Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial

 , ,
, ,
Abstract
:1. Introduction
2. Results
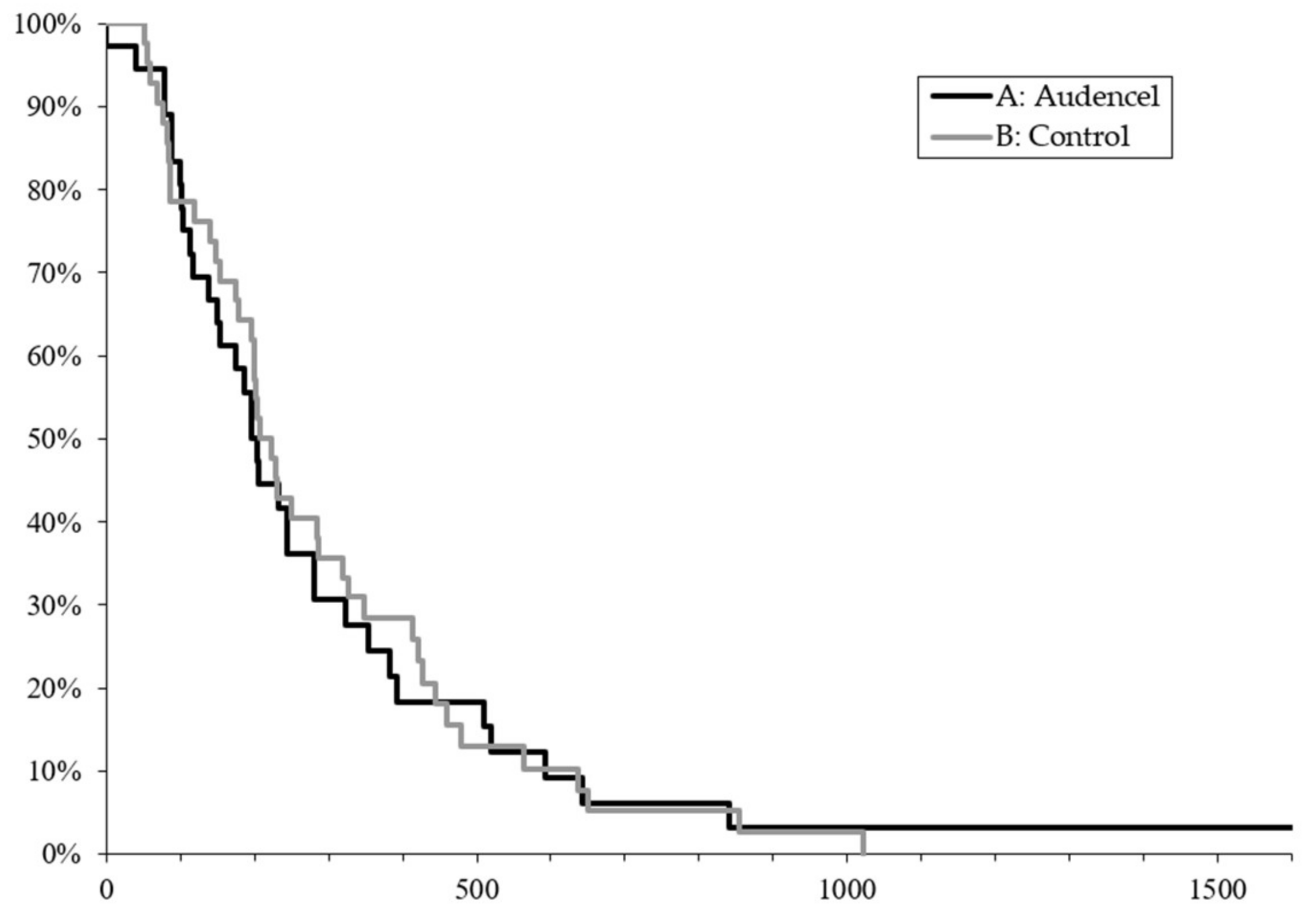
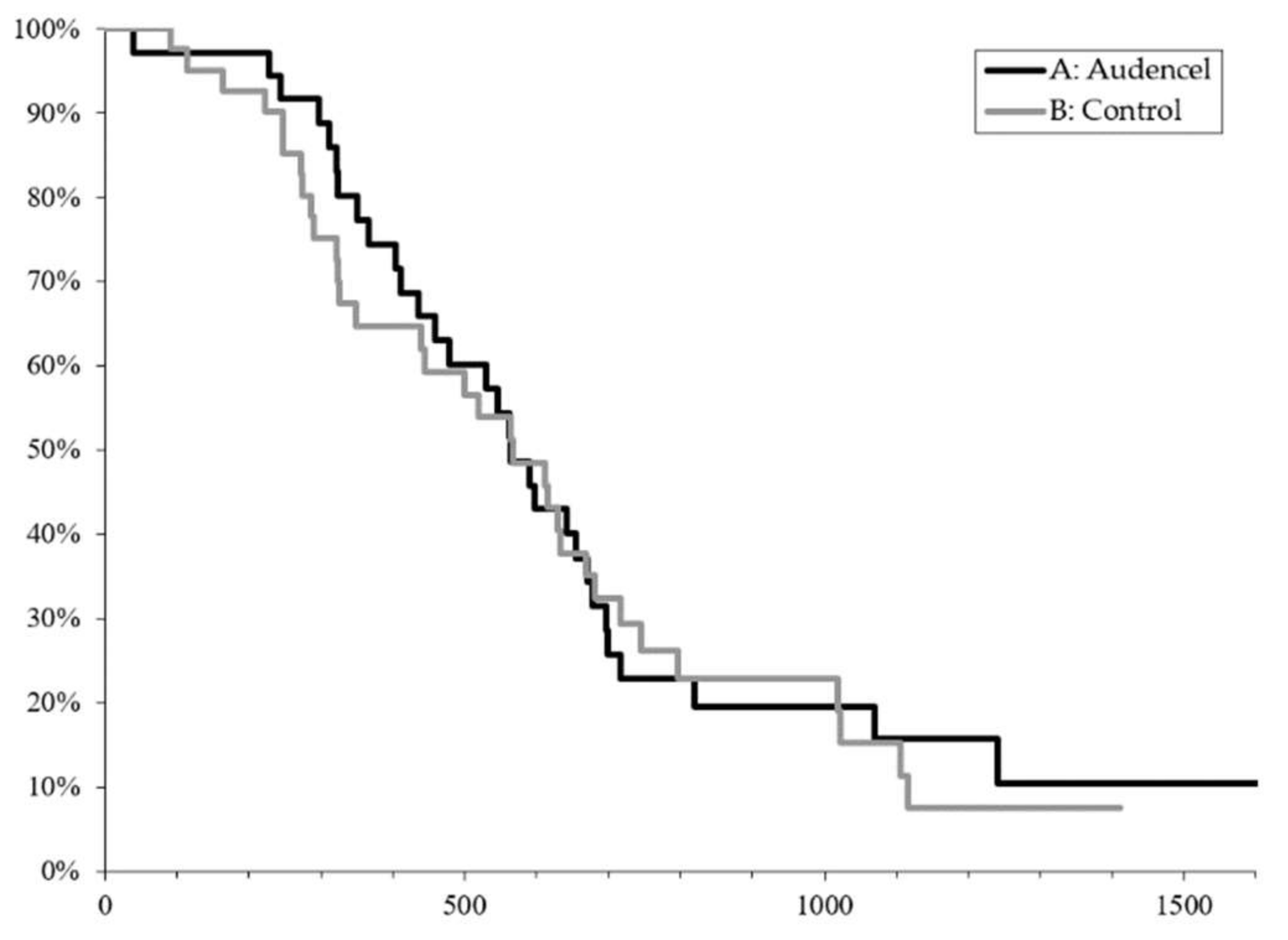
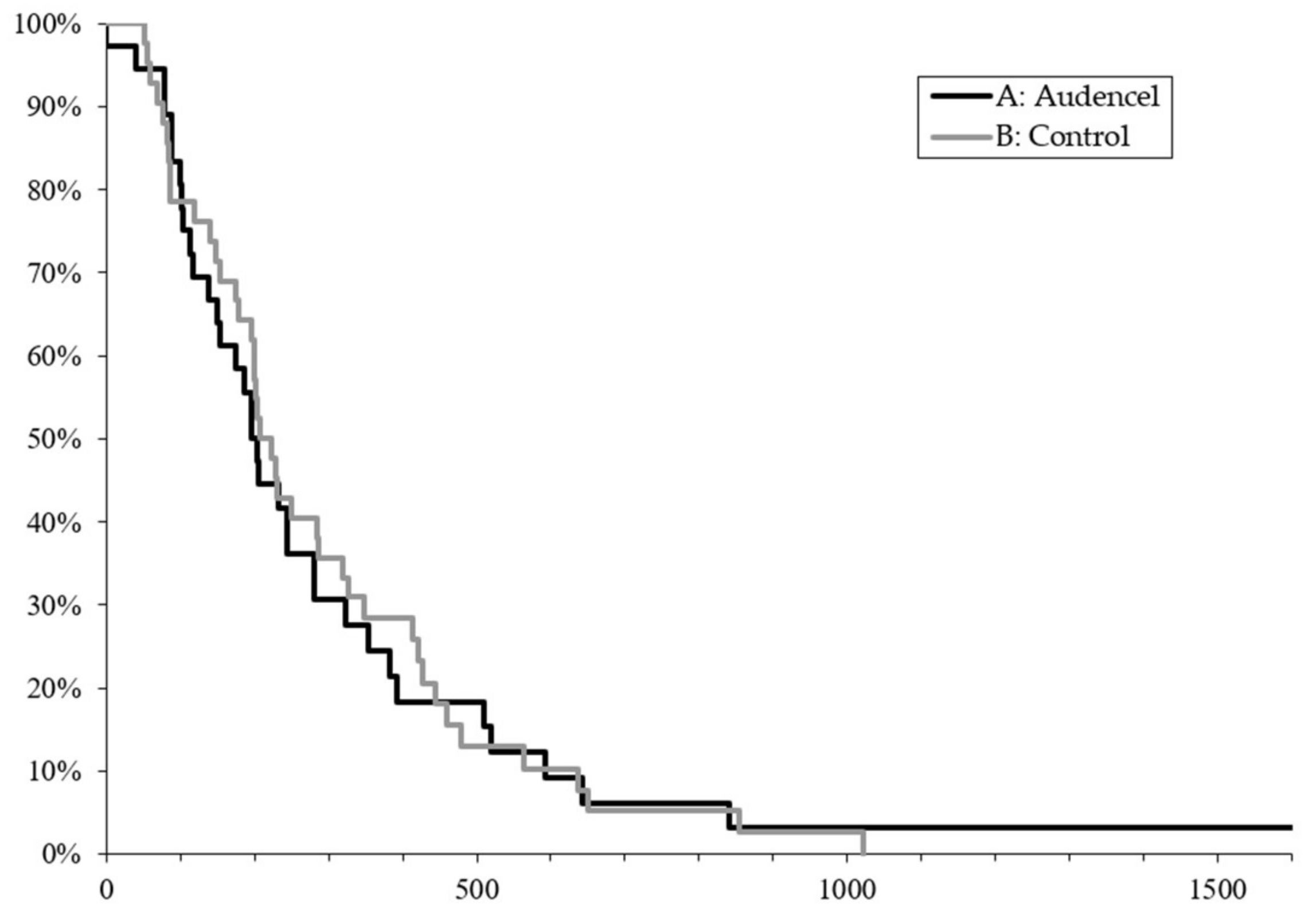
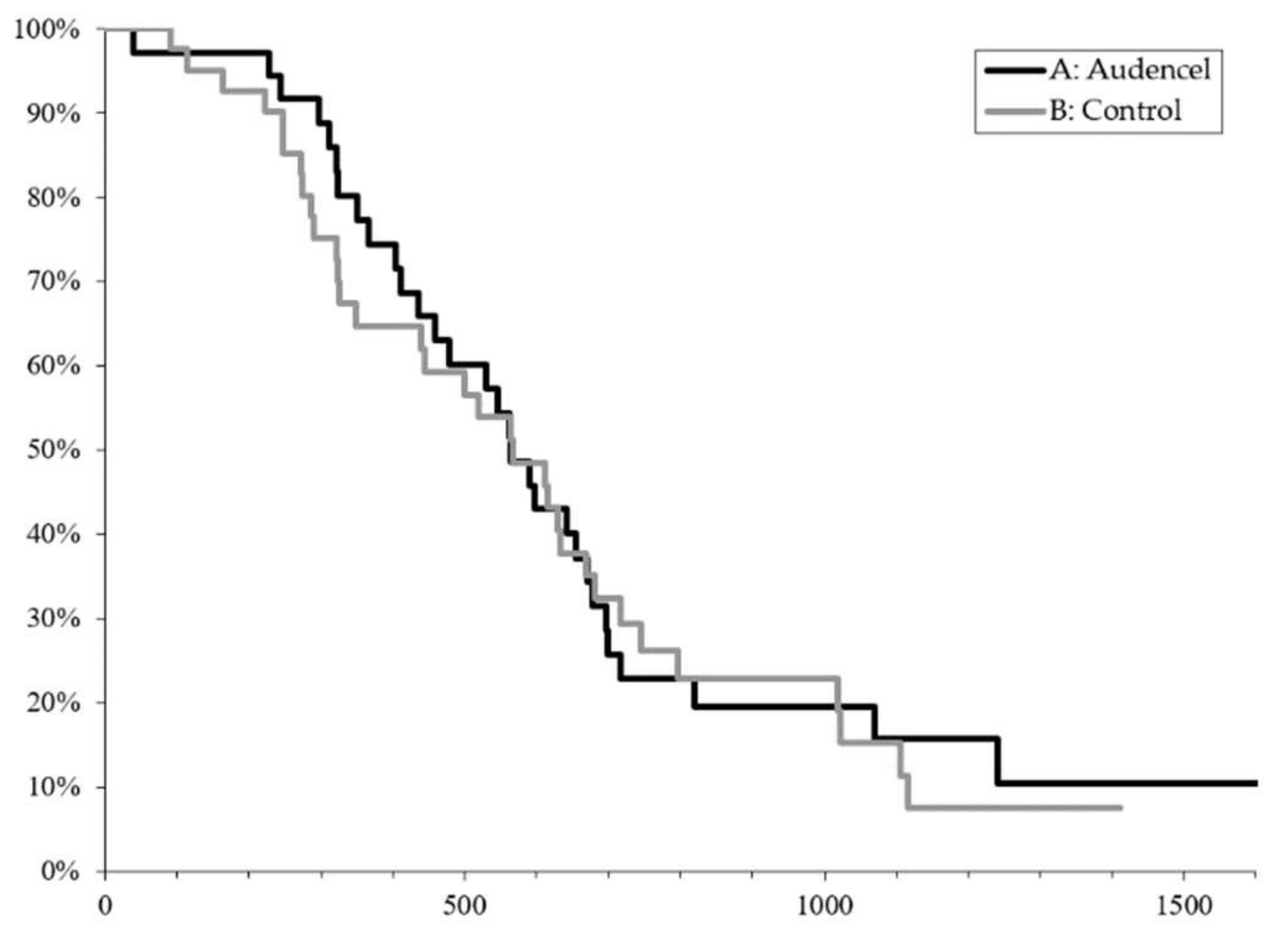
2.1. Survival Outcome
2.2. Adverse Events
2.3. Treatment after Relapse
2.4. Extent of Resection
2.5. DC Vaccine Quality
2.6. DC Vaccine Quantity
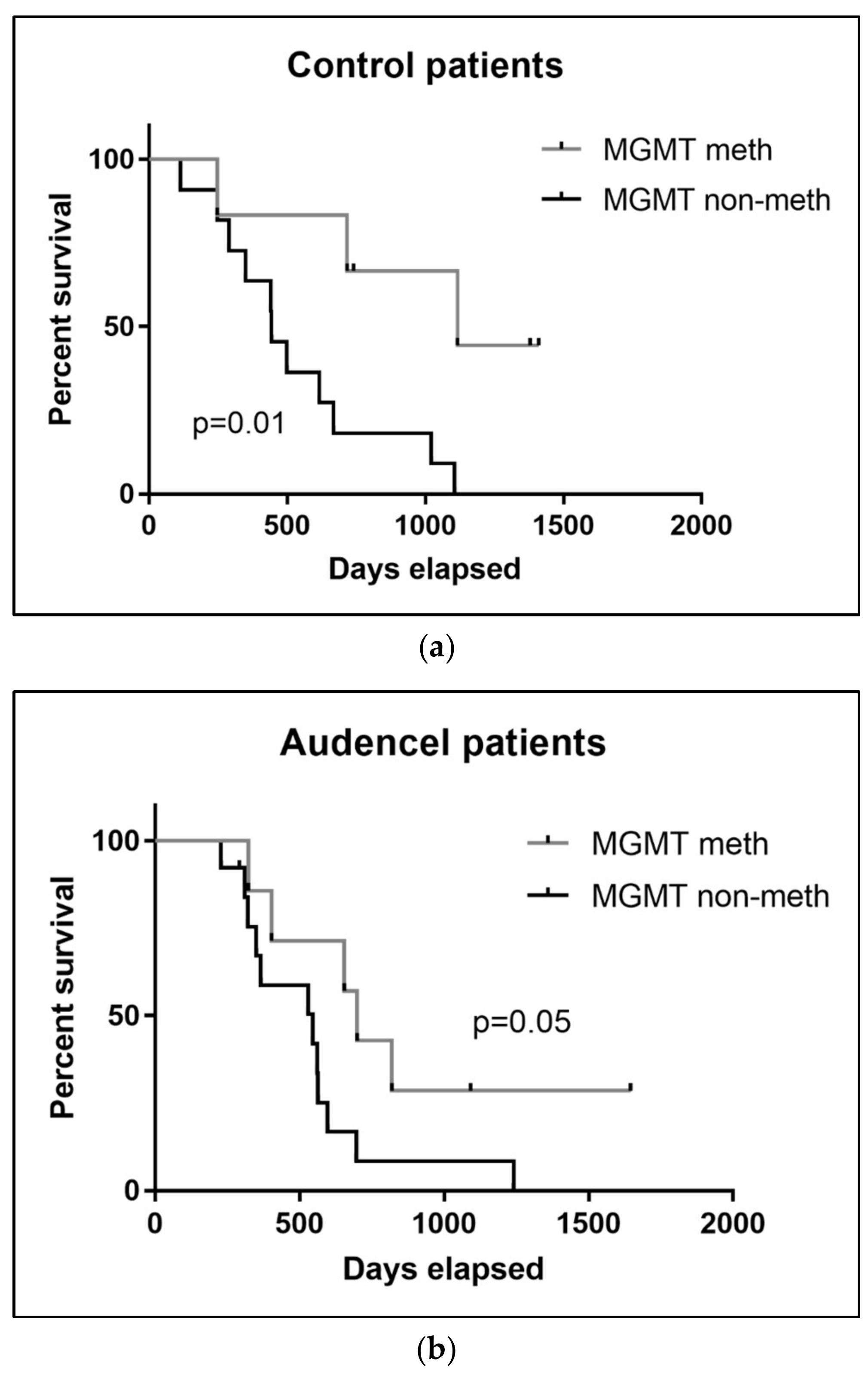
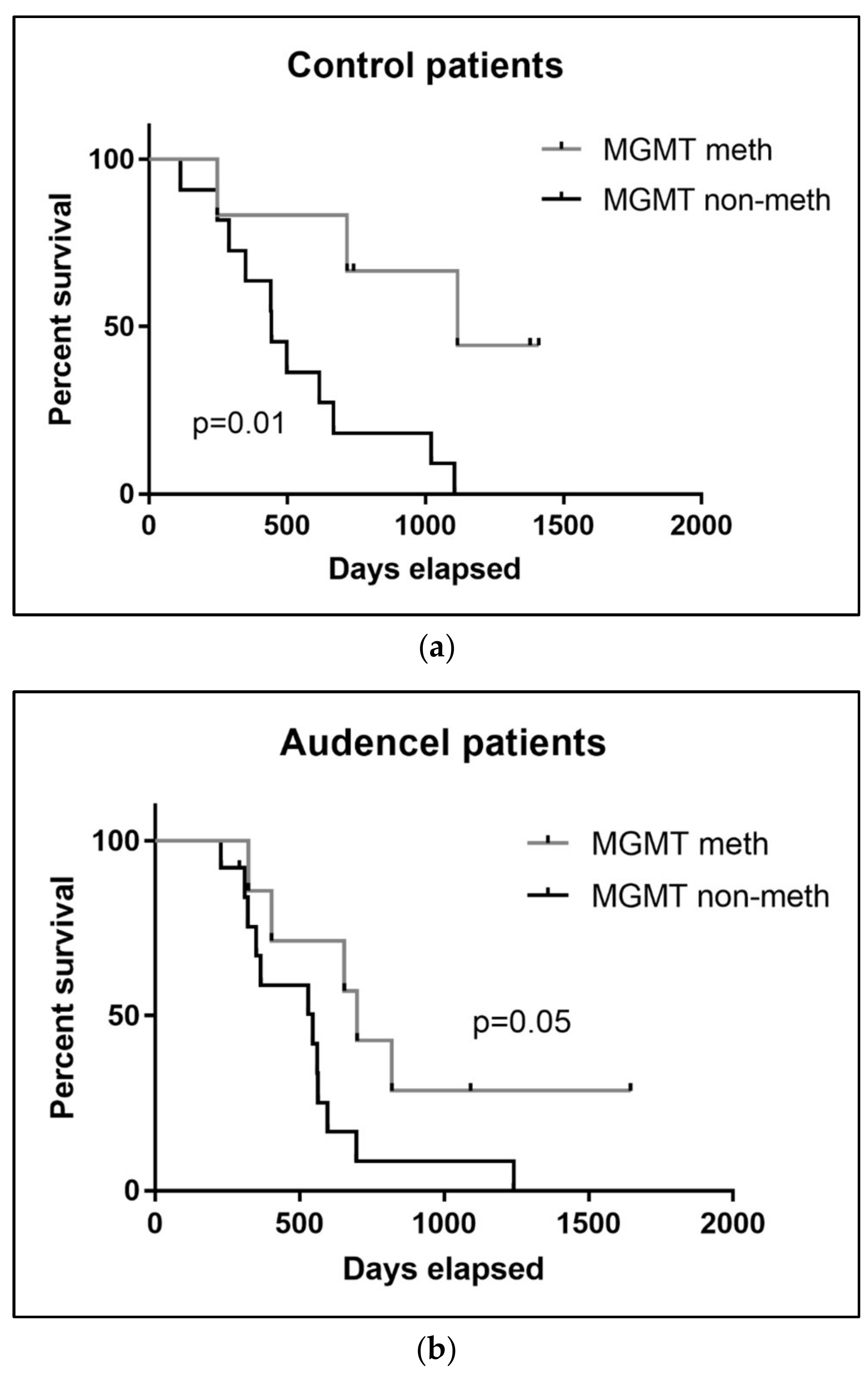
2.7. MGMT Promoter Methylation
3. Discussion
4. Materials and Methods
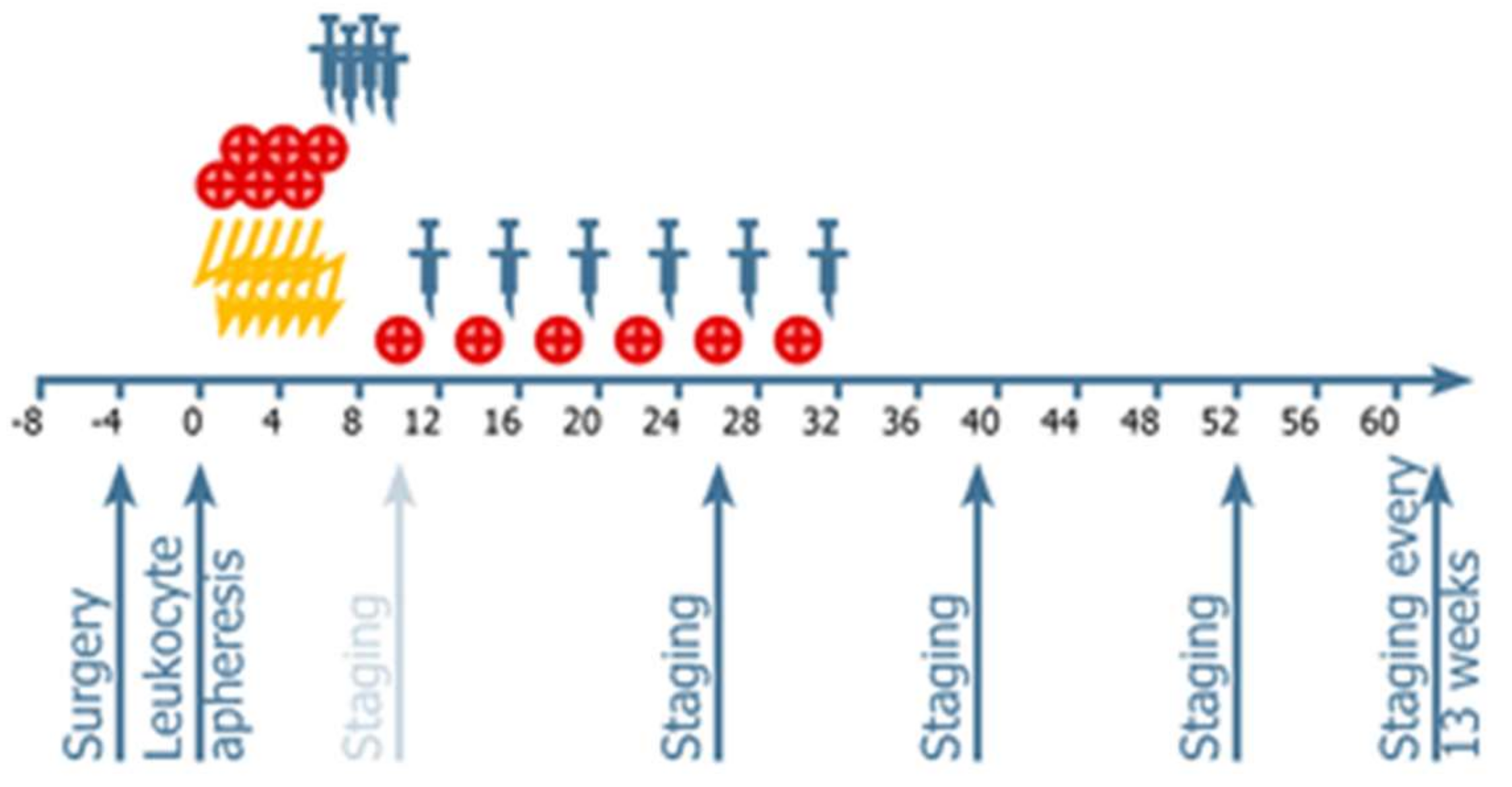
4.1. Randomization
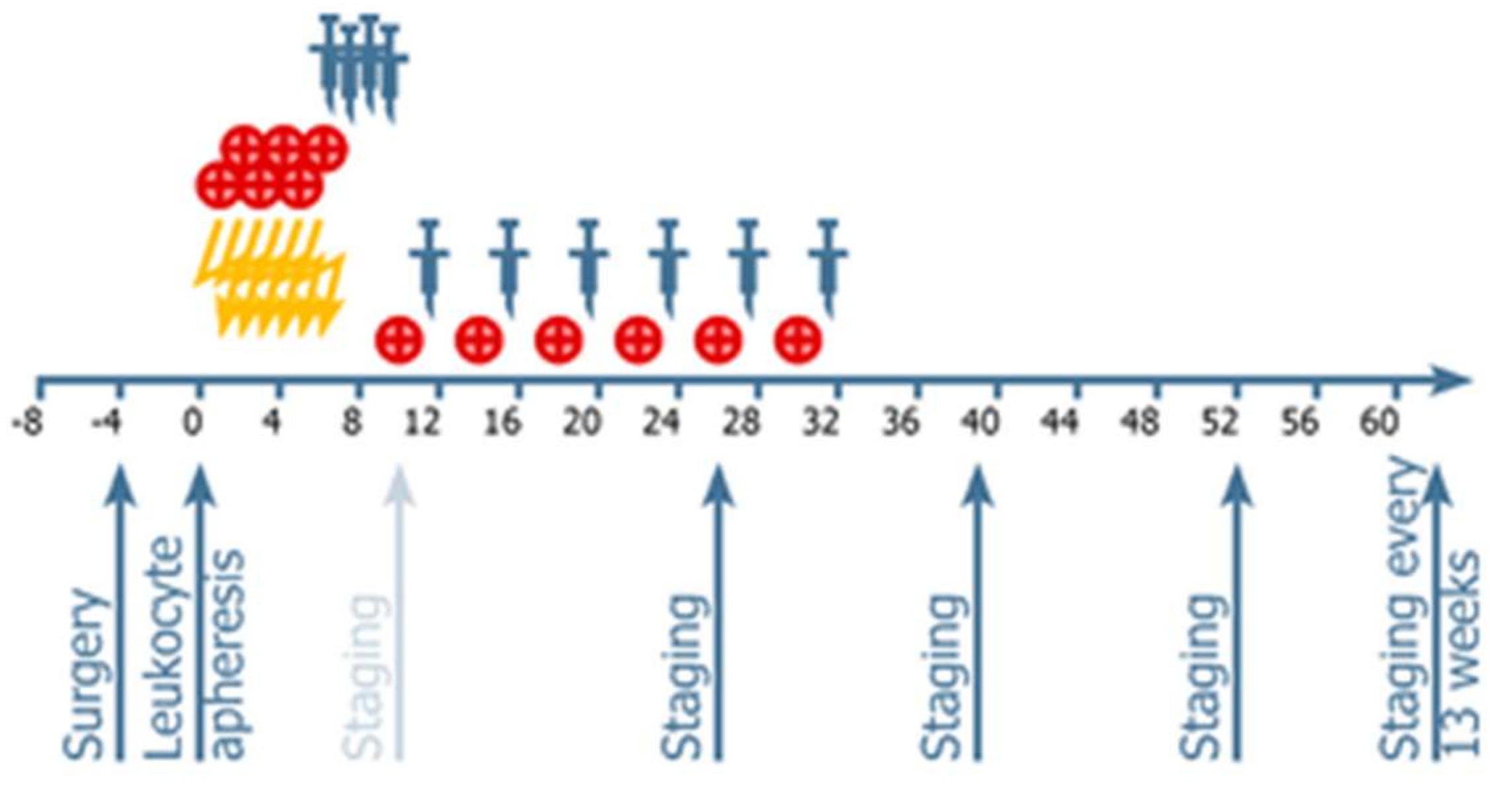
4.2. Procedures: Treatments Administered
4.3. Vaccine Administration
4.4. Neuroimaging and Response Assessment
4.5. Treatment after Relapse
4.6. DNA Extraction and MGMT Promoter Methylation Analysis
4.7. IDH1 Mutation Analysis
4.8. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Nabors, L.B.; Fink, K.L.; Mikkelsen, T.; Grujicic, D.; Tarnawski, R.; Nam, D.H.; Mazurkiewicz, M.; Salacz, M.; Ashby, L.; Zagonel, V.; et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: Results of the open-label, controlled, randomized phase II CORE study. Neuro Oncol. 2015, 17, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.-K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. CENTRIC study team Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Chinot, O.L.; Wick, W.; Cloughesy, T. Bevacizumab for newly diagnosed glioblastoma. N. Engl. J. Med. 2014, 370, e2049. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, M.R.; Dignam, J.J.; Armstrong, T.S.; Wefel, J.S.; Blumenthal, D.T.; Vogelbaum, M.A.; Colman, H.; Chakravarti, A.; Pugh, S.; Won, M.; et al. A Randomized Trial of Bevacizumab for Newly Diagnosed Glioblastoma. N. Engl. J. Med. 2014, 370, 699–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert, M.R.; Wang, M.; Aldape, K.D.; Stupp, R.; Hegi, M.E.; Jaeckle, K.A.; Armstrong, T.S.; Wefel, J.S.; Won, M.; Blumenthal, D.T.; et al. Dose-Dense Temozolomide for Newly Diagnosed Glioblastoma: A Randomized Phase III Clinical Trial. J. Clin. Oncol. 2013, 31, 4085–4091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stupp, R.; Taillibert, S.; Kanner, A.A.; Kesari, S.; Steinberg, D.M.; Toms, S.A.; Taylor, L.P.; Lieberman, F.; Silvani, A.; Fink, K.L.; et al. Maintenance Therapy With Tumor-Treating Fields Plus Temozolomide vs Temozolomide Alone for Glioblastoma: A Randomized Clinical Trial. JAMA 2015, 314, 2535–2543. [Google Scholar] [CrossRef] [PubMed]
- Pieczonka, C.M.; Telonis, D.; Mouraviev, V.; Albala, D. Sipuleucel-T for the treatment of patients with metastatic castrate-resistant prostate cancer: considerations for clinical practice. Rev. Urol. 2015, 17, 203–210. [Google Scholar] [PubMed]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, T.; Hori, A.; Kami, M. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 1967–1968. [Google Scholar]
- Nabhan, C. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 1966–1967. [Google Scholar] [PubMed]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol. 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Inogés, S.; Tejada, S.; de Cerio, A.L.-D.; Gállego Pérez-Larraya, J.; Espinós, J.; Idoate, M.A.; Domínguez, P.D.; de Eulate, R.G.; Aristu, J.; Bendandi, M.; et al. A phase II trial of autologous dendritic cell vaccination and radiochemotherapy following fluorescence-guided surgery in newly diagnosed glioblastoma patients. J. Transl. Med. 2017, 15, e104. [Google Scholar] [CrossRef] [PubMed]
- Prins, R.M.; Soto, H.; Konkankit, V.; Odesa, S.K.; Eskin, A.; Yong, W.H.; Nelson, S.F.; Liau, L.M. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin. Cancer Res. 2011, 17, 1603–1615. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, e142. [Google Scholar] [CrossRef] [PubMed]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louveau, A. Cerebral lymphatic drainage: implication in the brain immune privilege. Med. Sci. (Paris) 2015, 31, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Louveau, A.; Harris, T.H.; Kipnis, J. Revisiting the Mechanisms of CNS Immune Privilege. Trends Immunol. 2015, 36, 569–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razavi, S.-M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Van Meir, E.G. Cytokines and tumors of the central nervous system. GLIA 1995, 15, 264–288. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.; Wei, J.; Kong, L.-Y.; Wang, Y.; Priebe, W.; Qiao, W.; Sawaya, R.; Heimberger, A.B. Glioma cancer stem cells induce immunosuppressive macrophages/microglia. Neuro. Oncol. 2010, 12, 1113–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doucette, T.A.; Kong, L.-Y.; Yang, Y.; Ferguson, S.D.; Yang, J.; Wei, J.; Qiao, W.; Fuller, G.N.; Bhat, K.P.; Aldape, K.; et al. Signal transducer and activator of transcription 3 promotes angiogenesis and drives malignant progression in glioma. Neuro. Oncol. 2012, 14, 1136–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinman, R.M.; Cohn, Z.A. Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Exp. Med. 1973, 137, 1142–1162. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Cohn, Z.A. Pillars Article: Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J. Immunol. 2007, 178, 5–25. [Google Scholar] [PubMed]
- Langenkamp, A.; Messi, M.; Lanzavecchia, A.; Sallusto, F. Kinetics of dendritic cell activation: impact on priming of TH1, TH2 and nonpolarized T cells. Nat. Immunol. 2000, 1, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Luger, R.; Valookaran, S.; Knapp, N.; Vizzardelli, C.; Dohnal, A.M.; Felzmann, T. Toll-like receptor 4 engagement drives differentiation of human and murine dendritic cells from a pro- into an anti-inflammatory mode. PLoS ONE 2013, 8, e54879. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C.A. Decoding the Patterns of Self and Nonself by the Innate Immune System. Science 2002, 296, 298–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hüttner, K.G.; Breuer, S.K.; Paul, P.; Majdic, O.; Heitger, A.; Felzmann, T. Generation of potent anti-tumor immunity in mice by interleukin-12-secreting dendritic cells. Cancer Immunol. Immunother. 2005, 54, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Felzmann, T.; Hüttner, K.G.; Breuer, S.K.; Wimmer, D.; Ressmann, G.; Wagner, D.; Paul, P.; Lehner, M.; Heitger, A.; Holter, W. Semi-mature IL-12 secreting dendritic cells present exogenous antigen to trigger cytolytic immune responses. Cancer Immunol. Immunother. 2005, 54, 769–780. [Google Scholar] [CrossRef] [PubMed]
- Dohnal, A.M.; Witt, V.; Hügel, H.; Holter, W.; Gadner, H.; Felzmann, T. Phase I study of tumor Ag-loaded IL-12 secreting semi-mature DC for the treatment of pediatric cancer. Cytotherapy 2007, 9, 755–770. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.; Bercovici, N.; Abastado, J.-P.; Nardin, A. Naive CD8+ T cell recruitment and proliferation are dependent on stage of dendritic cell maturation. Eur. J. Immunol. 2003, 33, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Camporeale, A.; Boni, A.; Iezzi, G.; Degl’Innocenti, E.; Grioni, M.; Mondino, A.; Bellone, M. Critical impact of the kinetics of dendritic cells activation on the in vivo induction of tumor-specific T lymphocytes. Cancer Res. 2003, 63, 3688–3694. [Google Scholar] [PubMed]
- Sabado, R.L.; Bhardwaj, N. Cancer immunotherapy: dendritic-cell vaccines on the move. Nature 2015, 519, 300–311. [Google Scholar] [CrossRef] [PubMed]
- Schuler, G.; Schuler-Thurner, B.; Steinman, R.M. The use of dendritic cells in cancer immunotherapy. Curr. Opin. Immunol. 2003, 15, 138–147. [Google Scholar] [CrossRef]
- Stummer, W.; Reulen, H.-J.; Meinel, T.; Pichlmeier, U.; Schumacher, W.; Tonn, J.-C.; Rohde, V.; Oppel, F.; Turowski, B.; Woiciechowsky, C.; et al. ALA-Glioma Study Group extent of resection and survival in glioblastoma multiforme. Neurosurgery 2008, 62, 564–576. [Google Scholar] [PubMed]
- Subklewe, M.; Geiger, C.; Lichtenegger, F.S.; Javorovic, M.; Kvalheim, G.; Schendel, D.J.; Bigalke, I. New generation dendritic cell vaccine for immunotherapy of acute myeloid leukemia. Cancer Immunol. Immunother. 2014, 63, 1093–1103. [Google Scholar] [CrossRef] [PubMed]
- Reinis, M.; Indrová, M.; Mendoza, L.; Mikysková, R.; Bieblová, J.; Bubeník, J.; Símová, J. HPV16-associated tumours: therapy of surgical minimal residual disease with dendritic cell-based vaccines. Int. J. Oncol. 2004, 25, 1165–1170. [Google Scholar] [PubMed]
- Fadul, C.E.; Fisher, J.L.; Hampton, T.H.; Lallana, E.C.; Li, Z.; Gui, J.; Szczepiorkowski, Z.M.; Tosteson, T.D.; Rhodes, C.H.; Wishart, H.A.; et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J. Immunother. 2011, 34, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Vik-Mo, E.O.; Nyakas, M.; Mikkelsen, B.V.; Moe, M.C.; Due-Tønnesen, P.; Suso, E.M.I.; Sæbøe-Larssen, S.; Sandberg, C.; Brinchmann, J.E.; Helseth, E.; et al. Therapeutic vaccination against autologous cancer stem cells with mRNA-transfected dendritic cells in patients with glioblastoma. Cancer Immunol. Immunother. 2013, 62, 1499–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Erhart, F.; Buchroithner, J.; Reitermaier, R.; Fischhuber, K.; Klingenbrunner, S.; Sloma, I.; Hibsh, D.; Kozol, R.; Efroni, S.; Ricken, G.; et al. Immunological analysis of phase II glioblastoma dendritic cell vaccine (Audencel) trial: immune system characteristics influence outcome and Audencel up-regulates Th1-related immunovariables. Acta Neuropath. Comm. Submitted for publication. 2018. [Google Scholar]
- Bregy, A.; Wong, T.M.; Shah, A.H.; Goldberg, J.M.; Komotar, R.J. Active immunotherapy using dendritic cells in the treatment of glioblastoma multiforme. Cancer Treat. Rev. 2013, 39, 891–907. [Google Scholar] [CrossRef] [PubMed]
- Ardon, H.; Van Gool, S.W.; Verschuere, T.; Maes, W.; Fieuws, S.; Sciot, R.; Wilms, G.; Demaerel, P.; Goffin, J.; Van Calenbergh, F.; et al. Integration of autologous dendritic cell-based immunotherapy in the standard of care treatment for patients with newly diagnosed glioblastoma: results of the HGG-2006 phase I/II trial. Cancer Immunol. Immunother. 2012, 61, 2033–2044. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-N.; Huang, Y.-C.; Yang, D.-M.; Kikuta, K.; Wei, K.-J.; Kubota, T.; Yang, W.-K. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J. Clin. Neurosci. 2011, 18, 1048–1054. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.-Y.; Yang, W.-K.; Lee, H.-C.; Hsu, D.-M.; Lin, H.-L.; Lin, S.-Z.; Chen, C.-C.; Harn, H.-J.; Liu, C.-L.; Lee, W.-Y.; et al. Adjuvant Immunotherapy with Whole-Cell Lysate Dendritic Cells Vaccine for Glioblastoma Multiforme: A Phase II Clinical Trial. World Neurosurg. 2012, 77, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.-W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F.; et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin. Cancer Res. 2005, 11, 5515–5525. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Homma, J.; Yajima, N.; Tsuchiya, N.; Sano, M.; Kobayashi, T.; Yoshida, S.; Abe, T.; Narita, M.; Takahashi, M.; et al. Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: Results of a clinical phase I/II trial. Clin. Cancer Res. 2005, 11, 4160–4167. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Black, K.L.; Liu, G.; Wheeler, C.J.; Wagenberg, M.; Das, A.; Mindlin, E.; Chu, R.M.; Luptrawan, A.; Badruddoja, M.A. 843 Results of a Phase II Trial of Tumor Lysate-pulsed Dendritic Cell Vaccination for Malignant Glioma. Neurosurgery 2005, 57, 409–410. [Google Scholar] [CrossRef]
- Phuphanich, S.; Wheeler, C.J.; Rudnick, J.D.; Mazer, M.; Wang, H.; Nuño, M.A.; Richardson, J.E.; Fan, X.; Ji, J.; Chu, R.M.; et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 2013, 62, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, C.J.; Black, K.L.; Liu, G.; Mazer, M.; Zhang, X.-X.; Pepkowitz, S.; Goldfinger, D.; Ng, H.; Irvin, D.; Yu, J.S. Vaccination Elicits Correlated Immune and Clinical Responses in Glioblastoma Multiforme Patients. Cancer Res. 2008, 68, 5955–5964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pallini, R.; Ricci-Vitiani, L.; Montano, N.; Mollinari, C.; Biffoni, M.; Cenci, T.; Pierconti, F.; Martini, M.; De Maria, R.; Larocca, L.M. Expression of the stem cell marker CD133 in recurrent glioblastoma and its value for prognosis. Cancer 2011, 117, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Zeppernick, F.; Ahmadi, R.; Campos, B.; Dictus, C.; Helmke, B.M.; Becker, N.; Lichter, P.; Unterberg, A.; Radlwimmer, B.; Herold-Mende, C.C. Stem Cell Marker CD133 Affects Clinical Outcome in Glioma Patients. Clin. Cancer Res. 2008, 14, 123–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esparza, R.; Azad, T.D.; Feroze, A.H.; Mitra, S.S.; Cheshier, S.H. Glioblastoma stem cells and stem cell-targeting immunotherapies. J. Neurooncol. 2015, 123, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Bonifant, C.L.; Jackson, H.J.; Brentjens, R.J.; Curran, K.J. Toxicity and management in CAR T-cell therapy. Mol. Ther. Oncolytics 2016, 3, e16011. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.S.; Liu, G.; Ying, H.; Yong, W.H.; Black, K.L.; Wheeler, C.J. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004, 64, 4973–4979. [Google Scholar] [CrossRef] [PubMed]
- Balss, J.; Meyer, J.; Mueller, W.; Korshunov, A.; Hartmann, C.; von Deimling, A. Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 2008, 116, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Von Deimling, A.; Korshunov, A.; Hartmann, C. The Next Generation of Glioma Biomarkers: MGMT Methylation, BRAF Fusions and IDH1 Mutations. Brain Pathol. 2011, 21, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Felzmann, T.; Witt, V.; Wimmer, D.; Ressmann, G.; Wagner, D.; Paul, P.; Hüttner, K.; Fritsch, G. Monocyte enrichment from leukapharesis products for the generation of DCs by plastic adherence, or by positive or negative selection. Cytotherapy 2003, 5, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, D.R.; Cascino, T.L.; Schold, S.C.; Cairncross, J.G. Response criteria for phase II studies of supratentorial malignant glioma. J. Clin. Oncol. 1990, 8, 1277–1280. [Google Scholar] [CrossRef] [PubMed]
- Preusser, M.; Capper, D.; Hartmann, C.; Euro-CNS Research, Committee. IDH testing in diagnostic neuropathology: Review and practical guideline article invited by the Euro-CNS research committee. Clin. Neuropathol. 2011, 30, 217–230. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Audencel Group | Control Group |
|---|---|---|
| N | 34 | 42 |
| Sex | ||
| female | 12 | 13 |
| male | 22 | 29 |
| Age (years) | 54.6 | 54 |
| ECOG | ||
| 0 | 14 (41%) | 14 (33%) |
| 1 | 20 (59%) | 21 (50%) |
| 2 | 0 | 7 (17%) |
| >2 | 0 | 0 |
| Surgery | ||
| Gross total resection | 24 (71%) | 35 (83%) |
| Partial resection | 10 (29%) | 7 (17%) |
| Number of target lesions at post OP screening | ||
| 0 | 24 (71%) | 35 (83%) |
| 1 | 10 (29%) | 7 (17%) |
| Number of non-target lesions at post-OP screening | ||
| 0 | 34 | 42 |
| >1 | 0 | 0 |
| Anti-epileptic drugs | ||
| Non enzyme-inducing AEDs | 11 | 18 |
| Enzyme inducing AEDs | 0 | 0 |
| MGMT promoter methylation | N = 20 samples measured | N = 17 samples measured |
| methylated | 7/20 35% | 6/17 35% |
| unmethylated | 13/20 65% | 11/17 65% |
| Toxicity | Control n = 42 | Audencel n = 34 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| WHO | II | % | III-IV | % | All Grades | II | % | III-IV | % | All Grades |
| Haematologic toxicity | ||||||||||
| Anemia | 0 | 0 | 0 | 0 | 0 | 3 | 8.2 | 0 | 0 | 3 |
| Leucopenia | 3 | 7.1 | 2 | 5 | 5 | 2 | 6 | 2 | 6 | 4 |
| Lymphopenia | 2 | 4.8 | 0 | 0 | 2 | 2 | 6 | 1 | 3 | 3 |
| Thrombopenia | 9 | 21 | 2 | 5 | 11 | 6 | 17 | 7 | 20.50 | 13 |
| Non-haematologic toxicity | ||||||||||
| Fatigue | 9 | 21 | 1 | 2 | 10 | 15 | 45 | 3 | 8 | 18 |
| Headache | 11 | 26 | 2 | 5 | 13 | 13 | 38 | 2 | 6 | 15 |
| Nausea | 7 | 17 | 0 | 0 | 7 | 13 | 38 | 1 | 3 | 14 |
| Vomiting | 4 | 9.5 | 0 | 0 | 4 | 4 | 12 | 0 | 0 | 4 |
| VTE event | 0 | 0 | 3 | 7 | 3 | 0 | 0 | 0 | 0 | 0 |
| Intracranial bleeding | 0 | 0 | 1 | 2 | 1 | 0 | 0 | 0 | 0 | 0 |
| Rash | 0 | 0 | 1 | 2 | 1 | 0 | 0 | 2 | 5 | 2 |
| Influenza like illness | 1 | 2.4 | 0 | 0 | 1 | 3 | 8.2 | 0 | 0 | 3 |
| Fever | 2 | 4.8 | 0 | 0 | 2 | 7 | 21 | 0 | 0 | 7 |
| Vaccination site reaction | not relevant | |||||||||
| erythema | 6 | 18 | 0 | 0 | 6 | |||||
| pruritus | 6 | 18 | 0 | 0 | 6 | |||||
| Pain, induration | 6 | 18 | 0 | 0 | 6 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buchroithner, J.; Erhart, F.; Pichler, J.; Widhalm, G.; Preusser, M.; Stockhammer, G.; Nowosielski, M.; Iglseder, S.; Freyschlag, C.F.; Oberndorfer, S.; et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Cancers 2018, 10, 372. https://doi.org/10.3390/cancers10100372
Buchroithner J, Erhart F, Pichler J, Widhalm G, Preusser M, Stockhammer G, Nowosielski M, Iglseder S, Freyschlag CF, Oberndorfer S, et al. Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial. Cancers. 2018; 10(10):372. https://doi.org/10.3390/cancers10100372
Chicago/Turabian StyleBuchroithner, Johanna, Friedrich Erhart, Josef Pichler, Georg Widhalm, Matthias Preusser, Günther Stockhammer, Martha Nowosielski, Sarah Iglseder, Christian F. Freyschlag, Stefan Oberndorfer, and et al. 2018. "Audencel Immunotherapy Based on Dendritic Cells Has No Effect on Overall and Progression-Free Survival in Newly Diagnosed Glioblastoma: A Phase II Randomized Trial" Cancers 10, no. 10: 372. https://doi.org/10.3390/cancers10100372