Nucleotide Weight Matrices Reveal Ubiquitous Mutational Footprints of AID/APOBEC Deaminases in Human Cancer Genomes

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
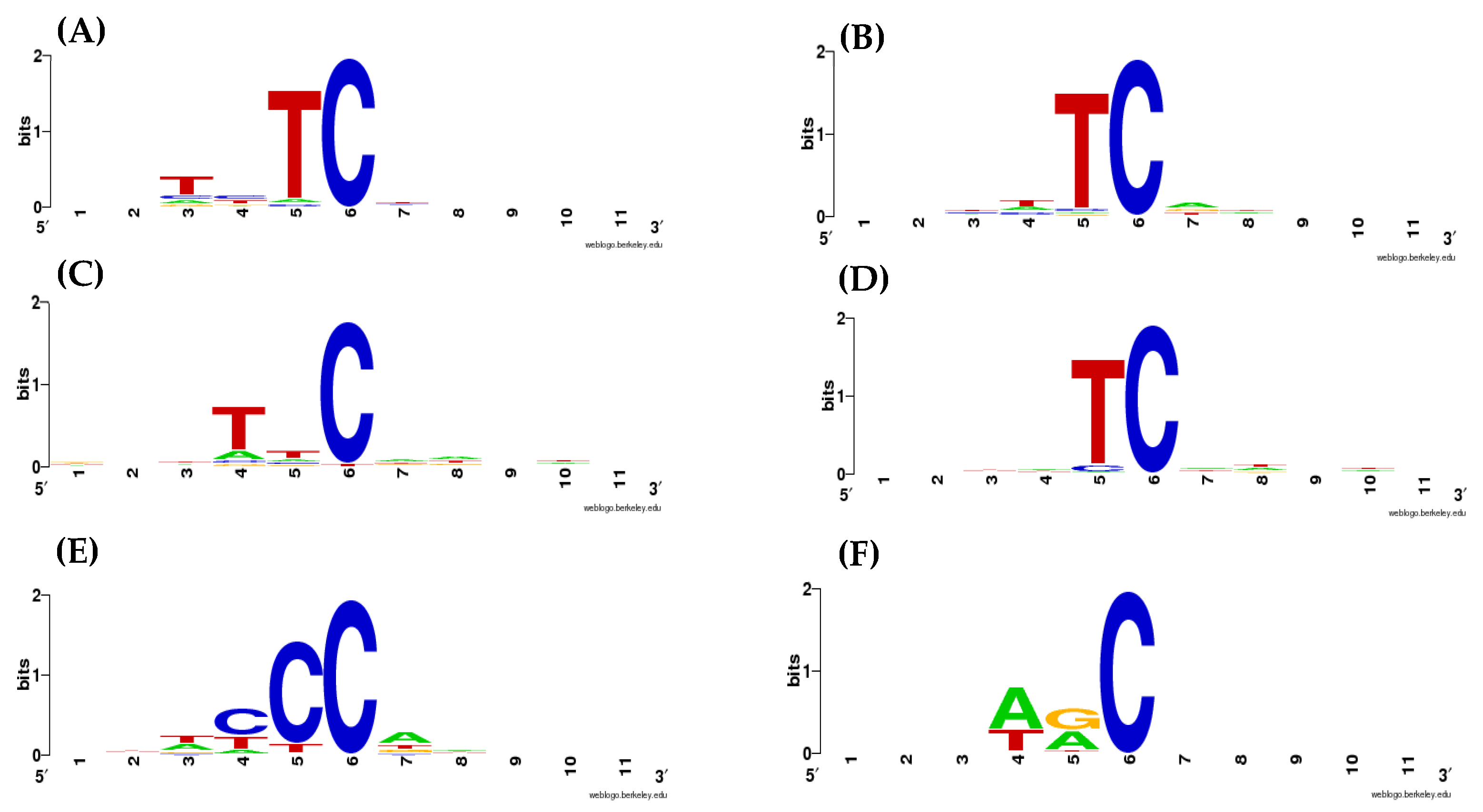
2.1. Weight Matrices of AID/APOBEC Mutable Motifs
2.2. Analysis of the Correlation between AID/APOBEC Mutable Motifs and Somatic Mutations in Cancer Cells: C:G>T:A Transitions
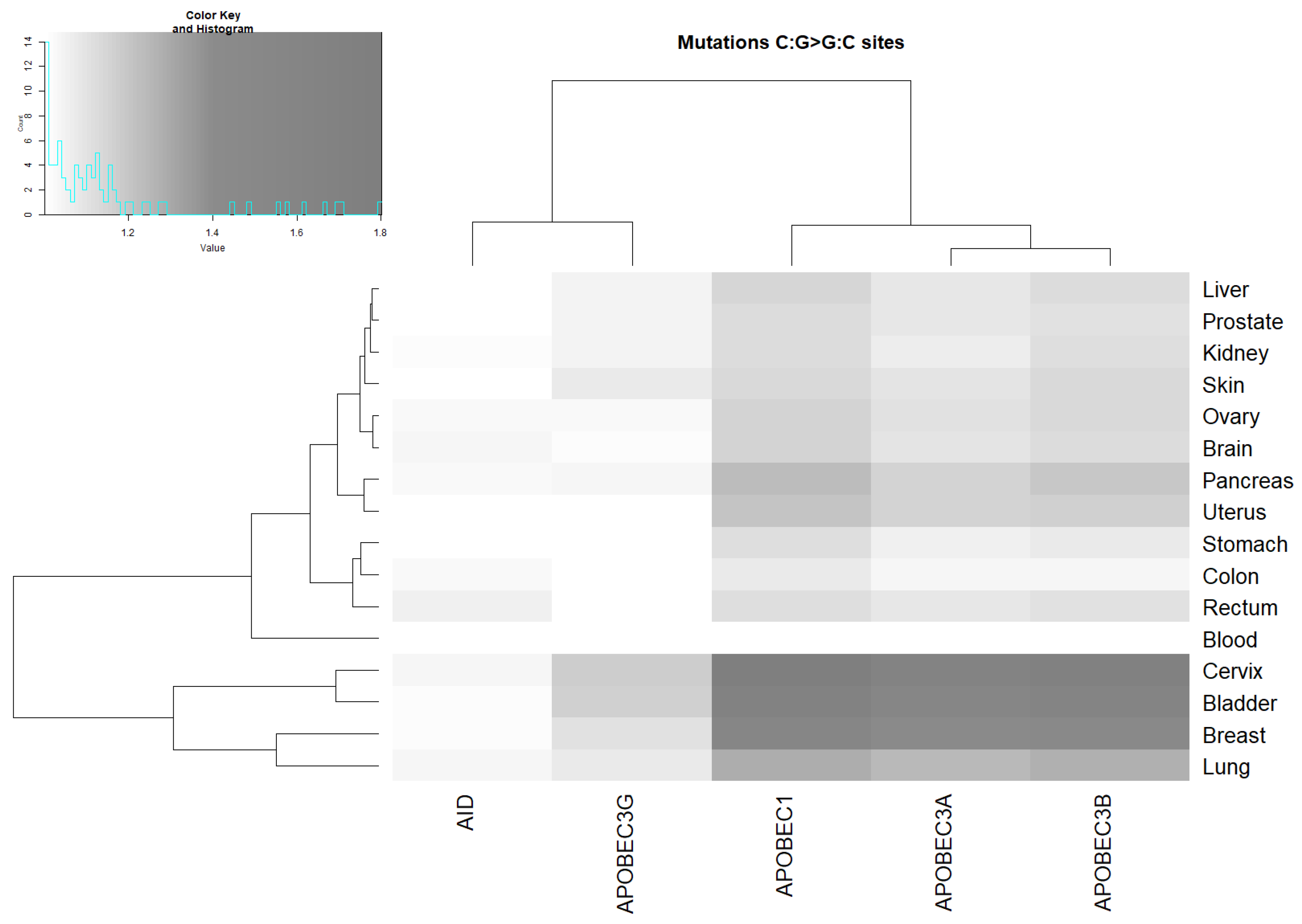
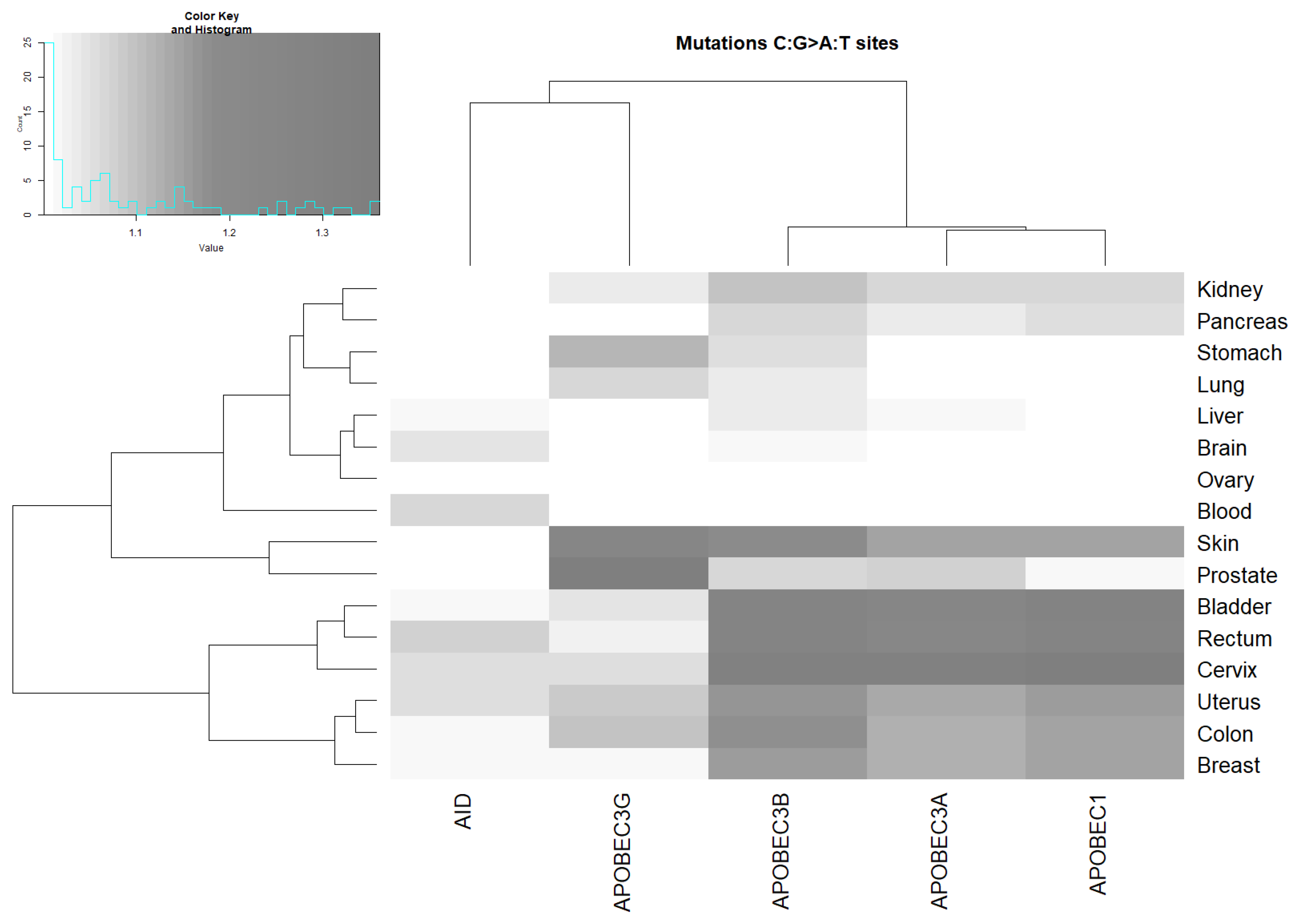
2.3. Analysis of the Correlation between AID/APOBEC Mutable Motifs and Somatic Mutations in Cancer Cells: C:G>G:C and C:G>A:T Transversions
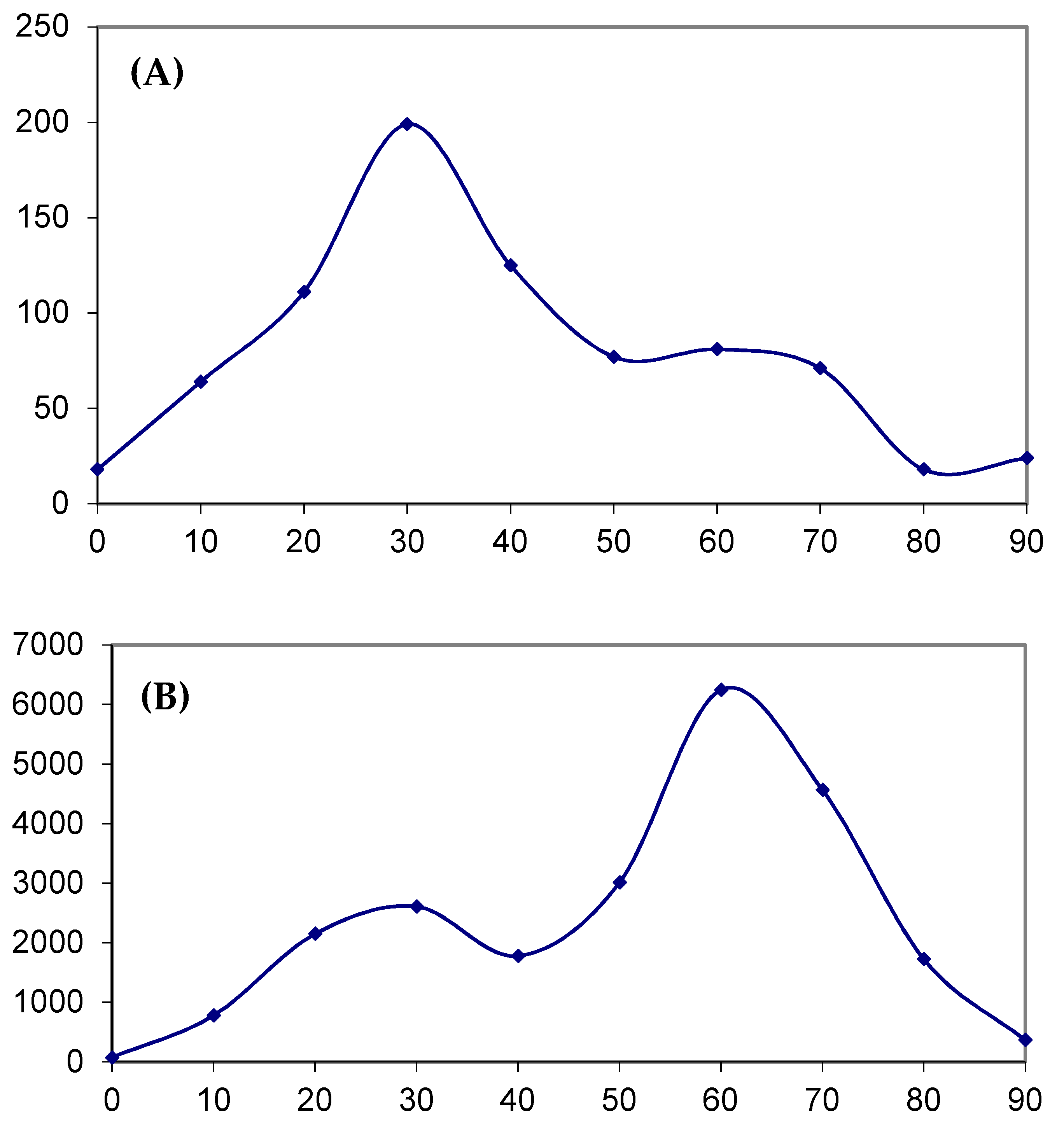
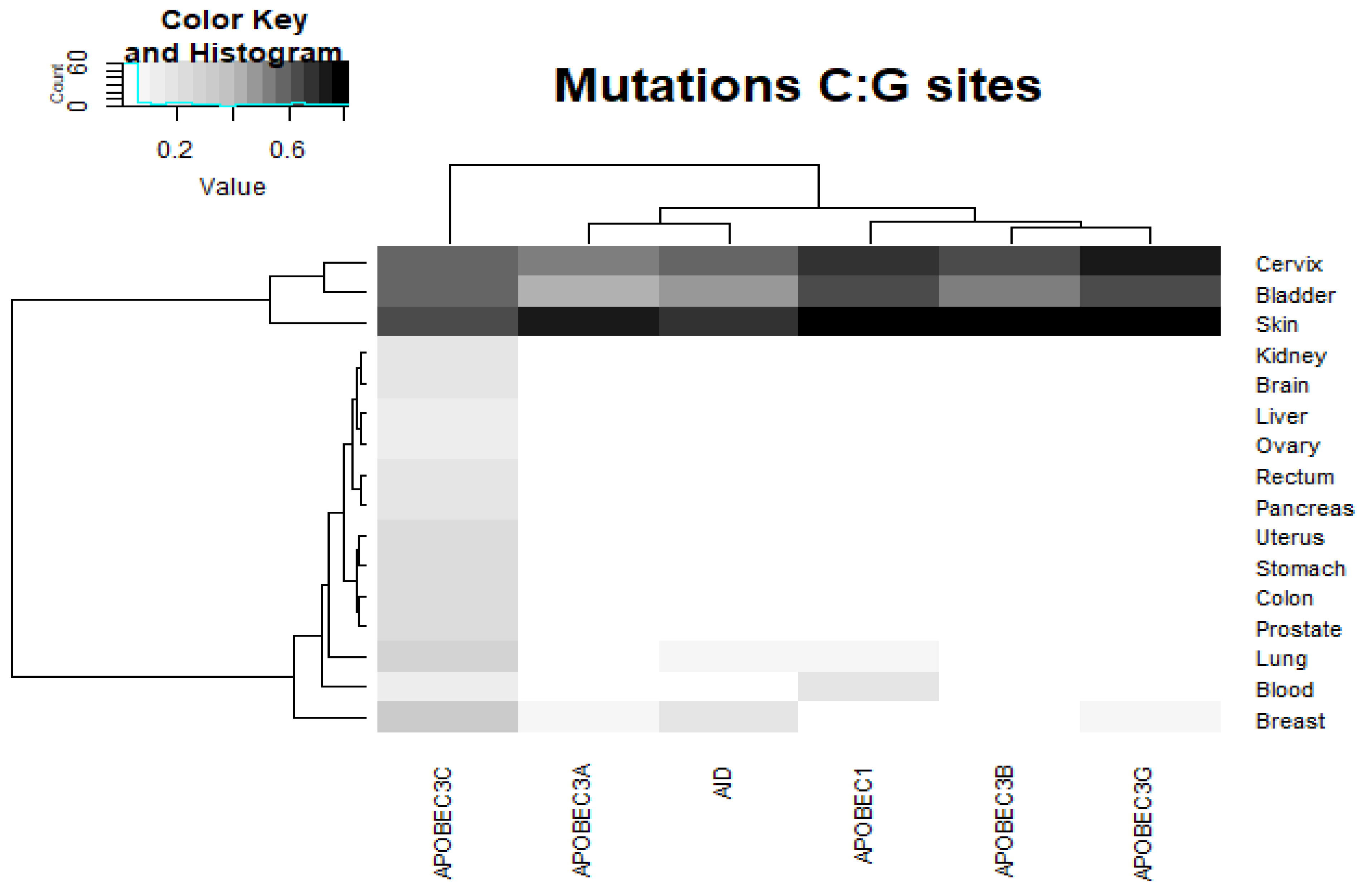
2.4. Analysis of Various Tumor Types in Blood and Skin
3. Discussion
4. Materials and Methods
4.1. Mutations in Yeast Genomes
4.2. Analysis of Mutable Motifs
S(b1, ..., bL) = Σ W(b,j)
j = 1
Smin = Σ MIN W(b,j) Smax = Σ MAX W(b,j)
j = 1 b j = 1 b
4.3. Datasets and Analysis of Somatic Mutations
4.4. Impact of AID/APOBEC Mutagenesis
4.5. Control Experiment 1: Analysis of Somatic Mutations in Mitochondrial DNA
4.6. Control Experiment 2: Correlation between the Matrices of Shuffled Sites of Mutations and the Sites of Somatic Mutation in Cancer Cells
4.7. Control Experiment 3: Correlation between Matrices of Randomly Sampled Sites from the Yeast Genome and Somatic Mutations in Cancer Cells
4.8. Control Experiment 4: Analysis of Somatic Mutations in Human Immunoglobulin Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, H.; Fujita, M. Whole genome sequencing analysis for cancer genomics and precision medicine. Cancer Sci. 2018, 109, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Springgate, C.F.; Battula, N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974, 34, 2311–2321. [Google Scholar] [PubMed]
- Roberts, S.A.; Gordenin, D.A. Hypermutation in human cancer genomes: Footprints and mechanisms. Nat. Rev. Cancer 2014, 14, 786–800. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Erratum: Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 174, 1034–1035. [Google Scholar] [CrossRef]
- Martincorena, I.; Roshan, A.; Gerstung, M.; Ellis, P.; Van Loo, P.; McLaren, S.; Wedge, D.C.; Fullam, A.; Alexandrov, L.B.; Tubio, J.M.; et al. Tumor evolution. High burden and pervasive positive selection of somatic mutations in normal human skin. Science 2015, 348, 880–886. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal patterns of selection in cancer and somatic tissues. Cell 2017, 171, 1029–1041. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef]
- Hutchinson, L. Genetics: Signatures of mutational processes in cancer-a big step closer. Nat. Rev. Clin. Oncol. 2013, 10, 545. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Stratton, M.R. Mutational signatures: The patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014, 24, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Goncearenco, A.; Rager, S.L.; Li, M.; Sang, Q.X.; Rogozin, I.B.; Panchenko, A.R. Exploring background mutational processes to decipher cancer genetic heterogeneity. Nucleic Acids Res. 2017, 45, W514–W522. [Google Scholar] [CrossRef]
- Burns, M.B.; Temiz, N.A.; Harris, R.S. Evidence for APOBEC3B mutagenesis in multiple human cancers. Nat. Genet. 2013, 45, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.V.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Pham, P.; Bransteitter, R.; Petruska, J.; Goodman, M.F. Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 2003, 424, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Gordenin, D.A. Clusters of multiple mutations: Incidence and molecular Mechanisms. Annu. Rev. Genet. 2015, 49, 243–267. [Google Scholar] [CrossRef]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef]
- Kuong, K.J.; Loeb, L.A. APOBEC3B mutagenesis in cancer. Nat. Genet. 2013, 45, 964–965. [Google Scholar] [CrossRef]
- Franchini, D.M.; Schmitz, K.M.; Petersen-Mahrt, S.K. 5-Methylcytosine DNA demethylation: More than losing a methyl group. Annu. Rev. Genet. 2012, 46, 419–441. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Lada, A.G.; Goncearenco, A.; Green, M.R.; De, S.; Nudelman, G.; Panchenko, A.R.; Koonin, E.V.; Pavlov, Y.I. Activation induced deaminase mutational signature overlaps with CpG methylation sites in follicular lymphoma and other cancers. Sci. Rep. 2016, 6, 38133. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Campbell, P.J.; Stratton, M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Rogozin, I.B.; Pavlov, Y.I.; Goncearenco, A.; De, S.; Lada, A.G.; Poliakov, E.; Panchenko, A.R.; Cooper, D.N. Mutational signatures and mutable motifs in cancer genomes. Brief. Bioinform. 2017, 19, 1085–1101. [Google Scholar] [CrossRef] [PubMed]
- Seplyarskiy, V.B.; Soldatov, R.A.; Popadin, K.Y.; Antonarakis, S.E.; Bazykin, G.A.; Nikolaev, S.I. APOBEC-induced mutations in human cancers are strongly enriched on the lagging DNA strand during replication. Genome Res. 2016, 26, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Haradhvala, N.J.; Polak, P.; Stojanov, P.; Covington, K.R.; Shinbrot, E.; Hess, J.M.; Rheinbay, E.; Kim, J.; Maruvka, Y.E.; Braunstein, L.Z.; et al. Mutational strand asymmetries in cancer genomes reveal mechanisms of DNA damage and repair. Cell 2016, 164, 538–549. [Google Scholar] [CrossRef] [PubMed]
- Temiz, N.A.; Donohue, D.E.; Bacolla, A.; Vasquez, K.M.; Cooper, D.N.; Mudunuri, U.; Ivanic, J.; Cer, R.Z.; Yi, M.; Stephens, R.M.; et al. The somatic autosomal mutation matrix in cancer genomes. Hum. Genet. 2015, 134, 851–864. [Google Scholar] [CrossRef] [PubMed]
- Staden, R. Computer methods to locate signals in nucleic acid sequences. Nucleic Acids Res. 1984, 12, 505–519. [Google Scholar] [CrossRef]
- Gelfand, M.S. Prediction of function in DNA sequence analysis. J. Comput. Biol.: J. Comput. Mol. Cell Biol. 1995, 2, 87–115. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Milanesi, L. Analysis of donor splice sites in different eukaryotic organisms. J. Mol. Evol. 1997, 45, 50–59. [Google Scholar] [CrossRef]
- Bishop, K.N.; Holmes, R.K.; Sheehy, A.M.; Davidson, N.O.; Cho, S.J.; Malim, M.H. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 2004, 14, 1392–1396. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Pavlov, Y.I. The cytidine deaminase AID exhibits similar functional properties in yeast and mammals. Mol. Immunol. 2006, 43, 1481–1484. [Google Scholar] [CrossRef] [PubMed]
- Ju, Y.S.; Alexandrov, L.B.; Gerstung, M.; Martincorena, I.; Nik-Zainal, S.; Ramakrishna, M.; Davies, H.R.; Papaemmanuil, E.; Gundem, G.; Shlien, A.; et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife 2014, 3, e02935. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B (Methodol.) 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Milstein, C.; Neuberger, M.S.; Staden, R. Both DNA strands of antibody genes are hypermutation targets. Proc. Natl. Acad. Sci. USA 1998, 95, 8791–8794. [Google Scholar] [CrossRef] [PubMed]
- Mayorov, V.I.; Rogozin, I.B.; Adkison, L.R.; Frahm, C.R.; Kunkel, T.A.; Pavlov, Y.I. Expression of human AID in yeast induces mutations in context similar to the context of somatic hypermutation at G-C pairs in immunoglobulin genes. BMC Immunol. 2005, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Zanotti, K.J.; Gearhart, P.J. Antibody diversification caused by disrupted mismatch repair and promiscuous DNA polymerases. DNA Repair 2016, 38, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.J.; Nik-Zainal, S.; Wu, Y.L.; Stebbings, L.A.; Raine, K.; Campbell, P.J.; Rada, C.; Stratton, M.R.; Neuberger, M.S. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. eLife 2013, 2, e00534. [Google Scholar] [CrossRef]
- Neuberger, M.S.; Rada, C. Somatic hypermutation: Activation-induced deaminase for C/G followed by polymerase eta for A/T. J. Exp. Med. 2007, 204, 7–10. [Google Scholar] [CrossRef]
- Rogozin, I.B.; Goncearenco, A.; Lada, A.G.; De, S.; Yurchenko, V.; Nudelman, G.; Panchenko, A.R.; Cooper, D.N.; Pavlov, Y.I. DNA polymerase eta mutational signatures are found in a variety of different types of cancer. Cell Cycle 2018, 17, 348–355. [Google Scholar] [CrossRef]
- Saini, N.; Roberts, S.A.; Klimczak, L.J.; Chan, K.; Grimm, S.A.; Dai, S.; Fargo, D.C.; Boyer, J.C.; Kaufmann, W.K.; Taylor, J.A.; et al. The impact of environmental and endogenous damage on somatic mutation load in human skin fibroblasts. PLoS Genet. 2016, 12, e1006385. [Google Scholar] [CrossRef]
- Pham, P.; Landolph, A.; Mendez, C.; Li, N.; Goodman, M.F. A biochemical analysis linking APOBEC3A to disparate HIV-1 restriction and skin cancer. J. Biol. Chem. 2013, 288, 29294–29304. [Google Scholar] [CrossRef] [PubMed]
- Cho, R.J.; Alexandrov, L.B.; den Breems, N.Y.; Atanasova, V.S.; Farshchian, M.; Purdom, E.; Nguyen, T.N.; Coarfa, C.; Rajapakshe, K.; Prisco, M.; et al. APOBEC mutation drives early-onset squamous cell carcinomas in recessive dystrophic epidermolysis bullosa. Sci. Transl. Med. 2018, 10, eaas9668. [Google Scholar] [CrossRef] [PubMed]
- Fickett, J.W.; Guigo, R. Estimation of protein coding density in a corpus of DNA sequence data. Nucleic Acid Res. 1993, 21, 2837–2844. [Google Scholar] [CrossRef] [PubMed]
- Glazko, G.B.; Milanesi, L.; Rogozin, I.B. The subclass approach for mutational spectrum analysis: Application of the SEM algorithm. J. Theor. Biol. 1998, 192, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Siriwardena, S.U.; Perera, M.L.W.; Senevirathne, V.; Stewart, J.; Bhagwat, A.S. A tumor promoting phorbol ester causes a large increase in APOBEC3A and a moderate increase in APOBEC3B expression in a normal human keratinocyte cell line without increasing genomic uracils. Mol. Cell. Biol. 2018, 39, e00238-18. [Google Scholar] [CrossRef] [PubMed]
- Neuberger, M.S.; Harris, R.S.; Di Noia, J.; Petersen-Mahrt, S.K. Immunity through DNA deamination. Trends Biochem. Sci. 2003, 28, 305–312. [Google Scholar] [CrossRef]
- Harris, R.S.; Petersen-Mahrt, S.K.; Neuberger, M.S. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 2002, 10, 1247–1253. [Google Scholar] [CrossRef]
- Petersen-Mahrt, S.K.; Harris, R.S.; Neuberger, M.S. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 2002, 418, 99–103. [Google Scholar] [CrossRef]
- Bhagwat, A.S. DNA-cytosine deaminases: From antibody maturation to antiviral defense. DNA Repair 2004, 3, 85–89. [Google Scholar] [CrossRef]
- Franchini, D.M.; Petersen-Mahrt, S.K. AID and APOBEC deaminases: Balancing DNA damage in epigenetics and immunity. Epigenomics 2014, 6, 427–443. [Google Scholar] [CrossRef]
- Rebhandl, S.; Huemer, M.; Greil, R.; Geisberger, R. AID/APOBEC deaminases and cancer. Oncoscience 2015, 2, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Landry, S.; Narvaiza, I.; Linfesty, D.C.; Weitzman, M.D. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. Embo Rep. 2011, 12, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.J.; Wu, Y.L.; Rada, C. Active RNAP pre-initiation sites are highly mutated by cytidine deaminases in yeast, with AID targeting small RNA genes. eLife 2014, 3, e03553. [Google Scholar] [CrossRef] [PubMed]
- Lada, A.G.; Kliver, S.F.; Dhar, A.; Polev, D.E.; Masharsky, A.E.; Rogozin, I.B.; Pavlov, Y.I. Disruption of transcriptional coactivator Sub1 leads to genome-wide re-distribution of clustered mutations induced by APOBEC in active yeast genes. PLoS Genet. 2015, 11, e1005217. [Google Scholar] [CrossRef] [PubMed]
- Lada, A.G.; Krick, C.F.; Kozmin, S.G.; Mayorov, V.I.; Karpova, T.S.; Rogozin, I.B.; Pavlov, Y.I. Mutator effects and mutation signatures of editing deaminases produced in bacteria and yeast. Biochemistry 2011, 76, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Lada, A.G.; Stepchenkova, E.I.; Zhuk, A.S.; Kliver, S.F.; Rogozin, I.B.; Polev, D.E.; Dhar, A.; Pavlov, Y.I. Recombination is responsible for the increased recovery of drug-resistant mutants with hypermutated genomes in resting yeast diploids expressing APOBEC deaminases. Front. Genet. 2017, 8, 202. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Carpenter, M.A.; Banerjee, S.; Shaban, N.M.; Kurahashi, K.; Salamango, D.J.; McCann, J.L.; Starrett, G.J.; Duffy, J.V.; Demir, O.; et al. Structural basis for targeted DNA cytosine deamination and mutagenesis by APOBEC3A and APOBEC3B. Nat. Struct. Mol. Biol. 2017, 24, 131–139. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Wedge, D.C.; Alexandrov, L.B.; Petljak, M.; Butler, A.P.; Bolli, N.; Davies, H.R.; Knappskog, S.; Martin, S.; Papaemmanuil, E.; et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat. Genet. 2014, 46, 487–491. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AID | APOBEC3G | APOBEC3C | APOBEC3B | APOBEC3A | |
|---|---|---|---|---|---|
| APOBEC1 | 1986.8 | 2299.2 | 203.2 | 378.6 | 344.1 |
| APOBEC3A | 1674.4 | 2057.0 | 138.4 | 175.7 | |
| APOBEC3B | 1764.5 | 2316.8 | 175.7 | ||
| APOBEC3C | 237.2 | 327.5 | |||
| APOBEC3G | 2711.8 |
| Cancer Tissue Type | Number of Mutations | Test | APOBEC1 | APOBEC3A | APOBEC3B | APOBEC3G | AID |
|---|---|---|---|---|---|---|---|
| Blood: acute myeloid leukemia | 6844 | Ratio | 0.920 | 0.978 | 0.958 | 0.977 | 1.031 |
| t-test | NSE # | NSE | NSE | NSE | 6.5 * | ||
| MC test | <0.001 | ||||||
| Fraction | |||||||
| Blood: GCB lymphomas | 2747 | Ratio | 0.967 | 1.030 | 0.979 | 0.980 | 1.091 |
| t-test | NSE | 3.4 * | NSE | NSE | 12.3 * | ||
| MC test | <0.001 | <0.001 | |||||
| Fraction | 0.208 | ||||||
| Skin: cutaneous melanoma | 235043 | Ratio | 1.388 | 1.308 | 1.334 | 1.138 | 1.026 |
| t-test | 321.3 * | 292.8 * | 344.6* | 176.2 * | 35.8 * | ||
| MC test | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | ||
| Fraction | 0.608 | 0.508 | 0.982 | 0.687 | |||
| Skin: adeno-carcinoma | 780 | Ratio | 1.045 | 1.073 | 1.088 | 1.075 | 1.025 |
| t-test | NSE | 4.4 * | 4.8 * | 4.6 * | NSE | ||
| MC test | <0.001 | <0.001 | <0.001 | ||||
| Fraction | 0.213 |
| Locus | Number of Mutations | Test | APOBEC1 | APOBEC3A | APOBEC3B | APOBEC3G | AID |
|---|---|---|---|---|---|---|---|
| VH26 | 708 | Ratio | 0.931 | 0.986 | 0.919 | 0.908 | 1.162 |
| t-test | NSE # | NSE | NSE | NSE | 11.1 * | ||
| MC test | <0.001 | ||||||
| Fraction | 0.477 | ||||||
| JH4 intron, control individuals | 177 | Ratio | 0.927 | 0.957 | 0.887 | 0.870 | 1.331 |
| t-test | NSE | NSE | NSE | NSE | 11.9 * | ||
| MC test | <0.001 | ||||||
| Fraction | 0.559 | ||||||
| JH4 intron, XP-V patients | 235 | Ratio | 0.981 | 1.008 | 0.957 | 0.930 | 1.266 |
| t-test | NSE | NSE | NSE | NSE | 9.6 * | ||
| MC test | <0.001 | ||||||
| Fraction | 0.366 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rogozin, I.B.; Roche-Lima, A.; Lada, A.G.; Belinky, F.; Sidorenko, I.A.; Glazko, G.V.; Babenko, V.N.; Cooper, D.N.; Pavlov, Y.I. Nucleotide Weight Matrices Reveal Ubiquitous Mutational Footprints of AID/APOBEC Deaminases in Human Cancer Genomes. Cancers 2019, 11, 211. https://doi.org/10.3390/cancers11020211
Rogozin IB, Roche-Lima A, Lada AG, Belinky F, Sidorenko IA, Glazko GV, Babenko VN, Cooper DN, Pavlov YI. Nucleotide Weight Matrices Reveal Ubiquitous Mutational Footprints of AID/APOBEC Deaminases in Human Cancer Genomes. Cancers. 2019; 11(2):211. https://doi.org/10.3390/cancers11020211
Chicago/Turabian StyleRogozin, Igor B., Abiel Roche-Lima, Artem G. Lada, Frida Belinky, Ivan A. Sidorenko, Galina V. Glazko, Vladimir N. Babenko, David N. Cooper, and Youri I. Pavlov. 2019. "Nucleotide Weight Matrices Reveal Ubiquitous Mutational Footprints of AID/APOBEC Deaminases in Human Cancer Genomes" Cancers 11, no. 2: 211. https://doi.org/10.3390/cancers11020211