Obatoclax and Paclitaxel Synergistically Induce Apoptosis and Overcome Paclitaxel Resistance in Urothelial Cancer Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
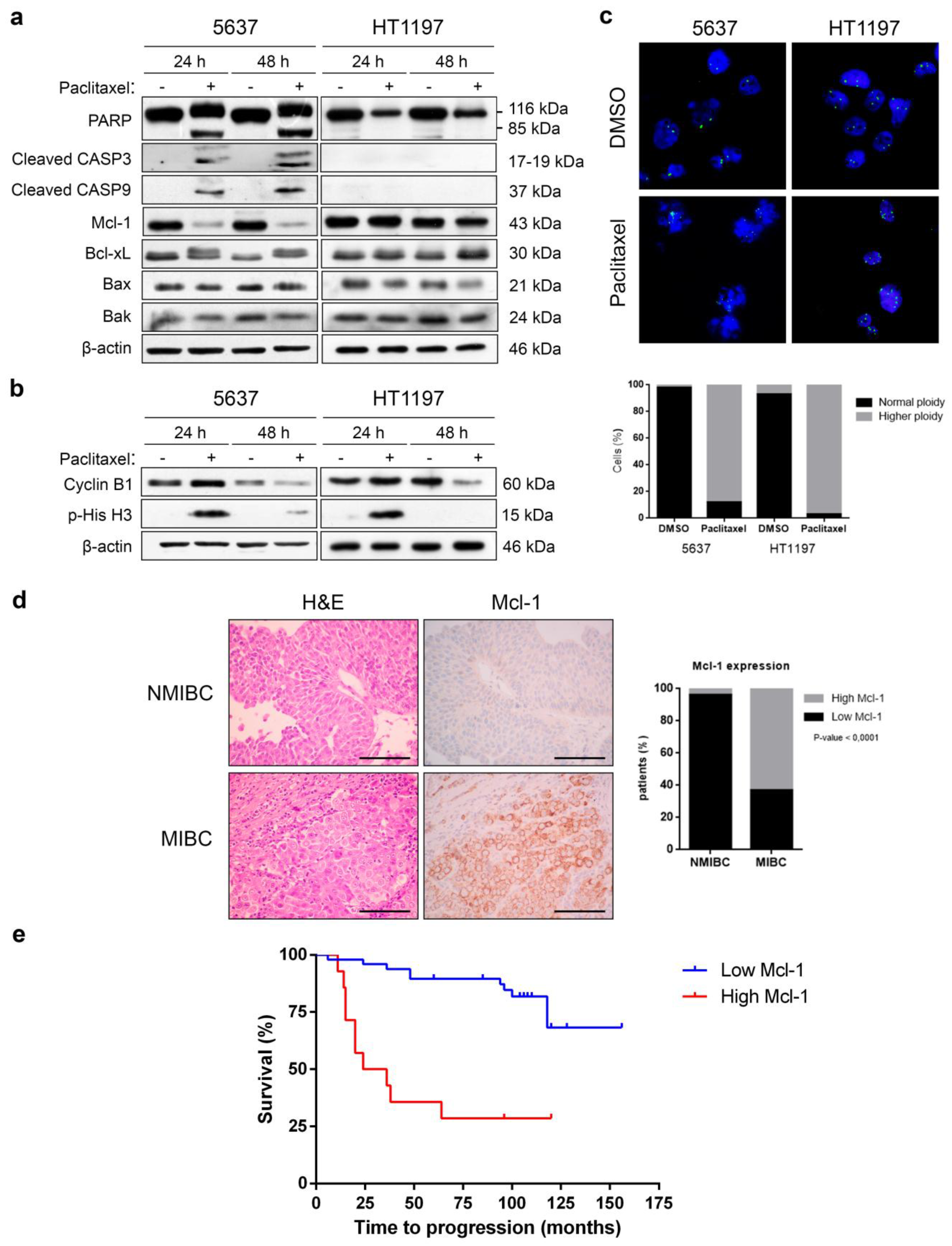
2.1. Decrease of Mcl-1 and Apoptosis Concur in Paclitaxel-Treated 5637, but Not in HT1197 Bladder Cancer Cells
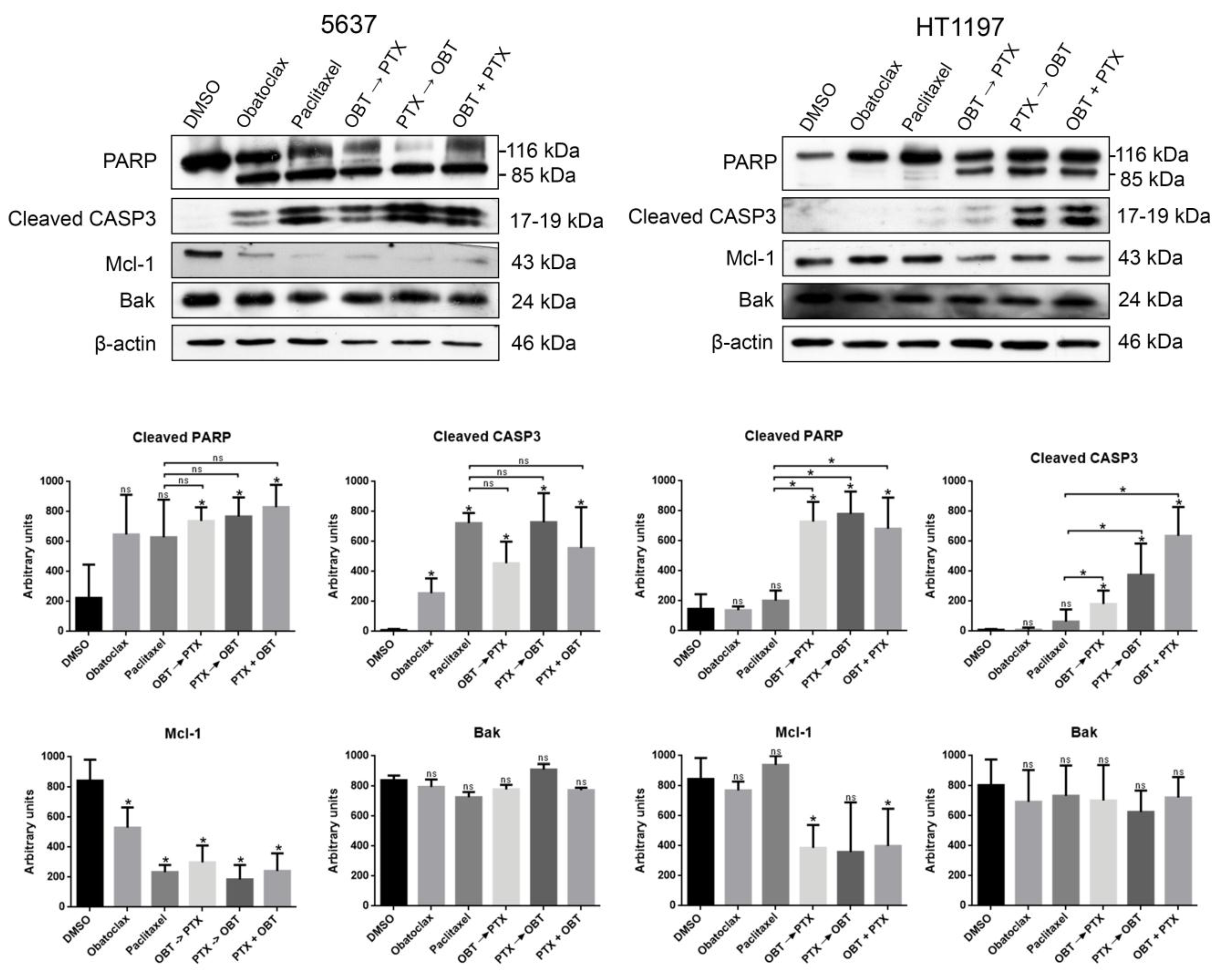
2.2. The Combination of Paclitaxel and Mcl-1 Antagonist Obatoclax Induces Apoptosis in Resistant HT1197 Cells
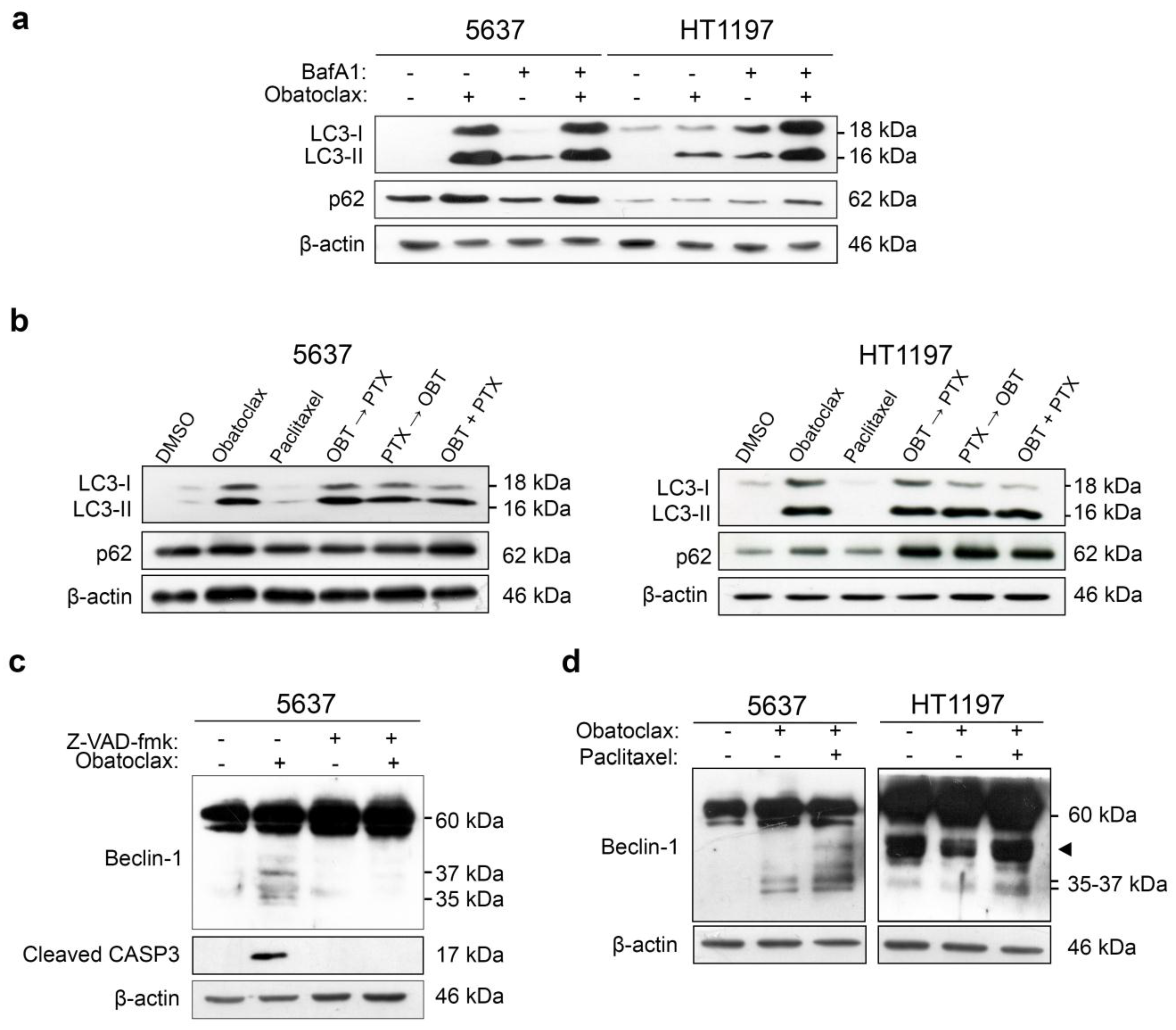
2.3. LC3-II and p62 Accumulate in 5637 Cells Treated with Obatoclax and HT1197 Cells Treated with Combinations of Obatoclax and Paclitaxel
2.4. Caspase-Dependent Cleavage of Beclin-1 Associates with Autophagy Blockade and Apoptosis in HT1197 Cells Treated with a Combination of Obatoclax and Paclitaxel
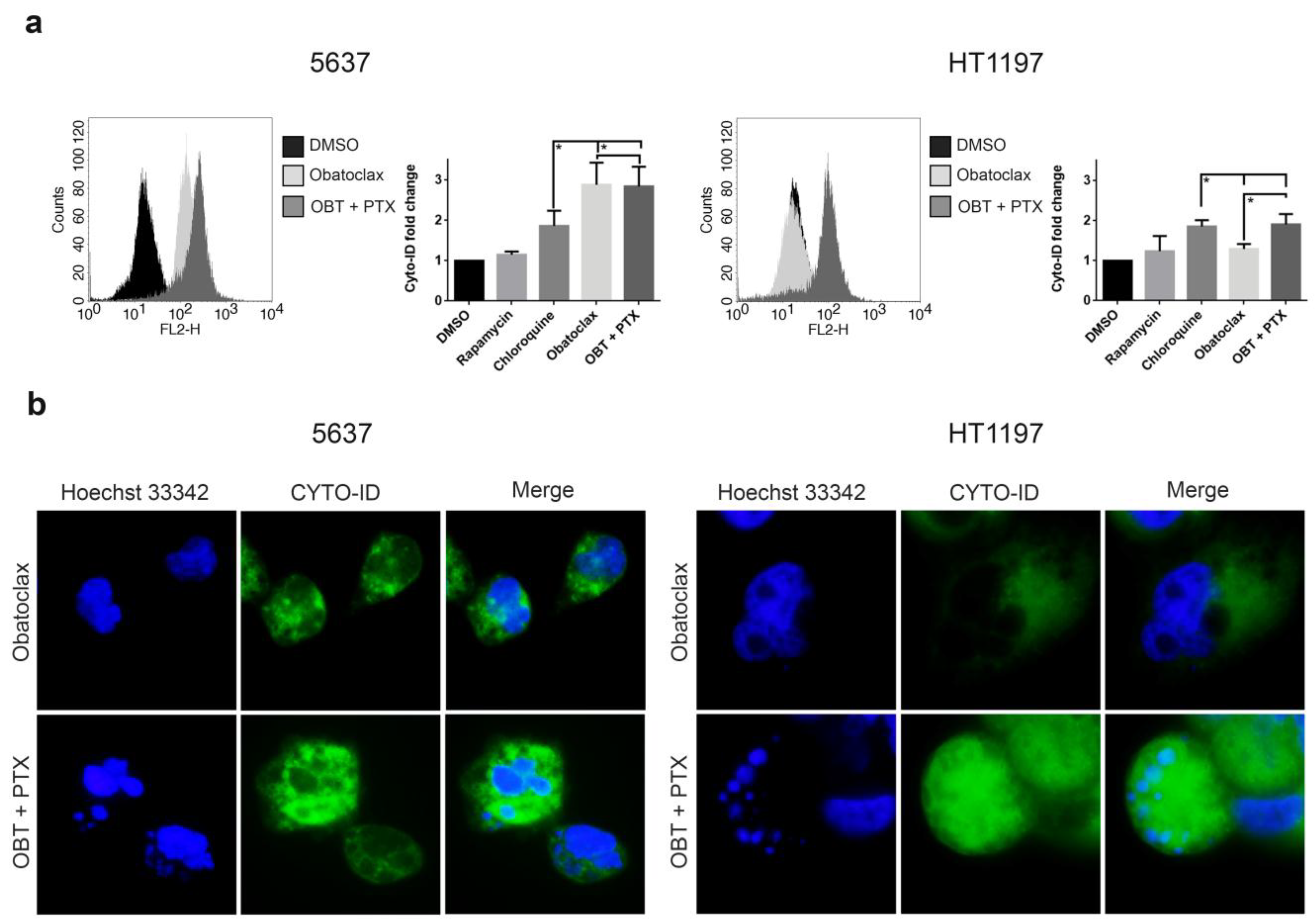
2.5. Blockade of the Autophagic Flux Correlates with Apoptotic Cell Death in HT1197 Cells Treated with a Combination of Obatoclax and Paclitaxel
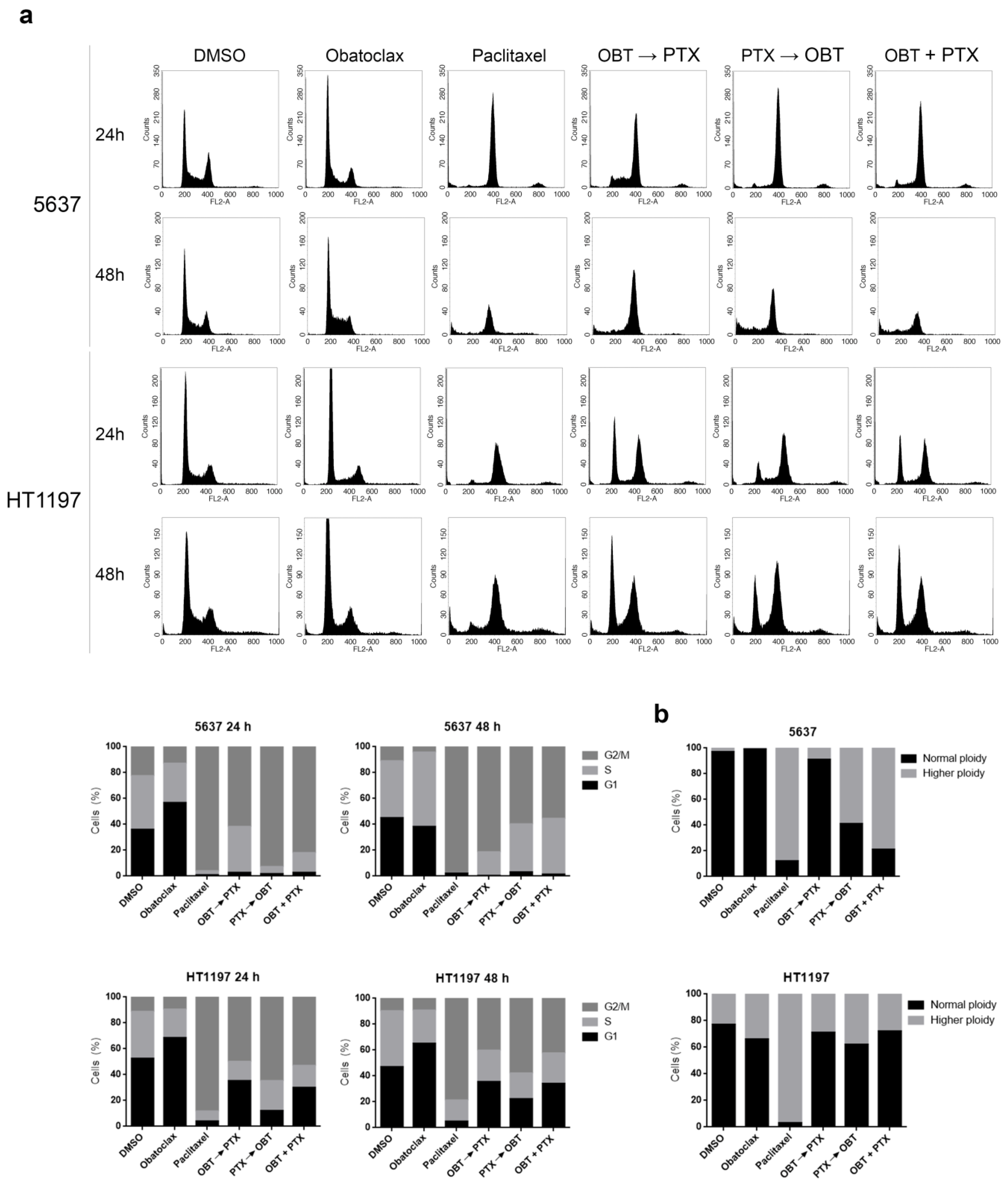
2.6. Obatoclax Retards the Cell Cycle of Paclitaxel-Treated HT1197 Cells and Favors Inhibition of Mitotic Slippage
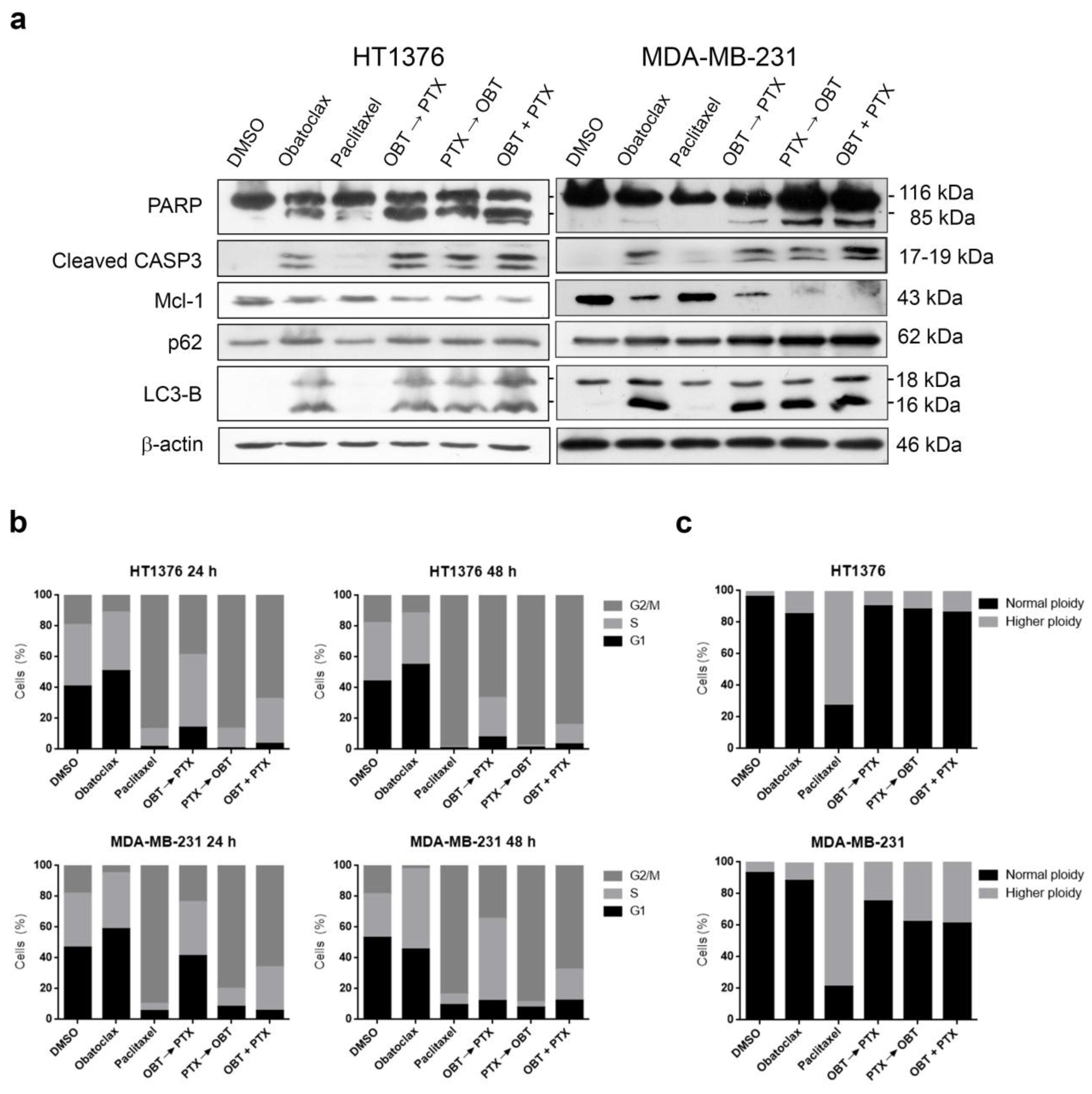
2.7. Combinations of Obatoclax and Paclitaxel Promote Apoptosis in Paclitaxel-Resistant HT1376 and MDA-MB-231 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Drugs
4.2. Antibodies
4.3. Western Blot
4.4. Flow Cytometric Analysis of Cell Cycle
4.5. Fluorescence In Situ Hybridization
4.6. Immunohistochemistry
4.7. Fluorescent Autophagy Detection Assay
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, D.S.; Shipley, W.U.; Feldman, A.S. Bladder cancer. Lancet 2009, 374, 239–249. [Google Scholar] [CrossRef]
- Narayanan, S.; Harshman, L.C.; Srinivas, S. Second-line therapies in metastatic urothelial carcinoma. Hematol. Oncol. Clin. North Am. 2015, 29, 341–359. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [Green Version]
- McGrogan, B.T.; Gilmartin, B.; Carney, D.N.; McCann, A. Taxanes, microtubules and chemoresistant breast cancer. Biochim. Biophys. Acta 2008, 1785, 96–132. [Google Scholar] [CrossRef] [PubMed]
- Castilla, C.; Flores, M.L.; Medina, R.; Pérez-Valderrama, B.; Romero, F.; Tortolero, M.; Japón, M.A.; Sáez, C. Prostate cancer cell response to paclitaxel is affected by abnormally expressed securin PTTG1. Mol. Cancer Ther. 2014, 13, 2372–2383. [Google Scholar] [CrossRef] [PubMed]
- Flores, M.L.; Castilla, C.; Gasca, J.; Medina, R.; Pérez-Valderrama, B.; Romero, F.; Japón, M.A.; Sáez, C. Loss of PKCδ induces prostate cancer resistance to paclitaxel through activation of Wnt/β-Catenin pathway and Mcl-1 accumulation. Mol. Cancer Ther. 2016, 15, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Gasca, J.; Flores, M.L.; Giráldez, S.; Ruiz-Borrego, M.; Tortolero, M.; Romero, F.; Japón, M.A.; Sáez, C. Loss of FBXW7 and accumulation of MCL1 and PLK1 promote paclitaxel resistance in breast cancer. Oncotarget 2016, 7, 52751–52765. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, M.; Marcellus, R.C.; Roulston, A.; Watson, M.; Serfass, L.; Murthy Madiraju, S.R.; Goulet, D.; Viallet, J.; Bélec, L.; Billot, X.; et al. Small molecule obatoclax (GX15-070) antagonizes MCL-1 and overcomes MCL-1-mediated resistance to apoptosis. Proc. Natl. Acad. Sci. USA 2007, 104, 19512–19517. [Google Scholar] [CrossRef] [Green Version]
- Trudel, S.; Li, Z.H.; Rauw, J.; Tiedemann, R.E.; Wen, X.Y.; Stewart, A.K. Preclinical studies of the pan-Bcl inhibitor obatoclax (GX015-070) in multiple myeloma. Blood 2007, 109, 5430–5438. [Google Scholar] [CrossRef] [Green Version]
- Schimmer, A.D.; O’Brien, S.; Kantarjian, H.; Brandwein, J.; Cheson, B.D.; Minden, M.D.; Yee, K.; Ravandi, F.; Giles, F.; Schuh, A.; et al. A phase I study of the pan bcl-2 family inhibitor obatoclaxmesylate in patients with advanced hematologic malignancies. Clin. Cancer Res. 2008, 14, 8295–8301. [Google Scholar] [CrossRef]
- Oki, Y.; Copeland, A.; Hagemeister, F.; Fayad, L.E.; Fanale, M.; Romaguera, J.; Younes, A. Experience with obatoclaxmesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist in patients with relapsed or refractory classical Hodgkin lymphoma. Blood 2012, 119, 2171–2172. [Google Scholar] [CrossRef] [PubMed]
- Konopleva, M.; Watt, J.; Contractor, R.; Tsao, T.; Harris, D.; Estrov, Z.; Bornmann, W.; Kantarjian, H.; Viallet, J.; Samudio, I.; et al. Mechanisms of antileukemic activity of the novel BH3 mimetic GX15-070 (obatoclax). Cancer Res. 2008, 68, 3413–3420. [Google Scholar] [CrossRef] [PubMed]
- Heidari, N.; Hicks, M.A.; Harada, H. GX15-070 (obatoclax) overcomes glucocorticoid resistance in acute lymphoblastic leukemia through induction of apoptosis and autophagy. Cell Death Dis. 2010, 16, e76. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.P.; Mitchell, C.; Rahmani, M.; Nephew, K.P.; Grant, S.; Dent, P. Inhibition of MCL-1 enhances lapatinib toxicity and overcomes lapatinib resistance via BAK-dependent autophagy. Cancer Biol. Ther. 2009, 8, 2084–2096. [Google Scholar] [CrossRef]
- Wei, Y.; Kadia, T.; Tong, W.; Zhang, M.; Jia, Y.; Yang, H.; Hu, Y.; Tambaro, F.P.; Viallet, J.; O’Brien, S.; et al. The combination of a histone deacetylase inhibitor with the Bcl-2 homology domain-3 mimetic GX15-070 has synergistic antileukemia activity by activating both apoptosis and autophagy. Clin. Cancer Res. 2010, 16, 3923–3932. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Cheng, C.; Verstovsek, S.; Chen, Q.; Jin, Y.; Cao, Q. The BH3-mimetic GX15-070 induces autophagy, potentiates the cytotoxicity of carboplatin and 5-fluorouracil in esophageal carcinoma cells. Cancer Lett. 2010, 293, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Liang, LZ.; Ma, B.; Liang, Y.J.; Liu, H.C.; Zhang, T.H.; Zheng, G.S.; Su, Y.X.; Liao, G.Q. Obatoclax induces Beclin 1- and ATG5-dependent apoptosis and autophagy in adenoid cystic carcinoma cells. Oral Dis. 2015, 21, 470–477. [Google Scholar] [CrossRef]
- Champa, D.; Orlacchio, A.; Patel, B.; Ranieri, M.; Shemetov, A.A.; Verkhusha, V.V.; Cuervo, A.M.; Di Cristofano, A. Obatoclax kills anaplastic thyroid cancer cells by inducing lysosome neutralization and necrosis. Oncotarget 2016, 7, 34453–34471. [Google Scholar] [CrossRef] [Green Version]
- Stamelos, V.A.; Fisher, N.; Bamrah, H.; Voisey, C.; Price, J.C.; Farrell, W.E.; Redman, C.W.; Richardson, A. The BH3 Mimetic Obatoclax Accumulates in Lysosomes and Causes Their Alkalinization. PLoS ONE 2016, 11, e0150696. [Google Scholar] [CrossRef]
- Yu, L.; Wu, W.K.; Gu, C.; Zhong, D.; Zhao, X.; Kong, Y.; Lin, Q.; Chan, M.T.; Zhou, Z.; Liu, S. Obatoclax impairs lysosomal function to block autophagy in cisplatin-sensitive and -resistant esophageal cancer cells. Oncotarget 2016, 7, 14693–14707. [Google Scholar] [CrossRef] [Green Version]
- Bonapace, L.; Bornhauser, B.C.; Schmitz, M.; Cario, G.; Ziegler, U.; Niggli, F.K.; Schäfer, B.W.; Schrappe, M.; Stanulla, M.; Bourquin, J.P. Induction of autophagy-dependent necroptosis is required for childhood acute lymphoblastic leukemia cells to overcome glucocorticoid resistance. J. Clin. Investig. 2010, 120, 1310–1323. [Google Scholar] [CrossRef] [Green Version]
- Basit, F.; Cristofanon, S.; Fulda, S. Obatoclax (GX15-070) triggers necroptosis by promoting the assembly of the necrosome on autophagosomal membranes. Cell Death Differ. 2013, 20, 1161–1173. [Google Scholar] [CrossRef] [Green Version]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef]
- Luo, S.; Rubinsztein, D.C. BCL2L11/BIM: A novel molecular link between autophagy and apoptosis. Autophagy 2013, 9, 104–105. [Google Scholar] [CrossRef] [Green Version]
- Norman, J.M.; Cohen, G.M.; Bampton, E.T. The in vitro cleavage of the hAtg proteins by cell death proteases. Autophagy 2010, 6, 1042–1056. [Google Scholar] [CrossRef] [Green Version]
- Wirawan, E.; VandeWalle, L.; Kersse, K.; Cornelis, S.; Claerhout, S.; Vanoverberghe, I.; Roelandt, R.; De Rycke, R.; Verspurten, J.; Declercq, W.; et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. 2010, 1, e18. [Google Scholar] [CrossRef]
- van Delft, M.F.; Wei, A.H.; Mason, K.D.; Vandenberg, C.J.; Chen, L.; Czabotar, P.E.; Willis, S.N.; Scott, C.L.; Day, C.L.; Cory, S.; et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell 2006, 10, 389–399. [Google Scholar] [CrossRef]
- Subramanian, A.; Andronache, A.; Li, Y.C.; Wade, M. Inhibition of MARCH5 ubiquitin ligase abrogates MCL1-dependent resistance to BH3 mimetics via NOXA. Oncotarget 2016, 7, 15986–16002. [Google Scholar] [CrossRef] [Green Version]
- Song, T.; Chai, G.; Liu, Y.; Xie, M.; Chen, Q.; Yu, X.; Sheng, H.; Zhang, Z. Mechanism of synergy of BH3 mimetics and paclitaxel in chronic myeloid leukemia cells: Mcl-1 inhibition. Eur. J. Pharm. Sci. 2015, 70, 64–71. [Google Scholar] [CrossRef]
- Lieber, J.; Ellerkamp, V.; Wenz, J.; Kirchner, B.; Warmann, S.W.; Fuchs, J.; Armeanu-Ebinger, S. Apoptosis sensitizers enhance cytotoxicity in hepatoblastoma cells. Pediatr. Surg. Int. 2012, 28, 149–159. [Google Scholar] [CrossRef]
- Tang, Y.; Hamed, H.A.; Cruickshanks, N.; Fisher, P.B.; Grant, S.; Dent, P. Obatoclax and Lapatinib Interact to Induce Toxic Autophagy through NOXA. Mol. Pharmacol. 2012, 81, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Luo, S.; Rubinsztein, D.C. Apoptosis blocks Beclin 1-dependent autophagosome synthesis: An effect rescued by Bcl-xL. Cell Death Differ. 2010, 17, 268–277. [Google Scholar] [CrossRef]
- Wirawan, E.; VandenBerghe, T.; Lippens, S.; Agostinis, P. Autophagy: For better or for worse. Cell Res. 2012, 22, 43–61. [Google Scholar] [CrossRef]
- De la Cruz-Morcillo, M.A.; Valero, M.L.; Callejas-Valera, J.L.; Arias-González, L.; Melgar-Rojas, P.; Galán-Moya, E.M.; García-Gil, E.; García-Cano, J.; Sánchez-Prieto, R. P38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluouracil: Implication in resistance. Oncogene 2012, 31, 1073–1085. [Google Scholar] [CrossRef]
- O’Donovan, T.R.; O’Sullivan, G.C.; McKennan, S.L. Induction of autophagy by drug-resistant esophageal cancer cells promotes their survival and recovery following treatment with chemotherapeutics. Autophagy 2011, 7, 509–524. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Yu, D.; Lum, J.J.; Bui, T.; Christophorou, M.A.; Evan, G.I.; Thomas-Tikhonenko, A.; Thompson, C.B. Autophagy inhibition enhances therapy-induced apoptosis in a Myc-induced model of lymphoma. J. Clin. Investig. 2007, 117, 326–336. [Google Scholar] [CrossRef] [Green Version]
- Jung, H.J.; Seo, I.; Casciello, F.; Jacquellin, S.; Lane, S.W.; Suh, S.-I.; Suh, M.H.; Lee, J.S.; Baek, W.K. The anticancer effect of chaetocin is enhanced by inhibition of autophagy. Cell Death Dis. 2016, 7, e2098. [Google Scholar] [CrossRef]
- Boya, P.; Gonzalez-Polo, R.A.; Casares, N.; Perfettini, J.L.; Dessen, P.; Larochette, N.; Métivier, D.; Meley, D.; Souquere, S.; Yoshimori, T.; et al. Inhibition of macroautophagy triggers apoptosis. Mol. Cell. Biol. 2005, 25, 1025–1040. [Google Scholar] [CrossRef]
- Sharma, A.; Singh, K.; Mazumder, S.; Hill, B.T.; Kalaycio, M.; Almasan, A. BECN1 and BIM interactions with MCL-1 determine fludarabine resistance in leukemic B cells. Cell Death Dis. 2013, 4, e628. [Google Scholar] [CrossRef]
- Li, H.; Wang, P.; Yu, J.; Zhang, L. Cleaving Beclin 1 to suppress autophagy in chemotherapy-induced apoptosis. Autophagy 2011, 7, 1239–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koehler, B.C.; Scherr, A.-L.; Lorenz, S.; Elssner, C.; Kautz, N.; Welte, S.; Jaeger, D.; Urbanik, T.; Schulze-Bergkamen, H. Pan-Bcl-2 Inhibitor Obatoclax Delays Cell Cycle Progression and Blocks Migration of Colorectal Cancer Cells. PLoS ONE 2014, 9, e106571. [Google Scholar] [CrossRef] [PubMed]
- Zhong, D.; Gu, C.; Shi, L.; Xun, T.; Li, X.; Liu, S.; Yu, L. Obatoclax Induces G1/G0-Phase Arrest via p38/p21waf1/Cip1 Signaling Pathway in Human Esophageal Cancer Cells. J. Cell Biochem. 2014, 115, 1624–1635. [Google Scholar] [CrossRef] [PubMed]
- Inuzuka, H.; Shaik, S.; Onoyama, I.; Gao, D.; Tseng, A.; Maser, R.S.; Zhai, B.; Wan, L.; Gutierrez, A.; Lau, A.W.; et al. SCF (Fbw7) regulates cellular apoptosis by targeting MCL1 for ubiquitination and destruction. Nature 2011, 471, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiménez-Guerrero, R.; Gasca, J.; Flores, M.L.; Pérez-Valderrama, B.; Tejera-Parrado, C.; Medina, R.; Tortolero, M.; Romero, F.; Japón, M.A.; Sáez, C. Obatoclax and Paclitaxel Synergistically Induce Apoptosis and Overcome Paclitaxel Resistance in Urothelial Cancer Cells. Cancers 2018, 10, 490. https://doi.org/10.3390/cancers10120490
Jiménez-Guerrero R, Gasca J, Flores ML, Pérez-Valderrama B, Tejera-Parrado C, Medina R, Tortolero M, Romero F, Japón MA, Sáez C. Obatoclax and Paclitaxel Synergistically Induce Apoptosis and Overcome Paclitaxel Resistance in Urothelial Cancer Cells. Cancers. 2018; 10(12):490. https://doi.org/10.3390/cancers10120490
Chicago/Turabian StyleJiménez-Guerrero, Rocío, Jessica Gasca, M. Luz Flores, Begoña Pérez-Valderrama, Cristina Tejera-Parrado, Rafael Medina, María Tortolero, Francisco Romero, Miguel A. Japón, and Carmen Sáez. 2018. "Obatoclax and Paclitaxel Synergistically Induce Apoptosis and Overcome Paclitaxel Resistance in Urothelial Cancer Cells" Cancers 10, no. 12: 490. https://doi.org/10.3390/cancers10120490