Targeting the Hippo Pathway Is a New Potential Therapeutic Modality for Malignant Mesothelioma

1
Division of Molecular Oncology, Aichi Cancer Center Research Institute, 1-1 Kanokoden, Chikusa-ku, Nagoya 464-8681, Japan
2
Department of Cancer Genetics, Nagoya University Graduate School of Medicine, Nagoya 464-8681, Japan
Cancers 2018, 10(4), 90; https://doi.org/10.3390/cancers10040090
Submission received: 28 February 2018
/
Revised: 20 March 2018
/
Accepted: 21 March 2018
/
Published: 22 March 2018
(This article belongs to the Special Issue Hippo Pathway in Cancer, towards Realization of the Hippo-Targeted Therapy)
{kind=link}
{kind=link}
{kind=link}
Abstract
:Malignant mesothelioma (MM) constitutes a very aggressive tumor that arises from the pleural or peritoneal cavities and is highly refractory to conventional therapies. Several key genetic alterations are associated with the development and progression of MM including mutations of the CDKN2A/ARF, NF2, and BAP1 tumor-suppressor genes. Notably, activating oncogene mutations are very rare; thus, it is difficult to develop effective inhibitors to treat MM. The NF2 gene encodes merlin, a protein that regulates multiple cell-signaling cascades including the Hippo pathway. MMs also exhibit inactivation of Hippo pathway components including LATS1/2, strongly suggesting that merlin-Hippo pathway dysregulation plays a key role in the development and progression of MM. Furthermore, Hippo pathway inactivation has been shown to result in constitutive activation of the YAP1/TAZ transcriptional coactivators, thereby conferring malignant phenotypes to mesothelial cells. Critical YAP1/TAZ target genes, including prooncogenic CCDN1 and CTGF, have also been shown to enhance the malignant phenotypes of MM cells. Together, these data indicate the Hippo pathway as a therapeutic target for the treatment of MM, and support the development of new strategies to effectively target the activation status of YAP1/TAZ as a promising therapeutic modality for this formidable disease.
1. Introduction
Malignant mesothelioma (MM) constitutes an aggressive neoplasm that arises primarily from pleura or peritoneum serosal cells, such that almost 80% of cases are pleural in origin [1,2]. However, current radiological diagnostic methods for MM, which consist of chest X-rays and computed tomography (CT) scans, are not effective at detecting malignant pleural mesothelioma (MPM) during the early stages of disease progression. Furthermore, despite continuous efforts to develop more sensitive and specific MM blood biomarkers, such as the recent demonstration of elevated high mobility group box protein 1 (HMGB1) levels in the serum of patients with MM [3], suitable blood or pleural effusion biomarkers for this disease have not yet been established [4]. Thus, MM is usually diagnosed at an advanced stage. In addition, as the anatomical characteristics of the body cavities where MM initially develops enable MM cells to easily disseminate via body fluids, it is also very difficult to completely surgically remove MM tumors without leaving residual microscopic disease.
MMs are pathologically classified as either epithelioid, sarcomatoid, or biphasic (i.e., consisting of both epithelioid and sarcomatoid components) [5]. As they are often refractory to conventional therapies, they often incur a poor patient prognosis, with the median overall survival (OS) time for patients with MPM being 12–18 months after diagnosis, regardless of trimodality therapy consisting of induction chemotherapy followed by surgery and post-operative radiation therapy [6]. The expected OS time is reduced for patients with non-epithelioid (i.e., sarcomatoid or biphasic) compared to epithelioid MM [7]; likewise, patients that exhibit an advanced overall tumor stage, higher T or N category, Eastern Cooperative Oncology Group (ECOG) performance status of 1 or higher, and/or are male also experience a worse overall prognosis [7].
Recent investigations into the genetic factors that underlie MM pathogenesis have identified frequent mutations in CDKN2A/ARF, NF2, BRCA1-associated protein 1 (BAP1), and TP53, albeit only very rare mutations in other well-known oncogenes [8]. A relatively small number of somatic mutations have also been observed in MM cases, whereas chromosome loss and/or mutations that affect genes involved in histone modification (SETDB1 and SETD2) and RNA processing (DDX3X, DDX51, and SF3B1), appear to be more common [8]. Epidemiologically, MM is most often caused by exposure to asbestos, with onset occurring after a long period of latency [9]. Notably, however, it can also be caused by a familial cancer syndrome arising from germline BAP1 mutations [10].
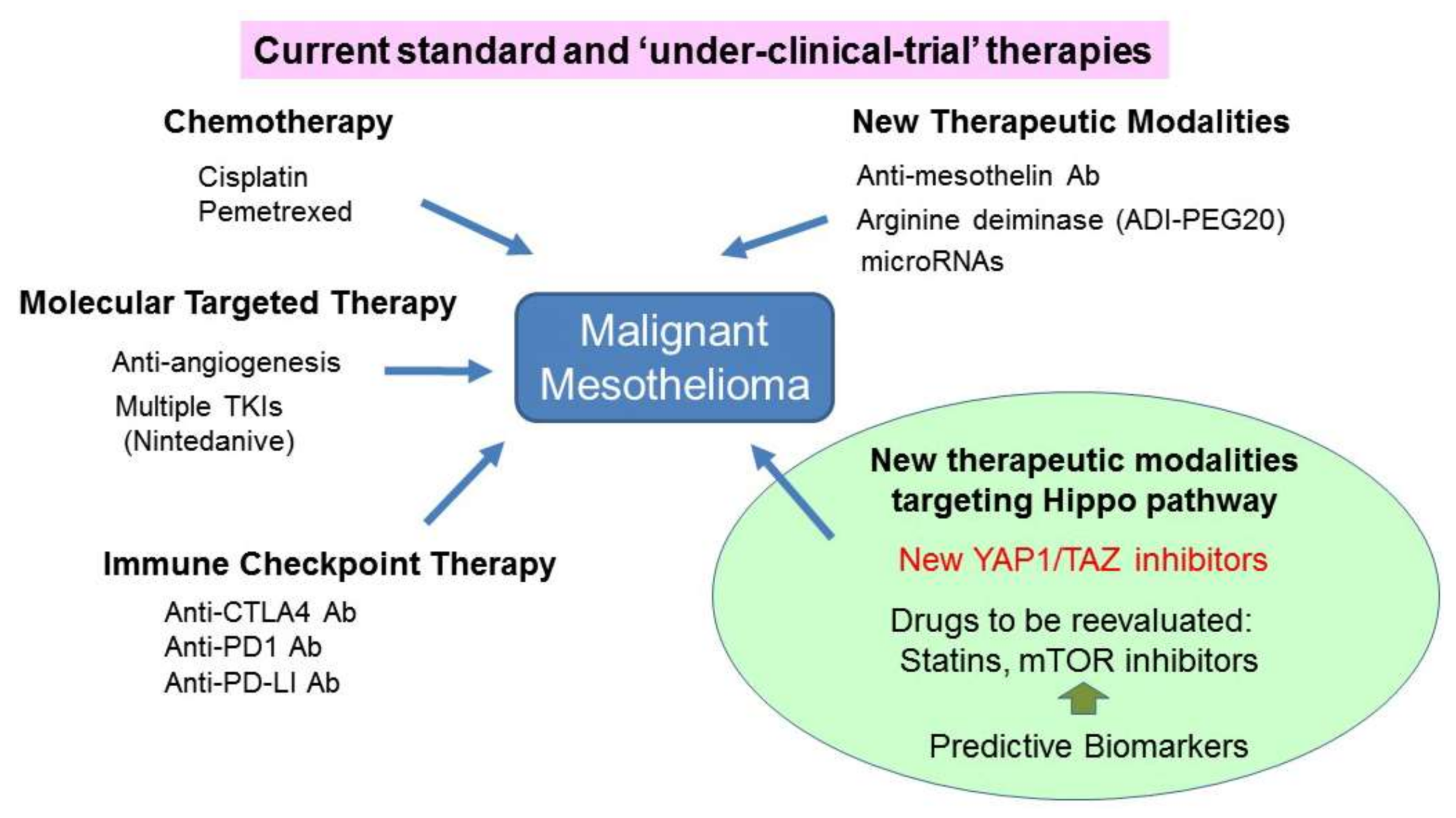
2. Current Standard, and ‘Under-Clinical-Trial’ Therapeutic Modalities for MM
Owing to its characteristic diffuse growth pattern and the resultant lack of surgical margins, it is theoretically impossible to achieve complete microscopic resection of MPM. Thus, two types of curative (intent) surgery are currently available to patients; extrapleural pneumonectomy (EPP) and pleurectomy/decortication (P/D). Maximal surgical cytoreduction treatments for MPM are currently performed in combination with chemotherapy, with or without radiation therapy [11].
Only two chemotherapy drugs, cisplatin, and the anti-folate drug pemetrexed, are currently approved and used as part of the first-line regimen for patients with advanced MM. Notably, administering a combination of these drugs alone has only been shown to slightly increase patient OS [12]. In addition, although novel molecularly targeted drugs have been recently shown to stabilize MM disease progression, none are currently recommended as standard MM treatments [13].
The first-generation tyrosine kinase inhibitors erlotinib and gefitinib, which target the epidermal growth factor receptor (EGFR), were shown not display any significant activity in MM cases [14]. Similarly, the multi-targeted small-molecule tyrosine kinase inhibitors cediranib, dasatinib, sorafenib, and sunitinib each failed to show adequate clinical activity as second-line treatments when administered as monotherapies [15,16,17,18]. In contrast, a recent phase-II trial found that an angiokinase inhibitor termed nintedanib, which targets vascular endothelial growth factor receptors (VEGFRs), platelet-derived growth factor receptors (PDGFRs), fibroblast growth factor receptors (FGFRs), and Src and Abl-kinase signaling, improved the progression free survival (PFS) time for patients with MPM when administered in combination with pemetrexed and cisplatin [19]. This effect is currently being confirmed via an ongoing phase-III trial [19]. Another phase-III study recently showed that administering bevacizumab (Avastin®, Genentech, South San Francisco, CA, USA), a humanized anti-VEGF antibody, in combination with pemetrexed and cisplatin significantly increased patient OS [20]. However, further investigation of this effect was halted in 2017 to allow the drug manufacturer the opportunity to seek approval from global health authorities to pioneer Avastin® as a treatment for MPM.
Immune checkpoint inhibitors including anti-CTLA4 (tremelimumab and ipilimumab), anti-PD1 antibodies (nivolumab and pembrolizumab), and anti-PD-L1 antibodies (avelumab and durvalumab) are currently undergoing intensive investigation in relevant MM clinical trials [21,22,23]. Thus far, tremelimumab treatment has been shown to not significantly prolong the OS of patients previously treated for MM compared to placebo [22]. Although the definitive conclusions of the anti-PD1/PD-L1 antibody studies have not yet been reported, administering immune checkpoint anti-PD1 or anti-PD-L1 antibodies either alone or in combination with the alternative-type inhibitor such as an CTLA-4 antibody appears to confer some benefits to a subset of patients with MM [23]. Thus, a combination of different types of immune checkpoint inhibitors may elicit a better patient response to treatment; although notably, the incurred side-effects may also be exacerbated, and may therefore require careful management.
Epigenetic MM therapies have also been tested [24]; however, the DNA methyl transferase (DNMT) inhibitor dihydro-5-azacytidine, and the histone deacetylase (HDAC) inhibitors vorinostat and belinostat showed only a modest [25], and no clinical effects [14,26], respectively. As BAP1 loss has been found to increase both the activity of EZH2 (which is a component of the polycomb repressor complex 2 (PRC-2)) and the levels of trimethylated histone H3 lysine 27 (H3K27me3), a recent study assessed the effects of EZH2 inhibition on MM progression. The results of this study demonstrated that inhibiting EZH2 suppressed the proliferation of BAP1-mutant MM cell lines, suggesting that EZH2 inhibitors may represent promising candidate therapeutic agents for MM [27].
Other promising antitumor agents include monoclonal antibody-based drugs against mesothelin [28] and CD26 [29]. MMs often exhibit reduced expression of arginosuccinate synthetase-1 (ASS1); thus, a synthetic lethal approach with pegylated-arginine deiminase ADI-PEG20, which depletes available arginine, showed a significant PFS improvement for patients with MM [30]. Additional novel MM treatment strategies based on alterations of tumor suppressor genes, including BAP1, are currently under development [31,32].
In terms of radiotherapy, intensity-modulated radiation therapy (IMRT) has been shown to potentially confer a survival benefit to a subset of patients with MM [33]. In addition, novel innovative approaches with pleural and induction-accelerated hemithoracic radiotherapy followed by surgery have been recently shown to be both feasible and safe [33]. Accordingly, the most recent American Society of Clinical Oncology Clinical Practice Guidelines provided evidence-based recommendations for the diagnosis and staging of patients with MPM, as well as for the subsequent administration of chemotherapeutic, surgical cytoreduction, radiation, and/or multimodal therapies [34].
3. Hippo Pathway Dysregulation in MM Cells
3.1. NF2 and the Hippo Pathway
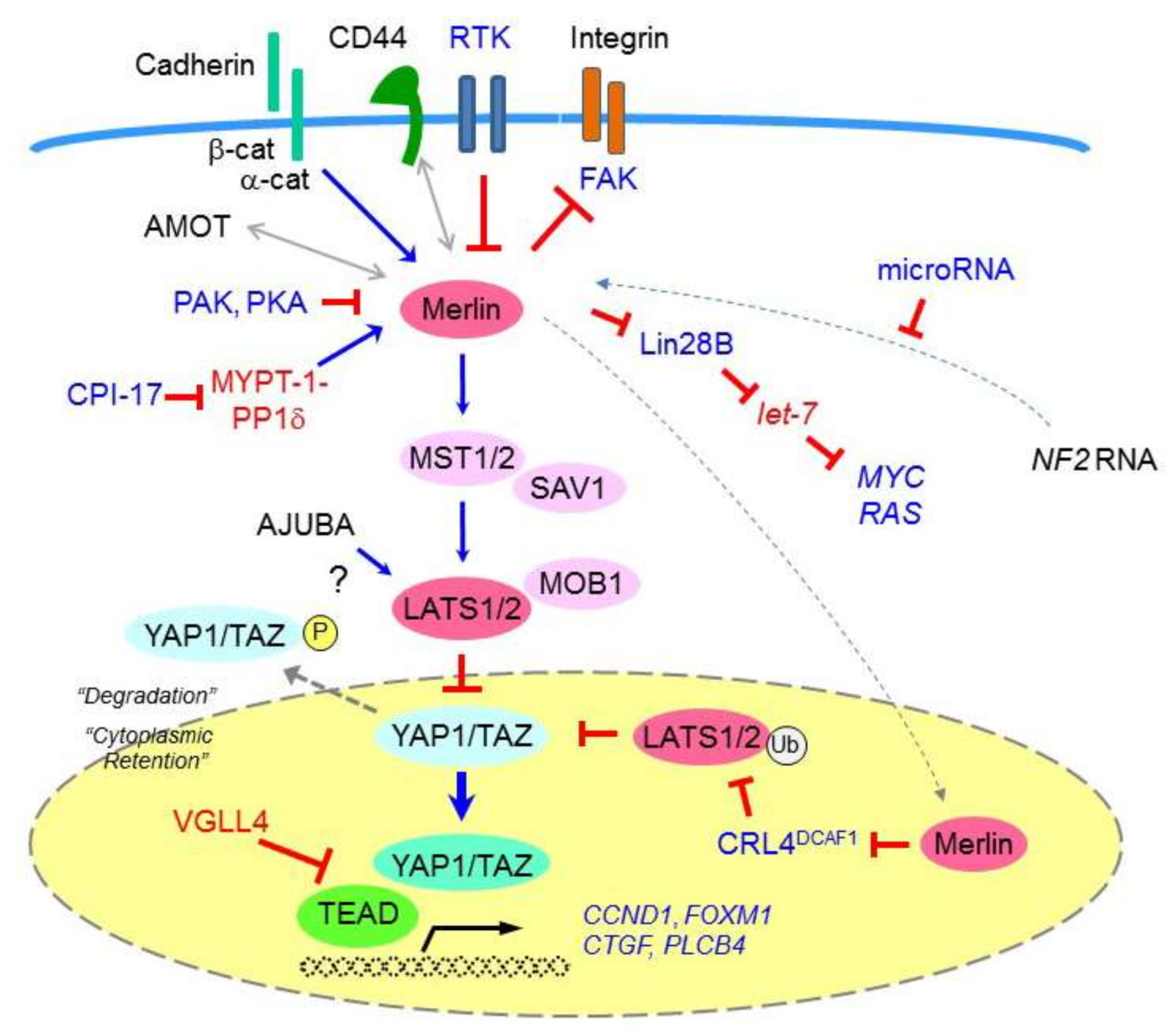
The neurofibromatosis type 2 (NF2) gene encodes the tumor suppressor protein moesin-ezrin-radixin-like protein (merlin), which is a member of the Band 4.1 family of cytoskeletal linker proteins [35] (Figure 1). One of the important downstream signaling cascades that are regulated by merlin is the Hippo pathway, which is involved in critical biological processes including organ-size control, development, differentiation, tissue regeneration (via cell-growth restriction), cell division regulation, apoptosis, and cancer development [36,37]. The four core components in this pathway comprise MST1 and MST2 kinases, SAV1 (also termed WW45), MOB1, and LATS1 and LATS2 kinases, all of which have been shown to act as tumor suppressors (Figure 1). Upon receiving upstream signaling, MST1/2 kinases (in a complex formed with the SAV1 scaffold protein) phosphorylate and activate LATS1/2. The latter is activated by the MOB1 scaffold protein to then phosphorylate and inactivate the YAP1 and TAZ transcriptional coactivators. The phosphorylated YAP1/TAZ are then directed to either be retained within the cytoplasm (via an interaction with 14-3-3) or degraded (via poly-ubiquitination). Conversely, when the Hippo pathway is not active, underphosphorylated YAP1/TAZ enter the nucleus and act as transcriptional coactivators. YAP1/TAZ interact with several distinct transcription factors including TEA domain (TEAD) transcription factors, SMADs, p73, Runt related transcription factor (RUNX), T-Box 5 (TBX5), the carboxyl-terminal fragment of Erb-B2 receptor tyrosine kinase 4 (ERBB4), and early growth response-1 (EGR1) [38].
Merlin also activates the Hippo pathway and suppresses YAP1/TAZ via its effects on cell junction-associated proteins known to modulate Hippo signaling [39]. Specifically (in addition to other ERM proteins and the intracellular CD44 domain), merlin binds to α-catenin and angiomotin at maturing adherens and tight junctions, respectively, to suppress cell growth. Although the relationship between angiomotin and merlin in the Hippo pathway is not yet well understood, angiomotin is thought to serve as a scaffold for MST1/2 and LATS1/2, and to directly bind and inhibit YAP1. Angiomotin is also speculated to bind and activate merlin, and thereby promote the binding of merlin to LATS1/2 [40].
3.2. NF2 Inactivation
NF2 was shown more than two decades ago to constitute the target gene located within the 22q12 chromosomal region that is very frequently deleted in MM cells [41,42,43]. In fact, an estimated 40–50% of MM cases harbor an NF2-inactivating mutation or deletion, resulting in bi-allelic loss of function. NF2 gene rearrangements have also been found to be frequently associated with MM. Of these, gene fusions have been demonstrated to be mutually exclusive with other types of mutations (e.g., point mutations) [8].
Merlin can also be inactivated via other mechanisms, such as the increased expression of protein kinase C-potentiated phosphatase inhibitor of 17 kDa (CPI-17) [44]. In particular, CPI-17 has been shown to inhibit MYPT-1-PP1δ, which in turn dephosphorylates merlin Ser518, thereby inactivating merlin to induce neoplastic cell transformations in vitro [45]. A previous study showed that a carboxyl-terminus NF2 (isoform 2) splicing variant does not exert any tumor suppressive activity [46], suggesting that functional merlin inactivation might also be caused by the expression of such variants [44]; however, more recent studies conversely reported that both merlin isoforms 1 and 2 act as tumor suppressors [47]. Merlin inactivation may also be caused via the upregulated expression of potential NF2-targeting microRNAs, such as hsa-miR-885-3p [48], although further research is required to elucidate how such mechanisms mediate MM pathogenesis.
Biologically, merlin re-expression has been confirmed to markedly inhibit the motility, spread, and invasiveness of NF2-deficient MM cells [49], whereas conversely, siRNA-mediated merlin silencing has been shown to result in the enhanced spreading and invasion of mouse embryonic fibroblasts (MEFs). Specifically, merlin expression attenuates the phosphorylation of focal adhesion kinase (FAK) at the critical Tyr397 phosphorylation site, and thereby disrupts the interaction between FAK and its binding partners, Src and p85, the regulatory subunit of phosphatidylinositol-3-kinase (PI3K) [49]. These findings suggested that merlin inactivation is likely related, at least in part, to the upregulation of FAK activity.
As NF2 mutations are frequently detected in MM cases, genetically-engineered Nf2-knockout mouse models have been developed to evaluate the impact of NF2 inactivation on MM pathogenesis. Asbestos-exposed Nf2 (+/−) knockout mice have resultantly been shown to exhibit markedly accelerated MM tumor formation compared to their asbestos-treated wild-type littermates [50,51]. Furthermore, loss of the wild-type Nf2 allele, leading to biallelic inactivation, was observed in all and 50% of asbestos-induced MMs from Nf2 (+/−) and wild-type mice, respectively [51]. Similar to human MMs, the tumors developed by the Nf2 (+/−) mice exhibited frequent homologous deletions of the Cdkn2a/Arf locus and adjacent Cdkn2b tumor-suppressor gene, as well as reciprocal inactivation of Tp53 in a subset of tumors that retained the Arf locus [51]. Moreover, conditional (mesothelium-specific) Nf2, Ink4a/Arf, and/or Tp53 knockout mouse models have also been shown to exhibit an increased incidence of MM development [52].
Notably, the underphosphorylated form of merlin has been shown to translocate to the nucleus, bind to the E3 ubiquitin ligase CRL4DCAF1, and inhibit the capacity of CRL4DCAF1 to ubiquitinate its target proteins; thus, merlin likely also functions as a negative regulator of CRL4DCAF1 [53]. Accordingly, merlin has been confirmed to exert CRL4DCAF1-mediated tumor-suppressive activity in both MM and a MeT-5A immortalized mesothelial-cell lines [53]. As CRL4DCAF1 directly binds to LATS1/2 to direct their conjugation to ubiquitin [54], it is likely that in merlin-deficient cells, de-repressed CRL4DCAF1 promotes LATS1/2 ubiquitination and degradation, and thus activates YAP1 by suppressing YAP1 phosphorylation.
Merlin also exhibits a cell-density-dependent, albeit Hippo-independent tumor-suppression activity via its downstream target, Lin28B (a let-7 microRNA inhibitor) [55]. Specifically, merlin is phosphorylated and thus does not bind to Lin28B at low cell densities; thus, Lin28B is able to enter the nucleus and both bind to and inhibit the maturation of pri-let-7 microRNAs. As let-7 microRNAs act as tumor suppressors by silencing the expression of critical oncogenes such as MYC and RAS, inhibiting pri-let-7 maturation promotes cell growth.
Finally, TRAF7, which encodes an atypical member of the tumor necrosis factor (TNF) receptor-associated factor family, is occasionally mutated in MM. A previous study showed that four of five identified MM-associated TRAF7 mutations were mutually exclusive with NF2 mutations [8]. Notably, a similar pattern of exclusivity has been observed in TRAF7-mutated meningioma cases, which also frequently exhibit NF2 mutations [56]. These observations suggest that TRAF7 and merlin may function in a common signaling pathway.
3.3. Hippo Pathway Component Inactivation
Hippo pathway dysregulation has been indicated in a broad range of human carcinoma types including lung, colorectal, ovarian, and liver cancers. Accordingly, the Hippo pathway has been investigated in these contexts as a potential therapeutic target [57,58,59,60]; however, these commonly occurring tumors only rarely exhibit genetic mutations in Hippo pathway components. In contrast, MM appears to be the only tumor type that is frequently associated with Hippo pathway mutations. In addition to NF2 mutations, genetic LATS2 inactivation is observed in MM cases, such that LATS2 inactivation, and its effects tumor suppression, were initially identified in seven of 20 MM cell lines analyzed in vitro [61]. Similarly, 11% of 61 MPM primary cultures were found to harbor point mutations and/or large exon deletions that inactivated LATS2 [62]. Together, these findings indicate that NF2 and LATS2 mutations can be coincident in a given MM tumor. A homozygous SAV1 deletion was also detected in an MM cell line [61], and one of 16 analyzed MM cell lines that were subjected to whole exome sequencing was previously shown to harbor a LATS1-inactivating LATS1-PSEN1 fusion gene [63]. Finally, a comprehensive genomic analysis of MM samples revealed frequent copy number loss among various Hippo pathway genes, including MST1 and LATS1 [8].
MM cells frequently exhibit downregulation of the LIM-domain protein AJUBA, which is a LATS2 binding partner [64]. Consistent with this finding, MM cell lines in which AJUBA is downregulated have been shown to exhibit higher levels of YAP1 dephosphorylation, whereas conversely, transducing MM cells with AJUBA has been demonstrated to significantly suppress YAP1 activity. Together, these data indicate that AJUBA negatively regulates YAP1 via its interactions with LATS1/2 [64]. In contrast, AJUBA has been suggested to exert oncogenic effects and to regulate the Hippo pathway in the context of other malignancies [65]; thus, the effects of AJUBA family LIM proteins on tumor development and progression require further clarification [66].
4. YAP1/TAZ
4.1. YAP1/TAZ Activation in MM Cells
As discussed, merlin-Hippo signaling inactivation leads to constitutive YAP1/TAZ activation. Moreover, YAP1 expression was observed in more than 70% of the analyzed primary MM tissues, with most positive cases showing greater YAP1-staining in the nucleus than the cytoplasm [61].
Gene amplification comprises one of several mechanisms of oncogene activation; accordingly, the chromosome 11q22 region where YAP1 is located has been shown to be amplified in a variety of human malignances [67]. In addition to YAP1, this region includes a cluster of matrix metalloproteinase (MMP) genes, two members of the BIRC family of caspase inhibitors (BIRC2 and BIRC3), and the progesterone receptor (PGR) [67]. Although 11q22 amplification is an infrequent event in MMs, YAP1 amplification has been detected in a subset of MM cases, and YAP1 has been shown to be an important effector of MM cell proliferation in vitro [68].
4.2. YAP1/TAZ Enhance the Transcription of Pro-Oncogenic Genes
Activation of the YAP1 transcriptional coactivator has been shown to induce the transcription of multiple cancer-promoting genes in MM cells (Figure 1), including cell cycle promoting genes such as cyclin D1 (CCND1) and forkhead box M1 (FOXM1) [69], connective tissue growth factor (CTGF) [70], and phospholipase-C beta 4 (PLCB4) [71].
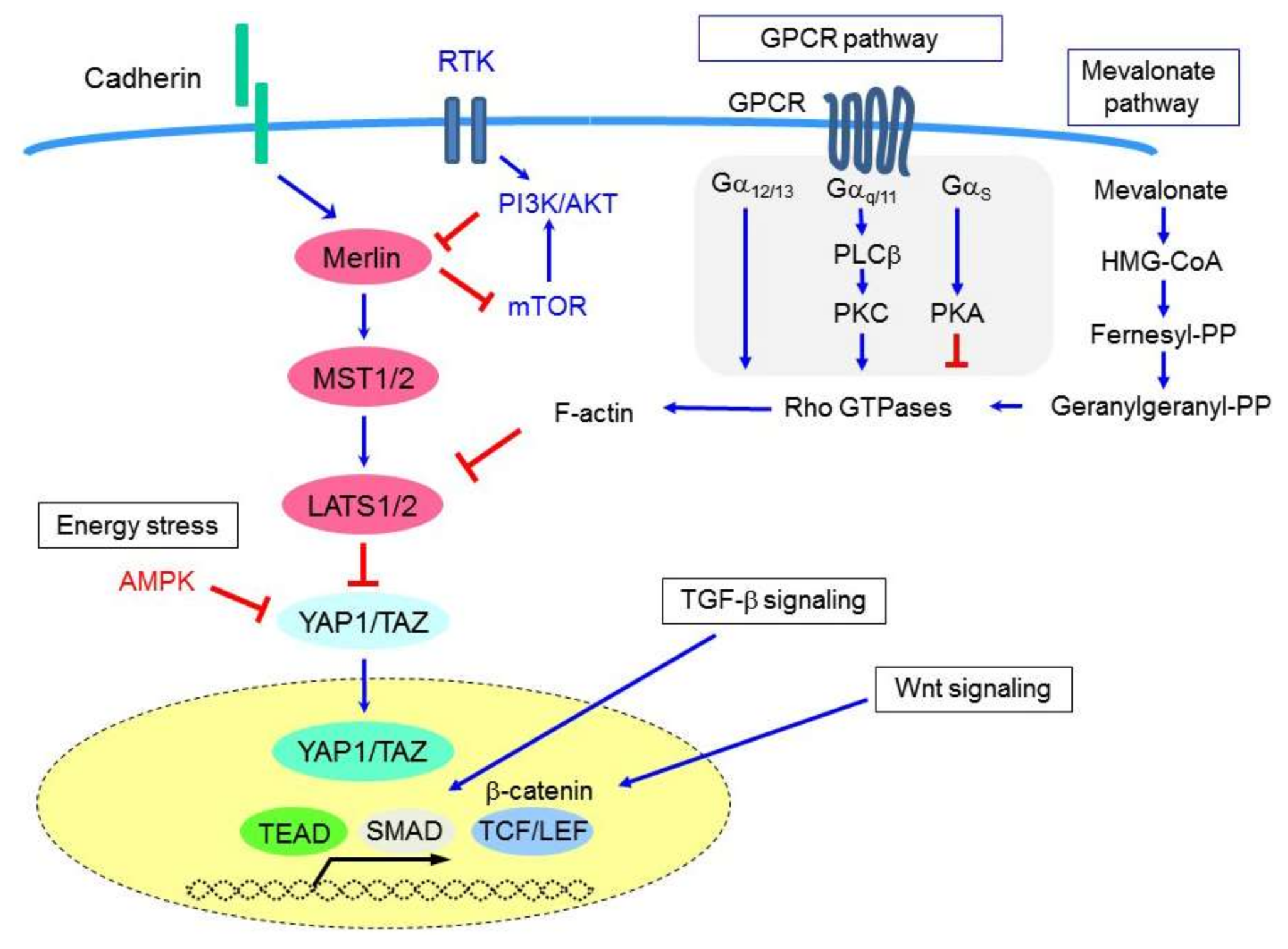
CTGF (or CCN2) is a member of the CCN protein family, which includes six members of secretory extracellular matrix-associated matricellular proteins. CTGF is a cysteine-rich protein that regulates cell-extracellular matrix (ECM) interactions and exerts multicellular functions including the regulation of cell proliferation, adhesion, migration, fibrosis, and inflammation. Notably, CTGF expression has been shown to be associated with abundant extracellular matrix formation in MM tissues [70]. Furthermore, CTGF expression was found to be significantly enhanced in MM cells in response to Hippo signaling inactivation, TGF-β stimulation [70], and/or β-catenin-TCF-LEF signaling [72] (Figure 2). CTGF has also been suggested to be highly expressed in sarcomatoid-type MMs and has been shown to mediate the epithelial-mesenchymal transition (EMT) in MM [72]. Similarly, another CCN family member, CYR61 (or CCN1), is also a well-known YAP1 target; however, its role in MM development and progression has not yet been determined.
The Hippo pathway is also regulated by G-protein-coupled receptor (GPCR) signaling [73] (Figure 2). Phospholipase-C, the downstream effector of Gαq signaling, is required to enable inositol 1,4,5-triphosphate and diacylglycerol to be formed from phosphatidylinositol 4,5-biphosphate, and thus also for signal transduction. PLCB4 (which encodes phospholipase-C β4) was shown to be a YAP1-target gene [71], as inducing PLCB4 knockdown attenuated the growth of YAP1-transduced immortalized mesothelial cells, and YAP1-active (but not YAP1-non-active) MM cells [71].
Similarly, uveal melanoma has been reported to frequently harbor activating mutations in GNAQ and GNA11. These genes encode two highly homologous Gαq/11 heterotrimeric G-protein α subunits, and their activation has been shown to activate YAP1 [74,75]. PLCB4 is also occasionally mutated in uveal melanoma [76], suggesting that a positive feedback loop may link YAP1 activation with GPCR signaling in a subset of human malignancies. Notably, uveal melanomas are also frequently associated with BAP1 mutations, in the same manner as MM [10,77,78,79,80].
BAP1 encodes a protein that regulates transcription, histone modification, DNA repair [81], and Ca2+ flux from the endoplasmic reticulum to mitochondria [82]; thus, there may be an as yet unknown functional relationship between the YAP1 activation and BAP1 inactivation that occurs during the development of both of these tumor types.
4.3. YAP1/TAZ Enhance the Tumorigenicity of Immortalized Mesothelial Cells
Cancer develops via the accumulation of genetic and epigenetic alterations that promote malignant cell phenotypes [83]. Although the processes by which these occur during MM pathogenesis are not well understood, changes to critical MM-associated tumor suppressor genes are thought to confer one or more malignant phenotypes to normal mesothelial cells [84].
Immortalized mesothelial cells that were generated via the transduction of human papilloma virus E6/E7 and hTERT genes were used to investigate the mechanisms underlying the effects of YAP1 activation on malignancy. Treatment of the cells with exogenously transduced wild-type YAP1, and even more so, with an activated mutant YAP1 S127A, stimulated the mesothelial cells to form mesothelioma-like tumors. The effect was more obvious when the immortalized cells were inoculated into the thoracic cavities of nude mice [71].
5. Diagnosis and Prognostic/Predictive Biomarkers Based on Hippo Pathway Dysregulation
As NF2 is frequently inactivated in MM cells, its deletion status has been investigated as a potential MM diagnostic marker. Such markers are needed to aid the diagnosis of MPM in the clinical setting, because it is sometimes difficult to differentiate between MPM and other malignancies (such as lung cancer and sarcoma) that develop in the thoracic cavity, and/or reactive mesothelial hyperplasia (RMH), which is a benign condition that essentially requires no further treatment. Adding genetic NF2 screening to the standard methods of using fluorescence in situ hybridization (FISH) techniques to identify CDKN2A (p16Ink4a/p14Arf) deletions did not significantly improve the sensitivity or specificity of MM diagnosis. Currently, CDKN2A FISH and BAP1 immuno-histochemical staining are considered to be the most effective methods of differentiating between MMs, other malignancies, and RMH [85]. A more convenient immunohistochemical analysis method for detecting the product of the MTAP gene, (which is adjacent to and often co-deleted with CDKN2A), has also been recently shown to be useful in diagnosing MM [85].
Both a low level of merlin expression and a high survivin labeling index have been shown to be indicators of a poor prognosis in patients with MPM [86], suggesting their potential as representing possible prognostic markers for MM. Likewise, a combination of homozygous CDKN2A deletions and hemizygous NF2 loss in peritoneal mesotheliomas has been shown to be an independent negative prognostic factor for both PFS and OS [87].
Recent studies have shown that a FAK inhibitor termed VS-4718 inhibits proliferation and induces apoptosis in MM cells that lack merlin expression [88]. This preferential effect of VS-4718 in merlin-deficient cells suggests that merlin may constitute a potential predictive biomarker for the enhanced response of MM cells to VS-4718 treatment. However, a phase-II (registration-directed, double-blind, placebo-controlled study (COMMAND)) trial of another selective FAK inhibitor, VS-6063 (or defactinib), for use in patients with mesothelioma was recently halted, owing to a lack of any observed benefits (despite correcting for merlin deficiency). Thus, whether FAK inhibitors can provide a clinical benefit for patients with merlin-negative MM cells remains unclear. Notably, another study recently reported that E-cadherin expression was correlated with resistance of the merlin-negative MM cells to FAK inhibitor treatment [89].
6. Therapeutic Applications Based on Hippo Pathway Dysregulation
Given that MM is highly refractory to current therapeutic modalities, new drugs, and novel therapeutic strategies based on the molecular and cellular characteristics of MM are urgently required to improve patient outcomes. Based upon the current literature, targeting the Hippo pathway is likely to be a very promising therapeutic approach for this disease (Figure 3) [90].
6.1. Targeting YAP1/TAZ
The significant impact of Hippo pathway dysregulation and constitutive YAP1/TAZ activation on MM cells renders these events highly attractive as molecular targets for the development of novel therapeutic approaches to MM. Restoring upstream regulatory Hippo pathway components would restore Hippo pathway function; however, reintroducing tumor suppressor gene expression and activation to whole cells in tumor tissues is likely to be technically very difficult.
An alternative approach is to block YAP1/TAZ interactions with their target transcription factors. For example, TEADs are thought to be the factors primarily expressed and involved in the prooncogenic functions of YAP1/TAZ in MM cells; thus, the disruption of YAP1/TAZ and TEAD interaction may represent a very promising approach. In this regard, several molecules have been developed and tested in an attempt to develop new therapeutic agents against human malignancies. The first small molecule shown to function as a YAP1-TEAD binding inhibitor was verteporfin (Visudyne), which is used clinically as a photosensitizer in photodynamic therapy for neovascular macular degeneration [91]. Notably, verteporfin has been shown not to require light activation to inhibit the growth and motility of cultured breast-cancer cells [91], and has been demonstrated to inhibit other human cancer-cell types including ovarian, hepatoma, and glioblastoma cancer cells in vitro [92]. Moreover, verteporfin treatment was also shown to suppress YAP1 activity along with the viability, invasion, and tumor sphere formation of MM cell lines in vitro [62,93].
Other YAP1-TEAD inhibitors have also been developed including the bioengineer-designed, cyclic YAP1-like peptides (17 mer), which have been demonstrated to interrupt the YAP-TEAD interaction [94]. Mammalian vestigial-like 4 (VGLL4) was also previously identified as a natural YAP1 antagonist that binds to TEADs via its Tondu (TDU) domains, such that the VGLL4 TDU region is sufficient to inhibit YAP1 activity [95,96]. Based on these observations, a synthetic peptide (48 mer) that mimicks the VGLL4 TDU domain (Super-TDU) was synthesized and shown to suppress gastric and lung cancer cells by competing with YAP1 to bind to TEADs [95]. Recently, VGLL4 was also shown to target a TCF4-TEAD4 complex, suggesting that it may also function as a regulator of Wnt/β-catenin signaling [97]. In support of this mechanism, Super-TDU treatment has been shown to reduce the number of colorectal adenomas exhibited by APCmin/− mice [97]. In addition, TIAM1, a component of the Wnt-regulated destruction complex, has recently been shown to antagonize TAZ and YAP1 in colorectal cancer cells by promoting TAZ cytoplasmic degradation and suppressing YAP1/TAZ interactions with TEADs [98]. As MM cells have also been reported to exhibit Wnt signaling activation [99], these inhibitors should be further investigated to determine their effects on both YAP1/TAZ and Wnt activated MM cells.
6.2. Targeting YAP1 Using Metabolic-Disorder Drugs
The cellular metabolic status, including cellular responses to glucose deprivation (energy stress) and the mevalonate cascade, has been shown to be linked to Hippo signaling [100]. Glucose deprivation reduces cellular ATP levels and induces activation of the AMP-dependent protein kinase (AMPK), which is a sensor of cellular energy stress. AMPK has been found to attenuate YAP1 activity via multiple mechanisms, such as reducing nuclear YAP1 levels and inhibiting YAP1-TEAD interactions [101,102,103]. The widely-used anti-diabetic drug metformin, which reduces blood glucose levels and activates AMPK, has previously been shown to inhibit YAP1. Moreover, it has been suggested that metformin functions as an anticancer agent, predominantly via mTOR pathway inhibition; thus, its administration has been reported to be associated with improved survival in the treatment of patients with diabetes plus several types of cancers [104]. However, a retrospective study of 300 patients with type 2 diabetes and MPM did not shown any improvement to patient survival [105]. Meanwhile, metformin has been recently shown to inhibit MM cell proliferation when administered in combination with nutlin-3a (an inhibitor of ubiquitin-mediated p53 degradation) [106].
Statins (i.e., inhibitors of the rate-limiting enzyme (HMG-CoA reductase) of the mevalonate cholesterol biosynthesis pathway) are currently used to treat hypercholesterolemia and prevent cardiovascular diseases. They have also been found to exert anticancer effects in many cancer types, with lovastatin in particular having been shown to induce apoptosis in human MM cell lines [107]. Statins have also been found to be effective against MM in vivo when combined with doxorubicin chemotherapy, likely by reducing the ability of the MM cells to acquire resistance to doxorubicin treatment [108]. In contrast, both a mouse model and human cohort study of 1738 patients with a history of asbestos exposure reported that statins do not moderate mesothelioma development or progression [109]. Although the putative preventive and therapeutic effects of statins in the treatment of various cancers, including mesothelioma, require further investigation, their effects on the mevalonate pathway have been confirmed to control YAP1/TAZ activity [110], such that statin treatment inhibits both YAP1/TAZ nuclear localization and transcriptional responses [110]. Mechanistically, the geranylgeranyl pyrophosphate produced by the mevalonate cascade is required for the activation of Rho GTPases that, in turn, activate YAP1/TAZ, likely via actin cytoskeleton rearrangements [110,111]. Consistent with these data, statins have been demonstrated to exert greater antiproliferative activity in MM cells that exhibit Hippo pathway inactivation [112]. Conversely, they exert a weaker inhibitory effect in MM cells that also harbor BAP1 mutations [112]; thus, the effectiveness of statin treatments in MM cells appears to vary dependent upon the cellular genetic/epigenetic background.
6.3. Targeting Molecules Activated by Merlin Deficiency
Merlin regulates mitogenic signaling by suppressing mTORC1 in MM cells, as demonstrated by the fact that merlin-deficient MM cells have been shown to be selectively sensitive to the growth-inhibiting effects of the allosteric mTOR inhibitor, rapamycin [113]. Although the mechanisms by which merlin suppresses mTOR signaling remain elusive, NF2 inactivation has been found to confer sensitivity to rapalogs in bladder cancer [114], suggesting that mTORC1 signaling likely sustains the expansion of merlin-deficient cancer cells.
A phase-II study of an mTOR inhibitor everolimus was conducted in advanced MPM, in which a total of 59 patients were evaluable for 4-month PFS [115]. The results of the trial suggested that everolimus had limited clinical activity, and that its use as a single-agent therapy for advanced MPM was not warranted. In comparison, a recent phase-I study of the dual class-I PI3K and mTOR kinase inhibitor apitolisib (GDC-0980) showed a partial response in patients with pleural and peritoneal mesothelioma [116].
As discussed, merlin loss (at least in part) drives tumorigenesis via E3 ubiquitin ligase CRL4DCAF1 activation and subsequent LATS1/2 inhibition. MLN4924, a NEDD8-activating enzyme (NAE) inhibitor, has been shown to suppress CRL4DCAF1 and attenuate YAP1 activation in NF2-mutant tumor cells [117]. Although MLN4924 alone did not exhibit significant preclinical activity, administering MLN4924 with the mTOR/PI3K inhibitor GCD-0980 suppressed the growth of NF2-mutant tumor cells in vitro as well as in mouse and patient-derived xenografts [117].
6.4. Targeting CTGF
CTGF is a well-known YAP1 target gene that encodes an ECM-associated protein. Deregulated CTGF expression has been observed in many types of human malignancies including MM, and has been shown to be associated with increased cell proliferation, drug resistance, angiogenesis, adhesion/migration, and metastasis [118]. Although CTGF transcription is induced by YAP1 activation, its expression in MM cells has also been demonstrated to be enhanced by both TGF-β and Wnt signaling [70,72].
CTGF levels are also elevated in patients with fibrotic lung disease including idiopathic pulmonary fibrosis (IPF). The human anti-CTGF monoclonal antibody pamrevlumab (FG-3019) is currently undergoing clinical testing for IPF and other indications [119]. As FG-3019 has been shown to be effective against high-grade serous ovarian cancer [120], it should also be investigated as a potential therapeutic agent for MM. Furthermore, a murine pancreatic ductal adenocarcinoma (PDA) model indicated that the efficacy of FG-3019 was increased when it was administered in combination with gemcitabine [121]. As both PDA and desmoplastic-type MM (a rare MM subtype characterized by a very poor patient prognosis) are characterized by excess ECM deposition, they may comprise good candidates for CTGF inhibitor treatment.
6.5. Extracellular Stimuli
Numerous extracellular stimuli regulate the Hippo pathway including cell-cell adhesion, cell-ECM contact, mechanical stresses (e.g., stiffness), and soluble factors [37].
Cell attachment to the ECM regulates Hippo pathway activity. During detachment from the ECM, cells normally undergo anoikis (programmed cell death), which has been shown (in non-transformed cells) to be mediated by YAP1 inactivation. Conversely, expression of constitutively active YAP1 has been found to promote the survival of detached cells [122]. Characteristically, mesothelial cells are thought to be relatively anoikis-resistant. When serosal injury occurs on the mesothelium, mesothelial cells not only migrate onto the wound surface from the wound edge but also detach from opposing surfaces and migrate to a distant site, where they settle on the wound surface to initiate wound repair [123]. Although it is unclear how YAP1/TAZ is involved in these mesothelial repair processes, the anoikis-resistant phenotype characteristic of MM cells may be innate and/or enhanced by merlin-Hippo pathway inactivation.
As discussed, GPCR signaling regulates the Hippo pathway, such that many mitogenic hormones and growth factors act via GPCRs to induce cell proliferation [37]. For example, ligand signals such as lysophosphatidic acid (LPA), sphingosine-1-phosphate (S1P), and estrogen can activate YAP1/TAZ through GPCRs coupled to Gα12/13 or Gαq/11. In addition, ligand signals such as epinephrine and glucagon can repress YAP1/TAZ activity through Gαs-coupled GPCRs and protein kinase A (PKA). Targeting GPCRs is now recognized as promising therapeutic approach for cancer [124] that has been evaluated in clinical trials. One such agent is sonepcizumab, a humanized monoclonal antibody directed at S1P, for treatment of metastatic renal cell carcinoma [125]. With regard to MM, LPA was previously shown to stimulate the proliferation and motility of MPM cells [126]. Although it remains unclear how these soluble factors influence MM cell growth and progression, and/or whether they influence YAP1/TAZ activity in MM cells, recurrent GRCR mutations (including GRM3, GPR149, and GPR98) have also been identified in MM cells [8]. These data support the need for further study to clarify the association between GPCR signaling and YAP1/TAZ activation in MM cells.
6.6. Other Possible Therapeutic MM Strategies
The two major families of kinases in the Hippo pathway, MST1/2 and LATS1/2, may also be promising therapeutic targets for MM, owing to their role in directing the phosphorylation and cytoplasmic sequestration of YAP1/TAZ. Thus, Hippo pathway agonists that activate MST1/2 and LATS1/2 kinases and inhibit YAP1 activation in cancer cells have been identified as candidate therapeutic agents [127]. For example, a small molecular compound termed C19 was recently shown to induce MST1/2 and LATS1/2 phosphorylation, and thereby stimulate TAZ degradation [128].
Several other distinct small molecules, including dasatinib, pazopanib, and ivermectin, have also been identified to inhibit YAP1/TAZ by drug screening using human cancer cell lines [129,130]. Dasatinib, a multi-targeted tyrosine kinase inhibitor against BCR/ABL, the Src family kinases, and PDGFRβ, was reported to suppress the proliferation, migration, and invasion of MM cells in vitro [131], and enhance pemetrexed-induced cytotoxicity [132]. Src phosphorylation was also shown to be suppressed by dasatinib treatment in surgically removed MM tissues [133]. Notably, however, the administration of dasatinib as a single-agent was not effective as a second-line treatment for MM during a recent phase II trial [16]; likewise, administration of dasatinib as a neoadjuvant treatment was not shown to confer any benefit to patients with MPM. In contrast, a recent study reported the ability of the anti-parasitic agent ivermectin to inhibit YAP1, and thereby exert significant effects on gastric cancer [134], hepatocellular carcinoma, and MM cells [130].
Additionally, a recent study showed that a TEAD4 splicing variant (TEDA4-S, in which exon 3 is skipped by the tumor suppressor RBM4 protein) acts as a dominant negative TEAD4 isoform, and thereby both attenuates YAP activity and inhibits tumor-cell proliferation [135]. It is possible that splicing dysregulation that affects other Hippo pathway genes may also mediate MM pathogenesis; thus, this process should be further investigated to identify novel MM therapeutic targets [135].
Finally, the induction of synthetic lethality constitutes another promising therapeutic strategy. For example, the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib induces enhanced cytotoxicity in breast cancer cells that harbor BRCA1/2 mutations, as their endogenous DNA damage repair systems are suppressed by both BRCA1 inactivation and PARP inhibition. Notably, when PARP inhibitors have been used to treat MM cell lines in vitro, they unexpectedly exerted inhibitory effects on all analyzed cell lines, regardless of their BAP1 mutation status [136]. In contrast (as discussed above), MM treatment with a FAK inhibitor was expected to induce synthetic lethality when combined with merlin deficiency [88], but failed in a clinical trial. Nevertheless, inducing synthetic lethality is a new approach in the context of MM research and should be further developed to identify novel candidate agents/genes that effectively induce a synthetic-lethal phenotype when combined with merlin-Hippo deficiency or BAP1 inactivation.
6.7. Rationale and Challenges of Targeting Hippo Signaling to Treat MM
To date, very few studies have investigated the effects of YAP1/TAZ-inhibition on MM either in vitro or in vivo; nevertheless, targeting YAP1/TAZ likely represents a very promising therapeutic strategy for patients with MM for several reasons. Firstly, in contrast to other human malignancies, MMs are frequently and occasionally associated with mutations in NF2, and genes encoding other components of the Hippo pathway, respectively, strongly suggesting that disruption of merlin-Hippo signaling is a key event during MM development and progression. Secondly, MMs also frequently exhibit YAP1/TAZ activation, which event has been previously shown to confer malignant phenotypes to mesothelial cells. Conversely, YAP1/TAZ suppression inhibits MM cell proliferation, migration, and invasion. Thirdly, MMs harbor a relatively small number of genetic changes, predominantly in tumor suppressor genes; however, as MM-associated mutations rarely affect targetable (e.g., kinase-encoding) oncogenes, new therapeutic targets and strategies are needed. Various Hippo pathway components appear to be promising targets for inhibiting protein-protein binding. Fourthly, although clear evidence for their therapeutic efficacy has not yet been demonstrated, some small molecules that are thought to impact Hippo pathway activation have been previously shown to inhibit MM cell proliferation and progression. As most of these were initially tested without regard to the merlin-Hippo pathway dysregulation status of the analyzed MM cells, they require careful reevaluation. Finally, the current literature suggests that a combination strategy, comprising YAP1/TAZ suppression and the inhibition of an additional relevant pathway, may be more effective in treating MM than any single strategy alone.
However, the potential that targeting YAP1/TAZ might also induce serious and unexpected adverse effects in patients should be considered when pursuing this strategy, as the Hippo pathway is essential for various aspects of normal cell functions. Thus, possible side effects for any agents or strategies targeting the Hippo pathway as described above should be carefully evaluated in both preclinical studies and clinical trials.
Additionally, although immunohistochemical analyses of merlin and YAP1/TAZ expression are often used to evaluate the merlin-Hippo pathway status in MM cells, the current methodologies and antibodies are neither very sensitive nor specific. More precise and rapid assays, along with more effective MM biomarkers, are needed to accurately determine which MM cases are associated with activated YAP1/TAZ (e.g., for drug development or patient screening) in the context of both laboratory and clinical trial-based research.
7. Conclusions
MM is a highly aggressive disease, which, along with pancreatic and bile-duct cancer, is recognized as one of the most formidable cancer types. The development of new therapies for this disease has lagged behind that achieved for other common malignancies, so that only a limited number of therapeutic strategies are currently available to patients. Although immune-checkpoint and anti-angiogenesis therapies have shown some promising results during preliminary testing, new and effective therapeutic modalities are still urgently needed [137]. The current literature supports the development of a novel approach based on the Hippo pathway dysregulation observed in MM cells as likely to be effective against MM. However, additional studies are required to elucidate and characterize the mechanisms underlying, and the impact of Hippo pathway inactivation, on the pathogenesis of this disease.
Acknowledgments
This work was supported in part by JSPS KAKENHI (16H04706, 17K19628), and AMED PRIME grants. I am aware of the vast number of excellent scientific contributions to this field, and apologize to all colleagues whose work could not be cited owing to space limitations.
Conflicts of Interest
The author received a collaboration grant from Kyowa Hakko Kirin Co., Ltd. (Tokyo, JAPAN), and Eisai Co., Ltd. (Tokyo, JAPAN). The founding sponsors had no role in the writing of the manuscript.
Abbreviations
| Ab | antibody |
| AMPK | AMP-dependent protein kinase |
| ASS1 | arginosuccinate synthetase-1 |
| BAP1 | BRCA1-associated protein 1 |
| BIRC | baculoviral IPA repeat containing |
| CCDN1 | cyclin D1 |
| CCN | CYR61, CTGF, NOV |
| CDKN2A | cyclin dependent kinase inhibitor 2A |
| CTGF | connective tissue growth factor |
| CPI-17 | C-potentiated phosphatase inhibitor |
| CRL | cullin-RING E3 ubiquitin ligase |
| DCAF1 | DDB1 and CUL4 associated factor 1 |
| DNMT | DNA methyltransferase |
| ECM | extracellular matrix |
| EGFR | epidermal growth factor receptor |
| EMT | epithelial-mesenchymal transition |
| ERM | ezrin/radixin/moesin |
| EZH2 | enhancer of Zeste homolog 2 |
| FAK | focal adhesion kinase |
| FGFR | fibroblast growth factor receptor |
| FISH | fluorescence in situ hybridization |
| FOXM1 | forkhead box M1 |
| GPCR | G-protein-coupled receptor |
| HDAC | histone deacetylase |
| HMG-CoA | hydroxymethylglutaryl-CoA |
| IPF | idiopathic pulmonary fibrosis |
| LATS | large tumor suppressor kinase |
| LEF | lymphoid enhancer-binding factor |
| MM | malignant mesothelioma |
| MPM | malignant pleural mesothelioma |
| MST | mammalian sterile 20-like protein kinase |
| MTAP | methylthioadenosine phosphorylase |
| mTOR | mammalian/mechanistic target of rapamycin |
| mTORC1 | mTOR complex 1 |
| MYPT-1-PP1δ | myosin phosphatase targeting subunit 1-protein phosphatase 1δ |
| NF2 | neurofibromatosis type 2 |
| OS | overall survival |
| PAK | p21-acivated kinase |
| PARP | poly(ADP-ribose) polymerase |
| PDA | pancreatic ductal adenocarcinoma |
| PDGFR | platelet-derived growth factor receptor |
| PD1 | programmed cell death 1 |
| PD-L1 | programmed cell death ligand 1 |
| PFS | progression free survival |
| PI3K | phosphoinositide 3-kinase |
| PKA | protein kinase A |
| PKC | protein kinase C |
| PLCB4 | phospholipase-C β 4 |
| RTK | receptor tyrosine kinase |
| RMH | reactive mesothelial hyperplasia |
| TAZ | transcriptional coactivator with PDZ-binding motif |
| TCF | transcription factor |
| TEAD | TEA domain transcription factor |
| TERT | telomerase reverse transcriptase |
| TGF-β | transforming growth factor β |
| TIAM1 | T-cell lymphoma invasion and metastasis 1 |
| TKI | tyrosine kinase inhibitor |
| TRAF7 | TNF receptor associated factor 7 |
| VEGFR | vascular endothelial growth factor receptor |
| VGLL4 | vestigial-like 4 |
| YAP1 | Yes-associated protein 1 |
References
- Robinson, B.W.; Lake, R.A. Advances in malignant mesothelioma. N. Engl. J. Med. 2005, 353, 1591–1603. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.S.; Wistuba, I.; Roth, J.A.; Kindler, H.L. Malignant pleural mesothelioma. J. Clin. Oncol. 2009, 27, 2081–2090. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, A.; Antoine, D.J.; Pellegrini, L.; Baumann, F.; Pagano, I.; Pastorino, S.; Goparaju, C.M.; Prokrym, K.; Canino, C.; Pass, H.I.; et al. HMGB1 and Its Hyperacetylated Isoform are Sensitive and Specific Serum Biomarkers to Detect Asbestos Exposure and to Identify Mesothelioma Patients. Clin. Cancer Res. 2016, 22, 3087–3096. [Google Scholar] [CrossRef] [PubMed]
- Lagniau, S.; Lamote, K.; van Meerbeeck, J.P.; Vermaelen, K.Y. Biomarkers for early diagnosis of malignant mesothelioma: Do we need another moonshot? Oncotarget 2017, 8, 53751–53762. [Google Scholar] [CrossRef] [PubMed]
- Husain, A.N.; Colby, T.V.; Ordonez, N.G.; Krausz, T.; Borczuk, A.; Cagle, P.T.; Chirieac, L.R.; Churg, A.; Galateau-Salle, F.; Gibbs, A.R.; et al. Guidelines for pathologic diagnosis of malignant mesothelioma: A consensus statement from the International Mesothelioma Interest Group. Arch. Pathol. Lab. Med. 2009, 133, 1317–1331. [Google Scholar] [PubMed]
- Nelson, D.B.; Rice, D.C.; Niu, J.; Atay, S.; Vaporciyan, A.A.; Antonoff, M.; Hofstetter, W.L.; Walsh, G.L.; Swisher, S.G.; Roth, J.A.; et al. Long-Term Survival Outcomes of Cancer-Directed Surgery for Malignant Pleural Mesothelioma: Propensity Score Matching Analysis. J. Clin. Oncol. 2017, 35, 3354–3362. [Google Scholar] [CrossRef] [PubMed]
- Rusch, V.W.; Giroux, D.; Kennedy, C.; Ruffini, E.; Cangir, A.K.; Rice, D.; Pass, H.; Asamura, H.; Waller, D.; Edwards, J.; et al. Initial analysis of the international association for the study of lung cancer mesothelioma database. J. Thorac. Oncol. 2012, 7, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Bueno, R.; Stawiski, E.W.; Goldstein, L.D.; Durinck, S.; De Rienzo, A.; Modrusan, Z.; Gnad, F.; Nguyen, T.T.; Jaiswal, B.S.; Chirieac, L.R.; et al. Comprehensive genomic analysis of malignant pleural mesothelioma identifies recurrent mutations, gene fusions and splicing alterations. Nat. Genet. 2016, 48, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; van Gerwen, M.; Bonassi, S.; Taioli, E. Epidemiology of Environmental Exposure and Malignant Mesothelioma. J. Thorac. Oncol. 2017, 12, 1031–1045. [Google Scholar] [CrossRef] [PubMed]
- Testa, J.R.; Cheung, M.; Pei, J.; Below, J.E.; Tan, Y.; Sementino, E.; Cox, N.J.; Dogan, A.U.; Pass, H.I.; Trusa, S.; et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011, 43, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, T.; Hasegawa, S. Current surgical strategies for malignant pleural mesothelioma. Surg. Today 2016, 46, 887–894. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, N.J.; Rusthoven, J.J.; Symanowski, J.; Denham, C.; Kaukel, E.; Ruffie, P.; Gatzemeier, U.; Boyer, M.; Emri, S.; Manegold, C.; et al. Phase III study of pemetrexed in combination with cisplatin versus cisplatin alone in patients with malignant pleural mesothelioma. J. Clin. Oncol. 2003, 21, 2636–2644. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, J.N.; Sorensen, J.B. Review on clinical trials of targeted treatments in malignant mesothelioma. Cancer Chemother. Pharmacol. 2011, 68, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Stahel, R.A.; Weder, W.; Felley-Bosco, E.; Petrausch, U.; Curioni-Fontecedro, A.; Schmitt-Opitz, I.; Peters, S. Searching for targets for the systemic therapy of mesothelioma. Ann. Oncol. 2015, 26, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Campbell, N.P.; Kunnavakkam, R.; Leighl, N.; Vincent, M.D.; Gandara, D.R.; Koczywas, M.; Gitlitz, B.J.; Agamah, E.; Thomas, S.P.; Stadler, W.M.; et al. Cediranib in patients with malignant mesothelioma: A phase II trial of the University of Chicago Phase II Consortium. Lung Cancer 2012, 78, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Dudek, A.Z.; Pang, H.; Kratzke, R.A.; Otterson, G.A.; Hodgson, L.; Vokes, E.E.; Kindler, H.L. Phase II study of dasatinib in patients with previously treated malignant mesothelioma (cancer and leukemia group B 30601): A brief report. J. Thorac. Oncol. 2012, 7, 755–759. [Google Scholar] [CrossRef] [PubMed]
- Papa, S.; Popat, S.; Shah, R.; Prevost, A.T.; Lal, R.; McLennan, B.; Cane, P.; Lang-Lazdunski, L.; Viney, Z.; Dunn, J.T.; et al. Phase 2 study of sorafenib in malignant mesothelioma previously treated with platinum-containing chemotherapy. J. Thorac. Oncol. 2013, 8, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Laurie, S.A.; Gupta, A.; Chu, Q.; Lee, C.W.; Morzycki, W.; Feld, R.; Foo, A.H.; Seely, J.; Goffin, J.R.; Laberge, F.; et al. Brief report: A phase II study of sunitinib in malignant pleural mesothelioma. the NCIC Clinical Trials Group. J. Thorac. Oncol. 2011, 6, 1950–1954. [Google Scholar] [CrossRef] [PubMed]
- Grosso, F.; Steele, N.; Novello, S.; Nowak, A.K.; Popat, S.; Greillier, L.; John, T.; Leighl, N.B.; Reck, M.; Taylor, P.; et al. Nintedanib Plus Pemetrexed/Cisplatin in Patients With Malignant Pleural Mesothelioma: Phase II Results From the Randomized, Placebo-Controlled LUME-Meso Trial. J. Clin. Oncol. 2017, 35, 3591–3600. [Google Scholar] [CrossRef] [PubMed]
- Zalcman, G.; Mazieres, J.; Margery, J.; Greillier, L.; Audigier-Valette, C.; Moro-Sibilot, D.; Molinier, O.; Corre, R.; Monnet, I.; Gounant, V.; et al. Bevacizumab for newly diagnosed pleural mesothelioma in the Mesothelioma Avastin Cisplatin Pemetrexed Study (MAPS): A randomised, controlled, open-label, phase 3 trial. Lancet 2016, 387, 1405–1414. [Google Scholar] [CrossRef]
- Calabro, L.; Morra, A.; Fonsatti, E.; Cutaia, O.; Amato, G.; Giannarelli, D.; Di Giacomo, A.M.; Danielli, R.; Altomonte, M.; Mutti, L.; et al. Tremelimumab for patients with chemotherapy-resistant advanced malignant mesothelioma: An open-label, single-arm, phase 2 trial. Lancet Oncol. 2013, 14, 1104–1111. [Google Scholar] [CrossRef]
- Maio, M.; Scherpereel, A.; Calabro, L.; Aerts, J.; Perez, S.C.; Bearz, A.; Nackaerts, K.; Fennell, D.A.; Kowalski, D.; Tsao, A.S.; et al. Tremelimumab as second-line or third-line treatment in relapsed malignant mesothelioma (DETERMINE): A multicentre, international, randomised, double-blind, placebo-controlled phase 2b trial. Lancet Oncol. 2017, 18, 1261–1273. [Google Scholar] [CrossRef]
- Alley, E.W.; Lopez, J.; Santoro, A.; Morosky, A.; Saraf, S.; Piperdi, B.; van Brummelen, E. Clinical safety and activity of pembrolizumab in patients with malignant pleural mesothelioma (KEYNOTE-028): Preliminary results from a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2017, 18, 623–630. [Google Scholar] [CrossRef]
- McLoughlin, K.C.; Kaufman, A.S.; Schrump, D.S. Targeting the epigenome in malignant pleural mesothelioma. Transl. Lung Cancer Res. 2017, 6, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Yogelzang, N.J.; Herndon, J.E., 2nd.; Cirrincione, C.; Harmon, D.C.; Antman, K.H.; Corson, J.M.; Suzuki, Y.; Citron, M.L.; Green, M.R. Dihydro-5-azacytidine in malignant mesothelioma. A phase II trial demonstrating activity accompanied by cardiac toxicity. Cancer and Leukemia Group B. Cancer 1997, 79, 2237–2242. [Google Scholar] [CrossRef]
- Krug, L.M.; Kindler, H.L.; Calvert, H.; Manegold, C.; Tsao, A.S.; Fennell, D.; Ohman, R.; Plummer, R.; Eberhardt, W.E.; Fukuoka, K.; et al. Vorinostat in patients with advanced malignant pleural mesothelioma who have progressed on previous chemotherapy (VANTAGE-014): A phase 3, double-blind, randomised, placebo-controlled trial. Lancet Oncol. 2015, 16, 447–456. [Google Scholar] [CrossRef]
- LaFave, L.M.; Beguelin, W.; Koche, R.; Teater, M.; Spitzer, B.; Chramiec, A.; Papalexi, E.; Keller, M.D.; Hricik, T.; Konstantinoff, K.; et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat. Med. 2015, 21, 1344–1349. [Google Scholar] [CrossRef] [PubMed]
- Hassan, R.; Thomas, A.; Alewine, C.; Le, D.T.; Jaffee, E.M.; Pastan, I. Mesothelin Immunotherapy for Cancer: Ready for Prime Time? J. Clin. Oncol. 2016, 34, 4171–4179. [Google Scholar] [CrossRef] [PubMed]
- Angevin, E.; Isambert, N.; Trillet-Lenoir, V.; You, B.; Alexandre, J.; Zalcman, G.; Vielh, P.; Farace, F.; Valleix, F.; Podoll, T.; et al. First-in-human phase 1 of YS110, a monoclonal antibody directed against CD26 in advanced CD26-expressing cancers. Br. J. Cancer 2017, 116, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Szlosarek, P.W.; Steele, J.P.; Nolan, L.; Gilligan, D.; Taylor, P.; Spicer, J.; Lind, M.; Mitra, S.; Shamash, J.; Phillips, M.M.; et al. Arginine Deprivation With Pegylated Arginine Deiminase in Patients With Argininosuccinate Synthetase 1-Deficient Malignant Pleural Mesothelioma: A Randomized Clinical Trial. JAMA Oncol. 2017, 3, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Aerts, J.G.; Popat, S.; Fennell, D.A. Novel insights into mesothelioma biology and implications for therapy. Nat. Rev. Cancer 2017, 17, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Schunselaar, L.M.; Zwart, W.; Baas, P. Targeting BAP1: A new paradigm for mesothelioma. Lung Cancer 2017, 109, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Perrot, M.d.; Wu, L.; Wu, M.; Cho, B.C.J. Radiotherapy for the treatment of malignant pleural mesothelioma. Lancet Oncol. 2017, 18, e532–e542. [Google Scholar] [CrossRef]
- Kindler, H.L.; Ismaila, N.; Armato, S.G., 3rd.; Bueno, R.; Hesdorffer, M.; Jahan, T.; Jones, C.M.; Miettinen, M.; Pass, H.; Rimner, A.; et al. Treatment of Malignant Pleural Mesothelioma: American Society of Clinical Oncology Clinical Practice Guideline. J. Clin. Oncol. 2018, JCO2017766394. [Google Scholar]
- Petrilli, A.M.; Fernandez-Valle, C. Role of Merlin/NF2 inactivation in tumor biology. Oncogene 2016, 35, 537–548. [Google Scholar] [CrossRef] [PubMed]
- Pan, D. The hippo signaling pathway in development and cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Guan, K.L. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef] [PubMed]
- Varelas, X. The Hippo pathway effectors TAZ and YAP in development, homeostasis and disease. Development 2014, 141, 1614–1626. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Giancotti, F.G. Molecular insights into NF2/Merlin tumor suppressor function. FEBS Lett. 2014, 588, 2743–2752. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, H.; Li, F.; Chan, S.W.; Lin, Z.; Wei, Z.; Yang, Z.; Guo, F.; Lim, C.J.; Xing, W.; et al. Angiomotin binding-induced activation of Merlin/NF2 in the Hippo pathway. Cell Res. 2015, 25, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Sekido, Y.; Pass, H.I.; Bader, S.; Mew, D.J.; Christman, M.F.; Gazdar, A.F.; Minna, J.D. Neurofibromatosis type 2 (NF2) gene is somatically mutated in mesothelioma but not in lung cancer. Cancer Res. 1995, 55, 1227–1231. [Google Scholar] [PubMed]
- Bianchi, A.B.; Mitsunaga, S.I.; Cheng, J.Q.; Klein, W.M.; Jhanwar, S.C.; Seizinger, B.; Kley, N.; Klein-Szanto, A.J.; Testa, J.R. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc. Natl. Acad. Sci. USA 1995, 92, 10854–10858. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.Q.; Lee, W.C.; Klein, M.A.; Cheng, G.Z.; Jhanwar, S.C.; Testa, J.R. Frequent mutations of NF2 and allelic loss from chromosome band 22q12 in malignant mesothelioma: Evidence for a two-hit mechanism of NF2 inactivation. Genes Chromosomes Cancer 1999, 24, 238–242. [Google Scholar] [CrossRef]
- Thurneysen, C.; Opitz, I.; Kurtz, S.; Weder, W.; Stahel, R.A.; Felley-Bosco, E. Functional inactivation of NF2/merlin in human mesothelioma. Lung Cancer 2009, 64, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Sperka, T.; Herrlich, P.; Morrison, H. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature 2006, 442, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Gutmann, D.H.; Sherman, L.; Seftor, L.; Haipek, C.; Hoang Lu, K.; Hendrix, M. Increased expression of the NF2 tumor suppressor gene product, merlin, impairs cell motility, adhesionand spreading. Hum. Mol. Genet. 1999, 8, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Zoch, A.; Mayerl, S.; Schulz, A.; Greither, T.; Frappart, L.; Rubsam, J.; Heuer, H.; Giovannini, M.; Morrison, H. Merlin Isoforms 1 and 2 Both Act as Tumour Suppressors and Are Required for Optimal Sperm Maturation. PLoS ONE 2015, 10, e0129151. [Google Scholar] [CrossRef] [PubMed]
- Guled, M.; Lahti, L.; Lindholm, P.M.; Salmenkivi, K.; Bagwan, I.; Nicholson, A.G.; Knuutila, S. CDKN2A, NF2, and JUN are dysregulated among other genes by miRNAs in malignant mesothelioma-A miRNA microarray analysis. Genes Chromosomes Cancer 2009, 48, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Poulikakos, P.I.; Xiao, G.H.; Gallagher, R.; Jablonski, S.; Jhanwar, S.C.; Testa, J.R. Re-expression of the tumor suppressor NF2/merlin inhibits invasiveness in mesothelioma cells and negatively regulates FAK. Oncogene 2006, 25, 5960–5968. [Google Scholar] [CrossRef] [PubMed]
- Fleury-Feith, J.; Lecomte, C.; Renier, A.; Matrat, M.; Kheuang, L.; Abramowski, V.; Levy, F.; Janin, A.; Giovannini, M.; Jaurand, M.C. Hemizygosity of Nf2 is associated with increased susceptibility to asbestos-induced peritoneal tumours. Oncogene 2003, 22, 3799–3805. [Google Scholar] [CrossRef] [PubMed]
- Altomare, D.A.; Vaslet, C.A.; Skele, K.L.; De Rienzo, A.; Devarajan, K.; Jhanwar, S.C.; McClatchey, A.I.; Kane, A.B.; Testa, J.R. A mouse model recapitulating molecular features of human mesothelioma. Cancer Res. 2005, 65, 8090–8095. [Google Scholar] [CrossRef] [PubMed]
- Jongsma, J.; van Montfort, E.; Vooijs, M.; Zevenhoven, J.; Krimpenfort, P.; van der Valk, M.; van de Vijver, M.; Berns, A. A conditional mouse model for malignant mesothelioma. Cancer Cell 2008, 13, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; You, L.; Cooper, J.; Schiavon, G.; Pepe-Caprio, A.; Zhou, L.; Ishii, R.; Giovannini, M.; Hanemann, C.O.; Long, S.B.; et al. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell 2010, 140, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Cooper, J.; Zhou, L.; Yang, C.; Erdjument-Bromage, H.; Zagzag, D.; Snuderl, M.; Ladanyi, M.; Hanemann, C.O.; Zhou, P.; et al. Merlin/NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 2014, 26, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Hikasa, H.; Sekido, Y.; Suzuki, A. Merlin/NF2-Lin28B-let-7 Is a Tumor-Suppressive Pathway that Is Cell-Density Dependent and Hippo Independent. Cell Rep. 2016, 14, 2950–2961. [Google Scholar] [CrossRef] [PubMed]
- Clark, V.E.; Erson-Omay, E.Z.; Serin, A.; Yin, J.; Cotney, J.; Ozduman, K.; Avsar, T.; Li, J.; Murray, P.B.; Henegariu, O.; et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013, 339, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Harvey, K.F.; Zhang, X.; Thomas, D.M. The Hippo pathway and human cancer. Nat. Rev. Cancer 2013, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Kim, S.M.; Lee, H. The Hippo signaling pathway provides novel anti-cancer drug targets. Oncotarget 2017, 8, 16084–16098. [Google Scholar] [CrossRef] [PubMed]
- Andl, T.; Zhou, L.; Yang, K.; Kadekaro, A.L.; Zhang, Y. YAP and WWTR1: New targets for skin cancer treatment. Cancer Lett. 2017, 396, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Li, T.; Zheng, S.; Wang, H. The Hippo pathway as a drug target in gastric cancer. Cancer Lett. 2018, 420, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Murakami, H.; Mizuno, T.; Taniguchi, T.; Fujii, M.; Ishiguro, F.; Fukui, T.; Akatsuka, S.; Horio, Y.; Hida, T.; Kondo, Y.; et al. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res. 2011, 71, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Tranchant, R.; Quetel, L.; Tallet, A.; Meiller, C.; Renier, A.; de Koning, L.; de Reynies, A.; Le Pimpec-Barthes, F.; Zucman-Rossi, J.; Jaurand, M.C.; et al. Co-occurring Mutations of Tumor Suppressor Genes, LATS2 and NF2, in Malignant Pleural Mesothelioma. Clin. Cancer Res. 2017, 23, 3191–3202. [Google Scholar] [CrossRef] [PubMed]
- Miyanaga, A.; Masuda, M.; Tsuta, K.; Kawasaki, K.; Nakamura, Y.; Sakuma, T.; Asamura, H.; Gemma, A.; Yamada, T. Hippo pathway gene mutations in malignant mesothelioma: Revealed by RNA and targeted exon sequencing. J. Thorac. Oncol. 2015, 10, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, I.; Osada, H.; Fujii, M.; Fukatsu, A.; Hida, T.; Horio, Y.; Kondo, Y.; Sato, A.; Hasegawa, Y.; Tsujimura, T.; et al. LIM-domain protein AJUBA suppresses malignant mesothelioma cell proliferation via Hippo signaling cascade. Oncogene 2015, 34, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Jagannathan, R.; Schimizzi, G.V.; Zhang, K.; Loza, A.J.; Yabuta, N.; Nojima, H.; Longmore, G.D. AJUBA LIM Proteins Limit Hippo Activity in Proliferating Cells by Sequestering the Hippo Core Kinase Complex in the Cytosol. Mol. Cell. Biol. 2016, 36, 2526–2542. [Google Scholar] [CrossRef] [PubMed]
- Jia, L.; Gui, B.; Zheng, D.; Decker, K.F.; Tinay, I.; Tan, M.; Wang, X.; Kibel, A.S. Androgen receptor-regulated miRNA-193a-3p targets AJUBA to promote prostate cancer cell migration. Prostate 2017, 77, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Lorenzetto, E.; Brenca, M.; Boeri, M.; Verri, C.; Piccinin, E.; Gasparini, P.; Facchinetti, F.; Rossi, S.; Salvatore, G.; Massimino, M.; et al. YAP1 acts as oncogenic target of 11q22 amplification in multiple cancer subtypes. Oncotarget 2014, 5, 2608–2621. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, T.; Osada, H.; Murakami, H.; Tatematsu, Y.; Taniguchi, T.; Kondo, Y.; Yatabe, Y.; Hasegawa, Y.; Shimokata, K.; Horio, Y.; et al. YAP1 is involved in mesothelioma development and negatively regulated by Merlin through phosphorylation. Carcinogenesis 2008, 29, 2139–2146. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, T.; Murakami, H.; Fujii, M.; Ishiguro, F.; Tanaka, I.; Kondo, Y.; Akatsuka, S.; Toyokuni, S.; Yokoi, K.; Osada, H.; et al. YAP induces malignant mesothelioma cell proliferation by upregulating transcription of cell cycle-promoting genes. Oncogene 2012, 31, 5117–5122. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Toyoda, T.; Nakanishi, H.; Yatabe, Y.; Sato, A.; Matsudaira, Y.; Ito, H.; Murakami, H.; Kondo, Y.; Kondo, E.; et al. TGF-beta synergizes with defects in the Hippo pathway to stimulate human malignant mesothelioma growth. J. Exp. Med. 2012, 209, 479–494. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, T.; Takahara, T.; Kasugai, Y.; Arita, K.; Yoshida, N.; Karube, K.; Suguro, M.; Matsuo, K.; Nakanishi, H.; Kiyono, T.; et al. Modeling mesothelioma utilizing human mesothelial cells reveals involvement of phospholipase-C beta 4 in YAP-active mesothelioma cell proliferation. Carcinogenesis 2016, 37, 1098–1109. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Yamashita, Y.; Chew, S.H.; Akatsuka, S.; Ukai, S.; Wang, S.; Nagai, H.; Okazaki, Y.; Takahashi, T.; Toyokuni, S. Connective tissue growth factor and beta-catenin constitute an autocrine loop for activation in rat sarcomatoid mesothelioma. J. Pathol. 2014, 233, 402–414. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.X.; Luo, J.; Mo, J.S.; Liu, G.; Kim, Y.C.; Meng, Z.; Zhao, L.; Peyman, G.; Ouyang, H.; Jiang, W.; et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell 2014, 25, 822–830. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Aoude, L.G.; Wadt, K.; Glasson, W.J.; Warrier, S.K.; Hewitt, A.W.; Kiilgaard, J.F.; Heegaard, S.; Isaacs, T.; Franchina, M.; et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 2016, 7, 4624–4631. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, Y.; Sato, A.; Tsujimura, T.; Emi, M.; Morinaga, T.; Fukuoka, K.; Yamada, S.; Murakami, A.; Kondo, N.; Matsumoto, S.; et al. Frequent inactivation of BAP1 gene in epithelioid-type malignant mesothelioma. Cancer Sci. 2012, 103, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell 2017, 32, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Bononi, A.; Giorgi, C.; Patergnani, S.; Larson, D.; Verbruggen, K.; Tanji, M.; Pellegrini, L.; Signorato, V.; Olivetto, F.; Pastorino, S.; et al. BAP1 regulates IP3R3-mediated Ca(2+) flux to mitochondria suppressing cell transformation. Nature 2017, 546, 549–553. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Sekido, Y. Molecular pathogenesis of malignant mesothelioma. Carcinogenesis 2013, 34, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Hida, T.; Hamasaki, M.; Matsumoto, S.; Sato, A.; Tsujimura, T.; Kawahara, K.; Iwasaki, A.; Okamoto, T.; Oda, Y.; Honda, H.; et al. Immunohistochemical detection of MTAP and BAP1 protein loss for mesothelioma diagnosis: Comparison with 9p21 FISH and BAP1 immunohistochemistry. Lung Cancer 2017, 104, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Meerang, M.; Berard, K.; Friess, M.; Bitanihirwe, B.K.; Soltermann, A.; Vrugt, B.; Felley-Bosco, E.; Bueno, R.; Richards, W.G.; Seifert, B.; et al. Low Merlin expression and high Survivin labeling index are indicators for poor prognosis in patients with malignant pleural mesothelioma. Mol. Oncol. 2016, 10, 1255–1265. [Google Scholar] [CrossRef] [PubMed]
- Singhi, A.D.; Krasinskas, A.M.; Choudry, H.A.; Bartlett, D.L.; Pingpank, J.F.; Zeh, H.J.; Luvison, A.; Fuhrer, K.; Bahary, N.; Seethala, R.R.; et al. The prognostic significance of BAP1, NF2, and CDKN2A in malignant peritoneal mesothelioma. Mod. Pathol. 2016, 29, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, I.M.; Kolev, V.N.; Vidal, C.M.; Kadariya, Y.; Ring, J.E.; Wright, Q.; Weaver, D.T.; Menges, C.; Padval, M.; McClatchey, A.I.; et al. Merlin deficiency predicts FAK inhibitor sensitivity: A synthetic lethal relationship. Sci. Transl. Med. 2014, 6, 237ra268. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Sato, T.; Yokoi, K.; Sekido, Y. E-cadherin expression is correlated with focal adhesion kinase inhibitor resistance in Merlin-negative malignant mesothelioma cells. Oncogene 2017, 36, 5522–5531. [Google Scholar] [CrossRef] [PubMed]
- Woodard, G.A.; Yang, Y.L.; You, L.; Jablons, D.M. Drug development against the hippo pathway in mesothelioma. Transl. Lung Cancer Res. 2017, 6, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Liu-Chittenden, Y.; Huang, B.; Shim, J.S.; Chen, Q.; Lee, S.J.; Anders, R.A.; Liu, J.O.; Pan, D. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev. 2012, 26, 1300–1305. [Google Scholar] [CrossRef] [PubMed]
- Al-Moujahed, A.; Brodowska, K.; Stryjewski, T.P.; Efstathiou, N.E.; Vasilikos, I.; Cichy, J.; Miller, J.W.; Gragoudas, E.; Vavvas, D.G. Verteporfin inhibits growth of human glioma in vitro without light activation. Sci. Rep. 2017, 7, 7602. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.Q.; Dai, Y.Y.; Hsu, P.C.; Wang, H.; Cheng, L.; Yang, Y.L.; Wang, Y.C.; Xu, Z.D.; Liu, S.; Chan, G.; et al. Targeting YAP in malignant pleural mesothelioma. J. Cell Mol. Med. 2017, 21, 2663–2676. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hu, T.; Xu, Z.; Lin, Z.; Zhang, Z.; Feng, T.; Zhu, L.; Rong, Y.; Shen, H.; Luk, J.M.; et al. Targeting Hippo pathway by specific interruption of YAP-TEAD interaction using cyclic YAP-like peptides. FASEB J. 2015, 29, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Wang, H.; Shi, Z.; Dong, A.; Zhang, W.; Song, X.; He, F.; Wang, Y.; Zhang, Z.; Wang, W.; et al. A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 2014, 25, 166–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Gao, Y.; Li, P.; Shi, Z.; Guo, T.; Li, F.; Han, X.; Feng, Y.; Zheng, C.; Wang, Z.; et al. VGLL4 functions as a new tumor suppressor in lung cancer by negatively regulating the YAP-TEAD transcriptional complex. Cell Res. 2014, 24, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Jiao, S.; Li, C.; Hao, Q.; Miao, H.; Zhang, L.; Li, L.; Zhou, Z. VGLL4 targets a TCF4-TEAD4 complex to coregulate Wnt and Hippo signalling in colorectal cancer. Nat. Commun. 2017, 8, 14058. [Google Scholar] [CrossRef] [PubMed]
- Diamantopoulou, Z.; White, G.; Fadlullah, M.Z.H.; Dreger, M.; Pickering, K.; Maltas, J.; Ashton, G.; MacLeod, R.; Baillie, G.S.; Kouskoff, V.; et al. TIAM1 Antagonizes TAZ/YAP Both in the Destruction Complex in the Cytoplasm and in the Nucleus to Inhibit Invasion of Intestinal Epithelial Cells. Cancer Cell 2017, 31, 621–634. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.A.; Richards, A.K.; Kusumah, I.; Perumal, V.; Bolitho, E.M.; Mutsaers, S.E.; Dharmarajan, A.M. Expression profile and function of Wnt signaling mechanisms in malignant mesothelioma cells. Biochem. Biophys. Res. Commun. 2013, 440, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Santinon, G.; Pocaterra, A.; Dupont, S. Control of YAP/TAZ Activity by Metabolic and Nutrient-Sensing Pathways. Trends Cell Biol. 2016, 26, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xiao, Z.D.; Li, X.; Aziz, K.E.; Gan, B.; Johnson, R.L.; Chen, J. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 2015, 17, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.S.; Guan, K.L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef] [PubMed]
- DeRan, M.; Yang, J.; Shen, C.H.; Peters, E.C.; Fitamant, J.; Chan, P.; Hsieh, M.; Zhu, S.; Asara, J.M.; Zheng, B.; et al. Energy stress regulates Hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep. 2014, 9, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.R.; Morris, A.D. Metformin in cancer treatment and prevention. Annu. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Walker, J.; Damhuis, R.A.; Brewster, D.H.; Wild, S.H. Metformin and survival of people with type 2 diabetes and pleural mesothelioma: A population-based retrospective cohort study. Lung Cancer 2016, 99, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Shimazu, K.; Tada, Y.; Morinaga, T.; Shingyoji, M.; Sekine, I.; Shimada, H.; Hiroshima, K.; Namiki, T.; Tatsumi, K.; Tagawa, M. Metformin produces growth inhibitory effects in combination with nutlin-3a on malignant mesothelioma through a cross-talk between mTOR and p53 pathways. BMC Cancer 2017, 17, 309. [Google Scholar] [CrossRef] [PubMed]
- Rubins, J.B.; Greatens, T.; Kratzke, R.A.; Tan, A.T.; Polunovsky, V.A.; Bitterman, P. Lovastatin induces apoptosis in malignant mesothelioma cells. Am. J. Respir. Crit. Care Med. 1998, 157, 1616–1622. [Google Scholar] [CrossRef] [PubMed]
- Riganti, C.; Orecchia, S.; Pescarmona, G.; Betta, P.G.; Ghigo, D.; Bosia, A. Statins revert doxorubicin resistance via nitric oxide in malignant mesothelioma. Int. J. Cancer 2006, 119, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.; Alfonso, H.; Woo, S.; Walsh, A.; Olsen, N.; Musk, A.W.; Robinson, B.W.; Nowak, A.K.; Lake, R.A. Statins do not alter the incidence of mesothelioma in asbestos exposed mice or humans. PLoS ONE 2014, 9, e103025. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Specchia, V.; Cordenonsi, M.; Mano, M.; Dupont, S.; Manfrin, A.; Ingallina, E.; Sommaggio, R.; Piazza, S.; et al. Metabolic control of YAP and TAZ by the mevalonate pathway. Nat. Cell Biol. 2014, 16, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, Y.; Wang, H.; Zhang, Y.; Mei, L.; Fang, X.; Zhang, X.; Zhang, F.; Chen, H.; Liu, Y.; et al. Interplay of mevalonate and Hippo pathways regulates RHAMM transcription via YAP to modulate breast cancer cell motility. Proc. Natl. Acad. Sci. USA 2014, 111, E89–E98. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Osada, H.; Murakami-Tonami, Y.; Horio, Y.; Hida, T.; Sekido, Y. Statin suppresses Hippo pathway-inactivated malignant mesothelioma cells and blocks the YAP/CD44 growth stimulatory axis. Cancer Lett. 2016, 385, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lago, M.A.; Okada, T.; Murillo, M.M.; Socci, N.; Giancotti, F.G. Loss of the tumor suppressor gene NF2, encoding merlin, constitutively activates integrin-dependent mTORC1 signaling. Mol. Cell. Biol. 2009, 29, 4235–4249. [Google Scholar] [CrossRef] [PubMed]
- Iyer, G.; Hanrahan, A.J.; Milowsky, M.I.; Al-Ahmadie, H.; Scott, S.N.; Janakiraman, M.; Pirun, M.; Sander, C.; Socci, N.D.; Ostrovnaya, I.; et al. Genome sequencing identifies a basis for everolimus sensitivity. Science 2012, 338, 221. [Google Scholar] [CrossRef] [PubMed]
- Ou, S.H.; Moon, J.; Garland, L.L.; Mack, P.C.; Testa, J.R.; Tsao, A.S.; Wozniak, A.J.; Gandara, D.R. SWOG S0722: Phase II study of mTOR inhibitor everolimus (RAD001) in advanced malignant pleural mesothelioma (MPM). J. Thorac. Oncol. 2015, 10, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Dolly, S.O.; Wagner, A.J.; Bendell, J.C.; Kindler, H.L.; Krug, L.M.; Seiwert, T.Y.; Zauderer, M.G.; Lolkema, M.P.; Apt, D.; Yeh, R.F.; et al. Phase I Study of Apitolisib (GDC-0980), Dual Phosphatidylinositol-3-Kinase and Mammalian Target of Rapamycin Kinase Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2016, 22, 2874–2884. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.; Xu, Q.; Zhou, L.; Pavlovic, M.; Ojeda, V.; Moulick, K.; de Stanchina, E.; Poirier, J.T.; Zauderer, M.; Rudin, C.M.; et al. Combined Inhibition of NEDD8-Activating Enzyme and mTOR Suppresses NF2 Loss-Driven Tumorigenesis. Mol. Cancer Ther. 2017, 16, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.E.; Howlett, M.; Cole, C.H.; Kees, U.R. Deregulated expression of connective tissue growth factor (CTGF/CCN2) is linked to poor outcome in human cancer. Int. J. Cancer 2015, 137, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Wirkner, U.; Bickelhaupt, S.; Lopez Perez, R.; Tietz, A.; Lipson, K.E.; Seeley, T.W.; Huber, P.E. Radiation-induced pulmonary gene expression changes are attenuated by the CTGF antibody Pamrevlumab. Respir. Res. 2018, 19, 14. [Google Scholar] [CrossRef] [PubMed]
- Moran-Jones, K.; Gloss, B.S.; Murali, R.; Chang, D.K.; Colvin, E.K.; Jones, M.D.; Yuen, S.; Howell, V.M.; Brown, L.M.; Wong, C.W.; et al. Connective tissue growth factor as a novel therapeutic target in high grade serous ovarian cancer. Oncotarget 2015, 6, 44551–44562. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Frese, K.K.; Bapiro, T.E.; Nakagawa, T.; Sternlicht, M.D.; Seeley, T.W.; Pilarsky, C.; Jodrell, D.I.; Spong, S.M.; Tuveson, D.A. CTGF antagonism with mAb FG-3019 enhances chemotherapy response without increasing drug delivery in murine ductal pancreas cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 12325–12330. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Li, L.; Wang, L.; Wang, C.Y.; Yu, J.; Guan, K.L. Cell detachment activates the Hippo pathway via cytoskeleton reorganization to induce anoikis. Genes Dev. 2012, 26, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, S.E.; Prele, C.M.; Pengelly, S.; Herrick, S.E. Mesothelial cells and peritoneal homeostasis. Fertil. Steril. 2016, 106, 1018–1024. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; An, S.; Ward, R.; Yang, Y.; Guo, X.X.; Li, W.; Xu, T.R. G protein-coupled receptors as promising cancer targets. Cancer Lett. 2016, 376, 226–239. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Drabkin, H.A.; Reeves, J.A.; Hainsworth, J.D.; Hazel, S.E.; Paggiarino, D.A.; Wojciak, J.; Woodnutt, G.; Bhatt, R.S. A phase 2 study of the sphingosine-1-phosphate antibody sonepcizumab in patients with metastatic renal cell carcinoma. Cancer 2017, 123, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Yano, S.; Ogino, H.; Ikuta, K.; Kakiuchi, S.; Hanibuchi, M.; Kanematsu, T.; Taniguchi, T.; Sekido, Y.; Sone, S. Lysophosphatidic acid stimulates the proliferation and motility of malignant pleural mesothelioma cells through lysophosphatidic acid receptors, LPA1 and LPA2. Cancer Sci. 2008, 99, 1603–1610. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.; Halder, G. The two faces of Hippo: Targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat. Rev. Drug Discov. 2014, 13, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Basu, D.; Lettan, R.; Damodaran, K.; Strellec, S.; Reyes-Mugica, M.; Rebbaa, A. Identification, mechanism of action, and antitumor activity of a small molecule inhibitor of Hippo, TGF-beta, and Wnt signaling pathways. Mol. Cancer Ther. 2014, 13, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Oku, Y.; Nishiya, N.; Shito, T.; Yamamoto, R.; Yamamoto, Y.; Oyama, C.; Uehara, Y. Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 2015, 5, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Sugimachi, K.; Goto, H.; Wang, J.; Morikawa, T.; Miyachi, Y.; Takano, Y.; Hikasa, H.; Itoh, T.; Suzuki, S.O.; et al. Dysregulated YAP1/TAZ and TGF-beta signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice. Proc. Natl. Acad. Sci. USA 2016, 113, E71–E80. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.S.; He, D.; Saigal, B.; Liu, S.; Lee, J.J.; Bakkannagari, S.; Ordonez, N.G.; Hong, W.K.; Wistuba, I.; Johnson, F.M. Inhibition of c-Src expression and activation in malignant pleural mesothelioma tissues leads to apoptosis, cell cycle arrest, and decreased migration and invasion. Mol. Cancer Ther. 2007, 6, 1962–1972. [Google Scholar] [CrossRef] [PubMed]
- Monica, V.; Lo Iacono, M.; Bracco, E.; Busso, S.; Di Blasio, L.; Primo, L.; Peracino, B.; Papotti, M.; Scagliotti, G. Dasatinib modulates sensitivity to pemetrexed in malignant pleural mesothelioma cell lines. Oncotarget 2016, 7, 76577–76589. [Google Scholar] [CrossRef] [PubMed]
- Tsao, A.S.; Lin, H.; Carter, B.W.; Lee, J.J.; Rice, D.; Vaporcyan, A.; Swisher, S.; Mehran, R.; Heymach, J.; Nilsson, M.; et al. Biomarker-Integrated Neoadjuvant Dasatinib Trial in Resectable Malignant Pleural Mesothelioma. J. Thorac. Oncol. 2018, 13, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Nambara, S.; Masuda, T.; Nishio, M.; Kuramitsu, S.; Tobo, T.; Ogawa, Y.; Hu, Q.; Iguchi, T.; Kuroda, Y.; Ito, S.; et al. Antitumor effects of the antiparasitic agent ivermectin via inhibition of Yes-associated protein 1 expression in gastric cancer. Oncotarget 2017, 8, 107666–107677. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Yu, J.; Han, W.; Fan, X.; Qian, H.; Wei, H.; Tsai, Y.H.; Zhao, J.; Zhang, W.; Liu, Q.; et al. A splicing isoform of TEAD4 attenuates the Hippo-YAP signalling to inhibit tumour proliferation. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, G.; Sidhu, G.S.; Williamson, E.A.; Jaiswal, A.S.; Najmunnisa, N.; Wilcoxen, K.; Jones, D.; George, T.J., Jr.; Hromas, R. Synthetic lethality in malignant pleural mesothelioma with PARP1 inhibition. Cancer Chemother. Pharmacol. 2017, 80, 861–867. [Google Scholar] [CrossRef] [PubMed]
- McCambridge, A.J.; Napolitano, A.; Mansfield, A.S.; Fennell, D.A.; Sekido, Y.; Nowak, A.K.; Reungwetwattana, T.; Mao, W.; Pass, H.I.; Carbone, M.; et al. State of the Art: Advances in Malignant Pleural Mesothelioma in 2017. J. Thorac. Oncol. 2018. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation showing NF2/merlin-Hippo signaling cascade dysregulation in malignant mesothelioma (MM) cells. Signals from the extracellular environment, transduced via cell-cell contact (cadherin), cell-matrix contact (CD44 and integrin), and/or growth factor receptors (receptor tyrosine kinases, RTKs), affect merlin tumor-suppressive activity. Activated (underphosphorylated) merlin regulates the Hippo signaling cascade and suppresses the activity of the YAP1/TAZ transcriptional coactivators. As MMs show frequent alteration of merlin (the NF2 gene product) and Hippo pathway components including LATS1/2, the resultant underphosphorylated (activated) YAP1/TAZ transcriptional coactivators enhance the expression of many pro-oncogenic genes including CCDN1, FOXM1, CTGF, and PLCB4 in MM cells. P: phosphorylation, Ub: ubiquitination.
Figure 1.
Schematic representation showing NF2/merlin-Hippo signaling cascade dysregulation in malignant mesothelioma (MM) cells. Signals from the extracellular environment, transduced via cell-cell contact (cadherin), cell-matrix contact (CD44 and integrin), and/or growth factor receptors (receptor tyrosine kinases, RTKs), affect merlin tumor-suppressive activity. Activated (underphosphorylated) merlin regulates the Hippo signaling cascade and suppresses the activity of the YAP1/TAZ transcriptional coactivators. As MMs show frequent alteration of merlin (the NF2 gene product) and Hippo pathway components including LATS1/2, the resultant underphosphorylated (activated) YAP1/TAZ transcriptional coactivators enhance the expression of many pro-oncogenic genes including CCDN1, FOXM1, CTGF, and PLCB4 in MM cells. P: phosphorylation, Ub: ubiquitination.

Figure 2.
Signaling cascades and molecules that affect Hippo pathway signaling and YAP1/TAZ activation in MM cells. In addition to cell-to-cell adhesion status and RTK signaling, the Hippo pathway is also regulated by multiple signals including from the GPCR pathway, mevalonate pathway, and energy stress. Upon the activation of other intracellular signaling such as through TGF-β and Wnt signaling pathways, the transcription of YAP1/TAZ-target genes is further enhanced, which induces more aggressive malignant phenotypes of MM cells including cell proliferation, invasion, and EMT.
Figure 2.
Signaling cascades and molecules that affect Hippo pathway signaling and YAP1/TAZ activation in MM cells. In addition to cell-to-cell adhesion status and RTK signaling, the Hippo pathway is also regulated by multiple signals including from the GPCR pathway, mevalonate pathway, and energy stress. Upon the activation of other intracellular signaling such as through TGF-β and Wnt signaling pathways, the transcription of YAP1/TAZ-target genes is further enhanced, which induces more aggressive malignant phenotypes of MM cells including cell proliferation, invasion, and EMT.

Figure 3.
Overall concept of the current, and “under-clinical-trial” therapies available to patients with malignant mesothelioma (MM), as well as potential novel therapeutic modalities that target the Hippo pathway. Ab, antibody; TKIs, tyrosine kinase inhibitors.
Figure 3.
Overall concept of the current, and “under-clinical-trial” therapies available to patients with malignant mesothelioma (MM), as well as potential novel therapeutic modalities that target the Hippo pathway. Ab, antibody; TKIs, tyrosine kinase inhibitors.

© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sekido, Y. Targeting the Hippo Pathway Is a New Potential Therapeutic Modality for Malignant Mesothelioma. Cancers 2018, 10, 90. https://doi.org/10.3390/cancers10040090
AMA Style
Sekido Y. Targeting the Hippo Pathway Is a New Potential Therapeutic Modality for Malignant Mesothelioma. Cancers. 2018; 10(4):90. https://doi.org/10.3390/cancers10040090
Chicago/Turabian StyleSekido, Yoshitaka. 2018. "Targeting the Hippo Pathway Is a New Potential Therapeutic Modality for Malignant Mesothelioma" Cancers 10, no. 4: 90. https://doi.org/10.3390/cancers10040090
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.