatic tumors show high levels of hypoxia. Int. J. Radiat. Oncol. Biol. Phys. 2000, 48, 919–922. [Google Scholar] [CrossRef]
- Akakura, N.; Kobayashi, M.; Horiuchi, I.; Suzuki, A.; Wang, J.; Chen, J.; Niizeki, H.; Kawamura, K.; Hosokawa, M.; Asaka, M. Constitutive expression of hypoxia-inducible factor-1alpha renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 2001, 61, 6548–6554. [Google Scholar]
- Reiser-Erkan, C.; Erkan, M.; Pan, Z.; Bekasi, S.; Giese, N.A.; Streit, S.; Michalski, C.W.; Friess, H.; Kleeff, J. Hypoxia-inducible proto-oncogene pim-1 is a prognostic marker in pancreatic ductal adenocarcinoma. Cancer Biol. Ther. 2008, 7, 1352–1359. [Google Scholar] [CrossRef]
- Schwartz, D.L.; Bankson, J.A.; Lemos, R., Jr.; Lai, S.Y.; Thittai, A.K.; He, Y.; Hostetter, G.; Demeure, M.J.; Von Hoff, D.D.; Powis, G. Radiosensitization and stromal imaging response correlates for the hif-1 inhibitor px-478 given with or without chemotherapy in pancreatic cancer. Mol. Cancer Ther. 2010, 9, 2057–2067. [Google Scholar] [CrossRef]
- Ruan, K.; Song, G.; Ouyang, G. Role of hypoxia in the hallmarks of human cancer. J. Cell. Biochem. 2009, 107, 1053–1062. [Google Scholar] [CrossRef]
- Kizaka-Kondoh, S.; Itasaka, S.; Zeng, L.; Tanaka, S.; Zhao, T.; Takahashi, Y.; Shibuya, K.; Hirota, K.; Semenza, G.L.; Hiraoka, M. Selective killing of hypoxia-inducible factor-1-active cells improves survival in a mouse model of invasive and metastatic pancreatic cancer. Clin. Cancer Res. 2009, 15, 3433–3441. [Google Scholar] [CrossRef]
- Apte, M.V.; Park, S.; Phillips, P.A.; Santucci, N.; Goldstein, D.; Kumar, R.K.; Ramm, G.A.; Buchler, M.; Friess, H.; McCarroll, J.A.; Keogh, G.; Merrett, N.; Pirola, R.; Wilson, J.S. Desmoplastic reaction in pancreatic cancer: Role of pancreatic stellate cells. Pancreas 2004, 29, 179–187. [Google Scholar] [CrossRef]
- Bachem, M.G.; Schunemann, M.; Ramadani, M.; Siech, M.; Beger, H.; Buck, A.; Zhou, S.; Schmid-Kotsas, A.; Adler, G. Pancreatic carcinoma cells induce fibrosis by stimulating proliferation and matrix synthesis of stellate cells. Gastroenterology 2005, 128, 907–921. [Google Scholar] [CrossRef]
- Erkan, M.; Kleeff, J.; Gorbachevski, A.; Reiser, C.; Mitkus, T.; Esposito, I.; Giese, T.; Buchler, M.W.; Giese, N.A.; Friess, H. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology 2007, 132, 1447–1464. [Google Scholar] [CrossRef]
- Zhang, W.; Erkan, M.; Abiatari, I.; Giese, N.A.; Felix, K.; Kayed, H.; Buchler, M.W.; Friess, H.; Kleeff, J. Expression of extracellular matrix metalloproteinase inducer (emmprin/cd147) in pancreatic neoplasm and pancreatic stellate cells. Cancer Biol. Ther. 2007, 6, 218–227. [Google Scholar] [CrossRef]
- Bao, S.; Ouyang, G.; Bai, X.; Huang, Z.; Ma, C.; Liu, M.; Shao, R.; Anderson, R.M.; Rich, J.N.; Wang, X.F. Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the akt/pkb pathway. Cancer Cell 2004, 5, 329–339. [Google Scholar] [CrossRef]
- Erkan, M.; Reiser-Erkan, C.; Michalski, C.W.; Deucker, S.; Sauliunaite, D.; Streit, S.; Esposito, I.; Friess, H.; Kleeff, J. Cancer-stellate cell interactions perpetuate the hypoxia-fibrosis cycle in pancreatic ductal adenocarcinoma. Neoplasia 2009, 11, 497–508. [Google Scholar]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; Frese, K.K.; Denicola, G.; Feig, C.; Combs, C.; Winter, S.P.; Ireland-Zecchini, H.; Reichelt, S.; Howat, W.J.; Chang, A.; Dhara, M.; Wang, L.; Ruckert, F.; Grutzmann, R.; Pilarsky, C.; Izeradjene, K.; Hingorani, S.R.; Huang, P.; Davies, S.E.; Plunkett, W.; Egorin, M.; Hruban, R.H.; Whitebread, N.; McGovern, K.; Adams, J.; Iacobuzio-Donahue, C.; Griffiths, J.; Tuveson, D.A. Inhibition of hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Moustakas, A.; Heldin, C.H. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007, 98, 1512–1520. [Google Scholar] [CrossRef]
- Lopez-Novoa, J.M.; Nieto, M.A. Inflammation and emt: An alliance towards organ fibrosis and cancer progression. EMBO Mol. Med. 2009, 1, 303–314. [Google Scholar] [CrossRef]
- Jiang, Y.G.; Luo, Y.; He, D.L.; Li, X.; Zhang, L.L.; Peng, T.; Li, M.C.; Lin, Y.H. Role of wnt/beta-catenin signaling pathway in epithelial-mesenchymal transition of human prostate cancer induced by hypoxia-inducible factor-1alpha. Int. J. Urol. 2007, 14, 1034–1039. [Google Scholar] [CrossRef]
- Yang, M.H.; Wu, M.Z.; Chiou, S.H.; Chen, P.M.; Chang, S.Y.; Liu, C.J.; Teng, S.C.; Wu, K.J. Direct regulation of twist by hif-1alpha promotes metastasis. Nat. Cell Biol. 2008, 10, 295–305. [Google Scholar] [CrossRef]
- Chen, J.; Imanaka, N.; Griffin, J.D. Hypoxia potentiates notch signaling in breast cancer leading to decreased e-cadherin expression and increased cell migration and invasion. Br. J. Cancer 2010, 102, 351–360. [Google Scholar] [CrossRef]
- Cannito, S.; Novo, E.; Compagnone, A.; Valfre di Bonzo, L.; Busletta, C.; Zamara, E.; Paternostro, C.; Povero, D.; Bandino, A.; Bozzo, F.; Cravanzola, C.; Bravoco, V.; Colombatto, S.; Parola, M. Redox mechanisms switch on hypoxia-dependent epithelial-mesenchymal transition in cancer cells. Carcinogenesis 2008, 29, 2267–2278. [Google Scholar] [CrossRef]
- Fujii, S.; Mitsunaga, S.; Yamazaki, M.; Hasebe, T.; Ishii, G.; Kojima, M.; Kinoshita, T.; Ueno, T.; Esumi, H.; Ochiai, A. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Sci. 2008, 99, 1813–1819. [Google Scholar]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. Cancer genes and the pathways they control. Nat. Med. 2004, 10, 789–799. [Google Scholar] [CrossRef]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; Diaz, L.A., Jr.; Velculescu, V.E.; Lengauer, C.; Kinzler, K.W.; Vogelstein, B.; Papadopoulos, N. Glucose deprivation contributes to the development of kras pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef]
- Kim, J.W.; Gao, P.; Dang, C.V. Effects of hypoxia on tumor metabolism. Cancer Metastasis Rev 2007, 26, 291–298. [Google Scholar] [CrossRef]
- Chen, Y.; Cairns, R.; Papandreou, I.; Koong, A.; Denko, N.C. Oxygen consumption can regulate the growth of tumors, a new perspective on the warburg effect. PLoS One 2009, 4, e7033. [Google Scholar]
- Bhardwaj, V.; Rizvi, N.; Lai, M.B.; Lai, J.C.; Bhushan, A. Glycolytic enzyme inhibitors affect pancreatic cancer survival by modulating its signaling and energetics. Anticancer Res. 2010, 30, 743–749. [Google Scholar]
- Kroemer, G.; Pouyssegur, J. Tumor cell metabolism: Cancer's achilles' heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Hezel, A.F.; Kimmelman, A.C.; Stanger, B.Z.; Bardeesy, N.; Depinho, R.A. Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 2006, 20, 1218–1249. [Google Scholar] [CrossRef]
- Hill, R.; Calvopina, J.H.; Kim, C.; Wang, Y.; Dawson, D.W.; Donahue, T.R.; Dry, S.; Wu, H. Pten loss accelerates krasg12d-induced pancreatic cancer development. Cancer Res. 2010, 70, 7114–7124. [Google Scholar]
- Mikuriya, K.; Kuramitsu, Y.; Ryozawa, S.; Fujimoto, M.; Mori, S.; Oka, M.; Hamano, K.; Okita, K.; Sakaida, I.; Nakamura, K. Expression of glycolytic enzymes is increased in pancreatic cancerous tissues as evidenced by proteomic profiling by two-dimensional electrophoresis and liquid chromatography-mass spectrometry/mass spectrometry. Int. J. Oncol. 2007, 30, 849–855. [Google Scholar]
- Dong, X.; Tang, H.; Hess, K.R.; Abbruzzese, J.L.; Li, D. Glucose metabolism gene polymorphisms and clinical outcome in pancreatic cancer. Cancer 2010. [Google Scholar]
- Liu, H.; Huang, D.; McArthur, D.L.; Boros, L.G.; Nissen, N.; Heaney, A.P. Fructose induces transketolase flux to promote pancreatic cancer growth. Cancer Res. 2010, 70, 6368–6376. [Google Scholar] [CrossRef]
- Jones, R.G.; Thompson, C.B. Tumor suppressors and cell metabolism: A recipe for cancer growth. Genes Dev 2009, 23, 537–548. [Google Scholar] [CrossRef]
- Dang, C.V. Glutaminolysis: Supplying carbon or nitrogen or both for cancer cells? Cell Cycle 2010, 9, 3884–3886. [Google Scholar] [CrossRef]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; Thompson, C.B. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; Dang, C.V. C-myc suppression of mir-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Tisdale, M.J. Mechanisms of cancer cachexia. Physiol. Rev. 2009, 89, 381–410. [Google Scholar] [CrossRef]
- Newsholme, E.A.; Crabtree, B.; Ardawi, M.S. Glutamine metabolism in lymphocytes: Its biochemical, physiological and clinical importance. Q. J. Exp. Physiol. 1985, 70, 473–489. [Google Scholar]
- DeBerardinis, R.J.; Cheng, T. Q's next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef]
- Smolkova, K.; Plecita-Hlavata, L.; Bellance, N.; Benard, G.; Rossignol, R.; Jezek, P. Waves of gene regulation suppress and then restore oxidative phosphorylation in cancer cells. Int. J. Biochem. Cell Biol. 2010. [Google Scholar]
- Soh, H.; Wasa, M.; Fukuzawa, M. Hypoxia upregulates amino acid transport in a human neuroblastoma cell line. J. Pediatr. Surg. 2007, 42, 608–612. [Google Scholar] [CrossRef]
- Sonveaux, P.; Vegran, F.; Schroeder, T.; Wergin, M.C.; Verrax, J.; Rabbani, Z.N.; De Saedeleer, C.J.; Kennedy, K.M.; Diepart, C.; Jordan, B.F.; Kelley, M.J.; Gallez, B.; Wahl, M.L.; Feron, O.; Dewhirst, M.W. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J. Clin. Invest. 2008, 118, 3930–3942. [Google Scholar]
- Walenta, S.; Wetterling, M.; Lehrke, M.; Schwickert, G.; Sundfor, K.; Rofstad, E.K.; Mueller-Klieser, W. High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 2000, 60, 916–921. [Google Scholar]
- Eden, E.; Edstrom, S.; Bennegard, K.; Schersten, T.; Lundholm, K. Glucose flux in relation to energy expenditure in malnourished patients with and without cancer during periods of fasting and feeding. Cancer Res. 1984, 44, 1718–1724. [Google Scholar]
- Yoshikawa, T.; Noguchi, Y.; Doi, C.; Makino, T.; Okamoto, T.; Matsumoto, A. Insulin resistance was connected with the alterations of substrate utilization in patients with cancer. Cancer Lett. 1999, 141, 93–98. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Giardiello, F.M.; Trimbath, J.D. Peutz-jeghers syndrome and management recommendations. Clin. Gastroenterol. Hepatol. 2006, 4, 408–415. [Google Scholar] [CrossRef]
- Morton, J.P.; Jamieson, N.B.; Karim, S.A.; Athineos, D.; Ridgway, R.A.; Nixon, C.; McKay, C.J.; Carter, R.; Brunton, V.G.; Frame, M.C.; Ashworth, A.; Oien, K.A.; Evans, T.R.; Sansom, O.J. Lkb1 haploinsufficiency cooperates with kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology 2010, 139, 586–597. [Google Scholar] [CrossRef]
- Hezel, A.F.; Gurumurthy, S.; Granot, Z.; Swisa, A.; Chu, G.C.; Bailey, G.; Dor, Y.; Bardeesy, N.; Depinho, R.A. Pancreatic lkb1 deletion leads to acinar polarity defects and cystic neoplasms. Mol. Cell Biol. 2008, 28, 2414–2425. [Google Scholar] [CrossRef]
- Bardeesy, N.; Sinha, M.; Hezel, A.F.; Signoretti, S.; Hathaway, N.A.; Sharpless, N.E.; Loda, M.; Carrasco, D.R.; DePinho, R.A. Loss of the lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature 2002, 419, 162–167. [Google Scholar] [CrossRef]
- Hardie, D.G. Amp-activated/snf1 protein kinases: Conserved guardians of cellular energy. Nat. Rev. Mol. Cell Biol. 2007, 8, 774–785. [Google Scholar] [CrossRef]
- Koh, H.J.; Arnolds, D.E.; Fujii, N.; Tran, T.T.; Rogers, M.J.; Jessen, N.; Li, Y.; Liew, C.W.; Ho, R.C.; Hirshman, M.F.; Kulkarni, R.N.; Kahn, C.R.; Goodyear, L.J. Skeletal muscle-selective knockout of lkb1 increases insulin sensitivity, improves glucose homeostasis, and decreases trb3. Mol. Cell. Biol. 2006, 26, 8217–8227. [Google Scholar] [CrossRef]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The kinase lkb1 mediates glucose homeostasis in liver and therapeutic effects of metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef]
- Godlewski, J.; Nowicki, M.O.; Bronisz, A.; Nuovo, G.; Palatini, J.; De Lay, M.; Van Brocklyn, J.; Ostrowski, M.C.; Chiocca, E.A.; Lawler, S.E. Microrna-451 regulates lkb1/ampk signaling and allows adaptation to metabolic stress in glioma cells. Mol. Cell 2010, 37, 620–632. [Google Scholar] [CrossRef]
- Carretero, J.; Shimamura, T.; Rikova, K.; Jackson, A.L.; Wilkerson, M.D.; Borgman, C.L.; Buttarazzi, M.S.; Sanofsky, B.A.; McNamara, K.L.; Brandstetter, K.A.; Walton, Z.E.; Gu, T.L.; Silva, J.C.; Crosby, K.; Shapiro, G.I.; Maira, S.M.; Ji, H.; Castrillon, D.H.; Kim, C.F.; Garcia-Echeverria, C.; Bardeesy, N.; Sharpless, N.E.; Hayes, N.D.; Kim, W.Y.; Engelman, J.A.; Wong, K.K. Integrative genomic and proteomic analyses identify targets for lkb1-deficient metastatic lung tumors. Cancer Cell 2010, 17, 547–559. [Google Scholar] [CrossRef]
- Li, D.; Yeung, S.C.; Hassan, M.M.; Konopleva, M.; Abbruzzese, J.L. Antidiabetic therapies affect risk of pancreatic cancer. Gastroenterology 2009, 137, 482–488. [Google Scholar] [CrossRef]
- Kisfalvi, K.; Eibl, G.; Sinnett-Smith, J.; Rozengurt, E. Metformin disrupts crosstalk between g protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009, 69, 6539–6545. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vasseur, S.; Tomasini, R.; Tournaire, R.; Iovanna, J.L. Hypoxia Induced Tumor Metabolic Switch Contributes to Pancreatic Cancer Aggressiveness. Cancers 2010, 2, 2138-2152. https://doi.org/10.3390/cancers2042138
Vasseur S, Tomasini R, Tournaire R, Iovanna JL. Hypoxia Induced Tumor Metabolic Switch Contributes to Pancreatic Cancer Aggressiveness. Cancers. 2010; 2(4):2138-2152. https://doi.org/10.3390/cancers2042138
Chicago/Turabian StyleVasseur, Sophie, Richard Tomasini, Roselyne Tournaire, and Juan L. Iovanna. 2010. "Hypoxia Induced Tumor Metabolic Switch Contributes to Pancreatic Cancer Aggressiveness" Cancers 2, no. 4: 2138-2152. https://doi.org/10.3390/cancers2042138