STAT3: A Promising Therapeutic Target in Multiple Myeloma


1
Cancer Science Institute of Singapore, National University of Singapore, Singapore 117599, Singapore
2
Department of Haematology-Oncology, National University Cancer Institute of Singapore, National University Health System, Singapore 119074, Singapore
*
Author to whom correspondence should be addressed.
Cancers 2019, 11(5), 731; https://doi.org/10.3390/cancers11050731
Submission received: 11 April 2019
/
Revised: 21 May 2019
/
Accepted: 22 May 2019
/
Published: 26 May 2019
Abstract
:Multiple myeloma (MM) is an incurable plasma cell malignancy for which novel treatment options are required. Signal Transducer and Activator of Transcription 3 (STAT3) overexpression in MM appears to be mediated by a variety of factors including interleukin-6 signaling and downregulation of Src homology phosphatase-1 (SHP-1). STAT3 overexpression in MM is associated with an adverse prognosis and may play a role in microenvironment-dependent treatment resistance. In addition to its pro-proliferative role, STAT3 upregulates anti-apoptotic proteins and leads to microRNA dysregulation in MM. Phosphatase of regenerating liver 3 (PRL-3) is an oncogenic phosphatase which is upregulated by STAT3. PRL-3 itself promotes STAT-3 phosphorylation resulting in a positive feedback loop. PRL-3 is overexpressed in a subset of MM patients and may cooperate with STAT3 to promote survival of MM cells. Indirectly targeting STAT3 via JAK (janus associated kinase) inhibition has shown promise in early clinical trials. Specific inhibitors of STAT3 showed in vitro efficacy but have failed in clinical trials while several STAT3 inhibitors derived from herbs have been shown to induce apoptosis of MM cells in vitro. Optimising the pharmacokinetic profiles of novel STAT3 inhibitors and identifying how best to combine these agents with existing anti-myeloma therapy are key questions to be addressed in future clinical trials.
1. Introduction
1.1. Multiple Myeloma
Multiple myeloma (MM) is the second most common haematological malignancy in adults worldwide [1]. MM is characterised by the accumulation of malignant plasma cells in the bone marrow and is usually associated with a monoclonal protein. Anaemia, lytic bone lesions, renal impairment and immune paresis are common clinical findings in MM [2]. A variety of genetic subtypes have been described which correlate with clinical behaviour as well as treatment response and prognosis [3]. Translocations (4;14), (14;16) as well as, 17p13 deletion and 1q21 amplification, are associated with an adverse outcome and the optimal management of this group of patients remains to be established [3,4]. Treatment for MM has evolved significantly over the last two decades and patients now have access to effective therapeutic agents with acceptable toxicity profiles [5]. In particular, the advent of proteasome inhibitors (PI), immuno-modulators (IMID), and monoclonal antibodies have resulted in significant improvements in survival for patients with MM [6]. Unfortunately MM remains incurable, with relapse occurring in virtually all patients despite novel agent-based therapy [1]. There is hence an urgent need to identify novel treatment options with the endeavour to find a cure for MM.
1.2. Overview of the STAT Transcription Factor Family in Oncogenesis
Signal transducers and activators of transcription (STAT) have been recognised for over two decades as a family of transcription factors critical for a variety of cellular functions including cell survival, proliferation, angiogenesis, invasion and metastasis [7]. Seven members of the STAT protein family (STAT-1,2,3,4,5a, 5b and 6) are currently known, they reside in the cytoplasm and are activated by extracellular factors to elicit an intracellular response through regulating gene expression [8]. Growth factor receptors or oncogenic kinases such as Janus Associated Kinases (JAK), epidermal growth factor receptor (EGFR), Src family members and Bcr-Abl have been reported to activate STAT proteins [9]. In MM, interleukin 6 (IL-6) signalling in the bone marrow leads to activation of the JAK-STAT pathway resulting in phosphorylation of STAT proteins [9]. Upon phosphorylation, the STAT dimers translocate to the nucleus and regulate transcription [10].
The STAT transcription factors play a physiologic role as mediators of cell proliferation and growth [8]. In particular, they play a pivotal role in normal haematopoiesis and immune development [11]. Deregulation of STAT signalling also plays an important part in the pathogenesis of a variety of malignancies such as breast and lung cancer as well as melanoma [12]. Among the STAT proteins, STAT3 and STAT5 are the most strongly implicated in the pathogenesis of cancer [13]. Not only are they transducers of many important oncogenic signalling pathways, through their role as transcription factors they also modulate the expression of a variety of genes involved in neoplasia [12]. STAT3 regulates genes by binding to their promoter region through recognizing the consensus site TTN5AA (i.e., TTNNNNNAA) [14]. Potent proto-oncogenes like c-myc, cyclin D1 and anti-apoptotic genes Mcl-1, Bcl-xL and Bcl-2 were identified as downstream targets of STAT3 [14]. There is also emerging evidence that STAT signalling impacts the tumour microenvironment and may play a role in tumours escaping immune surveillance [12,15,16]. CD8+ T effector cells are an important component of both the innate and adaptive anti-tumour response [15]. Activation of STAT3 has been shown to negatively regulate the cytotoxic T-cell mediated immune response [15]. In a mouse model of melanoma, it was further demonstrated that silencing STAT3 in the myeloid and B cells of the tumour microenvironment could circumvent the immunosuppressive effects of STAT3 and achieve clearance of the malignant cells [15].
1.3. STAT3 Activation Is an Unfavourable Prognostic Factor in MM
STAT3 expression has been shown to have an adverse prognostic impact in MM. In a cohort of newly diagnosed MM patients, tyrosine phosphorylation of STAT3, which is a marker for STAT3 activation, was detected by immunohistochemical staining (IHC) in 10 out of 94 patients (10.6%) [17]. Patients with STAT3 activation were found to have significantly shorter progression-free and overall survival. In another study, activation of STAT3 could be detected in almost half of 48 MM patients, survival data for this cohort were however not available [18]. Preclinical data also suggest that STAT3 activation may promote drug resistance [17,19].
The role of STAT3 in malignancy, along with its prognostic impact has generated significant interest in STAT3 as a therapeutic target in MM. In this review we will discuss the key aspects of STAT signalling in the pathogenesis of MM and highlight its potential as a novel therapeutic target for this incurable malignancy.
2. Mechanisms of STAT3 Activation in MM
The importance of STAT3 in MM was highlighted by the finding of constitutively activated STAT3 in CD138+ bone marrow cells isolated from the majority of MM patient samples, but not in healthy controls [20,21]. The finding of uniform STAT3 activation despite the known heterogeneity of MM supports its potential as a therapeutic target. Furthermore, the majority of MM cell lines are responsive to IL-6 treatment and consequently upregulate STAT3 phosphorylation [22] As mutations in the STAT3 gene were not observed in MM, the hyperactivation of STAT3 can be attributed to various genetic and epigenetic mechanisms. The mechanisms leading to STAT3 activation in MM are summarised in Figure 1 and provide alternative examples of strategies for indirect inhibition of STAT3.
The canonical IL-6 signalling is tightly regulated by the repressors Src homology containing protein 1 (SHP-1) and SHP-2 tyrosine phosphatases, together with suppressor of cytokine signalling 1 (SOCS1). The disruption of this negative feedback loop results in a hyper-stimulated JAK/STAT pathway. Indeed, the expression of these genes were found to be silenced in malignant plasma cells, and this correlated with increased JAK/STAT3 activity [23,24]. In a pivotal study, Chim et al. demonstrated that hypermethylation of SHP-1 is frequently observed in MM (79.4%, n = 34) [25]. Using the human MM cell line U266 which displayed complete methylation of SHP-1, they further demonstrated that 5-azacytidine could reverse the methylation and restore the expression of SHP-1. This corresponded with a down-regulation of STAT3 phosphorylation, demonstrating that epigenetic downregulation of SHP-1 results in activation of STAT3. Likewise, SOCS1 was also found to be silenced by hypermethylation in 62.9% (n = 35) of MM patient samples [24]. In contrast, hypermethylation of SOCS-1 was absent in normal plasma cells. Considering these findings, demethylating agents may warrant evaluation as a therapeutic modality in STAT3 overexpressing MM.
Nuclear receptors are a distinct class of transcription factors that respond to hormones and ligands, and have been reported as negative regulators of IL6-mediated STAT3 activity [26,27]. Binding of estrogen to estrogen receptors (ER) induced the transcription of PIAS3 (protein inhibitor of activated STAT3), which interacts with STAT3 at its DNA-binding domain to block STAT3 mediated activation of target genes [26], without affecting the activation of JAK2 or phosphorylation of STAT3. Treating MM cells with estrogen or estrogenic ligands abolished IL-6 induced MM cell proliferation. Another nuclear receptor, peroxisome proliferator-activated receptor (PPARγ) was also implicated as a regulator of IL6-STAT3 signalling. PPARγ ligands, but not PPARα, suppressed IL-6-mediated MM cell growth in PPARγ-positive MM cell lines [28]. Additionally, this inhibition could be observed in MM cell lines which are responsive but do not depend on IL-6 for survival. More importantly, PPARγ ligands inhibited the growth of patient derived MM cells. Mechanistically, the inhibition of cell growth was demonstrated to be associated with the downregulation of the expression of c-myc, mcl-1, bcl-xl, bcl-2, and upregulation of pro-apoptotic genes bax and bak. Ligand-activated PPARγ reduced binding of STAT3 to the SIE(c-sis-inducible element) as demonstrated by electrophoretic mobility shift assay (EMSA), and luciferase reporter assay further revealed the abrogation of the DNA binding ability of STAT3. Specifically, in the presence of PPARγ ligands, the occupancy of STAT3 on c-myc and mcl-1 promoters were abolished. The involvement of at least two different classes of nuclear receptors in supressing STAT3 function suggest potential therapeutic implications.
Constitutive activation of STAT3 could be the result of dysregulated upstream signalling from JAK2 [29]. A gain-of-function mutation at position 617 from valine to phenylalanine in the JH2 domain of JAK2 (V617F) leads to hypersensitivity towards cytokine stimulation and constitutive activity of the kinase. This mutation is well recognised in myeloproliferative neoplasms [30,31]. The JAK2V617F allele, however, was not found in lymphoid malignancies or multiple myeloma [31,32,33]. Despite the absence of mutations, overexpression of JAK2 was reported in 57% of MM patients [34]. JAK2 overexpression was also demonstrated in MM cells with miRNA-375 promoter hypermethylation [35]. Aberrant repression of miRNA-375 was found in plasma cells from monoclonal gammopathy of undetermined significance (MGUS), newly diagnosed and relapsed MM patients. Restoration of miRNA-375 expression using hypomethylating agents or histone deacetylase inhibitors could modify JAK2 levels [35].
Post-translational modification through reversible methylation of STAT3 by histone-modifying enzymes has been reported in other cancers [35,36,37]. Dimethylation at lysine 140 is catalysed by SET9 (SET domain containing lysine methyltransferase 9) and lysine 49 by EZH2 (enhancer of zeste homolog 2), while trimethylation of lysine 180 is mediated by phospho-EZH2 [35,36,37]. These methylation events were demonstrated to modulate the IL-6 response and transcription of target genes [36]. Apart from phosphorylation and methylation, other post-translational modifications like acetylation, SUMOylation, S-nitrosylation and ubiquitination are reported to regulate the functions of STAT3 in multiple cancer types [38]. Acetylation of STAT3 has been reported to promote its transcriptional activity, especially on lysine 685 [39,40]. STAT3 also binds to protein inhibitor of activated STAT(PIAS) proteins (SUMO E3 ligases), and the interaction can be promoted by cytokines such as IL-6, implicating a role of SUMOylation in cytokine signaling [41]. In MM cell lines, S-nitrosylation of STAT3 negatively regulates its activities resulting in cell cycle arrest and could rescue MM cells from melphalan induced cytotoxicity [42]. Activation of the E3 ubiquitin ligase c-Cbl leads to the proteasomal degradation of SOCS3 (suppressor of cytokine signalling3), promoting STAT3 activity [43]. The relevance of these modifications in MM should be evaluated in future studies.
While the canonical functions of STAT3 as a transcriptional regulator are well-studied, findings that STAT3 can localize to the mitochondria (mitoStat3) in multiple cell types, including MM, revealed its regulatory activities in cellular metabolism [44]. The interacting partners of STAT3 in the mitochondria are implicated in mitochondrial translation and electron transport chain (ETC) pathways. It was also reported that mitoSTAT3 is essential for Ras-mediated cellular transformation, and blockade of mitoSTAT3 leads to anti-tumor activity. Moreover, these processes were dependent on the phosphorylation of STAT3 on serine 727, which can be induced by IL-6. Given the importance of the IL6-STAT3 signaling pathway in the survival of myeloma cells, it will be interesting to study mitoSTAT3 in the context of myeloma. Additionally, the role of STAT3 in mediating drug resistance was also suggested to be attributed to mitoSTAT3 through resistance to oxidative stress. Collectively, these findings suggest that targeting mitoSTAT3 should also be considered in addition to targeting the transcriptional activities of STAT3. The finding that miR551b and high glutamate levels were associated with activation of STAT3 in invasive ovarian cancer further support the hypothesis that STAT3 has a role in cellular metabolism [45].
3. Downstream Targets of STAT3 Implicated in Myeloma Cell Survival
STAT3 is a central regulator of IL-6 signalling, activating both immediate/early IL-6 responsive genes, as well as genes involved in the prolonged response towards IL-6 [22]. STAT3 elicits a pro-survival status in MM cells largely through suppression of apoptosis. Hence, members of the Bcl-2 anti-apoptotic family have been implicated as important mediators of IL-6-dependent survival [21,46,47,48]. STAT3 binding motifs were found in the promoter region of Bcl-xL [21] and Mcl-1 [49], indicating that STAT3 could regulate their expression as a transcription factor. The U266 human MM cell line, which displays high levels of activated STAT-3 and Bcl-xL, is intrinsically resistant to Fas-mediated apoptosis [21]. Further studies have suggested that Mcl-1 may play a more essential role than Bcl-xL or Bcl-2 in MM cells dependent on IL-6 [46,47].
While upregulation of anti-apoptotic genes is important for myeloma cell survival, it does not fully account for the survival benefits conferred by IL-6 through STAT3. In the microarray dataset reported by Brocke-Heidrich et al. it is interesting to note that PTP4A3, a gene encoding for the phosphatase of regenerating liver 3 (PRL-3), was identified as an IL-6-responsive gene [22]. PRL-3 is a potent oncogenic phosphatase with an important role in metastasis and disease progression [50]. The overexpression of PRL-3, driven by numerous regulatory mechanisms, could be observed across different solid tumours and haematological malignancies [51]. The oncogenic properties of PRL-3 are attributed to its ability to dephosphorylate a large number of signalling proteins, through its conserved C(X5)R (e.g., CXXXXXR) catalytic domain [52]. In addition to IL-6, IL-15 and IL-21 were also reported to induce PRL-3 expression, suggesting the importance of PRL-3 as a converging signal of different growth-promoting cytokines in MM [53]. Furthermore, ectopic expression of PRL-3 in INA-6 could promote cell survival and rendered the cells IL-6-independent [54]. In our previous report, we identified a conserved and functional STAT3 binding motif in the promoter of PRL-3, indicating that STAT3 is a direct upstream regulator of PRL-3 [55]. To further support our notion, PRL-3 expression significantly correlates with a STAT3 activation signature in acute myeloid leukaemia (AML) datasets [55]. On the other hand, we and others have observed that PRL-3 promotes STAT3 phosphorylation [54,55], suggesting that a positive feedback loop exists between PRL-3 and STAT3 to drive a potent pro-survival signal. While STAT3 directly regulates PRL-3 at the transcriptional level, how PRL-3 promotes the phosphorylation of STAT3 remains to be determined. Importantly, elevated expression of PTP4A3 was observed in a subset of patient derived MM cells. [56].
STAT3 was reported to play a role in the regulation of microRNA-21 (miR-21) and long non-coding RNAs (lncRNAs). miRNAs are a class of short, non-coding RNA that bind to target mRNAs and mediate their degradation at the post-transcriptional level. There is growing evidence implicating miRNA in the pathogenesis, progression and drug resistance of MM. [57,58,59,60,61]. The enhancer region of miR-21 contained two consensus STAT3 binding sites and is evolutionally conserved across all vertebrates. To determine whether the STAT3-binding site of miR-21 is functional, IL-6 treated cells were subjected to chromatin immunoprecipitation and reporter assay which demonstrated STAT3 recruitment to the upstream region of miR-21 to regulate its expression [57]. As miR-21 is an anti-apoptotic factor in cancer cells [61], STAT3-mediated expression of miR-21 could partly explain the IL-6-induced survival of myeloma cells. Additionally, increased expression of miR-21 could confer cytokine independence from IL-6.
Another novel class of non-coding RNAs, the lncRNAs, consist of >200 nucleotides with a functional role in gene regulation [62]. The emerging role of lncRNAs in MM was discovered when lncRNA metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) was found to be overexpressed in newly diagnosed MM, but not in post-treatment patients and healthy controls [63]. Another study was carried out to profile differentially-regulated lncRNAs in newly diagnosed MM, which led to the identification of ST3GAL6 (ST3 beta-galactoside alpha-2,3-sialyltransferase 6)-AS1, LAMA5-AS1 and RP11-175D17.3 [64]. Interestingly, tiling arrays revealed that IL-6 also induced the transcription of a number of lncRNAs through STAT3, and the authors termed these transcripts STAT3-induced ncRNAs (non-coding RNAs), in short STAiRs [64]. Specifically, STAiRs 1, 2, 6, 15 and 18 were described to be important lncRNAs mediated by STAT3. While STAiR18 is a potential epigenetic regulator, STAiR2 was found to regulate its host gene, the tumour suppressor named Deleted in Colorectal Cancer (DCC). The tissue-specific expression of these STAiRs were also analysed and STAiRs 1 and 2 could serve as myeloma-specific markers. Further studies are required to elucidate how the alterations in lncRNAs promote the progression of MM.
14-3-3 proteins bind to an array of serine/threonine phosphorylated proteins, playing the role of adaptor or scaffold proteins in signal transduction pathways [65]. Serine 727-phosphorylated STAT3 was found to physically associate with 14-3-3ζ in a proteomics screen for novel cancer-associated 14-3-3ζ-binding partners [66,67]. While phosphorylation of STAT3 on tyrosine 705 is well-elucidated and functionally important, 14-3-3ζ modulation of STAT3 activity through its serine 727 phosphosite is as important since concurrent phosphorylation of both tyrosine 705 and serine 727 is required for the full activation of STAT3 [68]. It was further shown that 14-3-3ζ protects STAT3 from protein phosphatase 2A (PP2A)-mediated dephosphorylation, prolonging the signalling response, and is required for the nuclear translocation and transactivation activities of STAT3. While therapeutic targeting of 14-3-3ζ is premature due to its involvement in cellular homeostasis, abrogation of the interaction between STAT3 and 14-3-3ζ can be considered as a potential therapeutic strategy.
4. Strategies for STAT3 Inhibition in MM
Modalities for targeting STAT3 in cancer may be classified into direct and indirect approaches [10]. Agents directly targeting STAT3 may either inhibit dimerization of the STAT proteins, inhibit STAT3 mRNA or interfere with STAT3 DNA binding [10]. Indirect approaches include preventing ligands binding to growth factor receptors, inhibiting upstream tyrosine kinases such as JAK or activating negative regulators of STAT3 [10]. An overview of the STAT3 inhibitors evaluated against MM is presented in Table 1.
4.1. Indirect STAT3 Inhibition
The JAK inhibitor ruxolitnib is approved for the treatment of patients with myelofibrosis [69]. Given the overexpression of JAK 1 and 2 in MM, there has been increasing interest in evaluating JAK inhibition as a therapeutic target. D’Oliveira and colleagues showed that ruxolitinib as a single agent had no effect on MM cell lines but induced apoptosis when combined with bortezomib [33]. Co-culture with stromal cells rescued the MM cells from apoptosis but this effect was ameliorated by the addition of lenalidomide [33]. These findings suggest that the microenvironment protects MM cells from JAK inhibition. This may be explained by the stromal cells providing an alternative means of STAT activation. The cytotoxic effect of lenalidomide in this setting maybe occurring through its immunomodulatory function and further studies are required to better understand the mechanisms involved.
Chen et al. demonstrated that ruxolitinib had single agent activity against MM cell lines as well as primary MM cells. There was also synergy with lenalidomide and dexamethasone. This combination was also effective in patient derived xenografts as well as a patient with concurrent polycythaemia vera and MM [70]. A phase 1 study has recently reported encouraging data that ruxolitinib may restore sensitivity to lenalidomide in MM patients who were previously lenalidomide refractory [71]. Larger trials are required to definitively assess the role of ruxolitinib in MM. An important question to be answered by these studies is whether sensitivity to ruxolitinib is limited to JAK overexpressing MM.
Through a drug repurposing screen, Lam et al. identified tofacitinib, a JAK inhibitor approved for rheumatoid arthritis to have activity against MM cells. The activity of tofacitinib appeared greater in an IL-6 dependent MM cell line and was particularly effective at inhibiting stromal cell derived growth signals [72]. Interestingly ruxolitinib was shown to have very limited activity against MM cell lines in this study. Intriguingly, tofacitinib synergised with venetoclax to induce apoptosis of MM cells in co culture with stromal cells but not in monoculture [72]. These data highlight the importance of stromal growth signalling in MM and call for further studies to evaluate the mechanisms behind this unexpected synergy.
The pan JAK inhibitor INCB20 has also shown promising in vitro efficacy against MM cell lines which was sustained in the presence of bone marrow stromal cells [73]. IL-6 is known to impair dexamethasone induced apoptosis of MM cells. Treatment with INCB20 abrogated the protective effect of IL-6 and sensitised MM cells to dexamethasone [73]. Importantly, STAT3 phosphorylation was markedly reduced in INCB20 treated cells. These data support the hypothesis that JAK signalling as well as IL-6 are important for MM cell survival and that STAT3 can be effectively targeted by inhibition of JAK. Given the broader spectrum of JAK inhibition by INCB20, clinical trials evaluating this compound are important to compare the toxicities and off target effects with more specific JAK inhibitors such as ruxolitinib.
A JAK inhibitor with greater specificity for JAK1 and 2 (INCB16562) was also shown to induce apoptosis in MM cell lines [74]. As seen with the INCB20, IL-6 mediated survival of MM cells was diminished and sensitivity to bortezomib and melphalan were increased by treatment with INCB16562 [74]. These findings were also confirmed in patient derived xenograft (PDX) models. This data suggests that JAK1 and JAK2 maybe the key JAK family kinases involved in MM survival and that pan JAK inhibition may not be required to achieve cytotoxicity. Further data supporting more selective JAK inhibitors were presented by Scuto, et al. who demonstrated that the selective JAK2 inhibitor AZD1480 was able to inhibit STAT3 phosphorylation and induce apoptosis in MM cell lines as well as in PDX [75]. Another novel JAK inhibitor NS-018 was not only effective at inhibiting IL-6 mediated survival of MM cells but also abrogated osteoclast activation in a murine MM model, likely through its activity against the Src kinase. These findings suggest that JAK inhibition has significant potential as a therapeutic strategy in MM. Identification of the genetic subtypes of MM most susceptible to JAK inhibition as well as studying how JAK inhibition may combine with current therapeutic modalities in MM are challenges to be addressed by future studies. Ramakrishnan et al. reported synergism between JAK2 inhibition and the pro-apoptotic caspase activator LCL-161 against MM cell lines [76]. Interestingly LCL-161 by itself did not induce apoptosis in all MM cell lines, suggesting there is an interplay between JAK signaling and apoptotic pathways in MM.
The antihelminth drug niclosamide was identified through a drug redeployment screen to have anti myeloma activity [77]. Niclosamide induced apoptosis in MM cell lines through multiple mechanisms including inhibiting IL-6 mediated STAT3 phosphorylation. Although synergy with known anti myeloma drugs was not assessed in this study, niclosamide showed a similar potency in vitro to some anti myeloma agents. The established safety profile of this drug in humans makes it an attractive option for evaluation in clinical trials.
4.2. Direct STAT3 Inhibition
Sinomenine is a derivative of a medicinal plant sinemeniumacatum which has been used to treat rheumatoid arthritis [78]. Wang et al. identified a derivative of sinomenine (YL064) as a potent inhibitor of STAT3 phosphorylation with anti-MM effects [79]. YL064 was shown to target the SH2 domain of STAT3 resulting in selective inhibition of Tyr705 but not Ser727 phosphorylation [79]. Importantly, YL064 continued to exert its pro-apoptotic effect when MM cells were co-cultured with stromal cells [79]. This data adds to the evidence supporting STAT3 inhibition as a means to overcome pro-survival signalling from the microenvironment.
Nifuroxazide was identified as a novel STAT3 inhibitor in a high throughput screen of candidate compounds [80]. This study showed nifuroxazide to be a selective inhibitor of STAT3 tyrosine phosphorylation which also inhibited the upstream kinases JAK2 and non receptor tyrosine protein kinase (TYK2). It was also shown that treatment with nifuroxazide leads to downregulation of MCL-1 [80]. It is not certain however whether the STAT3 inhibition reported here is a direct effect of nifuroxazide on STAT3 or a result of JAK2 inhibition.
Synthetic inhibitors targeting specific domains of STAT3 having been evaluated extensively against a variety of malignancies [81]. Among the synthetic STAT3 inhibitors, compounds targeting the SH2 domain have shown the most promise against MM [81]. LLL12, OPB-31121 and OPB-51602 showed potent in vitro activity against MM cells [82,83,84]. Unfortunately, they also showed significant toxicity and unfavourable pharmacokinetic profiles when evaluated in clinical trials which has hampered their development [81,83].
A number of other plant derived compounds have been reported to induce apoptosis in MM cells via inhibition of STAT3 [85,86,87,88,89,90,91,92,93,94,95]. Inhibition of upstream kinases JAK1,2, SRC and upregulation of SHP1 or other protein tyrosine phosphatases appear to be a common feature of these agents. Other protein tyrosine phosphatases activated include PIAS3 (induced by hydrocalamenene) phosphatase and tensin homolog (induced by Icariside II) and protein tyrosine phosphatase epsilon (induced by Arctiin) [89,92,93]. Among these agents, hydrocalaemenene, genipin, icariside II, arctiin and ursolic acid were shown to synergise in vitro with bortezomib and thalidomide while decursin synergised with doxorubicin [89,90,91,92,95]. The plant derived triterpenoid asiaticoside was shown to exert anti myeloma effects through induction of autophagy and activation of caspases in addition to reducing STAT3 phosphorylation [96]. Gossypol, a polyphenol derived from the cotton plant has also been shown to inhibit STAT3 phosphorylation via inhibition of IL-6 signalling [97]. Interestingly this compound also acts as a BCL2 homology domain 3 (BH3) mimetic, inducing apoptosis in MM cell lines, suggesting it has multiple mechanisms of anti MM activity. It is noteworthy that a number of these agents have been used historically in traditional medication, providing some evidence of their safety [88,89,91]. More systematic evaluation of the efficacy and toxicity profiles of these compounds in clinical trials is however called for. Given the promising in vitro data, their combination with proteasome inhibitor and IMIDs would be of interest.
5. Conclusions and Future Directions
Major advances have been made in our understanding of the role of STAT3 as well as JAK/STAT signalling in MM [10,81]. Although a variety of novel STAT3 inhibitors have been described, their lack of progress in clinical trials is disappointing. The chances of success with STAT3 inhibition in clinical trials maybe enhanced by better selection of patients who may benefit from STAT3 inhibition.
Given the rarity of STAT3 mutations in MM, developing a standardised biomarker to select patients for STAT3 inhibition maybe a challenge. Gene expression profiling to identify a STAT3 overexpression signature as described by Huang et al. in lymphoma maybe a useful strategy to identify patients for clinical trials [98].
The unfavourable pharmacokinetic profiles of some STAT3 inhibitors evaluated thus far has accounted for their failure in clinical trials [81]. The use of CD38 targeted nanoparticles loaded with STAT3 inhibitors shows promise as an innovative method to overcome this hurdle [99]. The development of more novel drug delivery techniques will be an important means of making STAT3 inhibition more clinically applicable.
Monoclonal antibodies against PRL-3 (PRL3-zumab) are currently in Phase I clinical trials [100], and targeting PRL-3 could be an attractive strategy to target STAT3 in MM. Synergism between PRL-3-zumab and PIs or immunomodulatory agents (IMIDs) also warrants further investigation.
Finally, the role of STAT3 in mediating microenvironment-derived survival signalling and treatment resistance provides an exciting opportunity as a therapeutic target. The acquisition of new genetic abnormalities and clonal evolution is key to the development of treatment resistance and may explain why MM remains incurable [101]. The prevalence of STAT3 overexpression in relation to the clonal architecture at serial time points during the patient’s disease would therefore be of great interest. As treatment resistance is often associated with loss of TP53, the question of whether STAT3 inhibition retains its potency in this setting would be particularly important to answer [102]. It is also interesting to speculate whether STAT3 inhibitors may augment the activity of current MM therapies in vivo. The role of STAT3 as a therapeutic target in MM is likely to evolve rapidly over the next few years. With more precise patient selection and optimisation of drug combinations, STAT3 inhibitors are likely to become a valuable addition to the expanding arsenal of drugs against MM.
Author Contributions
W.-J.C., S.d.M. and P.S.Y.C. conceptualised the review. P.S.Y.C. and S.d.M. wrote the manuscript. W.-J.C. critically reviewed and edited the manuscript. All authors have read and approved of the final version.
Acknowledgments
Phyllis S.Y. Chong is supported by Open Fund Young Individual Research Grant awarded by National Medical Research Council (NMRC/OFYIRG/0039/2017).
Conflicts of Interest
The authors declare that they have no competing interests.
References
- Palumbo, A.; Anderson, K. Multiple Myeloma. New Engl. J. Med. 2011, 364, 1046–1060. [Google Scholar] [CrossRef]
- Rajkumar, S.V.; A Dimopoulos, M.; Palumbo, A.; Bladé, J.; Merlini, G.; Mateos, M.-V.; Kumar, S.; Hillengass, J.; Kastritis, E.; Richardson, P.; et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014, 15, e538–e548. [Google Scholar] [CrossRef]
- Chng, W.J.; Dispenzieri, A.; Chim, C.S.; Fonseca, R.; Goldschmidt, H.; Lentzsch, S.; Munshi, N.; Palumbo, A.; Miguel, J.S.; Sonneveld, P.; et al. IMWG consensus on risk stratification in multiple myeloma. Leukemia 2014, 28, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Walker, B.A.; Mavrommatis, K.; Wardell, C.P.; Ashby, T.C.; Bauer, M.; Davies, F.; Rosenthal, A.; Wang, H.; Qu, P.; Hoering, A.; et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia 2019, 33, 159–170. [Google Scholar] [CrossRef]
- Moreau, P.; Attal, M.; Facon, T. Frontline therapy of multiple myeloma. Blood 2015, 125, 3076–3084. [Google Scholar] [CrossRef]
- Rajkumar, S.V. Multiple myeloma: 2018 update on diagnosis, risk-stratification, and management. Am. J. Hematol. 2018, 93, 1091–1110. [Google Scholar] [CrossRef] [Green Version]
- Darnell, J.E., Jr. The JAK-STAT pathway: Summary of initial studies and recent advances. Recent Prog. Horm. Res. 1996, 51, 391–403. [Google Scholar]
- Ihle, J.N. The Stat family in cytokine signaling. Cell Boil. 2001, 13, 211–217. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Arora, L.; Kumar, A.P.; Arfuso, F.; Chng, W.J.; Sethi, G. The Role of Signal Transducer and Activator of Transcription 3 (STAT3) and Its Targeted Inhibition in Hematological Malignancies. Cancers 2018, 10, 327. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, J.S.; Rosler, K.M.; Harrison, D.A. The JAK/STAT signaling pathway. J. Cell Sci. 2004, 117, 1281–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Lee, H.; Herrmann, A.; Buettner, R.; Jove, R. Revisiting STAT3 signalling in cancer: New and unexpected biological functions. Nat. Rev. Cancer 2014, 14, 736–746. [Google Scholar] [CrossRef] [PubMed]
- Buettner, R.; Mora, L.B.; Jove, R. Activated STAT signaling in human tumors provides novel molecular targets for therapeutic intervention. Clin. Res. 2002, 8, 945–954. [Google Scholar]
- Alvarez, J.V.; Frank, D.A. Genome-wide analysis of STAT target genes: Elucidating the mechanism of STAT-mediated oncogenesis. Cancer Biol. Ther. 2004, 3, 1045–1050. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, A.; Kortylewski, M.; Kujawski, M.; Zhang, C.; Reckamp, K.; Armstrong, B.; Wang, L.; Kowolik, C.; Deng, J.; Figlin, R.; et al. Targeting Stat3 in the myeloid compartment drastically improves the in vivo antitumor functions of adoptively transferred T cells. Cancer Res. 2010, 70, 7455–7464. [Google Scholar] [CrossRef]
- Kujawski, M.; Zhang, C.; Herrmann, A.; Reckamp, K.; Scuto, A.; Jensen, M.; Deng, J.; Forman, S.; Figlin, R.; Yu, H. Targeting STAT3 in adoptively transferred T cells promotes their in vivo expansion and antitumor effects. Cancer Res. 2010, 70, 9599–9610. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-H.; Ahn, S.-Y.; Choi, H.-W.; Shin, M.-G.; Lee, S.-S.; Yang, D.-H.; Ahn, J.-S.; Kim, Y.-K.; Kim, H.-J.; Lee, J.-J. STAT3 expression is associated with poor survival in non-elderly adult patients with newly diagnosed multiple myeloma. Blood Res. 2017, 52, 293–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintanilla-Martinez, L.; Kremer, M.; Specht, K.; Calzada-Wack, J.; Nathrath, M.; Schaich, R.; Hofler, H.; Fend, F. Analysis of signal transducer and activator of transcription 3 (Stat 3) pathway in multiple myeloma: Stat 3 activation and cyclin D1 dysregulation are mutually exclusive events. Am. J. Pathol. 2003, 162, 1449–1461. [Google Scholar] [CrossRef]
- Zhu, Y.X.; Shi, C.-X.; Bruins, L.A.; Wang, X.; Riggs, D.L.; Porter, B.; Ahmann, J.M.; De Campos, C.B.; Braggio, E.; Bergsagel, P.L.; et al. Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Bharti, A.C.; Shishodia, S.; Reuben, J.M.; Weber, D.; Alexanian, R.; Raj-Vadhan, S.; Estrov, Z.; Talpaz, M.; Aggarwal, B.B. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood 2004, 103, 3175–3184. [Google Scholar] [CrossRef] [PubMed]
- Catlett-Falcone, R.; Landowski, T.H.; Oshiro, M.M.; Turkson, J.; Levitzki, A.; Savino, R.; Ciliberto, G.; Moscinski, L.; Fernández-Luna, J.L.; Núñez, G.; et al. Constitutive Activation of Stat3 Signaling Confers Resistance to Apoptosis in Human U266 Myeloma Cells. Immunity 1999, 10, 105–115. [Google Scholar] [CrossRef] [Green Version]
- Brocke-Heidrich, K.; Kretzschmar, A.K.; Pfeifer, G.; Henze, C.; Löffler, D.; Koczan, D.; Thiesen, H.-J.; Burger, R.; Gramatzki, M.; Horn, F. Interleukin-6–dependent gene expression profiles in multiple myeloma INA-6 cells reveal a Bcl-2 family–independent survival pathway closely associated with Stat3 activation. Blood 2004, 103, 242–251. [Google Scholar] [CrossRef]
- Beldi-Ferchiou, A.; Skouri, N.; Ben Ali, C.; Safra, I.; Abdelkefi, A.; Ladeb, S.; Mrad, K.; Ben Othman, T.; Ben Ahmed, M. Abnormal repression of SHP-1, SHP-2 and SOCS-1 transcription sustains the activation of the JAK/STAT3 pathway and the progression of the disease in multiple myeloma. PLoS One 2017, 12, e0174835. [Google Scholar] [CrossRef] [PubMed]
- Galm, O.; Yoshikawa, H.; Esteller, M.; Osieka, R.; Herman, J.G. SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood 2003, 101, 2784–2788. [Google Scholar] [CrossRef] [Green Version]
- Chim, C.-S.; Fung, T.-K.; Cheung, W.-C.; Liang, R.; Kwong, Y.-L. SOCS1 and SHP1 hypermethylation in multiple myeloma: Implications for epigenetic activation of the Jak/STAT pathway. Blood 2004, 103, 4630–4635. [Google Scholar] [CrossRef]
- Wang, L.H. Activation of Estrogen Receptor Blocks Interleukin-6-inducible Cell Growth of Human Multiple Myeloma Involving Molecular Cross-talk between Estrogen Receptor and STAT3 Mediated by Co-regulator PIAS3. J. Biol. Chem. 2001, 276, 31839–31844. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.H.; Yang, X.Y.; Zhang, X.; Farrar, W.L. Nuclear receptors as negative modulators of STAT3 in multiple myeloma. Cell Cycle 2005, 4, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Yang, X.Y.; Zhang, X.; Huang, J.; Hou, J.; Li, J.; Xiong, H.; Mihalic, K.; Zhu, H.; Xiao, W.; et al. Transcriptional inactivation of STAT3 by PPARgamma suppresses IL-6-responsive multiple myeloma cells. Immun. 2004, 20, 205–218. [Google Scholar] [CrossRef]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. The Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Levine, R.L.; Loriaux, M.; Huntly, B.J.P.; Loh, M.L.; Beran, M.; Stoffregen, E.; Berger, R.; Clark, J.J.; Willis, S.G.; Nguyen, K.T.; et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood 2005, 106, 3377–3379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorini, A.; Farina, G.; Reddiconto, G.; Palladino, M.; Rossi, E.; Za, T.; Laurenti, L.; Giammarco, S.; Chiusolo, P.; Leone, G.; et al. Screening of JAK2 V617F mutation in multiple myeloma. Leukemia 2006, 20, 1912–1913. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.; Li, X.; Chen, W.; Weiss, L.M. Absence of JAK-2V617F point mutations in multiple myeloma. Leukemia 2007, 21, 813–814. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira, M.B.; Fook-Alves, V.L.; Eugenio, A.I.; Fernando, R.C.; Sanson, L.F.G.; De Carvalho, M.F.; Braga, W.M.; Davies, F.E.; Colleoni, G.W. Anti-myeloma effects of ruxolitinib combined with bortezomib and lenalidomide: A rationale for JAK/STAT pathway inhibition in myeloma patients. Cancer Lett. 2017, 403, 206–215. [Google Scholar] [CrossRef] [PubMed]
- Tatekawa, S.; Chinen, Y.; Ri, M.; Narita, T.; Shimura, Y.; Matsumura-Kimoto, Y.; Tsukamoto, T.; Kobayashi, T.; Kawata, E.; Uoshima, N.; et al. Epigenetic repression of miR-375 is the dominant mechanism for constitutive activation of the PDPK1/RPS6KA3 signalling axis in multiple myeloma. Br. J. Haematol. 2017, 7, 534–546. [Google Scholar] [CrossRef]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible methylation of promoter-bound STAT3 by histone-modifying enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, M.; Dermawan, J.K.T.; Willard, B.; Stark, G.R. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc. Natl. Acad. Sci. USA 2015, 112, 3985–3990. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Kim, M.; Woo, D.-H.; Shin, Y.; Shin, J.; Chang, N.; Oh, Y.T.; Kim, H.; Rheey, J.; Nakano, I.; et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell 2013, 23, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Laudisi, F.; Cherubini, F.; Monteleone, G.; Stolfi, C. STAT3 Interactors as Potential Therapeutic Targets for Cancer Treatment. Int. J. Mol. Sci. 2018, 19, 1787. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Cherukuri, P.; Luo, J. Activation of Stat3 Sequence-specific DNA Binding and Transcription by p300/CREB-binding Protein-mediated Acetylation. J. Biol. Chem. 2005, 280, 11528–11534. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.-L.; Guan, Y.-J.; Chatterjee, D.; Chin, Y.E. Stat3 Dimerization Regulated by Reversible Acetylation of a Single Lysine Residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Shuai, K. Regulation of cytokine signaling pathways by PIAS proteins. Cell Res. 2006, 16, 196–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Choi, S.; Saxena, N.; Singh, A.K.; Singh, I.; Won, J.-S. Regulation of STAT3 and NF-κB Activations by S-Nitrosylation in Multiple Myeloma. Radic. Biol. Med. 2017, 106, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Cao, L.; Liu, Y.; Wang, K.; Zhang, X.; Qin, X.; Zhao, D.; Hao, J.; Chang, Y.; Huang, X.; et al. B7-H3 promotes multiple myeloma cell survival and proliferation by ROS-dependent activation of Src/STAT3 and c-Cbl-mediated degradation of SOCS3. Leukemia 2018. [Google Scholar] [CrossRef]
- Yang, L.; Moss, T.; Mangala, L.S.; Marini, J.; Zhao, H.; Wahlig, S.; Armaiz-Pena, G.N.; Jiang, D.; Achreja, A.; Win, J.; et al. Metabolic shifts toward glutamine regulate tumor growth, invasion and bioenergetics in ovarian cancer. Mol. Syst. Biol. 2014, 10. [Google Scholar] [CrossRef]
- Chaluvally-Raghavan, P.; Jeong, K.J.; Pradeep, S.; Silva, A.M.; Yu, S.; Liu, W.; Moss, T.; Rodriguez-Aguayo, C.; Zhang, D.; Ram, P.; et al. Direct upregulation of STAT3 by microRNA-551b-3p deregulates growth and metastasis of ovarian cancer. Cell Rep. 2016, 15, 1493–1504. [Google Scholar] [CrossRef]
- Puthier, D.; Derenne, S.; Barillé, S.; Moreau, P.; Harousseau, J.L.; Bataille, R.; Amiot, M. Mcl-1 and Bcl-xL are co-regulated by IL-6 in human myeloma cells. Br. J. Haematol. 1999, 107, 392–395. [Google Scholar] [CrossRef]
- Derenne, S.; Monia, B.; Dean, N.M.; Taylor, J.K.; Rapp, M.-J.; Harousseau, J.-L.; Bataille, R.; Amiot, M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-xL is an essential survival protein of human myeloma cells. Blood 2002, 100, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Jourdan, M.; Veyrune, J.-L.; De Vos, J.; Redal, N.; Couderc, G.; Klein, B. A major role for Mcl-1 antiapoptotic protein in the IL-6-induced survival of human myeloma cells. Oncogene 2003, 22, 2950–2959. [Google Scholar] [CrossRef]
- Epling-Burnette, P.; Liu, J.H.; Catlett-Falcone, R.; Turkson, J.; Oshiro, M.; Kothapalli, R.; Li, Y.; Wang, J.-M.; Yang-Yen, H.-F.; Karras, J.; et al. Inhibition of STAT3 signaling leads to apoptosis of leukemic large granular lymphocytes and decreased Mcl-1 expression. J. Clin. Investig. 2001, 107, 351–362. [Google Scholar] [CrossRef]
- Al-Aidaroos, A.Q.O.; Zeng, Q.; Al-Aidaroos, A.Q.O. PRL-3 phosphatase and cancer metastasis. J. Cell. Biochem. 2010, 111, 1087–1098. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Vardy, L.A.; Tan, C.P.; Loo, J.M.; Guo, K.; Li, J.; Lim, S.G.; Zhou, J.; Chng, W.J.; Ng, S.B.; et al. PCBP1 Suppresses the Translation of Metastasis-Associated PRL-3 Phosphatase. Cancer Cell 2010, 18, 52–62. [Google Scholar] [CrossRef] [Green Version]
- Guo, K.; Li, J.; Tang, J.P.; Koh, V.; Gan, B.Q.; Zeng, Q. Catalytic domain of PRL-3 plays an essential role in tumor metastasis: Formation of PRL-3 tumors inside the blood vessels. Cancer Biol. Ther. 2004, 3, 945–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fagerli, U.-M.; Holt, R.U.; Holien, T.; Vaatsveen, T.K.; Zhan, F.; Egeberg, K.W.; Barlogie, B.; Waage, A.; Aarset, H.; Dai, H.Y.; et al. Overexpression and involvement in migration by the metastasis-associated phosphatase PRL-3 in human myeloma cells. Blood 2008, 111, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Slørdahl, T.S.; Abdollahi, P.; Vandsemb, E.N.; Rampa, C.; Misund, K.; Baranowska, K.A.; Westhrin, M.; Waage, A.; Rø, T.B.; Børset, M. The phosphatase of regenerating liver-3 (PRL-3) is important for IL-6-mediated survival of myeloma cells. Oncotarget 2016, 7, 27295–27306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Chong, P.S.; Lu, X.; Cheong, L.-L.; Bi, C.; Liu, S.-C.; Zhou, Y.; Tan, T.Z.; Yang, H.; Chung, T.-H.; et al. Phosphatase of regenerating liver-3 is regulated by signal transducer and activator of transcription 3 in acute myeloid leukemia. Exp. Hematol. 2014, 42, 1041–1052. [Google Scholar] [CrossRef]
- Broyl, A.; Hose, D.; Lokhorst, H.; De Knegt, Y.; Peeters, J.; Jauch, A.; Bertsch, U.; Buijs, A.; Stevens-Kroef, M.; Beverloo, H.B.; et al. Gene expression profiling for molecular classification of multiple myeloma in newly diagnosed patients. Blood 2010, 116, 2543–2553. [Google Scholar] [CrossRef] [Green Version]
- Löffler, D.; Brocke-Heidrich, K.; Pfeifer, G.; Stocsits, C.; Hackermüller, J.; Kretzschmar, A.K.; Bürger, R.; Gramatzki, M.; Blumert, C.; Bauer, K.; et al. Interleukin-6 dependent survival of multiple myeloma cells involves the Stat3-mediated induction of microRNA-21 through a highly conserved enhancer. Blood 2007, 110, 1330–1333. [Google Scholar] [CrossRef]
- Binder, S.; Hösler, N.; Riedel, D.; Zipfel, I.; Buschmann, T.; Kämpf, C.; Reiche, K.; Burger, R.; Gramatzki, M.; Hackermüller, J.; et al. STAT3-induced long noncoding RNAs in multiple myeloma cells display different properties in cancer. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- 5Benetatos, L.; Vartholomatos, G. Deregulated microRNAs in multiple myeloma. Cancer 2012, 118, 878–887. [Google Scholar] [CrossRef]
- Bi, C.; Chng, W.J. MicroRNA: Important Player in the Pathobiology of Multiple Myeloma. BioMed Int. 2014, 2014, 1–12. [Google Scholar] [CrossRef]
- Chan, J.A. MicroRNA-21 Is an Antiapoptotic Factor in Human Glioblastoma Cells. Cancer Res. 2005, 65, 6029–6033. [Google Scholar] [CrossRef]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Microbiol. 2009, 10, 155–159. [Google Scholar] [CrossRef]
- Cho, S.-F.; Chang, Y.C.; Chang, C.-S.; Lin, S.-F.; Liu, Y.-C.; Hsiao, H.-H.; Chang, J.-G.; Liu, T.-C. MALAT1 long non-coding RNA is overexpressed in multiple myeloma and may serve as a marker to predict disease progression. BMC Cancer 2014, 14. [Google Scholar] [CrossRef]
- Shen, Y.; Feng, Y.; Chen, H.; Huang, L.; Wang, F.; Bai, J.; Yang, Y.; Wang, J.; Zhao, W.; Jia, Y.; et al. Focusing on long non-coding RNA dysregulation in newly diagnosed multiple myeloma. Life Sci. 2018, 196, 133–142. [Google Scholar] [CrossRef]
- Morrison, D.K. The 14-3-3 proteins: Integrators of diverse signaling cues that impact cell fate and cancer development. Cancer Cell Biol. 2009, 19, 16–23. [Google Scholar] [CrossRef]
- Ge, F.; Li, W.-L.; Bi, L.-J.; Tao, S.-C.; Zhang, Z.-P.; Zhang, X.-E. Identification of Novel 14-3-3ζ Interacting Proteins by Quantitative Immunoprecipitation Combined with Knockdown (QUICK). J. Proteome 2010, 9, 5848–5858. [Google Scholar] [CrossRef]
- Zhang, J.; Chen, F.; Li, W.; Xiong, Q.; Yang, M.; Zheng, P.; Li, C.; Pei, J.; Ge, F. 14-3-3ζ Interacts with Stat3 and Regulates Its Constitutive Activation in Multiple Myeloma Cells. PLoS ONE 2012, 7. [Google Scholar] [CrossRef]
- Wen, Z.; Zhong, Z.; E Darnell, J. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 1995, 82, 241–250. [Google Scholar] [CrossRef]
- Verstovsek, S.; Mesa, R.A.; Gotlib, J.; Levy, R.S.; Gupta, V.; DiPersio, J.F.; Catalano, J.V.; Deininger, M.; Miller, C.; Silver, R.T.; et al. A Double-Blind Placebo-Controlled Trial of Ruxolitinib for Myelofibrosis. New Engl. J. Med. 2012, 366, 799–807. [Google Scholar] [CrossRef]
- Chen, C.; Sanchez, E.; Li, M.; Wang, C.; Gillespie, A.; Shvartsur, A.; Vardanyan, S.; Harutyunyan, N.; Garzio, G.; Tang, G.; et al. Anti-Myeloma Activity by the Combination of the JAK2 Inhibitor Ruxolitinib with Lenalidomide and Corticosteroids. Blood 2014, 124, 2114. [Google Scholar]
- Berenson, J.R.; Jennifer, T.; Spektor, T.M.; Turner, C.; Swift, R.; Eades, B.; Schwartz, G.; Eshaghian, S.; Stampleman, L.V.; Moss, R.A.; et al. A Phase 1 Trial of Ruxolitinib, Lenalidomide and Methylprednisolone for Relapsed/Refractory Multiple Myeloma Patients. Blood 2018, 132. [Google Scholar] [CrossRef]
- Lam, C.; Ferguson, I.D.; Mariano, M.C.; Lin, Y.-H.T.; Murnane, M.; Liu, H.; Smith, G.A.; Wong, S.W.; Taunton, J.; Liu, J.O.; et al. Repurposing tofacitinib as an anti-myeloma therapeutic to reverse growth-promoting effects of the bone marrow microenvironment. Haematologica 2018, 103, 1218–1228. [Google Scholar] [CrossRef] [Green Version]
- Burger, R.; Le Gouill, S.; Tai, Y.-T.; Shringarpure, R.; Tassone, P.; Neri, P.; Podar, K.; Catley, L.; Hideshima, T.; Chauhan, D.; et al. Janus kinase inhibitor INCB20 has antiproliferative and apoptotic effects on human myeloma cells in vitro and in vivo. Mol. Cancer Ther. 2009, 8, 26–35. [Google Scholar] [CrossRef]
- Li, J.; Favata, M.; Kelley, J.A.; Caulder, E.; Thomas, B.; Wen, X.; Sparks, R.B.; Arvanitis, A.; Rogers, J.D.; Combs, A.P.; et al. INCB16562, a JAK1/2 Selective Inhibitor, Is Efficacious against Multiple Myeloma Cells and Reverses the Protective Effects of Cytokine and Stromal Cell Support. Neoplasia 2010, 12, 28–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scuto, A.; Krejci, P.; Popplewell, L.; Wu, J.; Kujawski, M.; Kowolik, C.; Xin, H.; Chen, L.; Kretzner, L.; Yu, H.; et al. The novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression of human myeloma cell growth and survival. Leukemia 2010, 25, 538–550. [Google Scholar] [CrossRef]
- Ramakrishnan, V.; Painuly, U.; Kimlinger, T.; Haug, J.; Rajkumar, S.V.; Kumar, S. Inhibitor of apoptosis proteins as therapeutic targets in multiple myeloma. Leukemia 2014, 28, 1519–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanim, F.L.; Merrick, B.A.M.E.; Giles, H.V.; Jankute, M.; Jackson, J.B.; Giles, L.J.; Birtwistle, J.; Bunce, C.M.; Drayson, M.T. Redeployment-based drug screening identifies the anti-helminthic niclosamide as anti-myeloma therapy that also reduces free light chain production. Blood Cancer J. 2011, 1. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, L.; Cai, H.; Lei, H.; Ma, C.-M.; Yang, L.; Xu, H.; Zhu, Q.; Yao, Z.; Wu, Y. YL064 directly inhibits STAT3 activity to induce apoptosis of multiple myeloma cells. Cell Death Discov. 2018, 4. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, L.; Cai, H.; Lei, H.; Ma, C.-M.; Yang, L.; Xu, H.; Zhu, Q.; Yao, Z.; Wu, Y. Sinomenine derivative YL064: A novel STAT3 inhibitor with promising anti-myeloma activity. Cell Death 2018, 9. [Google Scholar] [CrossRef]
- Nelson, E.A.; Walker, S.R.; Kepich, A.; Gashin, L.B.; Hideshima, T.; Ikeda, H.; Chauhan, D.; Anderson, K.C.; Frank, D.A. Nifuroxazide inhibits survival of multiple myeloma cells by directly inhibiting STAT3. Blood 2008, 112, 5095–5102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beebe, J.D.; Liu, J.-Y.; Zhang, J.-T. Two decades of research in discovery of anticancer drugs targeting STAT3, how close are we? Pharmacol. Ther. 2018, 191, 74–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, L.; Benson, D.M., Jr.; DeAngelis, S.; Bakan, C.E.; Li, P.-K.; Li, C.; Lin, J. A small molecule, LLL12 inhibits constitutive STAT3 and IL-6-induced STAT3 signaling and exhibits potent growth suppressive activity in human multiple myeloma cells. Int. J. Cancer 2012, 130, 1459–1469. [Google Scholar] [CrossRef]
- Ogura, M.; Uchida, T.; Terui, Y.; Hayakawa, F.; Kobayashi, Y.; Taniwaki, M.; Takamatsu, Y.; Naoe, T.; Tobinai, K.; Munakata, W.; et al. Phase I study of OPB-51602, an oral inhibitor of signal transducer and activator of transcription 3, in patients with relapsed/refractory hematological malignancies. Cancer Sci. 2015, 106, 896–901. [Google Scholar] [CrossRef]
- Hayakawa, F.; Sugimoto, K.; Harada, Y.; Hashimoto, N.; Ohi, N.; Kurahashi, S.; Naoe, T. A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STAT-addictive oncokinases. Blood Cancer J. 2013, 3. [Google Scholar] [CrossRef]
- Takeda, T.; Tsubaki, M.; Tomonari, Y.; Kawashima, K.; Itoh, T.; Imano, M.; Satou, T.; Nishida, S. Bavachin induces the apoptosis of multiple myeloma cell lines by inhibiting the activation of nuclear factor kappa B and signal transducer and activator of transcription 3. Biomed. Pharmacother. 2018, 100, 486–494. [Google Scholar] [CrossRef]
- Padmavathi, G.; Rathnakaram, S.R.; Monisha, J.; Bordoloi, D.; Roy, N.K.; Kunnumakkara, A.B. Potential of butein, a tetrahydroxychalcone to obliterate cancer. Phytomedicine 2015, 22, 1163–1171. [Google Scholar] [CrossRef]
- Padmavathi, G.; Roy, N.K.; Bordoloi, D.; Arfuso, F.; Mishra, S.; Sethi, G.; Bishayee, A.; Kunnumakkara, A.B. Butein in health and disease: A comprehensive review. Phytomedicine 2017, 25, 118–127. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.Y.; Lee, J.H.; Nam, D.; Narula, A.S.; Namjoshi, O.A.; Blough, B.E.; Um, J.-Y.; Sethi, G.; Ahn, K.S. Anti-myeloma Effects of Icariin Are Mediated Through the Attenuation of JAK/STAT3-Dependent Signaling Cascade. Front. Pharmacol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Nam, D.; Song, J.; Kim, S.-M.; Chiang, S.Y.; Kim, J.-S.; Chung, W.-S.; Jang, H.-J.; Jung, S.H.; Na, Y.-S.; Kim, S.-H.; et al. 8-Hydrocalamenene, Derived from Reynoutria elliptica, Suppresses Constitutive STAT3 Activation, Inhibiting Proliferation and Enhancing Chemosensitization of Human Multiple Myeloma Cells. J. Med. Food 2014, 17, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.; Jeong, S.J.; Kwon, H.Y.; Jung, J.H.; Sohn, E.J.; Lee, H.J.; Kim, J.H.; Kim, S.H.; Kim, J.H.; Kim, S.H. Decursin and Doxorubicin Are in Synergy for the Induction of Apoptosis via STAT3 and/or mTOR Pathways in Human Multiple Myeloma Cells. Evid. Based Complement. Alternat. Med. 2013. [Google Scholar] [CrossRef]
- Lee, J.C.; Ahn, K.S.; Jeong, S.-J.; Jung, J.H.; Kwon, T.-R.; Rhee, Y.-H.; Kim, S.-H.; Kim, S.-Y.; Yoon, H.-J.; Zhu, S.; et al. Signal transducer and activator of transcription 3 pathway mediates genipin-induced apoptosis in U266 multiple myeloma cells. J. Cell. Biochem. 2011, 112, 1552–1562. [Google Scholar] [CrossRef]
- Kim, S.-H.; Ahn, K.S.; Jeong, S.-J.; Kwon, T.-R.; Jung, J.H.; Yun, S.-M.; Han, I.; Lee, S.-G.; Kim, D.K.; Kang, M.; et al. Janus activated kinase 2/signal transducer and activator of transcription 3 pathway mediates icariside II-induced apoptosis in U266 multiple myeloma cells. Eur. J. Pharmacol. 2011, 654, 10–16. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, C.; Lee, J.; Um, J.Y.; Sethi, G.; Ahn, K.S. Arctiin is a pharmacological inhibitor of STAT3 phosphorylation at tyrosine 705 residue and potentiates bortezomib-induced apoptotic and anti-angiogenic effects in human multiple myeloma cells. Phytomedicine 2019, 55, 282–292. [Google Scholar] [CrossRef]
- Rhee, Y.-H.; Jeong, S.-J.; Lee, H.-J.; Lee, H.-J.; Koh, W.; Jung, J.H.; Kim, S.-H.; Sung-Hoon, K.; Kim, S.-H. Inhibition of STAT3 signaling and induction of SHP1 mediate antiangiogenic and antitumor activities of ergosterol peroxide in U266 multiple myeloma cells. BMC Cancer. 2012, 12. [Google Scholar] [CrossRef] [PubMed]
- Pathak, A.K.; Bhutani, M.; Nair, A.S.; Ahn, K.S.; Chakraborty, A.; Kadara, H.; Guha, S.; Sethi, G.; Aggarwal, B.B. Ursolic Acid Inhibits STAT3 Activation Pathway Leading to Suppression of Proliferation and Chemosensitization of Human Multiple Myeloma Cells. Mol. Cancer Res. 2007, 5, 943–955. [Google Scholar] [CrossRef] [Green Version]
- Yingchun, L.; Huihan, W.; Rong, Z.; Guojun, Z.; Ying, Y.; Zhuogang, L. Antitumor Activity of Asiaticoside Against Multiple Myeloma Drug-Resistant Cancer Cells Is Mediated by Autophagy Induction, Activation of Effector Caspases, and Inhibition of Cell Migration, Invasion, and STAT-3 Signaling Pathway. Med Sci. 2019, 25, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Sadahira, K.; Sagawa, M.; Nakazato, T.; Uchida, H.; Ikeda, Y.; Okamoto, S.; Nakajima, H.; Kizaki, M. Gossypol induces apoptosis in multiple myeloma cells by inhibition of interleukin-6 signaling and Bcl-2/Mcl-1 pathway. Int. J. Oncol. 2014, 45, 2278–2286. [Google Scholar] [CrossRef]
- Huang, X.; Meng, B.; Iqbal, J.; Ding, B.B.; Perry, A.M.; Cao, W.; Smith, L.M.; Bi, C.; Jiang, C.; Greiner, T.C.; et al. Activation of the STAT3 Signaling Pathway Is Associated with Poor Survival in Diffuse Large B-Cell Lymphoma Treated With R-CHOP. J. Clin. Oncol. 2013, 31, 4520–4528. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.-H.; Vakili, M.R.; Molavi, O.; Morrissey, Y.; Wu, C.; Paiva, I.; Soleimani, A.H.; Sanaee, F.; Lavasanifar, A.; Lai, R. Decoration of Anti-CD38 on Nanoparticles Carrying a STAT3 Inhibitor Can Improve the Therapeutic Efficacy Against Myeloma. Cancers 2019, 11, 248. [Google Scholar] [CrossRef]
- Thura, M.; Al-Aidaroos, A.Q.O.; Yong, W.P.; Kono, K.; Gupta, A.; Bin Lin, Y.; Mimura, K.; Thiery, J.P.; Goh, B.C.; Tan, P.; et al. PRL3-zumab, a first-in-class humanized antibody for cancer therapy. JCI Insight 2016. [Google Scholar] [CrossRef]
- Keats, J.J.; Chesi, M.; Egan, J.B.; Garbitt, V.M.; Palmer, S.E.; Braggio, E.; Van Wier, S.; Blackburn, P.R.; Baker, A.S.; Dispenzieri, A.; et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012, 120, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Chin, M.; I Sive, J.; Allen, C.; Roddie, C.; Chavda, S.J.; Smith, D.; Blombery, P.; Jones, K.; Ryland, G.L.; Popat, R.; et al. Prevalence and timing of TP53 mutations in del(17p) myeloma and effect on survival. Blood Cancer J. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
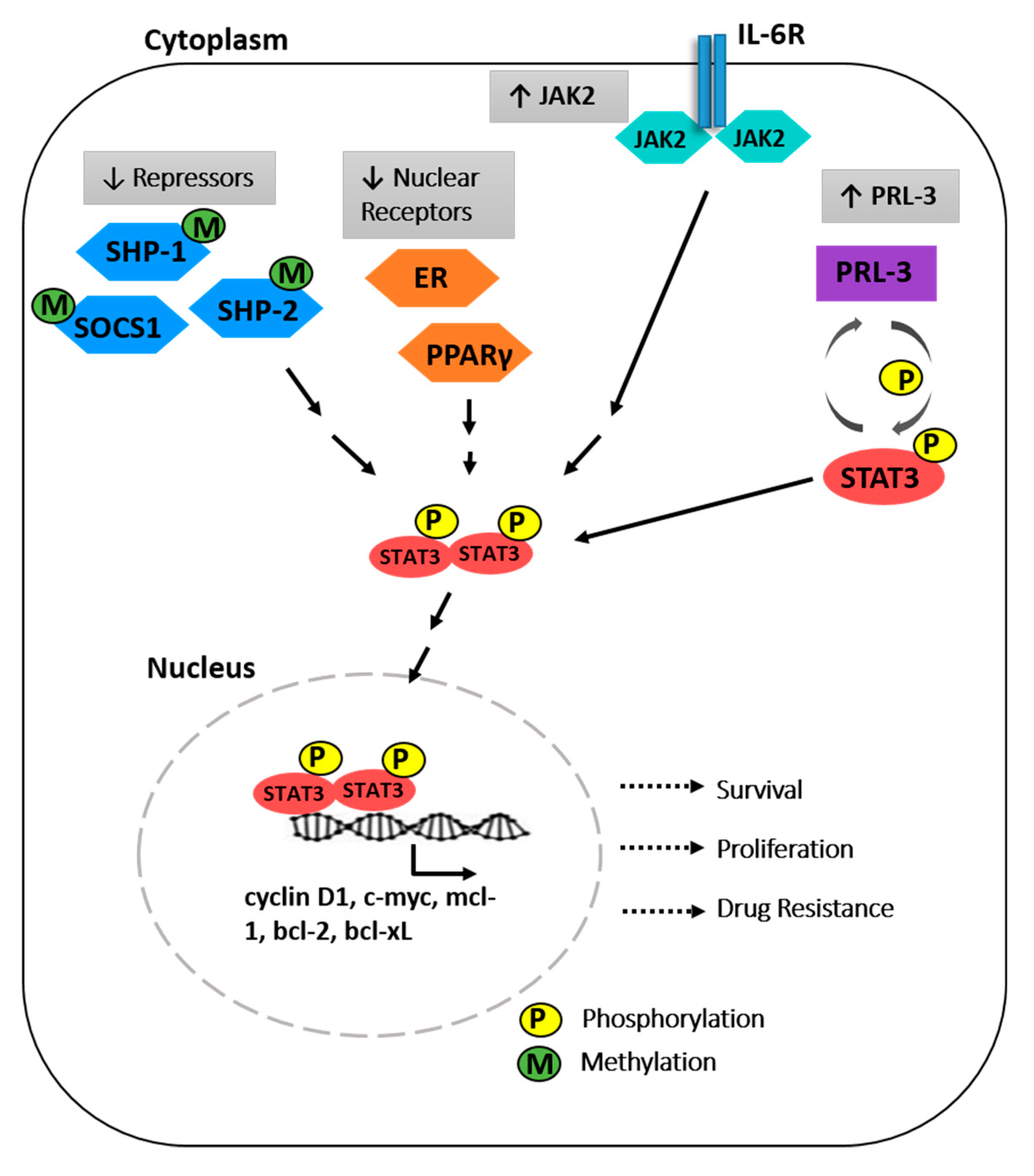
Figure 1.
The molecular mechanisms driving constitutive signal transducers and activators of transcription (STAT3) activation in myeloma cells. Phosphorylated STAT3 translocate to the nucleus to mediate transcription of target genes, resulting in increased survival, proliferation and drug resistance of myeloma cells. Src homology containing protein 1 (SHP-1), suppressor of cytokine signalling 1 (SOCS1), phosphatase of regenerating liver 3 (PRL3), Janus Associated kinase (JAK), peroxisome proliferator activated receptors (PPAR), estrogen receptor (ER).
Figure 1.
The molecular mechanisms driving constitutive signal transducers and activators of transcription (STAT3) activation in myeloma cells. Phosphorylated STAT3 translocate to the nucleus to mediate transcription of target genes, resulting in increased survival, proliferation and drug resistance of myeloma cells. Src homology containing protein 1 (SHP-1), suppressor of cytokine signalling 1 (SOCS1), phosphatase of regenerating liver 3 (PRL3), Janus Associated kinase (JAK), peroxisome proliferator activated receptors (PPAR), estrogen receptor (ER).


{kind=link}
Table 1.
Selected STAT3 inhibitors showing in vitro and/or clinical efficacy against multiple myeloma. The mechanism by which STAT3 is inhibited by the compound as well as evidence for in vitro synergy with known anti myeloma therapy and clinical evidence where applicable are presented. STAT3 (Signal transducers and activators of transcription 3), PIAS3 (protein inhibitor of activated STAT3), SHP-1 (SRC homology 2 domain containing phosphatase 1) PTEN (phosphatase and tensin homolog). MM (multiple myeloma), NA (data not available), SH2 (src homology 2), JAK (janus associated kinase), IL-6 (Interleukin 6).
Table 1.
Selected STAT3 inhibitors showing in vitro and/or clinical efficacy against multiple myeloma. The mechanism by which STAT3 is inhibited by the compound as well as evidence for in vitro synergy with known anti myeloma therapy and clinical evidence where applicable are presented. STAT3 (Signal transducers and activators of transcription 3), PIAS3 (protein inhibitor of activated STAT3), SHP-1 (SRC homology 2 domain containing phosphatase 1) PTEN (phosphatase and tensin homolog). MM (multiple myeloma), NA (data not available), SH2 (src homology 2), JAK (janus associated kinase), IL-6 (Interleukin 6).
| STAT-3 Inhibitor | Mechanism of STAT3 Inhibition | In Vitro Synergy with Known Anti MM Agents | Clinical Evidence of Efficacy | Reference |
|---|---|---|---|---|
| Ruxolitinib | Indirect, via JAK inhibition | Bortezomib Lenalidomide | Phase 1 clinical trial | Chen et al. 2014 [70] Berenson et al. 2018 [71] |
| Tofacitinib | Indirect, via JAK inhibition | Venetoclax | NA | Lam et al. 2018 [72] |
| INCB16562 | Indirect, via JAK1 inhibition | Bortezomib Melphalan | NA | Li et al. 2010 [74] |
| YL064 | Direct, STAT3 SH2 domain inhibitor | NA | NA | Wang et al. 2018 [78] |
| OPB51602 | Direct, STAT3 SH2 domain inhibitor | NA | Phase 1 clinical trial. Excessive toxicity and unfavourable pharmacokinetic profile | Ogura et al. 2015 [83] |
| Hydrocalamenene | Indirect, JAK1,2, SRC inhibition. Upregulation of PIAS3 | Bortezomib | NA | Nam et al. 2014 [89] |
| Genipin | Indirect, SRC inhibition, SHP-1 upregulation | Bortezomib, thalidomide, paclitaxel | NA | Lee et al. 2011 [91] |
| Icariside II | Indirect, JAK2, SRC inhibition. Upregulation of SHP-1 and PTEN | Bortezomib, Thalidomide | NA | Kim et al. 2011 [92] |
| Niclosamide | Indirect inhibition by inhibiting IL-6 mediated phosphorylation of STAT3 | NA | NA | Khanim et al. [77] |
| Asiaticoside | Reduced phosphorylation of STAT3, mechanism not known | NA | NA | Yingchun et al. [96] |
| Gossypol | Inhibition of IL-6 signalling | NA | NA | Sadahira et al. [97] |
| LCL161 | Not known, synergism with JAK2 inhibitor against MM cell lines | NA | NA | Ramakrishnan et al. [76] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chong, P.S.Y.; Chng, W.-J.; de Mel, S. STAT3: A Promising Therapeutic Target in Multiple Myeloma. Cancers 2019, 11, 731. https://doi.org/10.3390/cancers11050731
AMA Style
Chong PSY, Chng W-J, de Mel S. STAT3: A Promising Therapeutic Target in Multiple Myeloma. Cancers. 2019; 11(5):731. https://doi.org/10.3390/cancers11050731
Chicago/Turabian StyleChong, Phyllis S. Y., Wee-Joo Chng, and Sanjay de Mel. 2019. "STAT3: A Promising Therapeutic Target in Multiple Myeloma" Cancers 11, no. 5: 731. https://doi.org/10.3390/cancers11050731
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.