Dissecting the Mutational Landscape of Cutaneous Melanoma: An Omic Analysis Based on Patients from Greece

 , ,
, ,  , and
, and
Abstract
:1. Introduction
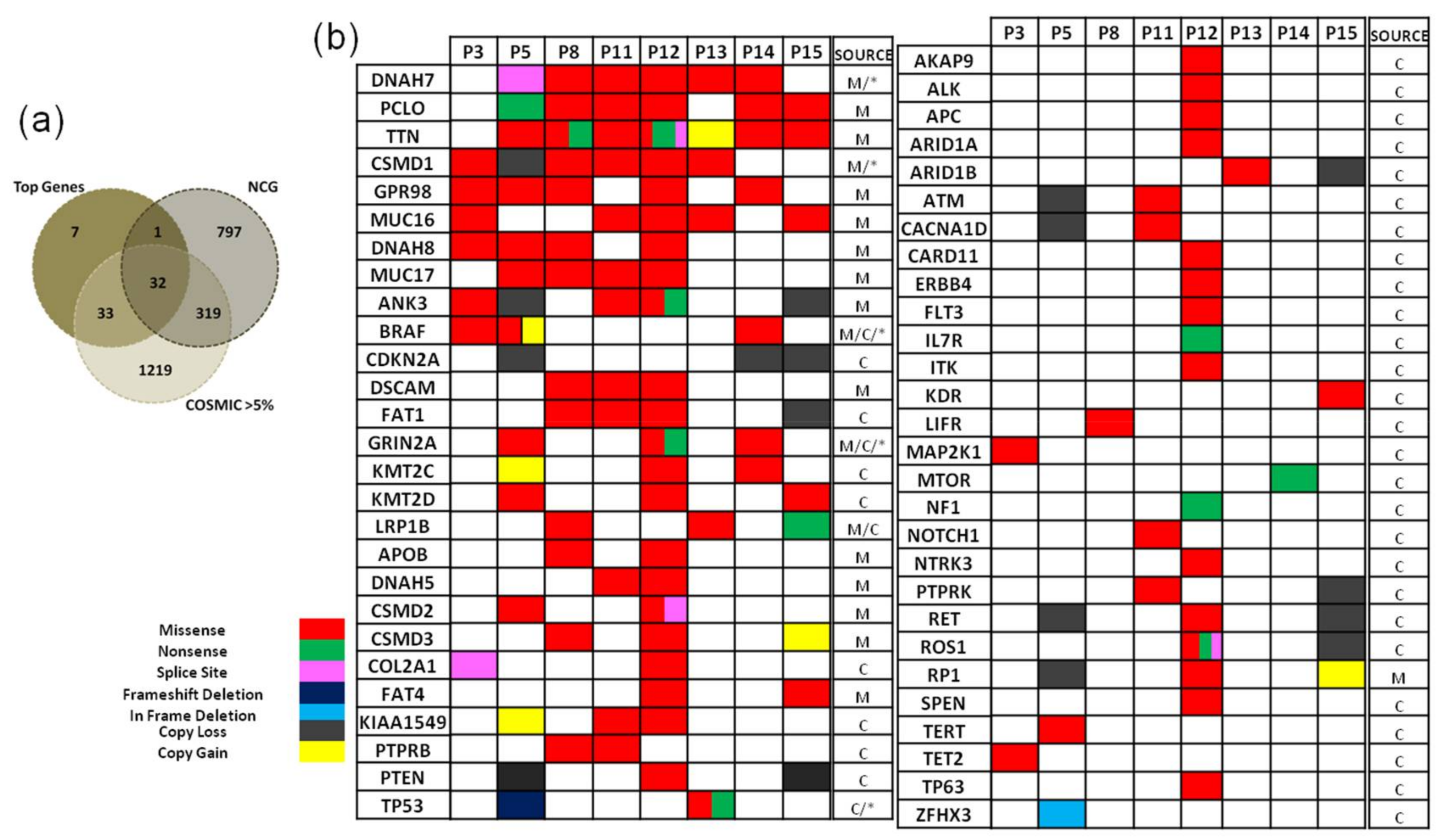
2. Results
2.1. Sequencing Data
2.2. Identification of Germ-Line Variation
2.3. Identification of Somatic Coding Mutations
2.3.1. Characterisation of Mutated Genes and Copy Number Variations
2.3.2. Bioinformatics Pathway Analysis
3. Discussion
4. Materials & Methods
4.1. Melanoma Samples
4.2. DNA Extraction and Exome Sequencing
4.3. Raw Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BMR | background mutation rate |
| CM | cutaneous melanoma |
| CNV | copy number variation |
| COSMIC | Catalogue of Somatic Mutations in Cancer |
| FFPE | formalin fixed paraffin embedded |
| GWAS | genome wide association study |
| Indel | insertion/deletion |
| NCG | Network of Cancer Genes |
| NGS | next generation sequencing |
| SNP | single nucleotide polymorphism |
| TCGA | The Cancer Genome Atlas |
| TPM | Transcripts per million |
| UTR | un-translated region |
| UVR | ultraviolet radiation |
| WES | whole exome sequencing |
| Nucleotides | A-adenine, C-cytosine, G-guanine, T-thymine |
| Amino-Acids | E-glutamic acid, V-valine |
References
- Uong, A.; Zon, L.I. Melanocytes in development and cancer. J. Cell. Physiol. 2010, 222, 38–41. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, V.; Stratigos, A. Emerging trends in the epidemiology of melanoma. Br. J. Dermatol. 2014, 170, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Garbe, C.; Leiter, U. Melanoma epidemiology and trends. Clin. Dermatol. 2009, 27, 3–9. [Google Scholar] [CrossRef] [PubMed]
- EUCAN (European Cancer Observatory). Available online: http://eco.iarc.fr (accessed on 1 January 2017).
- Forsea, A.M.; Del Marmol, V.; De Vries, E.; Bailey, E.; Geller, A. Melanoma incidence and mortality in Europe: new estimates, persistent disparities. Br. J. Dermatol. 2012, 167, 1124–1130. [Google Scholar] [CrossRef] [PubMed]
- Hussussian, C.J.; Struewing, J.P.; Goldstein, A.M.; Higgins, P.A.; Ally, D.S.; Sheahan, M.D.; Clark, W.H.; Tucker, M.A.; Dracopoli, N.C. Germline p16 mutations in familial melanoma. Nat. Genet. 1994, 8, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Kamb, A.; Shattuck-Eidens, D.; Eeles, R.; Liu, Q.; Gruis, N.A.; Ding, W.; Hussey, C.; Tran, T.; Miki, Y.; Weaver-Feldhaus, J.; et al. Analysis of the p16 gene (CDKN2) as a candidate for the chromosome 9p melanoma susceptibility locus. Nat. Genet. 1994, 8, 23–26. [Google Scholar] [CrossRef] [PubMed]
- Aoude, L.G.; Wadt, K.A.; Pritchard, A.L.; Hayward, N.K. Genetics of familial melanoma: 20 years after CDKN2A. Pigment Cell Melanoma Res. 2015, 28, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Athanasiadis, E.I.; Antonopoulou, K.; Chatzinasiou, F.; Lill, C.M.; Bourdakou, M.M.; Sakellariou, A.; Kypreou, K.; Stefanaki, I.; Evangelou, E.; Ioannidis, J.P.; et al. A Web-based database of genetic association studies in cutaneous melanoma enhanced with network-driven data exploration tools. Database J. Biol. Databases Curation 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Antonopoulou, K.; Stefanaki, I.; Lill, C.M.; Chatzinasiou, F.; Kypreou, K.P.; Karagianni, F.; Athanasiadis, E.; Spyrou, G.M.; Ioannidis, J.P.; Bertram, L.; et al. Updated field synopsis and systematic meta-analyses of genetic association studies in cutaneous melanoma: The MelGene database. J. Investig. Dermatol. 2015, 135, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Chatzinasiou, F.; Lill, C.M.; Kypreou, K.; Stefanaki, I.; Nicolaou, V.; Spyrou, G.; Evangelou, E.; Roehr, J.T.; Kodela, E.; Katsambas, A. Comprehensive field synopsis and systematic meta-analyses of genetic association studies in cutaneous melanoma. J. Natl. Cancer Inst. 2011, 103, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Law, M.H.; Bishop, D.T.; Lee, J.E.; Brossard, M.; Martin, N.G.; Moses, E.K.; Song, F.; Barrett, J.H.; Kumar, R.; Easton, D.F.; et al. Genome-wide meta-analysis identifies five new susceptibility loci for cutaneous malignant melanoma. Nat. Genet. 2015, 47, 987–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutton-Regester, K.; Hayward, N.K. Reviewing the somatic genetics of melanoma: from current to future analytical approaches. Pigment Cell Melanoma Res. 2012, 25, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Walia, V.; Mu, E.W.; Lin, J.C.; Samuels, Y. Delving into somatic variation in sporadic melanoma. Pigment Cell Melanoma Res. 2012, 25, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Walia, V.; Lin, J.C.; Teer, J.K.; Prickett, T.D.; Gartner, J.; Davis, S.; Stemke-Hale, K.; Davies, M.A.; Gershenwald, J.E. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat. Genet. 2011, 43, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, S.I.; Rimoldi, D.; Iseli, C.; Valsesia, A.; Robyr, D.; Gehrig, C.; Harshman, K.; Guipponi, M.; Bukach, O.; Zoete, V.; et al. Exome sequencing identifies recurrent somatic MAP2K1 and MAP2K2 mutations in melanoma. Nat. Genet. 2011, 44, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Krauthammer, M.; Kong, Y.; Ha, B.H.; Evans, P.; Bacchiocchi, A.; McCusker, J.P.; Cheng, E.; Davis, M.J.; Goh, G.; Choi, M.; et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat. Genet. 2012, 44, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.S.; Woods, S.L.; Gartside, M.G.; Bonazzi, V.F.; Dutton-Regester, K.; Aoude, L.G.; Chow, D.; Sereduk, C.; Niemi, N.M.; Tang, N.; et al. Frequent somatic mutations in MAP3K5 and MAP3K9 in metastatic melanoma identified by exome sequencing. Nat. Genet. 2012, 44, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Stark, M.S.; Klein, K.; Weide, B.; Haydu, L.E.; Pflugfelder, A.; Tang, Y.H.; Palmer, J.M.; Whiteman, D.C.; Scolyer, R.A.; Mann, G.J.; et al. The prognostic and predictive value of melanoma-related microRNAs using tissue and serum: A microRNA expression analysis. EBioMedicine 2015, 2, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodis, E.; Watson, I.R.; Kryukov, G.V.; Arold, S.T.; Imielinski, M.; Theurillat, J.P.; Nickerson, E.; Auclair, D.; Li, L.; Place, C.; et al. A landscape of driver mutations in melanoma. Cell 2012, 150, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Lopez-Bigas, N. Functional impact bias reveals cancer drivers. Nucleic Acids Res. 2012, 40, e169. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Tamborero, D.; Schroeder, M.P.; Jene-Sanz, A.; Santos, A.; Lopez-Bigas, N. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods 2013, 10, 1081–1082. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Dobson, J.R.; Oesper, L.; Vandin, F. Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine. Genome Med. 2014, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Dutton-Regester, K.; Brown, K.M.; Hayward, N.K. The genomic landscape of cutaneous melanoma. Pigment Cell Melanoma Res. 2016, 29, 266–283. [Google Scholar] [CrossRef] [PubMed]
- Pikor, L.A.; Enfield, K.S.; Cameron, H.; Lam, W.L. DNA extraction from paraffin embedded material for genetic and epigenetic analyses. J. Vis. Exp. 2011, 49, e2763. [Google Scholar] [CrossRef] [PubMed]
- Sengüven, B.; Baris, E.; Oygur, T.; Berktas, M. Comparison of methods for the extraction of DNA from formalin-fixed, paraffin-embedded archival tissues. Int. J. Med. Sci. 2014, 11, 494–499. [Google Scholar] [CrossRef] [PubMed]
- De Paoli-Iseppi, R.; Johansson, P.A.; Menzies, A.M.; Dias, K.R.; Pupo, G.M.; Kakavand, H.; Wilmott, J.S.; Mann, G.J.; Hayward, N.K.; Dinger, M.E.; et al. Comparison of whole-exome sequencing of matched fresh and formalin fixed paraffin embedded melanoma tumours: Implications for clinical decision making. Pathology 2016, 48, 261–266. [Google Scholar] [CrossRef] [PubMed]
- Menon, R.; Deng, M.; Boehm, D.; Braun, M.; Fend, F.; Boehm, D.; Biskup, S.; Perner, S. Exome enrichment and SOLiD sequencing of formalin fixed paraffin embedded (FFPE) prostate cancer tissue. Int. J. Mol. Sci. 2012, 13, 8933–8942. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Sehn, J.K.; Abel, H.J.; Watson, M.A.; Pfeifer, J.D.; Duncavage, E.J. Comparison of clinical targeted next-generation sequence data from formalin-fixed and fresh-frozen tissue specimens. J. Mol. Diagn. 2013, 15, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Wagle, N.; Stojanov, P.; Perrin, D.L.; Cibulskis, K.; Marlow, S.; Jane-Valbuena, J.; Friedrich, D.C.; Kryukov, G.; Carter, S.L.; et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat. Med. 2014, 20, 682–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, P.; Lehmann, B.D.; Shyr, Y.; Guo, Y. The Utilization of Formalin Fixed-Paraffin-Embedded Specimens in High Throughput Genomic Studies. Int. J. Genom. 2017, 2017, 1926304. [Google Scholar] [CrossRef] [PubMed]
- Kypreou, K.P.; Stefanaki, I.; Antonopoulou, K.; Karagianni, F.; Ntritsos, G.; Zaras, A.; Nikolaou, V.; Kalfa, I.; Chasapi, V.; Polydorou, D.; et al. Prediction of Melanoma Risk in a Southern European Population Based on a Weighted Genetic Risk Score. J. Investig. Dermatol. 2016, 136, 690–695. [Google Scholar] [CrossRef] [PubMed]
- Majewski, J.; Schwartzentruber, J.; Lalonde, E.; Montpetit, A.; Jabado, N. What can exome sequencing do for you? J. Med. Genet. 2011, 48, 580–589. [Google Scholar] [CrossRef]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2016, 45, D896–D901. [Google Scholar] [CrossRef] [PubMed]
- Barrett, J.H.; Iles, M.M.; Harland, M.; Taylor, J.C.; Aitken, J.F.; Andresen, P.A.; Akslen, L.A.; Armstrong, B.K.; Avril, M.F.; Azizi, E.; et al. Genome-wide association study identifies three new melanoma susceptibility loci. Nat. Genet. 2011, 43, 1108–1113. [Google Scholar] [CrossRef] [PubMed]
- Nan, H.; Kraft, P.; Hunter, D.J.; Han, J. Genetic variants in pigmentation genes, pigmentary phenotypes, and risk of skin cancer in Caucasians. Int. J. Cancer 2009, 125, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Aitken, J.; Welch, J.; Duffy, D.; Milligan, A.; Green, A.; Martin, N.; Hayward, N. CDKN2A variants in a population-based sample of Queensland families with melanoma. J. Natl. Cancer Inst. 1999, 91, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Schrama, D.; Scherer, D.; Schneider, M.; Zapatka, M.; Bröcker, E.B.; Schadendorf, D.; Ugurel, S.; Kumar, R.; Becker, J.C. ERCC5 p. Asp1104His and ERCC2 p. Lys751Gln polymorphisms are independent prognostic factors for the clinical course of melanoma. J. Investig. Dermatol. 2011, 131, 1280–1290. [Google Scholar] [CrossRef] [PubMed]
- Sturm, R.A.; Duffy, D.L.; Zhao, Z.Z.; Leite, F.P.; Stark, M.S.; Hayward, N.K.; Martin, N.G.; Montgomery, G.W. A single SNP in an evolutionary conserved region within intron 86 of the HERC2 gene determines human blue-brown eye color. Am. J. Hum. Genet. 2008, 82, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Gerstenblith, M.R.; Shi, J.; Landi, M.T. Genome-wide association studies of pigmentation and skin cancer: A review and meta-analysis. Pigment Cell Melanoma Res 2010, 23, 587–606. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; DiCarlo, J.; Satya, R.V.; Peng, Q.; Wang, Y. Comparison of somatic mutation calling methods in amplicon and whole exome sequence data. BMC Genom. 2014, 15, 244. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Brash, D.E. UV signature mutations. Photochem. Photobiol. 2015, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- An, O.; Dall’Olio, G.M.; Mourikis, T.P.; Ciccarelli, F.D. NCG 5.0: Updates of a manually curated repository of cancer genes and associated properties from cancer mutational screenings. Nucleic Acids Res. 2016, 44, D992–D999. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- COSMIC Database. Available online: http://cancer.sanger.ac.uk (accessed on 1 January 2017).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shain, A.H.; Garrido, M.; Botton, T.; Talevich, E.; Yeh, I.; Sanborn, J.Z.; Chung, J.; Wang, N.J.; Kakavand, H.; Mann, G.J.; et al. Exome sequencing of desmoplastic melanoma identifies recurrent NFKBIE promoter mutations and diverse activating mutations in the MAPK pathway. Nat. Genet. 2015, 47, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Hodis, E.; Heffernan, T.P.; Deribe, Y.L.; Lawrence, M.S.; Protopopov, A.; Ivanova, E.; Watson, I.R.; Nickerson, E.; Ghosh, P.; et al. Melanoma genome sequencing reveals frequent PREX2 mutations. Nature 2012, 485, 502–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastian, B.C. The molecular pathology of melanoma: An integrated taxonomy of melanocytic neoplasia. Annu. Rev. Pathol. 2014, 9, 239–271. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Alessi, D.R.; James, S.R.; Downes, C.P.; Holmes, A.B.; Gaffney, P.R.; Reese, C.B.; Cohen, P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr. Biol. 1997, 7, 261–269. [Google Scholar] [CrossRef]
- Gagliardi, P.A.; Puliafito, A.; Primo, L. PDK1: At the crossroad of cancer signaling pathways. Semin. Cancer Biol. 2018, 49, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Su, T.; Saal, L.H.; Koujak, S.; Hopkins, B.D.; Barkley, C.R.; Wu, J.; Nandula, S.; Dutta, B.; Xie, Y. 3-Phosphoinositide-dependent kinase 1 potentiates upstream lesions on the phosphatidylinositol 3-kinase pathway in breast carcinoma. Cancer Res. 2009, 69, 6299–6306. [Google Scholar] [CrossRef] [PubMed]
- Choucair, K.A.; Guérard, K.P.; Ejdelman, J.; Chevalier, S.; Yoshimoto, M.; Scarlata, E.; Fazli, L.; Sircar, K.; Squire, J.A.; Brimo, F. The 16p13. 3 (PDPK1) genomic gain in prostate cancer: a potential role in disease progression. Transl. Oncol. 2012, 5, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Scortegagna, M.; Ruller, C.; Feng, Y.; Lazova, R.; Kluger, H.; Li, J.L.; De, S.K.; Rickert, R.; Pellecchia, M.; Bosenberg, M. Genetic inactivation or pharmacological inhibition of Pdk1 delays development and inhibits metastasis of BrafV600E: Pten–/– melanoma. Oncogene 2014, 33, 4330–4339. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Marino-Enriquez, A.; Bennett, R.R.; Zhu, M.; Shen, Y.; Eilers, G.; Lee, J.C.; Henze, J.; Fletcher, B.S.; Gu, Z. Dystrophin is a tumor suppressor in human cancers with myogenic programs. Nat. Genet. 2014, 46, 601–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Körner, H.; Epanchintsev, A.; Berking, C.; Schuler-Thurner, B.; Speicher, M.R.; Menssen, A.; Hermeking, H. Digital karyotyping reveals frequent inactivation of the dystrophin/DMD gene in malignant melanoma. Cell Cycle 2007, 6, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Luce, L.N.; Abbate, M.; Cotignola, J.; Giliberto, F. Non-myogenic tumors display altered expression of dystrophin (DMD) and a high frequency of genetic alterations. Oncotarget 2017, 8, 145. [Google Scholar] [CrossRef] [PubMed]
- Kontogianni, G.; Papadodima, O.; Maglogiannis, I.; Frangia-Tsivou, K.; Chatziioannou, A. Integrative Bioinformatic Analysis of a Greek Epidemiological Cohort Provides Insight into the Pathogenesis of Primary Cutaneous Melanoma. In Proceedings of the IFIP International Conference on Artificial Intelligence Applications and Innovations, Thessaloniki, Greece, 16–18 Sepotember 2016; pp. 39–52. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Indelocator. Available online: http://archive.broadinstitute.org/cancer/cga/indelocator (accessed on 1 January 2016).
- Ramos, A.H.; Lichtenstein, L.; Gupta, M.; Lawrence, M.S.; Pugh, T.J.; Saksena, G.; Meyerson, M.; Getz, G. Oncotator: Cancer variant annotation tool. Hum. Mutat. 2015, 36, E2423–E2429. [Google Scholar] [CrossRef] [PubMed]
- Koutsandreas, T.; Binen0baum, I.; Pilalis, E.; Valavanis, I.; Papadodima, O.; Chatziioannou, A. Analyzing and visualizing genomic complexity for the derivation of the emergent molecular networks. Int. J. Monit. Surveill. Technol. Res. 2016, 4, 30–49. [Google Scholar] [CrossRef]
- Liu, X.; Jian, X.; Boerwinkle, E. dbNSFP v2.0: A database of human non-synonymous SNVs and their functional predictions and annotations. Hum. Mutat. 2013, 34, E2393–E2402. [Google Scholar] [CrossRef] [PubMed]
- Talevich, E.; Shain, A.H.; Botton, T.; Bastian, B.C. CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput. Biol. 2016, 12, e1004873. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Patients | 3 | 5 | 8 | 10 | 11 | 12 | 13 | 14 | 15 |
|---|---|---|---|---|---|---|---|---|---|
| Normal | |||||||||
| Alignment rate (%) | 96 | 84.2 | 65.6 | 93.8 | 96.8 | 96.8 | 97.4 | 87.9 | 95.6 |
| Average sequencing depth on target | 129.8 | 93.9 | 72.2 | 103.9 | 101.5 | 123.3 | 117.4 | 102.5 | 128.4 |
| Fraction of target covered by >20× (%) | 97.37 | 96.37 | 94.41 | 96.44 | 95.03 | 97.53 | 97 | 97.38 | 97.96 |
| Tumour | |||||||||
| Alignment rate (%) | 95.4 | 89.5 | 93.6 | 92.6 | 96.7 | 96.2 | 96.8 | 88.8 | 92 |
| Average sequencing depth on target | 118.9 | 100.8 | 102.2 | 111.5 | 111.4 | 104.8 | 111.9 | 102.2 | 120.7 |
| Fraction of target covered by >20× (%) | 97.4 | 95.86 | 96.48 | 96.75 | 96.74 | 96.62 | 96.16 | 96.3 | 96.66 |
| Average sequencing depth on target | 108.7 | ||||||||
| dbSNP_ID | Gene | Chr | Variant Classification | Ref. Allele | MA Allele | # Hom. Ref. | # Hom. MA | # Heter. |
|---|---|---|---|---|---|---|---|---|
| rs1801516 | ATM | 11 | Missense Mutation | G | A | 6 | 0 | 3 |
| rs11515 | CDKN2A | 9 | 3′UTR | C | G | 0 | 7 | 2 |
| rs16891982 | SLC45A2 | 5 | Missense Mutation | C | G | 0 | 9 | 0 |
| rs17655 | ERCC5 | 13 | Missense Mutation | G | C | 7 | 0 | 2 |
| rs1800407 | OCA2 | 15 | Missense Mutation | G | A | 8 | 0 | 1 |
| rs1042602 | TYR | 11 | Missense Mutation | C | A | 2 | 2 | 5 |
| Patients | 3 | 5 | 8 | 10 | 11 | 12 | 13 | 14 | 15 |
|---|---|---|---|---|---|---|---|---|---|
| Number of Mutations | 522 | 693 | 901 | 27 | 935 | 5324 | 511 | 589 | 528 |
| Frequency per Mb | 10.4 | 13.8 | 17.9 | 0.5 | 18.6 | 105.7 | 10.1 | 11.7 | 10.5 |
| Non-synonymous | 226 | 284 | 387 | 18 | 364 | 2036 | 222 | 224 | 193 |
| Genes with non-synonymous | 216 | 274 | 360 | 16 | 338 | 1634 | 201 | 218 | 185 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kontogianni, G.; Piroti, G.; Maglogiannis, I.; Chatziioannou, A.; Papadodima, O. Dissecting the Mutational Landscape of Cutaneous Melanoma: An Omic Analysis Based on Patients from Greece. Cancers 2018, 10, 96. https://doi.org/10.3390/cancers10040096
Kontogianni G, Piroti G, Maglogiannis I, Chatziioannou A, Papadodima O. Dissecting the Mutational Landscape of Cutaneous Melanoma: An Omic Analysis Based on Patients from Greece. Cancers. 2018; 10(4):96. https://doi.org/10.3390/cancers10040096
Chicago/Turabian StyleKontogianni, Georgia, Georgia Piroti, Ilias Maglogiannis, Aristotelis Chatziioannou, and Olga Papadodima. 2018. "Dissecting the Mutational Landscape of Cutaneous Melanoma: An Omic Analysis Based on Patients from Greece" Cancers 10, no. 4: 96. https://doi.org/10.3390/cancers10040096