The “Yin and Yang” of Natural Compounds in Anticancer Therapy of Triple-Negative Breast Cancers

 ,
, 
Abstract
:1. Background and Current Understanding
2. Types and Subtypes of Breast Cancer
3. Aberrant Pathways in Breast Cancer/TNBCs
3.1. Wnt/β-Catenin Signaling and TNBCs
3.2. Notch Signaling and TNBCs
3.3. NF-κB Signaling and TNBCs
3.4. PI3K/Akt/mTOR Signaling and TNBCs
3.5. MAPK (Ras/Raf/MEK/ERK) Signaling and TNBCs
3.6. Hedgehog Signaling and TNBCs
4. Phytochemicals as Anticancer Compounds Effective in the Treatment of TNBCs
4.1. Luteolin
4.2. Chalcones
4.3. Piperine
4.4. Deguelin
4.5. Quercetin
4.6. Rutin
4.7. Fisetin
4.8. Resveratrol
4.9. Curcumin
4.10. Maximiscin
4.11. Cyclopamine
4.12. Capsaicin
4.13. Genistein
5. Phytochemicals that Can Promote TNBC Growth, EMT and Metastasis
Asparagine
6. Observations, Inferences and Concluding Remarks
7. Future Perspective
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIF | Apoptosis-Inducing Factor |
| Akt | Serine/threonine-specific protein kinase or Protein Kinase B |
| AMPK | 5′ AMP-activated Protein Kinase |
| Ang1 | Angiopoietin-1 |
| APC | Adenomatous Polyposis Coli |
| AR | Androgen Receptor |
| BCRP | Breast Cancer Resistant Protein |
| BRCA1 | Breast Cancer 1, early onset |
| CD | Cardamonin |
| Chk 1/2 | Check point kinases 1/2 |
| CK1 | Casein Kinase-1 |
| COX2 | Cyclooxygenase-2 |
| CSCs | Cancer Stem Cells |
| CXCR4 | C-X-C chemokine Receptor type 4 |
| DHh | Desert Hedgehog |
| DLL | Delta-like Ligand |
| ECD | Extra Cellular Domain |
| EGFR | Extracellular Growth Factor Receptor |
| EMT | Epithelial-Mesenchymal Transition |
| ER | Estrogen Receptor |
| ER-α | Estrogen Receptor-alpha |
| ERK1/2 | Extracellular signal-Regulated Kinase-1/2 |
| FABP5 | Fatty Acid-Binding Protein 5 |
| FAK | Focal Adhesion Kinase |
| Fas | First apoptosis signal |
| FasL | Fas Ligand |
| FasR | Fas Receptor |
| FGF | Fibroblast Growth Factor |
| FZD7 | Frizzled Receptor 7 |
| GADD45 | Growth Arrest and DNA Damage-inducible 45 |
| Gli | Glioma-associated oncogene |
| GRB2 | Growth factor Receptor Binding protein 2 |
| GSK-3β | Glycogen Synthase Kinase-3β |
| GTPs | Green Tea Phenols |
| HER2 | Human Epidermal growth factor Receptor 2 |
| HGF | Hepatocyte Growth Factor |
| Hh | Hedgehog |
| HIF1 | Hypoxia Inducible Factor 1 |
| IHh | Indian Hedgehog |
| IL1 | Interleukin-1 |
| JAG1/2 | Jagged 1/2 |
| LAR | Luminal Androgen Receptor |
| LRP5/6 | Low-density lipoprotein Receptor-related Protein-5/6 |
| MAM | Mastermind |
| MAPK | Mitogen-Activated Protein Kinase |
| MAPKAP | Mitogen-Activated Protein Kinase Activated Protein |
| MAPKAP Kinase | Mitogen-Activated Protein Kinase Activated Protein Kinase |
| MCP | Monocyte Chemoattractant Protein |
| MDR1 | Multidrug Resistance Protein-1 |
| MEK1/2 | Mitogen-activated protein kinase ERK kinase 1/2 |
| MMP 2/7/9 | Matrix metalloproteinase 2/7/9 |
| NEXT | Notch Extracellular Truncation |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NICD | Notch Intracellular Domain |
| P-gp | P-glycoprotein |
| PARP | Poly (ADP-ribose) Polymerase |
| PDGF | Platelet-Derived Growth Factor |
| PIP2 | Phosphatidyl-Inositol biphosphate |
| PR | Progesterone Receptor |
| Ptch | Patched |
| PTEN | Phosphatase and tensin homolog |
| RHD | Rel Homology Domain |
| RSK | Ribosomal s6 kinase |
| SHh | Sonic Hedgehog |
| Smo | Smoothened |
| TCF/LEF | T-cell factor/Lymphoid Enhancing Factor |
| TGFβ | Transforming Growth Factor-b |
| TLR4 | Toll-like Receptor 4 |
| TNBCs | Triple-Negative Breast Cancers |
| TNF-α | Tumor Necrosis Factor-α |
| TNFR | Tumor Necrosis Factor Receptor |
| TRAIL | Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand |
| TSC 1/2 | Tuberous Sclerosis Complex-1/2 |
| Twist 1 | Twist-related protein-1 |
| VEGF | Vascular Endothelial Growth Factor |
| VEGFR2 | Vascular Endothelial Growth Factor Receptor-2 |
| VRK1 | Vaccinia-Related Kinase-1 |
| WISP3 | Wnt1-Inducible Signaling Protein-3 |
| XIAP | X-linked Inhibitor of Apoptosis Protein |
References
- Who Media Centre (Cancer—Fact Sheets). Available online: http://www.who.int/mediacentre/factsheets/fs297/en/ (accessed on 14 March 2018).
- DeSantis, C.E.; Lin, C.C.; Mariotto, A.B.; Siegel, R.L.; Stein, K.D.; Kramer, J.L.; Alteri, R.; Robbins, A.S.; Jemal, A. Cancer treatment and survivorship statistics. CA Cancer J. Clin. 2014, 64, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.P.; Quaresma, M.; Berrino, F.; Lutz, J.M.; De Angelis, R.; Capocaccia, R.; Baili, P.; Rachet, B.; Gatta, G.; Hakulinen, T.; et al. Cancer survival in five continents: A worldwide population-based study (concord). Lancet Oncol. 2008, 9, 730–756. [Google Scholar] [CrossRef]
- Bodai, B.I.; Tuso, P. Breast cancer survivorship: A comprehensive review of long-term medical issues and lifestyle recommendations. Perm. J. 2015, 19, 48–79. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Cheng, H.; Bai, Z.; Li, J. Breast cancer cell line classification and its relevance with breast tumor subtyping. J. Cancer 2017, 8, 3131–3141. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anders, C.K.; Carey, L.A. Biology, metastatic patterns, and treatment of patients with triple-negative breast cancer. Clin. Breast Cancer 2009, 9 (Suppl. 2), S73–S81. [Google Scholar] [CrossRef] [PubMed]
- Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Challenges and opportunities of a heterogeneous disease. Nat. Rev. Clin. Oncol. 2016, 13, 674–690. [Google Scholar] [CrossRef] [PubMed]
- Collignon, J.; Lousberg, L.; Schroeder, H.; Jerusalem, G. Triple-negative breast cancer: Treatment challenges and solutions. Breast Cancer 2016, 8, 93–107. [Google Scholar] [PubMed]
- Hudis, C.A.; Gianni, L. Triple-negative breast cancer: An unmet medical need. Oncologist 2011, 16 (Suppl. 1), 1–11. [Google Scholar] [CrossRef] [PubMed]
- Isakoff, S.J. Triple-negative breast cancer: Role of specific chemotherapy agents. Cancer J. 2010, 16, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Abotaleb, M.; Kubatka, P.; Caprnda, M.; Varghese, E.; Zolakova, B.; Zubor, P.; Opatrilova, R.; Kruzliak, P.; Stefanicka, P.; Busselberg, D. Chemotherapeutic agents for the treatment of metastatic breast cancer: An update. Biomed. Pharmacother. 2018, 101, 458–477. [Google Scholar] [CrossRef] [PubMed]
- Pem, D.; Jeewon, R. Fruit and vegetable intake: Benefits and progress of nutrition education interventions- narrative review article. Iran. J. Public Health 2015, 44, 1309–1321. [Google Scholar] [PubMed]
- Zhang, Y.J.; Gan, R.Y.; Li, S.; Zhou, Y.; Li, A.N.; Xu, D.P.; Li, H.B. Antioxidant phytochemicals for the prevention and treatment of chronic diseases. Molecules 2015, 20, 21138–21156. [Google Scholar] [CrossRef] [PubMed]
- Kapinova, A.; Stefanicka, P.; Kubatka, P.; Zubor, P.; Uramova, S.; Kello, M.; Mojzis, J.; Blahutova, D.; Qaradakhi, T.; Zulli, A.; et al. Are plant-based functional foods better choice against cancer than single phytochemicals? A critical review of current breast cancer research. Biomed. Pharmacother. 2017, 96, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Kubatka, P.; Kello, M.; Kajo, K.; Kruzliak, P.; Vybohova, D.; Mojzis, J.; Adamkov, M.; Fialova, S.; Veizerova, L.; Zulli, A.; et al. Oregano demonstrates distinct tumour-suppressive effects in the breast carcinoma model. Eur. J. Nutr. 2017, 56, 1303–1316. [Google Scholar] [CrossRef] [PubMed]
- Kubatka, P.; Uramova, S.; Kello, M.; Kajo, K.; Kruzliak, P.; Mojzis, J.; Vybohova, D.; Adamkov, M.; Jasek, K.; Lasabova, Z.; et al. Antineoplastic effects of clove buds (Syzygium aromaticum L.) in the model of breast carcinoma. J. Cell. Mol. Med. 2017, 21, 2837–2851. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 35. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.C.; Patchva, S.; Aggarwal, B.B. Therapeutic roles of curcumin: Lessons learned from clinical trials. AAPS J. 2013, 15, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Hatcher, H.; Planalp, R.; Cho, J.; Torti, F.M.; Torti, S.V. Curcumin: From ancient medicine to current clinical trials. Cell. Mol. Life Sci. 2008, 65, 1631–1652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bishayee, A. Cancer prevention and treatment with resveratrol: From rodent studies to clinical trials. Cancer Prev. Res. 2009, 2, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Cragg, G.M.; Newman, D.J. Natural products: A continuing source of novel drug leads. Biochim. Biophys. Acta 2013, 1830, 3670–3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.D.; Suto, M.J.; Li, Y. The Wnt/beta-catenin signaling pathway: A potential therapeutic target in the treatment of triple negative breast cancer. J. Cell. Biochem. 2012, 113, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Speiser, J.J.; Ersahin, C.; Osipo, C. The functional role of Notch signaling in triple-negative breast cancer. Vitam. Horm. 2013, 93, 277–306. [Google Scholar] [PubMed]
- Poma, P.; Labbozzetta, M.; D’Alessandro, N.; Notarbartolo, M. NF-κB is a potential molecular drug target in triple-negative breast cancers. OMICS 2017, 21, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Gordon, V.; Banerji, S. Molecular pathways: Pi3k pathway targets in triple-negative breast cancers. Clin. Cancer Res. 2013, 19, 3738–3744. [Google Scholar] [CrossRef] [PubMed]
- Giltnane, J.M.; Balko, J.M. Rationale for targeting the Ras/MAPK pathway in triple-negative breast cancer. Discov. Med. 2014, 17, 275–283. [Google Scholar] [PubMed]
- Habib, J.G.; O’Shaughnessy, J.A. The hedgehog pathway in triple-negative breast cancer. Cancer Med. 2016, 5, 2989–3006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jamdade, V.S.; Sethi, N.; Mundhe, N.A.; Kumar, P.; Lahkar, M.; Sinha, N. Therapeutic targets of triple-negative breast cancer: A review. Br. J. Pharmacol. 2015, 172, 4228–4237. [Google Scholar] [CrossRef] [PubMed]
- Rivenbark, A.G.; O’Connor, S.M.; Coleman, W.B. Molecular and cellular heterogeneity in breast cancer: Challenges for personalized medicine. Am. J. Pathol. 2013, 183, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Hong, R.; Ma, F.; Xu, B.; Li, Q.; Zhang, P.; Yuan, P.; Wang, J.; Fan, Y.; Cai, R. Efficacy of platinum-based chemotherapy in triple-negative breast cancer patients with metastases confined to the lungs: A single-institute experience. Anti-Cancer Drugs 2014, 25, 1089–1094. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.; Krajewski, K.; Cakar, B.; Ma, C.X. Targeted therapy for breast cancer. Am. J. Pathol. 2013, 183, 1096–1112. [Google Scholar] [CrossRef] [PubMed]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; Andre, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Tariq, K.; Rana, F. Tnbc vs. Non-tnbc: A five-year retrospective review of differences in mean age, family history, smoking history and stage at diagnosis at an inner city university program. World J. Oncol. 2013, 4, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Marotti, J.D.; de Abreu, F.B.; Wells, W.A.; Tsongalis, G.J. Triple-negative breast cancer. Am. J. Pathol. 2017, 187, 2133–2138. [Google Scholar] [CrossRef] [PubMed]
- Guestini, F.; McNamara, K.M.; Ishida, T.; Sasano, H. Triple negative breast cancer chemosensitivity and chemoresistance: Current advances in biomarkers indentification. Expert Opin. Ther. Targets 2016, 20, 705–720. [Google Scholar] [CrossRef] [PubMed]
- Neophytou, C.; Boutsikos, P.; Papageorgis, P. Molecular mechanisms and emerging therapeutic targets of triple-negative breast cancer metastasis. Front. Oncol. 2018, 8, 31. [Google Scholar] [CrossRef] [PubMed]
- Sledge, G.W., Jr.; Loehrer, P.J., Sr.; Roth, B.J.; Einhorn, L.H. Cisplatin as first-line therapy for metastatic breast cancer. J. Clin. Oncol. 1988, 6, 1811–1814. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Wu, X.; Wang, Y.; Zhang, K.; Wu, J.; Yuan, Y.C.; Deng, X.; Chen, L.; Kim, C.C.; Lau, S.; et al. Fzd7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 2011, 30, 4437–4446. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; Young, B.; Abramovitz, M.; Bouzyk, M.; Barwick, B.; De, P.; Leyland-Jones, B. Differential activation of Wnt-beta-catenin pathway in triple negative breast cancer increases mmp7 in a pten dependent manner. PLoS ONE 2013, 8, e77425. [Google Scholar] [CrossRef] [PubMed]
- Polakis, P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Kaidanovich-Beilin, O.; Woodgett, J.R. Gsk-3: Functional insights from cell biology and animal models. Front. Mol. Neurosci. 2011, 4, 40. [Google Scholar] [CrossRef] [PubMed]
- Prosperi, J.R.; Goss, K.H. A Wnt-ow of opportunity: Targeting the Wnt/beta-catenin pathway in breast cancer. Curr. Drug Targets 2010, 11, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Lindvall, C.; Zylstra, C.R.; Evans, N.; West, R.A.; Dykema, K.; Furge, K.A.; Williams, B.O. The Wnt co-receptor lrp6 is required for normal mouse mammary gland development. PLoS ONE 2009, 4, e5813. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Clevers, H. Mining the Wnt pathway for cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 997–1014. [Google Scholar] [CrossRef] [PubMed]
- Bayet-Robert, M.; Kwiatkowski, F.; Leheurteur, M.; Gachon, F.; Planchat, E.; Abrial, C.; Mouret-Reynier, M.A.; Durando, X.; Barthomeuf, C.; Chollet, P. Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol. Ther. 2010, 9, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pohl, S.G.; Brook, N.; Agostino, M.; Arfuso, F.; Kumar, A.P.; Dharmarajan, A. Wnt signaling in triple-negative breast cancer. Oncogenesis 2017, 6, e310. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Prosperi, J.R.; Choudhury, N.; Olopade, O.I.; Goss, K.H. Beta-catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS ONE 2015, 10, e0117097. [Google Scholar]
- Dey, N.; Barwick, B.G.; Moreno, C.S.; Ordanic-Kodani, M.; Chen, Z.; Oprea-Ilies, G.; Tang, W.; Catzavelos, C.; Kerstann, K.F.; Sledge, G.W., Jr.; et al. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer 2013, 13, 537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.C.; Prior, J.; Piwnica-Worms, D.; Bu, G. Lrp6 overexpression defines a class of breast cancer subtype and is a target for therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 5136–5141. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Ginther, C.; Kim, J.; Mosher, N.; Chung, S.; Slamon, D.; Vadgama, J.V. Expression of Wnt3 activates Wnt/beta-catenin pathway and promotes emt-like phenotype in trastuzumab-resistant her2-overexpressing breast cancer cells. Mol. Cancer Res. 2012, 10, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, X.; Zhang, H.; Li, X.; Li, X.; Cong, M.; Peng, F.; Yu, J.; Zhang, X.; Yang, Q.; Hu, G. Differential effects on lung and bone metastasis of breast cancer by Wnt signalling inhibitor dkk1. Nat. Cell Biol. 2017, 19, 1274–1285. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.Y.; Xia, W.; Wang, J.C.; Kwong, K.Y.; Spohn, B.; Wen, Y.; Pestell, R.G.; Hung, M.C. Beta-catenin, a novel prognostic marker for breast cancer: Its roles in cyclin d1 expression and cancer progression. Proc. Natl. Acad. Sci. USA 2000, 97, 4262–4266. [Google Scholar] [CrossRef] [PubMed]
- Geyer, F.C.; Lacroix-Triki, M.; Savage, K.; Arnedos, M.; Lambros, M.B.; MacKay, A.; Natrajan, R.; Reis-Filho, J.S. Beta-catenin pathway activation in breast cancer is associated with triple-negative phenotype but not with ctnnb1 mutation. Mod. Pathol. 2011, 24, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Khramtsov, A.I.; Khramtsova, G.F.; Tretiakova, M.; Huo, D.; Olopade, O.I.; Goss, K.H. Wnt/beta-catenin pathway activation is enriched in basal-like breast cancers and predicts poor outcome. Am. J. Pathol. 2010, 176, 2911–2920. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y.; Hong, I.S.; Nam, J.S. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef] [PubMed]
- De, P.; Carlson, J.H.; Leyland-Jones, B.; Dey, N. Wnt-β-catenin pathway regulates vascular mimicry in triple negative breast cancer. J. Cytol. Histol. 2013, 4, 198. [Google Scholar] [CrossRef]
- Liu, J.; Shen, J.X.; Wen, X.F.; Guo, Y.X.; Zhang, G.J. Targeting Notch degradation system provides promise for breast cancer therapeutics. Crit. Rev. Oncol. Hematol. 2016, 104, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Lamy, M.; Ferreira, A.; Dias, J.S.; Braga, S.; Silva, G.; Barbas, A. Notch-out for breast cancer therapies. New Biotechnol. 2017, 39, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Gazave, E.; Lapebie, P.; Richards, G.S.; Brunet, F.; Ereskovsky, A.V.; Degnan, B.M.; Borchiellini, C.; Vervoort, M.; Renard, E. Origin and evolution of the Notch signalling pathway: An overview from eukaryotic genomes. BMC Evol. Biol. 2009, 9, 249. [Google Scholar] [CrossRef] [PubMed]
- Al-Hussaini, H.; Subramanyam, D.; Reedijk, M.; Sridhar, S.S. Notch signaling pathway as a therapeutic target in breast cancer. Mol. Cancer Ther. 2011, 10, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Ilagan, M.X.; Kopan, R. Snapshot: Notch signaling pathway. Cell 2007, 128, 1246. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Liu, M.; Gonzalez-Perez, R.R. Role of Notch and its oncogenic signaling crosstalk in breast cancer. Biochim. Biophys. Acta 2011, 1815, 197–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, W.; Wang, G.; Guo, S. Regulation of angiogenesis via Notch signaling in breast cancer and cancer stem cells. Biochim. Biophys. Acta 2013, 1836, 304–320. [Google Scholar] [CrossRef] [PubMed]
- Soriano, J.V.; Uyttendaele, H.; Kitajewski, J.; Montesano, R. Expression of an activated Notch4(int-3) oncoprotein disrupts morphogenesis and induces an invasive phenotype in mammary epithelial cells in vitro. Int. J. Cancer 2000, 86, 652–659. [Google Scholar] [CrossRef]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res. 2004, 6, R605–R615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchor, L.; Smalley, M.J. Highway to heaven: Mammary gland development and differentiation. Breast Cancer Res. 2008, 10, 305. [Google Scholar] [CrossRef] [PubMed]
- Sansone, P.; Storci, G.; Giovannini, C.; Pandolfi, S.; Pianetti, S.; Taffurelli, M.; Santini, D.; Ceccarelli, C.; Chieco, P.; Bonafe, M. P66shc/Notch-3 interplay controls self-renewal and hypoxia survival in human stem/progenitor cells of the mammary gland expanded in vitro as mammospheres. Stem Cells 2007, 25, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Zang, S.; Ji, C.; Qu, X.; Dong, X.; Ma, D.; Ye, J.; Ma, R.; Dai, J.; Guo, D. A study on Notch signaling in human breast cancer. Neoplasma 2007, 54, 304–310. [Google Scholar] [PubMed]
- Nagamatsu, I.; Onishi, H.; Matsushita, S.; Kubo, M.; Kai, M.; Imaizumi, A.; Nakano, K.; Hattori, M.; Oda, Y.; Tanaka, M.; et al. Notch4 is a potential therapeutic target for triple-negative breast cancer. Anticancer Res. 2014, 34, 69–80. [Google Scholar] [PubMed]
- Speiser, J.; Foreman, K.; Drinka, E.; Godellas, C.; Perez, C.; Salhadar, A.; Ersahin, C.; Rajan, P. Notch-1 and Notch-4 biomarker expression in triple-negative breast cancer. Int. J. Surg. Pathol. 2012, 20, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Farnie, G.; Clarke, R.B. Mammary stem cells and breast cancer—role of Notch signalling. Stem Cell Rev. 2007, 3, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Bednarz-Knoll, N.; Efstathiou, A.; Gotzhein, F.; Wikman, H.; Mueller, V.; Kang, Y.; Pantel, K. Potential involvement of jagged1 in metastatic progression of human breast carcinomas. Clin. Chem. 2016, 62, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Li, C.F.; Chu, P.Y.; Lai, Y.S.; Chen, C.H.; Jiang, S.S.; Hou, M.F.; Hung, W.C. Lysine demethylase 2a promotes stemness and angiogenesis of breast cancer by upregulating jagged1. Oncotarget 2016, 7, 27689–27710. [Google Scholar] [CrossRef] [PubMed]
- Reedijk, M.; Odorcic, S.; Chang, L.; Zhang, H.; Miller, N.; McCready, D.R.; Lockwood, G.; Egan, S.E. High-level coexpression of jag1 and Notch1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005, 65, 8530–8537. [Google Scholar] [CrossRef] [PubMed]
- Orzechowska, M.; Jedroszka, D.; Bednarek, A.K. Common profiles of Notch signaling differentiate disease-free survival in luminal type A and triple negative breast cancer. Oncotarget 2017, 8, 6013–6032. [Google Scholar] [CrossRef] [PubMed]
- Pamarthy, S.; Jaiswal, M.K.; Kulshreshtha, A.; Katara, G.K.; Gilman-Sachs, A.; Beaman, K.D. The vacuolar atpase a2-subunit regulates Notch signaling in triple-negative breast cancer cells. Oncotarget 2015, 6, 34206–34220. [Google Scholar] [CrossRef] [PubMed]
- Borggrefe, T.; Lauth, M.; Zwijsen, A.; Huylebroeck, D.; Oswald, F.; Giaimo, B.D. The Notch intracellular domain integrates signals from Wnt, hedgehog, TGFbeta/bmp and hypoxia pathways. Biochim. Biophys. Acta 2016, 1863, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Pires, B.R.; IS, D.E.A.; Souza, L.D.; Rodrigues, J.A.; Mencalha, A.L. Targeting cellular signaling pathways in breast cancer stem cells and its implication for cancer treatment. Anticancer. Res. 2016, 36, 5681–5691. [Google Scholar] [CrossRef] [PubMed]
- Napetschnig, J.; Wu, H. Molecular basis of NF-κB signaling. Annu. Rev. Biophys. 2013, 42, 443–468. [Google Scholar] [CrossRef] [PubMed]
- Hoesel, B.; Schmid, J.A. The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 2013, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in NF-κB signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Shostak, K.; Chariot, A. NF-κB, stem cells and breast cancer: The links get stronger. Breast Cancer Res. 2011, 13, 214. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Sakamaki, T.; Casimiro, M.C.; Willmarth, N.E.; Quong, A.A.; Ju, X.; Ojeifo, J.; Jiao, X.; Yeow, W.S.; Katiyar, S.; et al. The canonical NF-κB pathway governs mammary tumorigenesis in transgenic mice and tumor stem cell expansion. Cancer Res. 2010, 70, 10464–10473. [Google Scholar] [CrossRef] [PubMed]
- Biswas, D.K.; Iglehart, J.D. Linkage between egfr family receptors and nuclear factor κB (NF-κB) signaling in breast cancer. J. Cell. Physiol. 2006, 209, 645–652. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Bhat-Nakshatri, P.; Martin, D.A.; Goulet, R.J., Jr.; Sledge, G.W., Jr. Constitutive activation of NF-κB during progression of breast cancer to hormone-independent growth. Mol. Cell. Biol. 1997, 17, 3629–3639. [Google Scholar] [CrossRef] [PubMed]
- Kendellen, M.F.; Bradford, J.W.; Lawrence, C.L.; Clark, K.S.; Baldwin, A.S. Canonical and non-canonical NF-κB signaling promotes breast cancer tumor-initiating cells. Oncogene 2014, 33, 1297–1305. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.M.; Lyu, Y.L.; Cai, L. NF-κB affects proliferation and invasiveness of breast cancer cells by regulating cd44 expression. PLoS ONE 2014, 9, e106966. [Google Scholar] [CrossRef] [PubMed]
- Gordon, A.H.; O'Keefe, R.J.; Schwarz, E.M.; Rosier, R.N.; Puzas, J.E. Nuclear factor-κB-dependent mechanisms in breast cancer cells regulate tumor burden and osteolysis in bone. Cancer Res. 2005, 65, 3209–3217. [Google Scholar] [CrossRef] [PubMed]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of triple-negative breast cancer cells relies upon coordinate autocrine expression of the proinflammatory cytokines il-6 and il-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef] [PubMed]
- Fusella, F.; Secli, L.; Busso, E.; Krepelova, A.; Moiso, E.; Rocca, S.; Conti, L.; Annaratone, L.; Rubinetto, C.; Mello-Grand, M.; et al. The IKK/NF-κB signaling pathway requires morgana to drive breast cancer metastasis. Nat. Commun. 2017, 8, 1636. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Angulo, A.M.; Blumenschein, G.R., Jr. Defining biomarkers to predict sensitivity to PI3K/Akt/mTOR pathway inhibitors in breast cancer. Cancer Treat. Rev. 2013, 39, 313–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massihnia, D.; Galvano, A.; Fanale, D.; Perez, A.; Castiglia, M.; Incorvaia, L.; Listi, A.; Rizzo, S.; Cicero, G.; Bazan, V.; et al. Triple negative breast cancer: Shedding light onto the role of PI3K/Akt/mTOR pathway. Oncotarget 2016, 7, 60712–60722. [Google Scholar] [CrossRef] [PubMed]
- Graupera, M.; Guillermet-Guibert, J.; Foukas, L.C.; Phng, L.K.; Cain, R.J.; Salpekar, A.; Pearce, W.; Meek, S.; Millan, J.; Cutillas, P.R.; et al. Angiogenesis selectively requires the p110alpha isoform of pi3k to control endothelial cell migration. Nature 2008, 453, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Saini, K.S.; Loi, S.; de Azambuja, E.; Metzger-Filho, O.; Saini, M.L.; Ignatiadis, M.; Dancey, J.E.; Piccart-Gebhart, M.J. Targeting the PI3K/Akt/mTOR and Raf/MEK/ERK pathways in the treatment of breast cancer. Cancer Treat. Rev. 2013, 39, 935–946. [Google Scholar] [CrossRef] [PubMed]
- Lauring, J.; Park, B.H.; Wolff, A.C. The phosphoinositide-3-kinase-Akt-mTOR pathway as a therapeutic target in breast cancer. J. Natl. Compr. Cancer Netw. 2013, 711, 670–678. [Google Scholar] [CrossRef]
- Yang, S.X.; Polley, E.; Lipkowitz, S. New insights on PI3k/Akt pathway alterations and clinical outcomes in breast cancer. Cancer Treat. Rev. 2016, 45, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Dey, N.; De, P.; Leyland-Jones, B. Pi3k-Akt-mTOR inhibitors in breast cancers: From tumor cell signaling to clinical trials. Pharmacol. Ther. 2017, 175, 91–106. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.S.; Banerji, U. Maximising the potential of Akt inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Loh, K.; Yap, Y.S. PI3K/Akt/mTOR inhibitors in breast cancer. Cancer Biol. Med. 2015, 12, 342–354. [Google Scholar] [PubMed]
- Yang, Y.; Gao, M.; Lin, Z.; Chen, L.; Jin, Y.; Zhu, G.; Wang, Y.; Jin, T. Dek promoted emt and angiogenesis through regulating PI3K/Akt/mTOR pathway in triple-negative breast cancer. Oncotarget 2017, 8, 98708–98722. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.U.; Sangai, T.; Akcakanat, A.; Chen, H.; Wei, C.; Meric-Bernstam, F. Vertical inhibition of the PI3K/Akt/mTOR pathway is synergistic in breast cancer. Oncogenesis 2017, 6, e385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O'Donnell, J.S.; Massi, D.; Teng, M.W.L.; Mandala, M. PI3k-Akt-mTOR inhibition in cancer immunotherapy, redux. Semin. Cancer Biol. 2018, 48, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ding, H.; Liu, X.; Li, X.; Li, L. Inpp4b overexpression enhances the antitumor efficacy of PARP inhibitor ag014699 in MDA-MB-231 triple-negative breast cancer cells. Tumor Biol. 2014, 35, 4469–4477. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Xie, J.; Zhang, M.; Zhao, Z.; Wan, Y.; Yao, Y. Mirna-21 promotes proliferation and invasion of triple-negative breast cancer cells through targeting pten. Am. J. Transl. Res. 2017, 9, 953–961. [Google Scholar] [PubMed]
- Sun, T.; Aceto, N.; Meerbrey, K.L.; Kessler, J.D.; Zhou, C.; Migliaccio, I.; Nguyen, D.X.; Pavlova, N.N.; Botero, M.; Huang, J.; et al. Activation of multiple proto-oncogenic tyrosine kinases in breast cancer via loss of the PTPN12 phosphatase. Cell 2011, 144, 703–718. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.Q.; Hu, P.; Gao, J.; Wei, W.D.; Xiao, X.S.; Tang, H.L.; Li, X.; Ge, Q.D.; Jia, W.H.; Liu, R.B.; et al. Low expression of tyrosine-protein phosphatase nonreceptor type 12 is associated with lymph node metastasis and poor prognosis in operable triple-negative breast cancer. Asian Pac. J. Cancer Prev. 2013, 14, 287–292. [Google Scholar] [CrossRef] [PubMed]
- LoRusso, P.M. Inhibition of the PI3K/Akt/mTOR pathway in solid tumors. J. Clin. Oncol. 2016, 34, 3803–3815. [Google Scholar] [CrossRef] [PubMed]
- Singel, S.M.; Cornelius, C.; Zaganjor, E.; Batten, K.; Sarode, V.R.; Buckley, D.L.; Peng, Y.; John, G.B.; Li, H.C.; Sadeghi, N.; et al. Kif14 promotes Akt phosphorylation and contributes to chemoresistance in triple-negative breast cancer. Neoplasia 2014, 16, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Ueng, S.H.; Chen, S.C.; Chang, Y.S.; Hsueh, S.; Lin, Y.C.; Chien, H.P.; Lo, Y.F.; Shen, S.C.; Hsueh, C. Phosphorylated mTOR expression correlates with poor outcome in early-stage triple negative breast carcinomas. Int. J. Clin. Exp. Pathol. 2012, 5, 806–813. [Google Scholar] [PubMed]
- Walsh, S.; Flanagan, L.; Quinn, C.; Evoy, D.; McDermott, E.W.; Pierce, A.; Duffy, M.J. mTOR in breast cancer: Differential expression in triple-negative and non-triple-negative tumors. Breast 2012, 21, 178–182. [Google Scholar] [CrossRef] [PubMed]
- Costa, R.L.B.; Han, H.S.; Gradishar, W.J. Targeting the PI3K/Akt/mTOR pathway in triple-negative breast cancer: A review. Breast Cancer Res. Treat. 2018, 169, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Fouque, A.; Jean, M.; Weghe, P.; Legembre, P. Review of PI3K/mTOR inhibitors entering clinical trials to treat triple negative breast cancers. Recent Patents Anti-Cancer Drug Discov. 2016, 11, 283–296. [Google Scholar] [CrossRef]
- Kim, S.B.; Dent, R.; Im, S.A.; Espie, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.J.; et al. Ipatasertib plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (lotus): A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2017, 18, 1360–1372. [Google Scholar] [CrossRef]
- Johnson, G.L.; Stuhlmiller, T.J.; Angus, S.P.; Zawistowski, J.S.; Graves, L.M. Molecular pathways: Adaptive kinome reprogramming in response to targeted inhibition of the BRAF-MEK-ERK pathway in cancer. Clin. Cancer Res. 2014, 20, 2516–2522. [Google Scholar] [CrossRef] [PubMed]
- Plotnikov, A.; Zehorai, E.; Procaccia, S.; Seger, R. The MAPK cascades: Signaling components, nuclear roles and mechanisms of nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1619–1633. [Google Scholar] [CrossRef] [PubMed]
- Cseh, B.; Doma, E.; Baccarini, M. “Raf” neighborhood: Protein-protein interaction in the Raf/MEK/ERK pathway. FEBS Lett. 2014, 588, 2398–2406. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Zabuawala, T.; Huang, H.; Zhang, J.; Gulati, P.; Fernandez, S.; Karlo, J.C.; Landreth, G.E.; Leone, G.; Ostrowski, M.C. ERK1 and ERK2 regulate endothelial cell proliferation and migration during mouse embryonic angiogenesis. PLoS ONE 2009, 4, e8283. [Google Scholar] [CrossRef] [PubMed]
- Galabova-Kovacs, G.; Kolbus, A.; Matzen, D.; Meissl, K.; Piazzolla, D.; Rubiolo, C.; Steinitz, K.; Baccarini, M. ERK and beyond: Insights from B-Raf and Raf-1 conditional knockouts. Cell Cycle 2006, 5, 1514–1518. [Google Scholar] [CrossRef] [PubMed]
- Giroux, S.; Tremblay, M.; Bernard, D.; Cardin-Girard, J.F.; Aubry, S.; Larouche, L.; Rousseau, S.; Huot, J.; Landry, J.; Jeannotte, L.; et al. Embryonic death of MEK1-deficient mice reveals a role for this kinase in angiogenesis in the labyrinthine region of the placenta. Curr. Biol. 1999, 9, 369–372. [Google Scholar] [CrossRef]
- Santen, R.J.; Song, R.X.; McPherson, R.; Kumar, R.; Adam, L.; Jeng, M.H.; Yue, W. The role of mitogen-activated protein (map) kinase in breast cancer. J. Steroid Biochem. Mol. Boil. 2002, 80, 239–256. [Google Scholar] [CrossRef]
- Poulikakos, P.I.; Solit, D.B. Resistance to MEK inhibitors: Should we co-target upstream? Sci. Signal. 2011, 4, pe16. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal growth factor receptor cell proliferation signaling pathways. Cancers 2017, 9, 52. [Google Scholar]
- Lowenstein, E.J.; Daly, R.J.; Batzer, A.G.; Li, W.; Margolis, B.; Lammers, R.; Ullrich, A.; Skolnik, E.Y.; Bar-Sagi, D.; Schlessinger, J. The sh2 and sh3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 1992, 70, 431–442. [Google Scholar] [CrossRef]
- Buday, L.; Downward, J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, GRB2 adapter protein, and SOS nucleotide exchange factor. Cell 1993, 73, 611–620. [Google Scholar] [CrossRef]
- Egan, S.E.; Giddings, B.W.; Brooks, M.W.; Buday, L.; Sizeland, A.M.; Weinberg, R.A. Association of SOS Ras exchange protein with GRB2 is implicated in tyrosine kinase signal transduction and transformation. Nature 1993, 363, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Batzer, A.; Daly, R.; Yajnik, V.; Skolnik, E.; Chardin, P.; Bar-Sagi, D.; Margolis, B.; Schlessinger, J. Guanine-nucleotide-releasing factor hsos1 binds to GRB2 and links receptor tyrosine kinases to Ras signalling. Nature 1993, 363, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Seger, R. The ERK signaling cascade—Views from different subcellular compartments. BioFactors 2009, 35, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Pokholok, D.K.; Zeitlinger, J.; Hannett, N.M.; Reynolds, D.B.; Young, R.A. Activated signal transduction kinases frequently occupy target genes. Science 2006, 313, 533–536. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Xie, Z.; Onishi, A.; Yu, X.; Jiang, L.; Lin, J.; Rho, H.S.; Woodard, C.; Wang, H.; Jeong, J.S.; et al. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 2009, 139, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Hoeflich, K.P.; O'Brien, C.; Boyd, Z.; Cavet, G.; Guerrero, S.; Jung, K.; Januario, T.; Savage, H.; Punnoose, E.; Truong, T.; et al. In vivo antitumor activity of MEK and phosphatidylinositol 3-kinase inhibitors in basal-like breast cancer models. Clin. Cancer Res. 2009, 15, 4649–4664. [Google Scholar] [CrossRef] [PubMed]
- Mirzoeva, O.K.; Das, D.; Heiser, L.M.; Bhattacharya, S.; Siwak, D.; Gendelman, R.; Bayani, N.; Wang, N.J.; Neve, R.M.; Guan, Y.; et al. Basal subtype and MAPK/ERK kinase (MEK)-phosphoinositide 3-kinase feedback signaling determine susceptibility of breast cancer cells to MEK inhibition. Cancer Res. 2009, 69, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.B.; Lin, C.C.; Farrugia, M.K.; McLaughlin, S.L.; Ellis, E.J.; Brundage, K.M.; Salkeni, M.A.; Ruppert, J.M. Micrornas 206 and 21 cooperate to promote Ras-extracellular signal-regulated kinase signaling by suppressing the translation of Rasa1 and spred1. Mol. Cell. Biol. 2014, 34, 4143–4164. [Google Scholar] [CrossRef] [PubMed]
- Gysin, S.; Salt, M.; Young, A.; McCormick, F. Therapeutic strategies for targeting Ras proteins. Genes Cancer 2011, 2, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Balmanno, K.; Chell, S.D.; Gillings, A.S.; Hayat, S.; Cook, S.J. Intrinsic resistance to the MEK1/2 inhibitor azd6244 (arry-142886) is associated with weak ERK1/2 signalling and/or strong PI3K signalling in colorectal cancer cell lines. Int. J. Cancer 2009, 125, 2332–2341. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORc1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Yacoub, R.; Taliaferro-Smith, L.D.; Sun, S.Y.; Graham, T.R.; Dolan, R.; Lobo, C.; Tighiouart, M.; Yang, L.; Adams, A.; et al. Combinatorial effects of lapatinib and rapamycin in triple-negative breast cancer cells. Mol. Cancer Ther. 2011, 10, 1460–1469. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.H.; Wilson, C.W.; Chuang, P.T. Snapshot: Hedgehog signaling pathway. Cell 2007, 130, 386. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Therond, P.P. The mechanisms of hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Cooper, M.K.; Maiti, T.; Beachy, P.A. Patched acts catalytically to suppress the activity of smoothened. Nature 2002, 418, 892–897. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Regl, G.; Frischauf, A.M.; Aberger, F. Gli transcription factors: Mediators of oncogenic hedgehog signalling. Eur. J. Cancer 2006, 42, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Moskowitz, M.A.; Sims, J.R. Sonic hedgehog inversely regulates the expression of angiopoietin-1 and angiopoietin-2 in fibroblasts. Int. J. Mol. Med. 2007, 19, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Adolphe, C.; Hetherington, R.; Ellis, T.; Wainwright, B. Patched1 functions as a gatekeeper by promoting cell cycle progression. Cancer Res. 2006, 66, 2081–2088. [Google Scholar] [CrossRef] [PubMed]
- Athar, M.; Li, C.; Tang, X.; Chi, S.; Zhang, X.; Kim, A.L.; Tyring, S.K.; Kopelovich, L.; Hebert, J.; Epstein, E.H., Jr.; et al. Inhibition of smoothened signaling prevents ultraviolet b-induced basal cell carcinomas through regulation of fas expression and apoptosis. Cancer Res. 2004, 64, 7545–7552. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Deng, W.; Nail, C.D.; Bailey, S.K.; Kraus, M.H.; Ruppert, J.M.; Lobo-Ruppert, S.M. Snail induction is an early response to gli1 that determines the efficiency of epithelial transformation. Oncogene 2006, 25, 609–621. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Mao, J.; Zhang, Q.; Li, L. Overexpression of hedgehog signaling molecules and its involvement in triple-negative breast cancer. Oncol. Lett. 2011, 2, 995–1001. [Google Scholar] [PubMed]
- Di Mauro, C.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Formisano, L.; De Falco, S.; Cicatiello, V.; et al. Hedgehog signalling pathway orchestrates angiogenesis in triple-negative breast cancers. Br. J. Cancer 2017, 116, 1425–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dholwani, K.K.; Saluja, A.K.; Gupta, A.R.; Shah, D.R. A review on plant-derived natural products and their analogs with anti-tumor activity. Indian J. Pharmacol. 2008, 40, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Reddy, L.; Odhav, B.; Bhoola, K.D. Natural products for cancer prevention: A global perspective. Pharmacol. Ther. 2003, 99, 1–13. [Google Scholar] [CrossRef]
- Singh, S.; Sharma, B.; Kanwar, S.S.; Kumar, A. Lead phytochemicals for anticancer drug development. Front. Plant. Sci. 2016, 7, 1667. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Shi, R.; Wang, X.; Shen, H.-M. Luteolin, a flavonoid with potentials for cancer prevention and therapy. Curr. Cancer Drug Targets 2008, 8, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Kamali, M.; Khosroyar, S.; Kamali, H.; Ahmadzadeh Sani, T.; Mohammadi, A. Phytochemical screening and evaluation of antioxidant activities of dracocephalum kotschyi and determination of its luteolin content. Avicenna J. Phytomed. 2016, 6, 425–433. [Google Scholar] [PubMed]
- Chen, D.; Bi, A.; Dong, X.; Jiang, Y.; Rui, B.; Liu, J.; Yin, Z.; Luo, L. Luteolin exhibits anti-inflammatory effects by blocking the activity of heat shock protein 90 in macrophages. Biochem. Biophys. Res. Commun. 2014, 443, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Seelinger, G.; Merfort, I.; Wölfle, U.; Schempp, C.M. Anti-carcinogenic effects of the flavonoid luteolin. Molecules 2008, 13, 2628–2651. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Kim, S.H.; Shin, J.; Harikishore, A.; Lim, J.K.; Jung, Y.; Lyu, H.N.; Baek, N.I.; Choi, K.Y.; Yoon, H.S.; et al. Luteolin suppresses cancer cell proliferation by targeting vaccinia-related kinase 1. PLoS ONE 2014, 9, e109655. [Google Scholar] [CrossRef] [PubMed]
- Cook, M.T.; Liang, Y.; Besch-Williford, C.; Hyder, S.M. Luteolin inhibits lung metastasis, cell migration, and viability of triple-negative breast cancer cells. Breast Cancer Targets Ther. 2017, 9, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Kuang, G.; Wan, J.; Zhang, X.; Li, H.; Gong, X.; Li, H. Luteolin suppresses the metastasis of triple-negative breast cancer by reversing epithelial-to-mesenchymal transition via downregulation of beta-catenin expression. Oncol. Rep. 2017, 37, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Reipas, K.M.; Law, J.H.; Couto, N.; Islam, S.; Li, Y.; Li, H.; Cherkasov, A.; Jung, K.; Cheema, A.S.; Jones, S.J.; et al. Luteolin is a novel p90 ribosomal s6 kinase (rsk) inhibitor that suppresses Notch4 signaling by blocking the activation of y-box binding protein-1 (yb-1). Oncotarget 2013, 4, 329–345. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.W.; Zhang, H.D.; Mao, L.; Mao, C.F.; Chen, W.; Cui, M.; Ma, R.; Cao, H.X.; Jing, C.W.; Wang, Z.; et al. Luteolin inhibits breast cancer development and progression in vitro and in vivo by suppressing Notch signaling and regulating mirnas. Cell. Physiol. Biochem. 2015, 37, 1693–1711. [Google Scholar] [CrossRef] [PubMed]
- Amin, A.R.; Wang, D.; Zhang, H.; Peng, S.; Shin, H.J.; Brandes, J.C.; Tighiouart, M.; Khuri, F.R.; Chen, Z.G.; Shin, D.M. Enhanced anti-tumor activity by the combination of the natural compounds (−)-epigallocatechin-3-gallate and luteolin: Potential role of p53. J. Biol. Chem. 2010, 285, 34557–34565. [Google Scholar] [CrossRef] [PubMed]
- Shih, Y.L.; Liu, H.C.; Chen, C.S.; Hsu, C.H.; Pan, M.H.; Chang, H.W.; Chang, C.H.; Chen, F.C.; Ho, C.T.; Yang, Y.Y.; et al. Combination treatment with luteolin and quercetin enhances antiproliferative effects in nicotine-treated MDA-MB-231 cells by down-regulating nicotinic acetylcholine receptors. J. Agric. Food Chem. 2010, 58, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Esmaeili, M.A. Synergistic inhibition of drug resistant breast cancer cells growth by the combination of luteolin and tamoxifen involves nrf2 downregulation. J. Chem. Pharm. Res. 2015, 7, 291–296. [Google Scholar]
- Miean, K.H.; Mohamed, S. Flavonoid (myricetin, quercetin, kaempferol, luteolin, and apigenin) content of edible tropical plants. J. Agric. Food Chem. 2001, 49, 3106–3112. [Google Scholar] [CrossRef] [PubMed]
- Sui, J.Q.; Xie, K.P.; Xie, M.J. Inhibitory effect of luteolin on the proliferation of human breast cancer cell lines induced by epidermal growth factor. Sheng Li Xue Bao 2016, 68, 27–34. [Google Scholar] [PubMed]
- Rozmer, Z.; Perjési, P. Naturally occurring chalcones and their biological activities. Phytochem. Rev. 2016, 15, 87–120. [Google Scholar] [CrossRef]
- Oh, Y.J.; Seo, Y.H. A novel chalcone-based molecule, BDP inhibits mdamb231 triple-negative breast cancer cell growth by suppressing hsp90 function. Oncol. Rep. 2017, 38, 2343–2350. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.; Jeengar, M.K.; Thummuri, D.; Koval, A.; Katanaev, V.L.; Marepally, S.; Naidu, V.G.M. Cardamonin, a chalcone, inhibits human triple negative breast cancer cell invasiveness by downregulation of Wnt/beta-catenin signaling cascades and reversal of epithelial-mesenchymal transition. BioFactors 2017, 43, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Greenshields, A.L.; Doucette, C.D.; Sutton, K.M.; Madera, L.; Annan, H.; Yaffe, P.B.; Knickle, A.F.; Dong, Z.; Hoskin, D.W. Piperine inhibits the growth and motility of triple-negative breast cancer cells. Cancer Lett. 2015, 357, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Abdelhamed, S.; Yokoyama, S.; Refaat, A.; Ogura, K.; Yagita, H.; Awale, S.; Saiki, I. Piperine enhances the efficacy of trail-based therapy for triple-negative breast cancer cells. Anticancer. Res. 2014, 34, 1893–1899. [Google Scholar] [PubMed]
- Rather, R.A.; Bhagat, M. Cancer chemoprevention and piperine: Molecular mechanisms and therapeutic opportunities. Front. Cell Dev. Boil. 2018, 6, 10. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.; Katta, H.; Alimirah, F.; Patel, R.; Murillo, G.; Peng, X.; Muzzio, M.; Mehta, R.G. Deguelin action involves c-Met and EGFR signaling pathways in triple negative breast cancer cells. PLoS ONE 2013, 8, e65113. [Google Scholar] [CrossRef] [PubMed]
- Sultan, A.S.; Khalil, M.I.M.; Sami, B.M.; Alkhuriji, A.F.; Sadek, O. Quercetin induces apoptosis in triple-negative breast cancer cells via inhibiting fatty acid synthase and β-catenin. Int. J. Clin. Exp. Pathol. 2017, 10, 156–172. [Google Scholar]
- Elsayed, H.E.; Ebrahim, H.Y.; Mohyeldin, M.M.; Siddique, A.B.; Kamal, A.M.; Haggag, E.G.; El Sayed, K.A. Rutin as a novel c-met inhibitory lead for the control of triple negative breast malignancies. Nutr. Cancer 2017, 69, 1256–1271. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.; Syed, D.N.; Ahmad, N.; Mukhtar, H. Fisetin: A dietary antioxidant for health promotion. Antioxid. Redox Signal. 2013, 19, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Ham, S.L.; Nasrollahi, S.; Shah, K.N.; Soltisz, A.; Paruchuri, S.; Yun, Y.H.; Luker, G.D.; Bishayee, A.; Tavana, H. Phytochemicals potently inhibit migration of metastatic breast cancer cells. Integr. Boil. Quant. Biosci. Nano Macro 2015, 7, 792–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alkhalaf, M. Resveratrol-induced growth inhibition in MDA-MB-231 breast cancer cells is associated with mitogen-activated protein kinase signaling and protein translation. Eur. J. Cancer Prev. 2007, 16, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Garvin, S.; Ollinger, K.; Dabrosin, C. Resveratrol induces apoptosis and inhibits angiogenesis in human breast cancer xenografts in vivo. Cancer Lett. 2006, 231, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Squires, M.S.; Hudson, E.A.; Howells, L.; Sale, S.; Houghton, C.E.; Jones, J.L.; Fox, L.H.; Dickens, M.; Prigent, S.A.; Manson, M.M. Relevance of mitogen activated protein kinase (MAPK) and phosphotidylinositol-3-kinase/protein kinase B (PI3K/PKB) pathways to induction of apoptosis by curcumin in breast cells. Biochem. Pharmacol. 2003, 65, 361–376. [Google Scholar] [CrossRef]
- Sun, X.D.; Liu, X.E.; Huang, D.S. Curcumin induces apoptosis of triple-negative breast cancer cells by inhibition of egfr expression. Mol. Med. Rep. 2012, 6, 1267–1270. [Google Scholar] [CrossRef] [PubMed]
- Guan, F.; Ding, Y.; Zhang, Y.; Zhou, Y.; Li, M.; Wang, C. Curcumin suppresses proliferation and migration of MDA-MB-231 breast cancer cells through autophagy-dependent Akt degradation. PLoS ONE 2016, 11, e0146553. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.J.; Du, L.; Cichewicz, R.H.; Mooberry, S.L. Maximiscin induces DNA damage, activates DNA damage response pathways and has selective cytotoxic activity against a subtype of triple-negative breast cancer. J. Nat. Prod. 2016, 79, 1822–1827. [Google Scholar] [CrossRef] [PubMed]
- Heller, E.; Hurchla, M.A.; Xiang, J.; Chen, S.; Schneider, J.; Joeng, K.S.; Vidal, M.; Goldberg, L.; Deng, H.; Hornick, M.C.; et al. Hedgehog signaling inhibition blocks growth of resistant tumors through effects on tumor microenvironment. Cancer Res. 2012, 72, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Che, J.; Zhang, F.Z.; Zhao, C.Q.; Hu, X.D.; Fan, S.J. Cyclopamine is a novel hedgehog signaling inhibitor with significant anti-proliferative, anti-invasive and anti-estrogenic potency in human breast cancer cells. Oncol. Lett. 2013, 5, 1417–1421. [Google Scholar] [CrossRef] [PubMed]
- Taş, S.; Avcı, O. Rapid clearance of psoriatic skin lesions induced by topical cyclopamine. Dermatology 2004, 209, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Li, B.H.; Yuan, L. Inhibitory effects of capsaicin on migration and invasion of breast cancer MDA-MB-231 cells and its mechanism. Sheng Li Xue Bao 2017, 69, 183–188. [Google Scholar] [PubMed]
- Thoennissen, N.H.; O'Kelly, J.; Lu, D.; Iwanski, G.B.; La, D.T.; Abbassi, S.; Leiter, A.; Karlan, B.; Mehta, R.; Koeffler, H.P. Capsaicin causes cell-cycle arrest and apoptosis in ER-positive and -negative breast cancer cells by modulating the egfr/her-2 pathway. Oncogene 2010, 29, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Polkowski, K.; Mazurek, A.P. Biological properties of genistein. A review of in vitro and in vivo data. Acta Pol. Pharm. Drug Res. 2000, 57, 135–155. [Google Scholar]
- Shao, Z.M.; Wu, J.; Shen, Z.Z.; Barsky, S.H. Genistein exerts multiple suppressive effects on human breast carcinoma cells. Cancer Res. 1998, 58, 4851–4857. [Google Scholar] [PubMed]
- Li, Y.; Upadhyay, S.; Bhuiyan, M.; Sarkar, F.H. Induction of apoptosis in breast cancer cells MDA-MB-231 by genistein. Oncogene 1999, 18, 3166–3172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, P.; Fan, S.; Wang, H.; Mao, J.; Shi, Y.; Ibrahim, M.M.; Ma, W.; Yu, X.; Hou, Z.; Wang, B.; et al. Genistein decreases the breast cancer stem-like cell population through hedgehog pathway. Stem Cell. Res. Ther. 2013, 4, 146. [Google Scholar] [CrossRef] [PubMed]
- Montales, M.T.E.; Rahal, O.M.; Kang, J.; Rogers, T.J.; Prior, R.L.; Wu, X.; Simmen, R.C.M. Repression of mammosphere formation of human breast cancer cells by soy isoflavone genistein and blueberry polyphenolic acids suggests diet-mediated targeting of cancer stem-like/progenitor cells. Carcinogenesis 2012, 33, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.M.; Wu, J.; Shen, Z.Z.; Barsky, S.H. Genistein inhibits both constitutive and EGF-stimulated invasion in ER-negative human breast carcinoma cell lines. Anticancer Res. 1998, 18, 1435–1439. [Google Scholar] [PubMed]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Anand, A.; Kumar, V. Recent developments in biological activities of chalcones: A mini review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Zhang, W.; Sheng, C.; Zhang, W.; Xing, C.; Miao, Z. Chalcone: A privileged structure in medicinal chemistry. Chem. Rev. 2017, 117, 7762–7810. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, D.K.; Bharti, S.K.; Asati, V. Anti-cancer chalcones: Structural and molecular target perspectives. Eur. J. Med. Chem. 2015, 98, 69–114. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.L.; Kuo, P.L.; Tzeng, W.S.; Lin, C.C. Chalcone inhibits the proliferation of human breast cancer cell by blocking cell cycle progression and inducing apoptosis. Food Chem. Toxicol. 2006, 44, 704–713. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.P.; Yearby, L.A.; Stoute, D.; Burow, M.E.; Rhodes, L.V.; Gray, M.; Carriere, P.; Tilghman, S.L.; McLachlan, J.A.; Ochieng, J. In vitro and in vivo evaluation of novel anticancer agents in triple negative breast cancer models. J. Health Care Poor Underserved 2013, 24, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Canela, M.D.; Noppen, S.; Bueno, O.; Prota, A.E.; Bargsten, K.; Saez-Calvo, G.; Jimeno, M.L.; Benkheil, M.; Ribatti, D.; Velazquez, S.; et al. Antivascular and antitumor properties of the tubulin-binding chalcone tub091. Oncotarget 2017, 8, 14325–14342. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.H.; Jiang, X.R.; Chen, G.L.; Guo, W.; Zhang, J.Y.; Cui, L.J.; Li, H.H.; Li, M.; Liu, X.; Yang, J.Y.; et al. Anti-tumor activity of sl4 against breast cancer cells: Induction of g2/m arrest through modulation of the MAPK-dependent p21 signaling pathway. Sci. Rep. 2016, 6, 36486. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lee, I.-S.; Moon, A. 2-hydroxychalcone and xanthohumol inhibit invasion of triple negative breast cancer cells. Chem. Boil. Interact. 2013, 203, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Lindamulage, I.K.; Vu, H.Y.; Karthikeyan, C.; Knockleby, J.; Lee, Y.F.; Trivedi, P.; Lee, H. Novel quinolone chalcones targeting colchicine-binding pocket kill multidrug-resistant cancer cells by inhibiting tubulin activity and mrp1 function. Sci. Rep. 2017, 7, 10298. [Google Scholar] [CrossRef] [PubMed]
- Doucette, C.D.; Hilchie, A.L.; Liwski, R.; Hoskin, D.W. Piperine, a dietary phytochemical, inhibits angiogenesis. J. Nutr. Biochem. 2013, 24, 231–239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Singhal, V.; Roshan, R.; Sharma, A.; Rembhotkar, G.W.; Ghosh, B. Piperine inhibits TNF-alpha induced adhesion of neutrophils to endothelial monolayer through suppression of NF-κB and IκB kinase activation. Eur. J. Pharmacol. 2007, 575, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Manayi, A.; Nabavi, S.M.; Setzer, W.N.; Jafari, S. Piperine as a potential anti-cancer agent: A review on preclinical studies. Curr. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Syed, S.B.; Arya, H.; Fu, I.H.; Yeh, T.K.; Periyasamy, L.; Hsieh, H.P.; Coumar, M.S. Targeting p-glycoprotein: Investigation of piperine analogs for overcoming drug resistance in cancer. Sci. Rep. 2017, 7, 7972. [Google Scholar] [CrossRef] [PubMed]
- Kakarala, M.; Brenner, D.E.; Korkaya, H.; Cheng, C.; Tazi, K.; Ginestier, C.; Liu, S.; Dontu, G.; Wicha, M.S. Targeting breast stem cells with the cancer preventive compounds curcumin and piperine. Breast Cancer Res. Treat. 2010, 122, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Sattarinezhad, E.; Bordbar, A.K.; Fani, N. Piperine derivatives as potential inhibitors of survivin: An in silico molecular docking. Comput. Biol. Med. 2015, 63, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, Y.T.; Fan, M.J.; Huang, A.C.; Lien, J.C.; Lin, J.J.; Chen, J.C.; Hsia, T.C.; Wu, R.S.; Chung, J.G. Deguelin impairs cell adhesion, migration and invasion of human lung cancer cells through the NF-(formula: See text)B signaling pathways. Am. J. Chin. Med. 2018, 46, 209–229. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Zheng, X.; Wang, P.; Guo, S. Deguelin exerts anticancer activity of human gastric cancer mgc-803 and mkn-45 cells in vitro. Int. J. Mol. Med. 2018, 41, 3157–3166. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhao, Z.; Yi, S.; Wen, L.; He, J.; Hu, J.; Ruan, J.; Fang, J.; Chen, Y. Deguelin induced differentiation of mutated npm1 acute myeloid leukemia in vivo and in vitro. Anti-Cancer Drugs 2017, 28, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.-H.; Karna, P.; Regan, R.M.; Liu, X.; Naithani, R.; Moriarty, R.M.; Wood, W.C.; Lee, H.-Y.; Yang, L. Down-regulation of inhibitor of apoptosis proteins by deguelin selectively induces apoptosis in breast cancer cells. Mol. Pharmacol. 2007, 71, 101. [Google Scholar] [CrossRef] [PubMed]
- Murillo, G.; Peng, X.; Torres, K.E.O.; Mehta, R.G. Deguelin inhibits growth of breast cancer cells by modulating the expression of key members of the Wnt signaling pathway. Cancer Prev. Res. 2009, 2, 942. [Google Scholar] [CrossRef] [PubMed]
- Mehta, R.R.; Katta, H.; Kalra, A.; Patel, R.; Gupta, A.; Alimirah, F.; Murillo, G.; Peng, X.; Unni, A.; Muzzio, M.; et al. Efficacy and mechanism of action of deguelin in suppressing metastasis of 4t1 cells. Clin. Exp. Metast. 2013, 30, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Robles, A.J.; Cai, S.; Cichewicz, R.H.; Mooberry, S.L. Selective activity of deguelin identifies therapeutic targets for androgen receptor-positive breast cancer. Breast Cancer Res. Treat. 2016, 157, 475–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, T.; Li, H.; Wang, X.; Wu, Z. Enhancement radiosensitization of breast cancer cells by deguelin. Cancer Biother. Radiopharm. 2008, 23, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Lee, Y.-H.; Sharma, A.R.; Park, J.-B.; Jagga, S.; Sharma, G.; Lee, S.-S.; Nam, J.-S. Quercetin induces apoptosis and cell cycle arrest in triple-negative breast cancer cells through modulation of foxo3a activity. Korean J. Physiol. Pharmacol. 2017, 21, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Duo, J.; Ying, G.G.; Wang, G.W.; Zhang, L. Quercetin inhibits human breast cancer cell proliferation and induces apoptosis via bcl-2 and bax regulation. Mol. Med. Rep. 2012, 5, 1453–1456. [Google Scholar] [PubMed]
- Deng, X.H.; Song, H.Y.; Zhou, Y.F.; Yuan, G.Y.; Zheng, F.J. Effects of quercetin on the proliferation of breast cancer cells and expression of survivin in vitro. Exp. Ther. Med. 2013, 6, 1155–1158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, L.; Hou, X.; Fan, F.; Wu, H. Quercetin stimulates mitochondrial apoptosis dependent on activation of endoplasmic reticulum stress in hepatic stellate cells. Pharm. Biol. 2016, 54, 3237–3243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, S.Y.; Wu, Y.C.; Chung, J.G.; Yang, J.S.; Lu, H.F.; Tsou, M.F.; Wood, W.G.; Kuo, S.J.; Chen, D.R. Quercetin-induced apoptosis acts through mitochondrial- and caspase-3-dependent pathways in human breast cancer MDA-MB-231 cells. Hum. Exp. Toxicol. 2009, 28, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.J.; Bae, S.M.; Ahn, W.S. Antiproliferative effects of quercetin through cell cycle arrest and apoptosis in human breast cancer mda-mb-453 cells. Arch. Pharm. Res. 2008, 31, 1281–1285. [Google Scholar] [CrossRef] [PubMed]
- Steiner, J.L.; Davis, J.M.; McClellan, J.L.; Enos, R.T.; Carson, J.A.; Fayad, R.; Nagarkatti, M.; Nagarkatti, P.S.; Altomare, D.; Creek, K.E.; et al. Dose-dependent benefits of quercetin on tumorigenesis in the c3(1)/sv40tag transgenic mouse model of breast cancer. Cancer Boil. Ther. 2014, 15, 1456–1467. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, A.; Thangavel, C.; Liu, Y.; Shoyele, S.; Den, R.B.; Selvakumar, P.; Lakshmikuttyamma, A. Quercetin regulates beta-catenin signaling and reduces the migration of triple negative breast cancer. Mol. Carcinog. 2016, 55, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Manouchehri, J.M.; Turner, K.A.; Kalafatis, M. Trail-induced apoptosis in trail-resistant breast carcinoma through quercetin cotreatment. Breast Cancer 2018, 12. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Miao, L.; Goodwin, T.J.; Li, J.; Liu, Q.; Huang, L. Quercetin remodels the tumor microenvironment to improve the permeation, retention, and antitumor effects of nanoparticles. ACS Nano 2017, 11, 4916–4925. [Google Scholar] [CrossRef] [PubMed]
- Murugan, C.; Rayappan, K.; Thangam, R.; Bhanumathi, R.; Shanthi, K.; Vivek, R.; Thirumurugan, R.; Bhattacharyya, A.; Sivasubramanian, S.; Gunasekaran, P.; et al. Combinatorial nanocarrier based drug delivery approach for amalgamation of anti-tumor agents in bresat cancer cells: An improved nanomedicine strategies. Sci. Rep. 2016, 6, 34053. [Google Scholar] [CrossRef] [PubMed]
- Demiroglu-Zergeroglu, A.; Basara-Cigerim, B.; Kilic, E.; Yanikkaya-Demirel, G. The investigation of effects of quercetin and its combination with cisplatin on malignant mesothelioma cells in vitro. J. Biomed. Biotechnol. 2010, 2010, 851589. [Google Scholar] [CrossRef] [PubMed]
- Demiroglu-Zergeroglu, A.; Ergene, E.; Ayvali, N.; Kuete, V.; Sivas, H. Quercetin and cisplatin combined treatment altered cell cycle and mitogen activated protein kinase expressions in malignant mesotelioma cells. BMC Complement. Altern. Med. 2016, 16, 281. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.; Han, H.; Ma, L.; Zhou, H.; Zhao, C. The chemosensitization effect of quercetin on cisplatin induces the apoptosis of human colon cancer ht-29 cell line. Int. J. Clin. Exp. Med. 2016, 9, 2285–2292. [Google Scholar]
- Shindikar, A.; Singh, A.; Nobre, M.; Kirolikar, S. Curcumin and resveratrol as promising natural remedies with nanomedicine approach for the effective treatment of triple negative breast cancer. J. Oncol. 2016, 2016, 9750785. [Google Scholar] [CrossRef] [PubMed]
- Kuhar, M.; Imran, S.; Singh, N. Curcumin and quercetin combined with cisplatin to induce apoptosis in human laryngeal carcinoma hep-2 cells through the mitochondrial pathway. J. Cancer Mol. 2007, 3, 121–128. [Google Scholar]
- Mohana, S.; Ganesan, M.; Agilan, B.; Karthikeyan, R.; Srithar, G.; Beaulah Mary, R.; Ananthakrishnan, D.; Velmurugan, D.; Rajendra Prasad, N.; Ambudkar, S.V. Screening dietary flavonoids for the reversal of p-glycoprotein-mediated multidrug resistance in cancer. Mol. Biosyst. 2016, 12, 2458–2470. [Google Scholar] [CrossRef] [PubMed]
- Perk, A.A.; Shatynska-Mytsyk, I.; Gercek, Y.C.; Boztas, K.; Yazgan, M.; Fayyaz, S.; Farooqi, A.A. Rutin mediated targeting of signaling machinery in cancer cells. Cancer Cell Int. 2014, 14, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Sghaier, M.; Pagano, A.; Mousslim, M.; Ammari, Y.; Kovacic, H.; Luis, J. Rutin inhibits proliferation, attenuates superoxide production and decreases adhesion and migration of human cancerous cells. Biomed. Pharmacother. 2016, 84, 1972–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Miao, Q.; Geng, M.; Liu, J.; Hu, Y.; Tian, L.; Pan, J.; Yang, Y. Anti-tumor effect of rutin on human neuroblastoma cell lines through inducing G2/m cell cycle arrest and promoting apoptosis. Sci. World J. 2013, 2013, 269165. [Google Scholar] [CrossRef] [PubMed]
- Iriti, M.; Kubina, R.; Cochis, A.; Sorrentino, R.; Varoni, E.M.; Kabala-Dzik, A.; Azzimonti, B.; Dziedzic, A.; Rimondini, L.; Wojtyczka, R.D. Rutin, a quercetin glycoside, restores chemosensitivity in human breast cancer cells. Phytother. Res. PTR 2017, 31, 1529–1538. [Google Scholar] [CrossRef] [PubMed]
- Sundarraj, K.; Raghunath, A.; Perumal, E. A review on the chemotherapeutic potential of fisetin: In vitro evidences. Biomed. Pharmacother. 2017, 97, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Youns, M.; Hegazy, WA.H. The natural flavonoid fisetin inhibits cellular proliferation of hepatic, colorectal, and pancreatic cancer cells through modulation of multiple signaling pathways. PLoS ONE 2017, 12, e0169335. [Google Scholar] [CrossRef] [PubMed]
- Pawar, A.; Singh, S.; Rajalakshmi, S.; Shaikh, K.; Bothiraja, C. Development of fisetin-loaded folate functionalized pluronic micelles for breast cancer targeting. Artif. Cells Nanomed. Biotechnol. 2018, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Murphy, K.; Doucette, C.D.; Greenshields, A.L.; Hoskin, D.W. The dietary flavonoid fisetin causes cell cycle arrest, caspase-dependent apoptosis, and enhanced cytotoxicity of chemotherapeutic drugs in triple-negative breast cancer cells. J. Cell. Biochem. 2016, 117, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, S.S.; Canto, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Hwang, K.A.; Choi, K.C. Anti-metastatic potential of resveratrol and its metabolites by the inhibition of epithelial-mesenchymal transition, migration, and invasion of malignant cancer cells. Phytomedicine 2016, 23, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Singh, C.K.; Ndiaye, M.A.; Ahmad, N. Resveratrol and cancer: Challenges for clinical translation. Biochim. Biophys. Acta 2015, 1852, 1178–1185. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Chen, K.; Cheng, L.; Yan, B.; Qian, W.; Cao, J.; Li, J.; Wu, E.; Ma, Q.; Yang, W. Resveratrol and cancer treatment: Updates. Ann. N. Y. Acad. Sci. 2017, 1403, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Tome-Carneiro, J.; Larrosa, M.; Gonzalez-Sarrias, A.; Tomas-Barberan, F.A.; Garcia-Conesa, M.T.; Espin, J.C. Resveratrol and clinical trials: The crossroad from in vitro studies to human evidence. Curr. Pharm. Des. 2013, 19, 6064–6093. [Google Scholar] [CrossRef] [PubMed]
- Varoni, E.M.; Lo Faro, A.F.; Sharifi-Rad, J.; Iriti, M. Anticancer molecular mechanisms of resveratrol. Front. Nutr. 2016, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Sinha, D.; Sarkar, N.; Biswas, J.; Bishayee, A. Resveratrol for breast cancer prevention and therapy: Preclinical evidence and molecular mechanisms. Semin. Cancer Biol. 2016, 40–41, 209–232. [Google Scholar] [CrossRef] [PubMed]
- Vinod, B.S.; Nair, H.H.; Vijayakurup, V.; Shabna, A.; Shah, S.; Krishna, A.; Pillai, K.S.; Thankachan, S.; Anto, R.J. Resveratrol chemosensitizes her-2-overexpressing breast cancer cells to docetaxel chemoresistance by inhibiting docetaxel-mediated activation of her-2-Akt axis. Cell Death Discov. 2015, 1, 15061. [Google Scholar] [CrossRef] [PubMed]
- Alayev, A.; Berger, S.M.; Kramer, M.Y.; Schwartz, N.S.; Holz, M.K. The combination of rapamycin and resveratrol blocks autophagy and induces apoptosis in breast cancer cells. J. Cell. Biochem. 2015, 116, 450–457. [Google Scholar] [CrossRef] [PubMed]
- Sprouse, A.A.; Herbert, B.S. Resveratrol augments paclitaxel treatment in MDA-MB-231 and paclitaxel-resistant MDA-MB-231 breast cancer cells. Anticancer Res. 2014, 34, 5363–5374. [Google Scholar] [PubMed]
- Bove, K.; Lincoln, D.W.; Tsan, M.F. Effect of resveratrol on growth of 4t1 breast cancer cells in vitro and in vivo. Biochem. Biophys. Res. Commun. 2002, 291, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Kotha, A.; Sekharam, M.; Cilenti, L.; Siddiquee, K.; Khaled, A.; Zervos, A.S.; Carter, B.; Turkson, J.; Jove, R. Resveratrol inhibits SRC and STAT3 signaling and induces the apoptosis of malignant cells containing activated STAT3 protein. Mol. Cancer Ther. 2006, 5, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Laux, M.T.; Aregullin, M.; Berry, J.P.; Flanders, J.A.; Rodriguez, E. Identification of a p53-dependent pathway in the induction of apoptosis of human breast cancer cells by the natural product, resveratrol. J. Altern. Complement. Med. 2004, 10, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.Y.; Shih, A.; Cao, H.J.; Davis, F.B.; Davis, P.J.; Lin, H.Y. Resveratrol-induced cyclooxygenase-2 facilitates p53-dependent apoptosis in human breast cancer cells. Mol. Cancer Ther. 2006, 5, 2034–2042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.H.; Mustafa, F.B.; Pervaiz, S.; Ng, F.S.; Lim, L.H. ERK1/2 activation is required for resveratrol-induced apoptosis in MDA-MB-231 cells. Int. J. Oncol. 2008, 33, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Shin, Y.J.; Won, A.J.; Lee, B.M.; Choi, W.S.; Jung, J.H.; Chung, H.Y.; Kim, H.S. Resveratrol enhances chemosensitivity of doxorubicin in multidrug-resistant human breast cancer cells via increased cellular influx of doxorubicin. Biochim. Biophys. Acta 2014, 1840, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.F.; Pan, M.H.; Chiou, Y.S.; Cheng, A.C.; Huang, H. Resveratrol modulates med28 (magicin/eg-1) expression and inhibits epidermal growth factor (EGF)-induced migration in MDA-MB-231 human breast cancer cells. J. Agric. Food Chem. 2011, 59, 11853–11861. [Google Scholar] [CrossRef] [PubMed]
- Kala, R.; Shah, H.N.; Martin, S.L.; Tollefsbol, T.O. Epigenetic-based combinatorial resveratrol and pterostilbene alters DNA damage response by affecting SIRT1 and DNMT enzyme expression, including sirt1-dependent gamma-H2AX and telomerase regulation in triple-negative breast cancer. BMC Cancer 2015, 15, 672. [Google Scholar] [CrossRef] [PubMed]
- Kala, R.; Tollefsbol, T.O. A novel combinatorial epigenetic therapy using resveratrol and pterostilbene for restoring estrogen receptor-alpha (ERα) expression in ERalpha-negative breast cancer cells. PLoS ONE 2016, 11, e0155057. [Google Scholar] [CrossRef] [PubMed]
- Schlachterman, A.; Valle, F.; Wall, K.M.; Azios, N.G.; Castillo, L.; Morell, L.; Washington, A.V.; Cubano, L.A.; Dharmawardhane, S.F. Combined resveratrol, quercetin, and catechin treatment reduces breast tumor growth in a nude mouse model. Transl. Oncol. 2008, 1, 19–27. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wang, Y.; Zhu, J.; Orloff, M.; Eng, C. Resveratrol enhances the anti-tumor activity of the mTOR inhibitor rapamycin in multiple breast cancer cell lines mainly by suppressing rapamycin-induced Akt signaling. Cancer Lett. 2011, 301, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Wu, X.N.; Chen, J.; Wang, W.X.; Lu, Z.F. Resveratrol reverses multidrug resistance in human breast cancer doxorubicin-resistant cells. Exp. Ther. Med. 2014, 7, 1611–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreani, C.; Bartolacci, C.; Wijnant, K.; Crinelli, R.; Bianchi, M.; Magnani, M.; Hysi, A.; Iezzi, M.; Amici, A.; Marchini, C. Resveratrol fuels HER2 and ERα-positive breast cancer behaving as proteasome inhibitor. Aging 2017, 9, 508–523. [Google Scholar] [PubMed] [Green Version]
- Fukui, M.; Yamabe, N.; Kang, K.S.; Zhu, B.T. Growth-stimulatory effect of resveratrol in human cancer cells. Mol. Carcinog. 2010, 49, 750–759. [Google Scholar] [CrossRef] [PubMed]
- Fukui, M.; Choi, H.J.; Zhu, B.T. Mechanism for the protective effect of resveratrol against oxidative stress-induced neuronal death. Free Radic. Biol. Med. 2010, 49, 800–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, M.; Yamabe, N.; Zhu, B.T. Resveratrol attenuates the anticancer efficacy of paclitaxel in human breast cancer cells in vitro and in vivo. Eur. J. Cancer 2010, 46, 1882–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Huyan, T.; Ye, L.J.; Li, J.; Shi, J.L.; Huang, Q.S. Concentration-dependent biphasic effects of resveratrol on human natural killer cells in vitro. J. Agric. Food Chem. 2014, 62, 10928–10935. [Google Scholar] [CrossRef] [PubMed]
- Szende, B.; Tyihak, E.; Kiraly-Veghely, Z. Dose-dependent effect of resveratrol on proliferation and apoptosis in endothelial and tumor cell cultures. Exp. Mol. Med. 2000, 32, 88–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calabrese, E.J.; Mattson, M.P.; Calabrese, V. Resveratrol commonly displays hormesis: Occurrence and biomedical significance. Hum. Exp. Toxicol. 2010, 29, 980–1015. [Google Scholar] [CrossRef] [PubMed]
- Prasad, S.; Aggarwal, B.B. Turmeric, the golden spice: From traditional medicine to modern medicine. In Herbal Medicine: Biomolecular and Clinical Aspects, 2nd ed.; Benzie, I.F.F., Wachtel-Galor, S., Eds.; CRC Press: Boca Raton, FL, USA, 2011. [Google Scholar]
- Kunnumakkara, A.B.; Bordoloi, D.; Padmavathi, G.; Monisha, J.; Roy, N.K.; Prasad, S.; Aggarwal, B.B. Curcumin, the golden nutraceutical: Multitargeting for multiple chronic diseases. Br. J. Pharmacol. 2017, 174, 1325–1348. [Google Scholar] [CrossRef] [PubMed]
- Kunnumakkara, A.B.; Bordoloi, D.; Harsha, C.; Banik, K.; Gupta, S.C.; Aggarwal, B.B. Curcumin mediates anticancer effects by modulating multiple cell signaling pathways. Clin. Sci. 2017, 131, 1781–1799. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, M.K.; Rane, G.; Kanchi, M.M.; Arfuso, F.; Chinnathambi, A.; Zayed, M.E.; Alharbi, S.A.; Tan, B.K.; Kumar, A.P.; Sethi, G. The multifaceted role of curcumin in cancer prevention and treatment. Molecules 2015, 20, 2728–2769. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, A.H.; Al Zohairy, M.A.; Aly, S.M.; Khan, M.A. Curcumin: A potential candidate in prevention of cancer via modulation of molecular pathways. BioMed Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, M.; Calaf, G.M. Curcumin inhibits invasive capabilities through epithelial mesenchymal transition in breast cancer cell lines. Int. J. Oncol. 2016, 49, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Palange, A.L.; Di Mascolo, D.; Singh, J.; De Franceschi, M.S.; Carallo, C.; Gnasso, A.; Decuzzi, P. Modulating the vascular behavior of metastatic breast cancer cells by curcumin treatment. Front. Oncol. 2012, 2, 161. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Liang, Y.; Jiang, B.; Li, X.; Xun, H.; Sun, J.; He, W.; Lau, H.T.; Ma, X. Curcumin inhibits intracellular fatty acid synthase and induces apoptosis in human breast cancer MDA-MB-231 cells. Oncol. Rep. 2016, 35, 2651–2656. [Google Scholar] [CrossRef] [PubMed]
- Chiu, T.L.; Su, C.C. Curcumin inhibits proliferation and migration by increasing the bax to bcl-2 ratio and decreasing NF-κBp65 expression in breast cancer MDA-MB-231 cells. Int. J. Mol. Med. 2009, 23, 469–475. [Google Scholar] [PubMed]
- Prasad, C.P.; Rath, G.; Mathur, S.; Bhatnagar, D.; Ralhan, R. Potent growth suppressive activity of curcumin in human breast cancer cells: Modulation of Wnt/beta-catenin signaling. Chem. Biol. Interact. 2009, 181, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Hua, W.F.; Fu, Y.S.; Liao, Y.J.; Xia, W.J.; Chen, Y.C.; Zeng, Y.X.; Kung, H.F.; Xie, D. Curcumin induces down-regulation of ezh2 expression through the MAPK pathway in mda-mb-435 human breast cancer cells. Eur. J. Pharmacol. 2010, 637, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.H.; Huang, H.C.; Huang, C.; Lin, J.K. Cycle arrest and apoptosis in MDA-MB-231/her2 cells induced by curcumin. Eur. J. Pharmacol. 2012, 690, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Loo, W.T.; Sze, S.C.; Tong, Y. Curcumin inhibits cell proliferation of MDA-MB-231 and bt-483 breast cancer cells mediated by down-regulation of NF-κB, cyclind and mmp-1 transcription. Phytomedicine 2009, 16, 916–922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vinod, B.S.; Antony, J.; Nair, H.H.; Puliyappadamba, V.T.; Saikia, M.; Narayanan, S.S.; Bevin, A.; Anto, R.J. Mechanistic evaluation of the signaling events regulating curcumin-mediated chemosensitization of breast cancer cells to 5-fluorouracil. Cell Death Dis. 2013, 4, e505. [Google Scholar] [CrossRef] [PubMed]
- Thulasiraman, P.; McAndrews, D.J.; Mohiudddin, I.Q. Curcumin restores sensitivity to retinoic acid in triple negative breast cancer cells. BMC Cancer 2014, 14, 724. [Google Scholar] [CrossRef] [PubMed]
- Falah, R.R.; Talib, W.H.; Shbailat, S.J. Combination of metformin and curcumin targets breast cancer in mice by angiogenesis inhibition, immune system modulation and induction of p53 independent apoptosis. Ther. Adv. Med. Oncol. 2017, 9, 235–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cridge, B.J.; Larsen, L.; Rosengren, R.J. Curcumin and its derivatives in breast cancer: Current developments and potential for the treatment of drug-resistant cancers. Oncol. Discov. 2013, 1, 6. [Google Scholar] [CrossRef]
- Lin, L.; Hutzen, B.; Ball, S.; Foust, E.; Sobo, M.; Deangelis, S.; Pandit, B.; Friedman, L.; Li, C.; Li, P.K.; et al. New curcumin analogues exhibit enhanced growth-suppressive activity and inhibit akt and signal transducer and activator of transcription 3 phosphorylation in breast and prostate cancer cells. Cancer Sci. 2009, 100, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, L.; Zhang, X.; Liang, Y.; Pu, Z.; Wang, L.; Mo, J. Curcumin: A calixarene derivative micelle potentiates anti-breast cancer stem cells effects in xenografted, triple-negative breast cancer mouse models. Drug Deliv. 2017, 24, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, L.; Bellini, M.; Vanna, R.; Morasso, C.; Zago, A.; Carcano, S.; Avvakumova, S.; Bertolini, J.A.; Rizzuto, M.A.; Colombo, M.; et al. H-ferritin enriches the curcumin uptake and improves the therapeutic efficacy in triple negative breast cancer cells. Biomacromolecules 2017, 18, 3318–3330. [Google Scholar] [CrossRef] [PubMed]
- Mittal, L.; Raman, V.; Camarillo, I.G.; Sundararajan, R. Ultra-microsecond pulsed curcumin for effective treatment of triple negative breast cancers. Biochem. Biophys. Res. Commun. 2017, 491, 1015–1020. [Google Scholar] [CrossRef] [PubMed]
- Taurin, S.; Nehoff, H.; Diong, J.; Larsen, L.; Rosengren, R.J.; Greish, K. Curcumin-derivative nanomicelles for the treatment of triple negative breast cancer. J. Drug Target. 2013, 21, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Robles, A.J.; King, J.B.; Powell, D.R.; Miller, A.N.; Mooberry, S.L.; Cichewicz, R.H. Crowdsourcing natural products discovery to access uncharted dimensions of fungal metabolite diversity. Angew. Chem. 2014, 126, 823–828. [Google Scholar] [CrossRef]
- Robles, A.J.; Du, L.; King, J.B.; Cichewicz, R.H.; Mooberry, S.L. Abstract 5519: Investigation of the cellular mechanisms of action of maximiscin, a novel natural product with selective cytotoxic activity against a triple negative breast cancer molecular subtype. Cancer Res. 2014, 74, 5519. [Google Scholar] [CrossRef]
- Lee, S.T.; Welch, K.D.; Panter, K.E.; Gardner, D.R.; Garrossian, M.; Chang, C.W. Cyclopamine: From cyclops lambs to cancer treatment. J. Agric. Food Chem. 2014, 62, 7355–7362. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Hallahan, A.R.; Pritchard, J.I.; Eberhart, C.G.; Watkins, D.N.; Chen, J.K.; Cooper, M.K.; Taipale, J.; Olson, J.M.; et al. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science 2002, 297, 1559–1561. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Jaks, V.; Fiaschi, M.; Toftgård, R. Hedgehog signalling in breast cancer. Carcinogenesis 2009, 30, 903–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.K.; Taipale, J.; Cooper, M.K.; Beachy, P.A. Inhibition of hedgehog signaling by direct binding of cyclopamine to smoothened. Genes Dev. 2002, 16, 2743–2748. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Harrington, N.; Moraes, R.C.; Wu, M.F.; Hilsenbeck, S.G.; Lewis, M.T. Cyclopamine inhibition of human breast cancer cell growth independent of smoothened (Smo). Breast Cancer Res. Treat. 2009, 115, 505–521. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Wang, W.; Liu, D.; Zhao, Y.; He, J.; Wang, X.; Dai, Z.; Zhang, H.; Li, X. Targeting of sonic hedgehog-gli signaling: A potential therapeutic target for patients with breast cancer. Oncol. Lett. 2016, 12, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Frolova, N.; Sadlonova, A.; Novak, Z.; Steg, A.; Page, G.P.; Welch, D.R.; Lobo-Ruppert, S.M.; Ruppert, J.M.; Johnson, M.R.; et al. Hedgehog signaling and response to cyclopamine differ in epithelial and stromal cells in benign breast and breast cancer. Cancer Biol. Ther. 2006, 5, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Chai, F.; Zhou, J.; Chen, C.; Xie, S.; Chen, X.; Su, P.; Shi, J. The hedgehog inhibitor cyclopamine antagonizes chemoresistance of breast cancer cells. Oncol. Targets Ther. 2013, 6, 1643–1647. [Google Scholar]
- Boamah, E.; Ibrahim, Q.; Kwinji, L.; Patel, R.; Ajayi, D.; Danquah, M. EGFR inhibitors in combination with cyclopamine as chemotherapeutic strategy for treating breast cancer. Synergy 2015, 2, 7–18. [Google Scholar] [CrossRef]
- Sabol, M.; Trnski, D.; Uzarevic, Z.; Ozretic, P.; Musani, V.; Rafaj, M.; Cindric, M.; Levanat, S. Combination of cyclopamine and tamoxifen promotes survival and migration of MCF-7 breast cancer cells-interaction of hedgehog-Gli and estrogen receptor signaling pathways. PLoS ONE 2014, 9, e114510. [Google Scholar] [CrossRef] [PubMed]
- Mahindroo, N.; Punchihewa, C.; Fujii, N. Hedgehog-Gli signaling pathway inhibitors as anticancer agents. J. Med. Chem. 2009, 52, 3829–3845. [Google Scholar] [CrossRef] [PubMed]
- Bode, A.M.; Dong, Z. The two faces of capsaicin. Cancer Res. 2011, 71, 2809–2814. [Google Scholar] [CrossRef] [PubMed]
- Clark, R.; Lee, S.H. Anticancer properties of capsaicin against human cancer. Anticancer Res. 2016, 36, 837–843. [Google Scholar] [PubMed]
- Pramanik, K.C.; Srivastava, S.K. Role of capsaicin in cancer prevention. In Role of Capsaicin in Oxidative Stress and Cancer: Diet and Cancer; Srivastava, S.K., Ed.; Springer Science & Business Media: Heidelberg, Germany; New York, NY, USA; London, UK, 2013; Volume 1, pp. 1–18. [Google Scholar]
- Bley, K.; Boorman, G.; Mohammad, B.; McKenzie, D.; Babbar, S. A comprehensive review of the carcinogenic and anticarcinogenic potential of capsaicin. Toxicol. Pathol. 2012, 40, 847–873. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhu, P.; Tao, Y.; Shen, C.; Wang, S.; Zhao, L.; Wu, H.; Fan, F.; Lin, C.; Chen, C.; et al. Cancer-promoting effect of capsaicin on DMBA/TPA-induced skin tumorigenesis by modulating inflammation, ERK and p38 in mice. Food Chem. Toxicol. 2015, 81, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hwang, M.K.; Bode, A.M.; Byun, S.; Song, N.R.; Lee, H.J.; Lee, K.W.; Dong, Z. Cocarcinogenic effect of capsaicin involves activation of EGFR signaling but not TRPV1. Cancer Res. 2010, 70, 6859–6869. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.; Ahmad, A.; Yang, H.; Sarkar, F.H. Tumor cell growth inhibition is correlated with levels of capsaicin present in hot peppers. Nutr. Cancer 2011, 63, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Min, J.K.; Han, K.Y.; Kim, E.C.; Kim, Y.M.; Lee, S.W.; Kim, O.H.; Kim, K.W.; Gho, Y.S.; Kwon, Y.G. Capsaicin inhibits in vitro and in vivo angiogenesis. Cancer Res. 2004, 64, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Chen, S.T.; Chien, S.Y.; Kuo, S.J.; Tsai, H.T.; Chen, D.R. Capsaicin may induce breast cancer cell death through apoptosis-inducing factor involving mitochondrial dysfunction. Hum. Exp. Toxicol. 2011, 30, 1657–1665. [Google Scholar] [CrossRef] [PubMed]
- Weber, L.V.; Al-Refae, K.; Wolk, G.; Bonatz, G.; Altmuller, J.; Becker, C.; Gisselmann, G.; Hatt, H. Expression and functionality of TRPV1 in breast cancer cells. Breast Cancer 2016, 8, 243–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, A.K.; Tavares, M.T.; Pasqualoto, K.F.; de Azevedo, R.A.; Teixeira, S.F.; Ferreira-Junior, W.A.; Bertin, A.M.; de-Sa-Junior, P.L.; Barbuto, J.A.; Figueiredo, C.R.; et al. Rpf151, a novel capsaicin-like analogue: In vitro studies and in vivo preclinical antitumor evaluation in a breast cancer model. Tumor Biol. 2015, 36, 7251–7267. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.T.; Lee, Y.K.; Shin, J.I.; Park, O.J. Anti-inflammatory and anticarcinogenic effect of genistein alone or in combination with capsaicin in TPA-treated rat mammary glands or mammary cancer cell line. Ann. N. Y. Acad. Sci. 2009, 1171, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Li, Y.; Wang, Z.; Sarkar, F.H. Multi-targeted therapy of cancer by genistein. Cancer Lett. 2008, 269, 226–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uifalean, A.; Schneider, S.; Ionescu, C.; Lalk, M.; Iuga, C.A. Soy isoflavones and breast cancer cell lines: Molecular mechanisms and future perspectives. Molecules 2016, 21, 13. [Google Scholar] [CrossRef] [PubMed]
- Gwin, J.; Drews, N.; Ali, S.; Stamschror, J.; Sorenson, M.; Rajah, T.T. Effect of genistein on P90RSK phosphorylation and cell proliferation in T47D breast cancer cells. Anticancer Res. 2011, 31, 209–214. [Google Scholar] [PubMed]
- Sotoca, A.M.; Ratman, D.; van der Saag, P.; Strom, A.; Gustafsson, J.A.; Vervoort, J.; Rietjens, I.M.; Murk, A.J. Phytoestrogen-mediated inhibition of proliferation of the human t47d breast cancer cells depends on the ERalpha/ERbeta ratio. J. Steroid Biochem. Mol. Boil. 2008, 112, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.F.; Huang, M.H.; Tzang, C.H.; Yang, M.; Wong, M.S. Inhibitory actions of genistein in human breast cancer (MCF-7) cells. Biochim. Biophys Acta 2003, 1638, 187–196. [Google Scholar] [CrossRef]
- Pan, H.; Zhou, W.; He, W.; Liu, X.; Ding, Q.; Ling, L.; Zha, X.; Wang, S. Genistein inhibits MDA-MB-231 triple-negative breast cancer cell growth by inhibiting NF-κB activity via the Notch-1 pathway. Int. J. Mol. Med. 2012, 30, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Zhang, Q.; Wang, X.; Yang, X.; Wang, X.; Huang, Z.; Jiao, Y.; Wang, J. Quantitative phosphoproteomics reveals genistein as a modulator of cell cycle and DNA damage response pathways in triple-negative breast cancer cells. Int. J. Oncol. 2016, 48, 1016–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, H.S.; Ju, J.H.; Jang, K.; Shin, I. Induction of apoptotic cell death by phytoestrogens by up-regulating the levels of phospho-p53 and p21 in normal and malignant estrogen receptor alpha-negative breast cells. Nutr. Res. 2011, 31, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Li, J.; Mo, B.; Hu, C.; Liu, H.; Qi, H.; Wang, X.; Xu, J. Genistein induces cell apoptosis in MDA-MB-231 breast cancer cells via the mitogen-activated protein kinase pathway. Toxicol. In Vitro 2008, 22, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Ziaei, S.; Halaby, R. Dietary isoflavones and breast cancer risk. Medicine 2017, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, R.M.; Al-Katib, A.; Aboukameel, A.; Doerge, D.R.; Sarkar, F.; Kucuk, O. Genistein sensitizes diffuse large cell lymphoma to chop (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy. Mol. Cancer Ther. 2003, 2, 1361–1368. [Google Scholar] [PubMed]
- Kaushik, S.; Shyam, H.; Sharma, R.; Balapure, A.K. Genistein synergizes centchroman action in human breast cancer cells. Indian J. Pharmacol. 2016, 48, 637–642. [Google Scholar] [PubMed]
- Hormann, V.; Kumi-Diaka, J.; Durity, M.; Rathinavelu, A. Anticancer activities of genistein-topotecan combination in prostate cancer cells. J. Cell. Mol. Med. 2012, 16, 2631–2636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, H.; Yamamoto, D.; Kiyozuka, Y.; Tsuta, K.; Uemura, Y.; Hioki, K.; Tsutsui, Y.; Tsubura, A. Effects of genistein and synergistic action in combination with eicosapentaenoic acid on the growth of breast cancer cell lines. J. Cancer Res. Clin. Oncol. 2000, 126, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Maginnes, A.; Apenten, R.O. Methotrexate combination effects with genistein and daidzein on MDA-MB-231 breast cancer cell viability. J. Appl. Life Sci. Int. 2017, 13, 1–9. [Google Scholar] [CrossRef]
- Rajah, T.T.; Du, N.; Drews, N.; Cohn, R. Genistein in the presence of 17beta-estradiol inhibits proliferation of ERbeta breast cancer cells. Pharmacology 2009, 84, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Bouker, K.B.; Hilakivi-Clarke, L. Genistein: Does it prevent or promote breast cancer? Environ. Health Perspect. 2000, 108, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Fung, M.K.L.; Chan, G.C. Drug-induced amino acid deprivation as strategy for cancer therapy. J. Hematol. Oncol. 2017, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Story, M.D.; Voehringer, D.W.; Stephens, L.C.; Meyn, R.E. L-asparaginase kills lymphoma cells by apoptosis. Cancer Chemother. Pharmacol. 1993, 32, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sarkissyan, M.; Vadgama, J.V. Epithelial-mesenchymal transition and breast cancer. J. Clin. Med. 2016, 5, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.Q.; Ma, C.; Wang, Q.; Song, Y.; Lv, T. The role of twist1 in epithelial-mesenchymal transition and cancers. Tumor Biol. 2016, 37, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; He, G.; Yan, S.; Chen, C.; Song, L.; Rosol, T.J.; Deng, X. Triple-negative breast cancer: Is there a treatment on the horizon? Oncotarget 2017, 8, 1913–1924. [Google Scholar] [CrossRef] [PubMed]
- Metzger-Filho, O.; Tutt, A.; de Azambuja, E.; Saini, K.S.; Viale, G.; Loi, S.; Bradbury, I.; Bliss, J.M.; Azim, H.A.; Ellis, P.; et al. Dissecting the heterogeneity of triple-negative breast cancer. J. Clin. Oncol. 2012, 30, 1879–1887. [Google Scholar] [CrossRef] [PubMed]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Boil. Med. 2015, 12, 106–116. [Google Scholar]
- Rapoport, B.L.; Nayler, S.; Demetriou, G.S.; Moodley, S.D.; Benn, C.A. Triple negative breast cancer pathologic diagnosis and current chemotherapy treatment options. Oncol. Hematol. Rev. 2014, 10, 25–32. [Google Scholar]
- Ho, J.W.; Cheung, M.W. Combination of phytochemicals as adjuvants for cancer therapy. Recent Patents Anti-Cancer Drug Discov. 2014, 9, 297–302. [Google Scholar] [CrossRef]
- Guestini, F.; McNamara, K.M.; Sasano, H. The use of chemosensitizers to enhance the response to conventional therapy in triple-negative breast cancer patients. Breast Cancer Manag. 2017, 6, 127–131. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, E.J.; Baldwin, L.A. Hormesis: The dose-response revolution. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 175–197. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Robles, A.J.; King, J.B.; Powell, D.R.; Miller, A.N.; Mooberry, S.L.; Cichewicz, R.H. Maximiscin, a novel shikimate-polyketide-nrps hybrid metabolite obtained from tolypocladium sp. with potent antitumor activities. Planta Med. 2013, 79, CL3. [Google Scholar] [CrossRef]
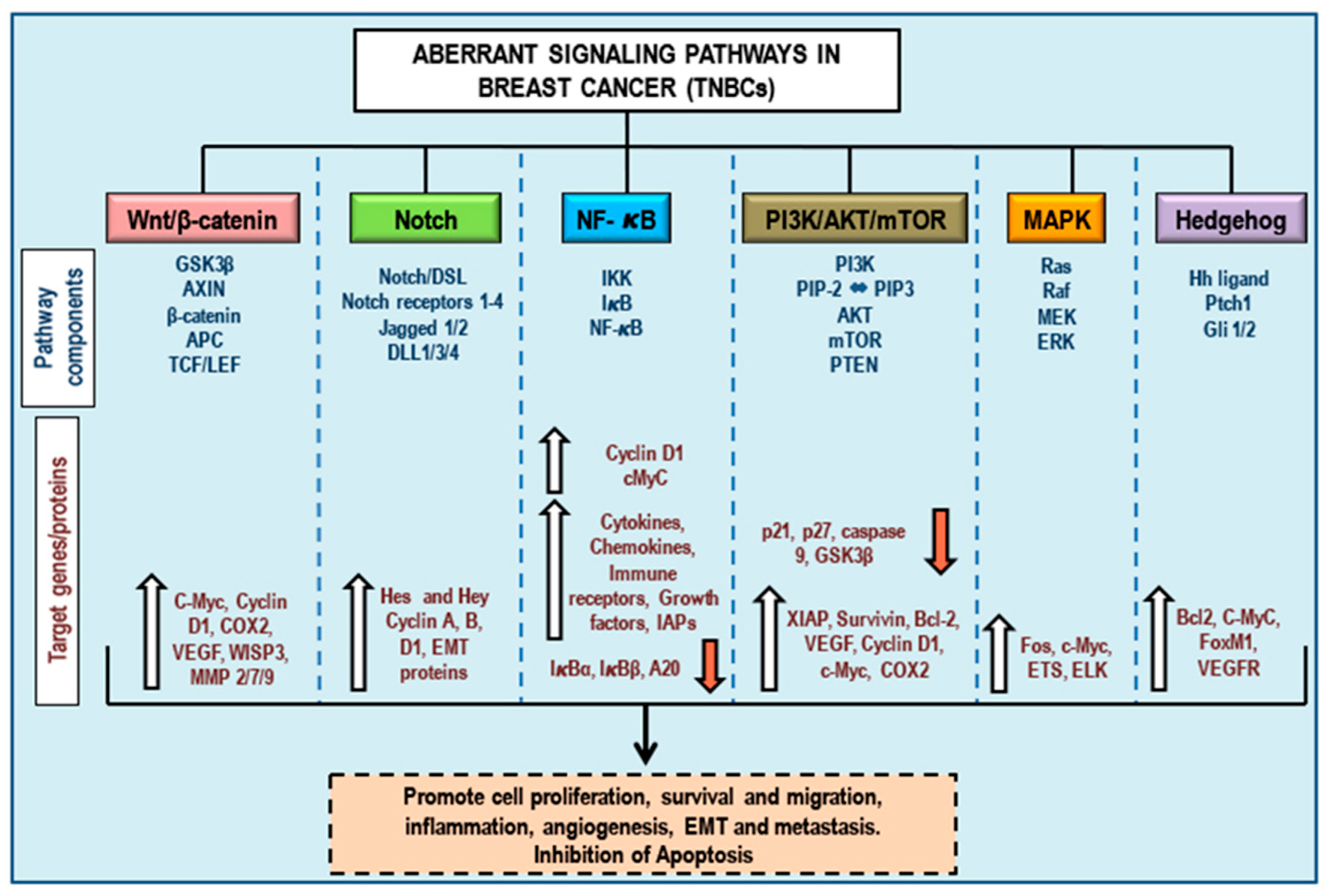
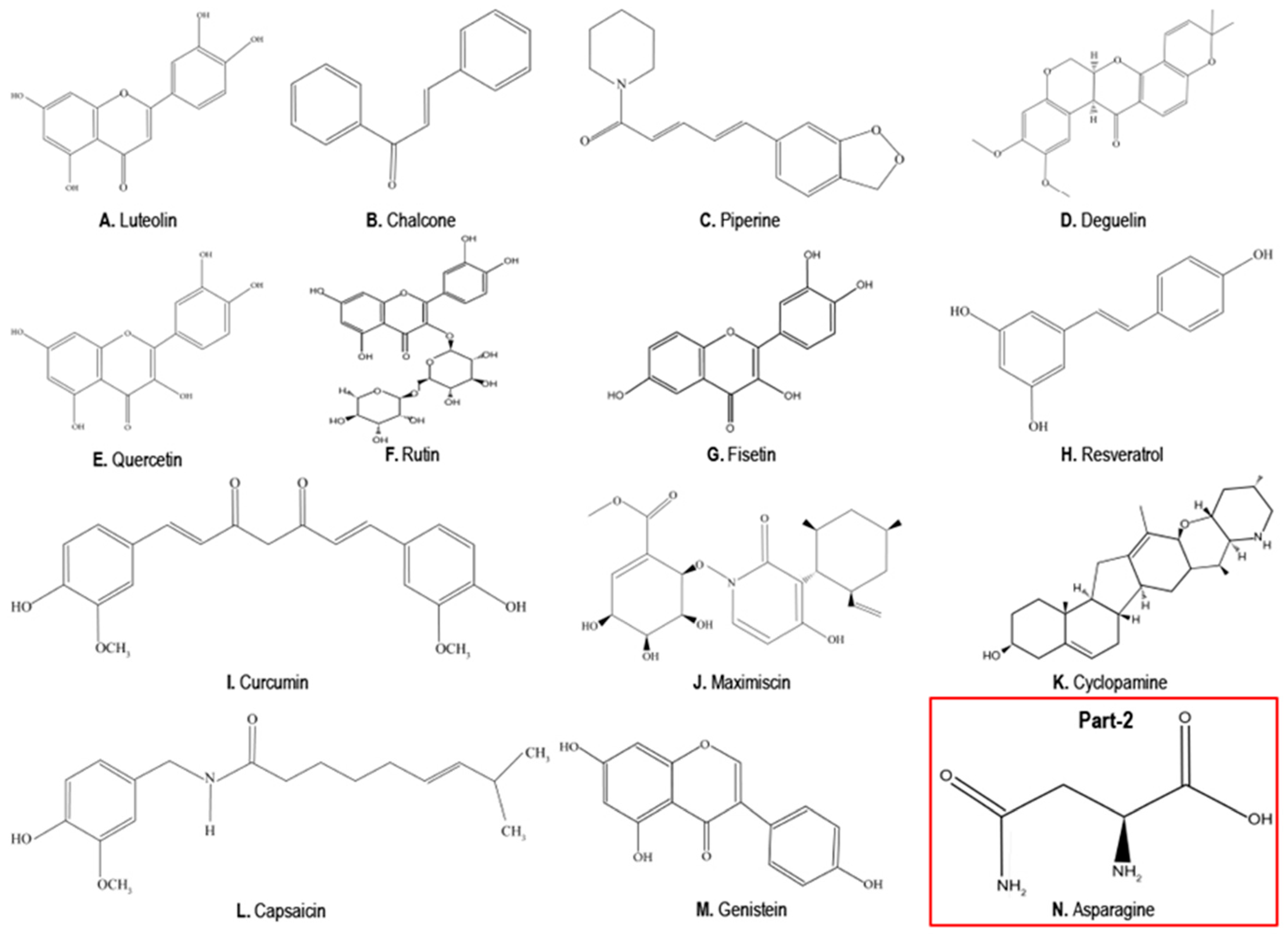

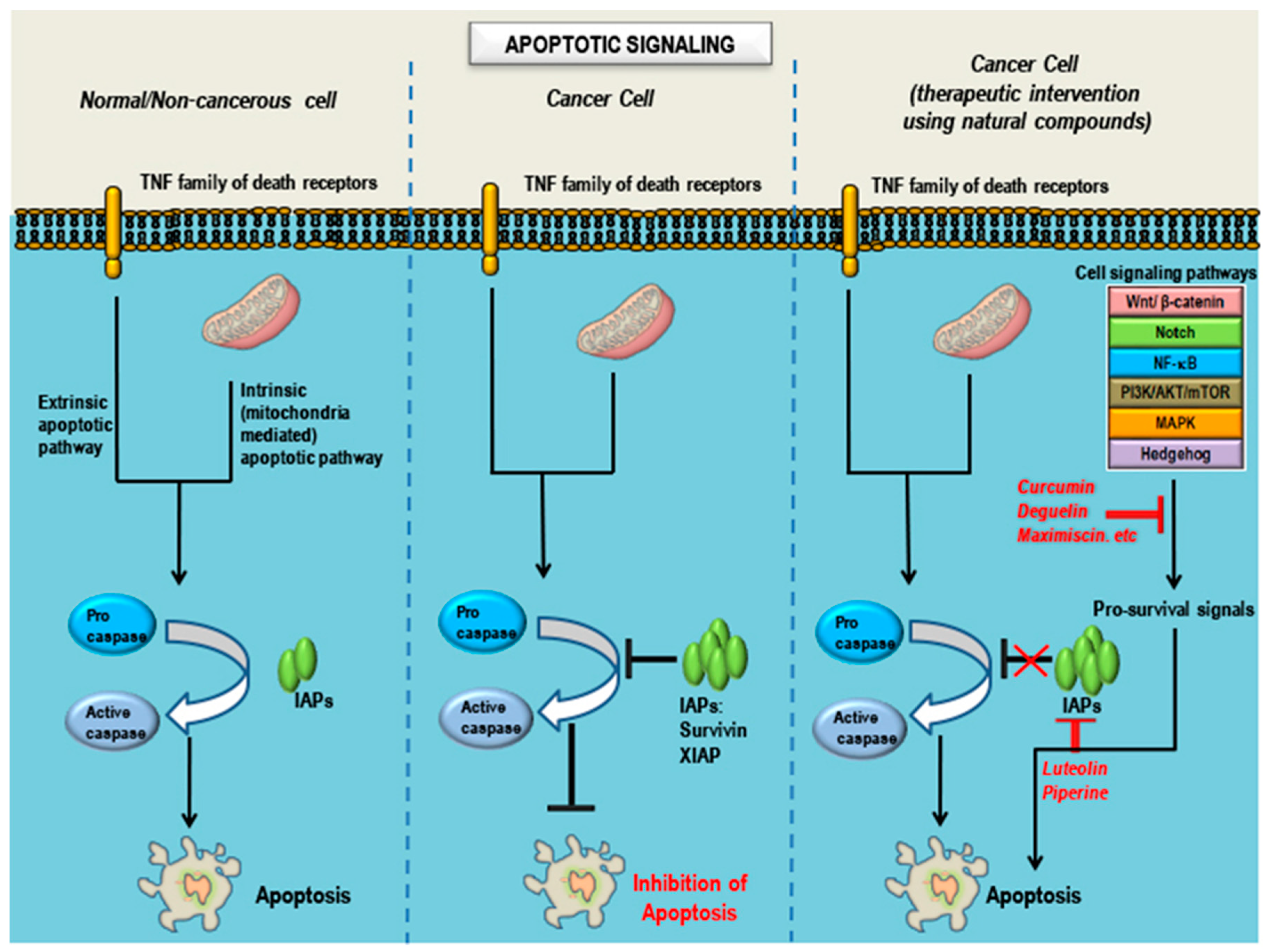
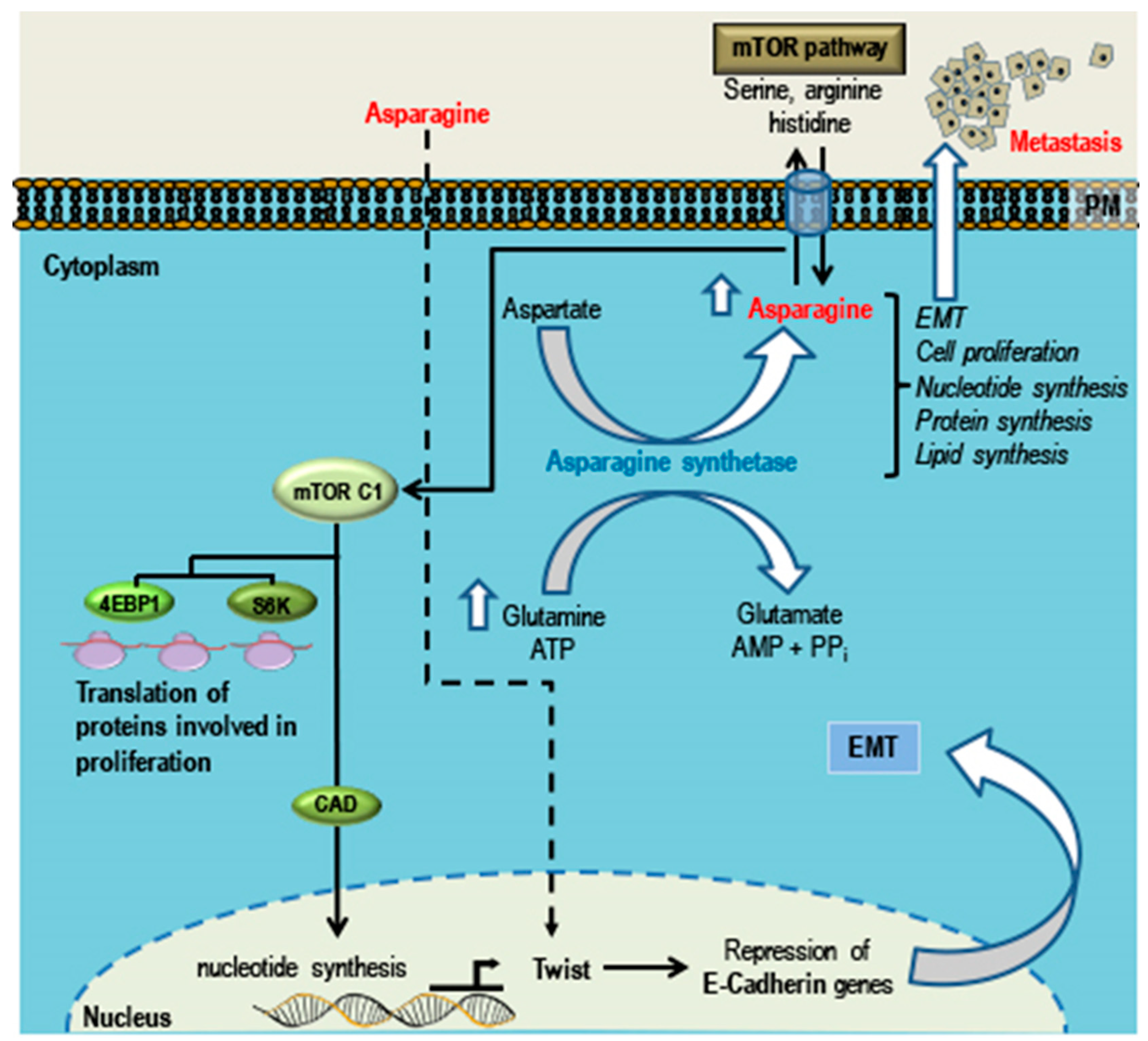

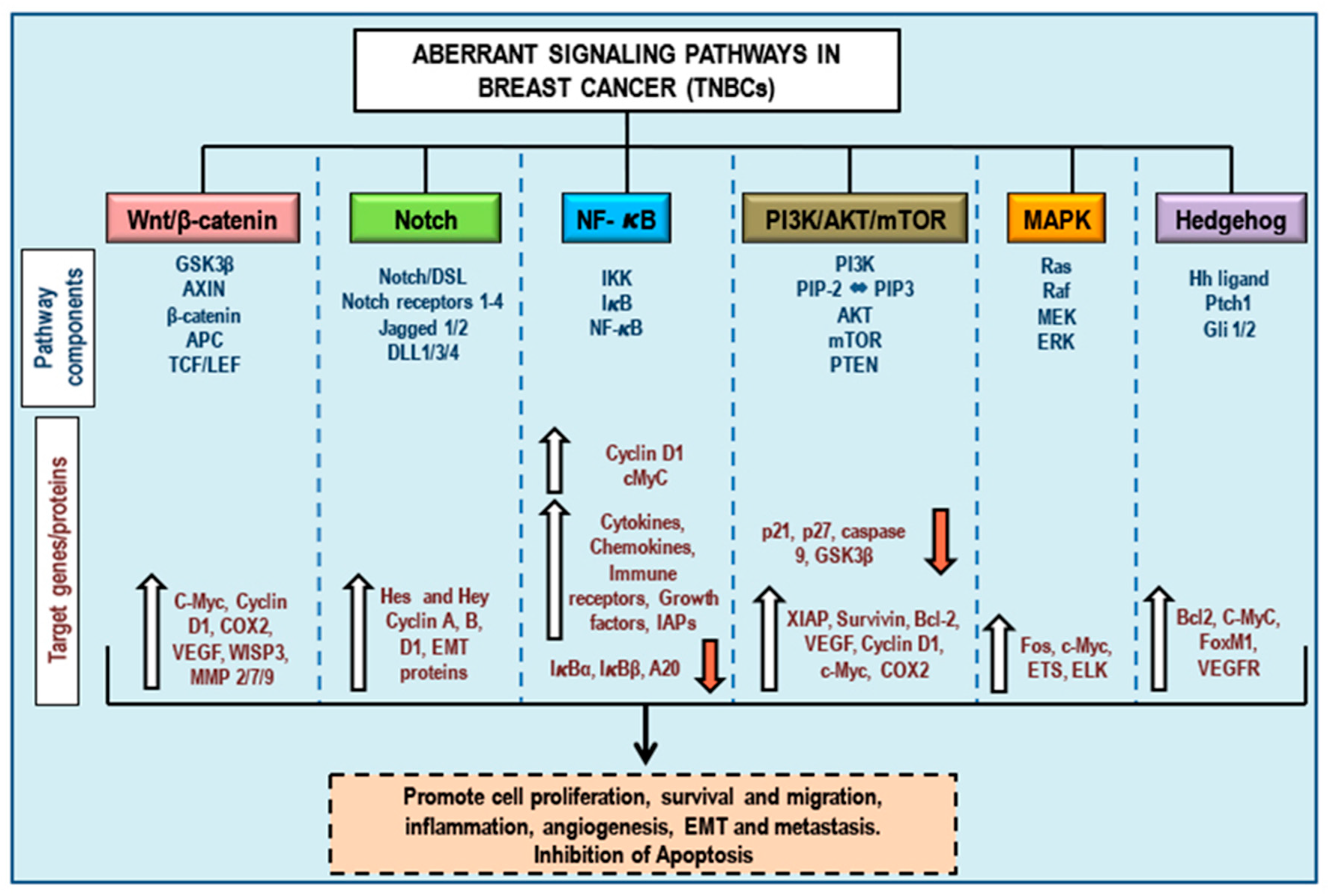{kind=link}
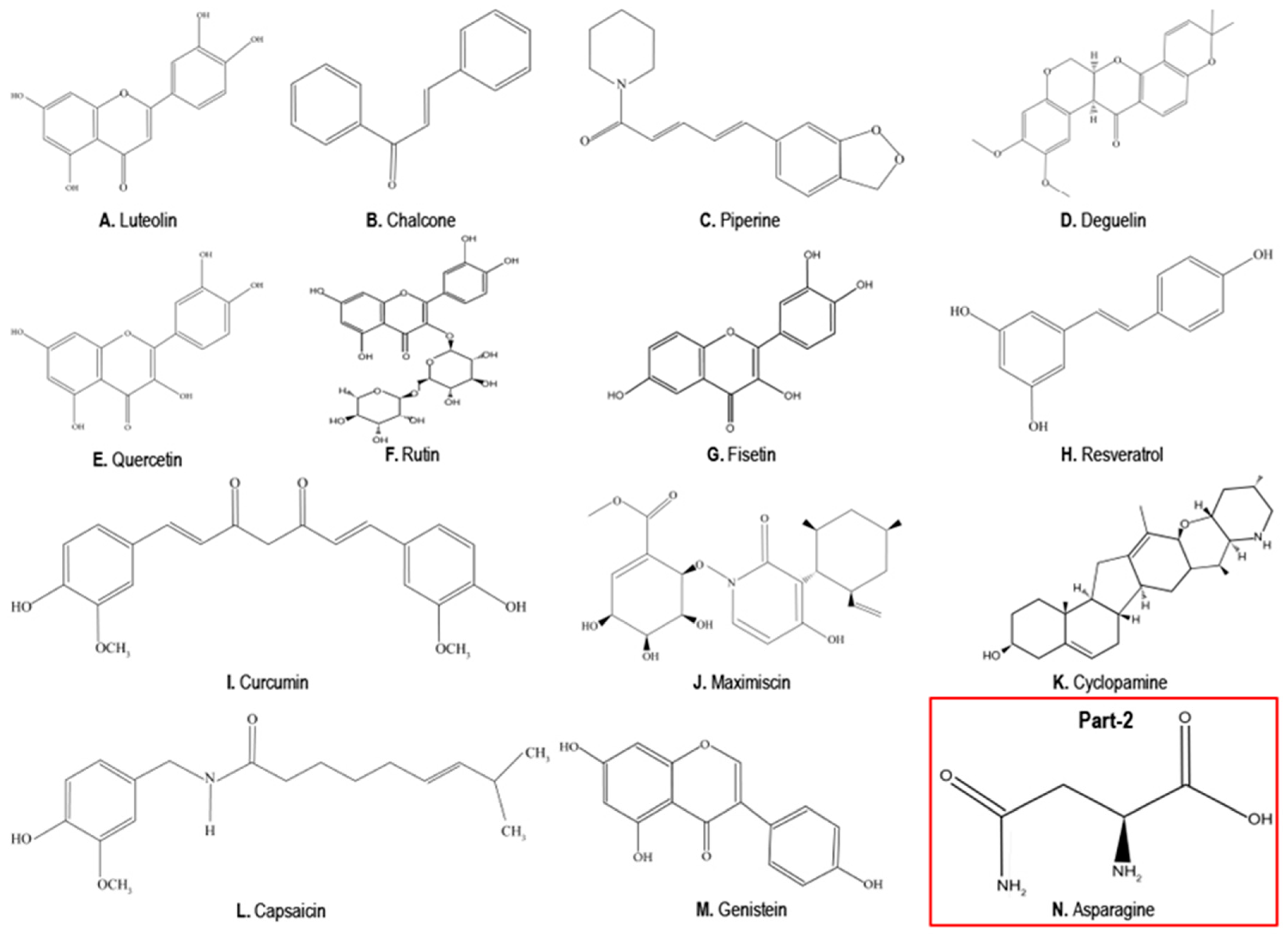{kind=link}
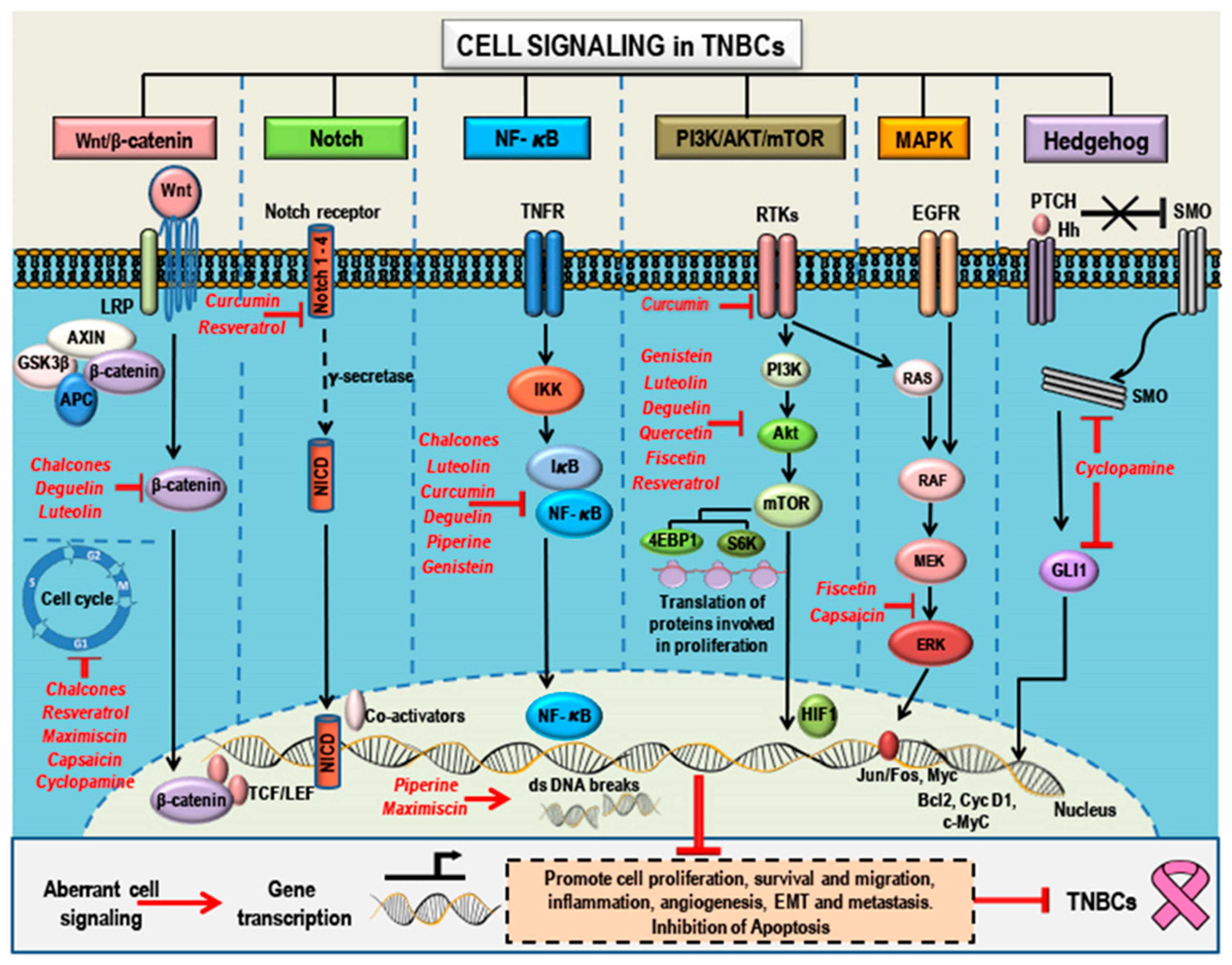{kind=link}
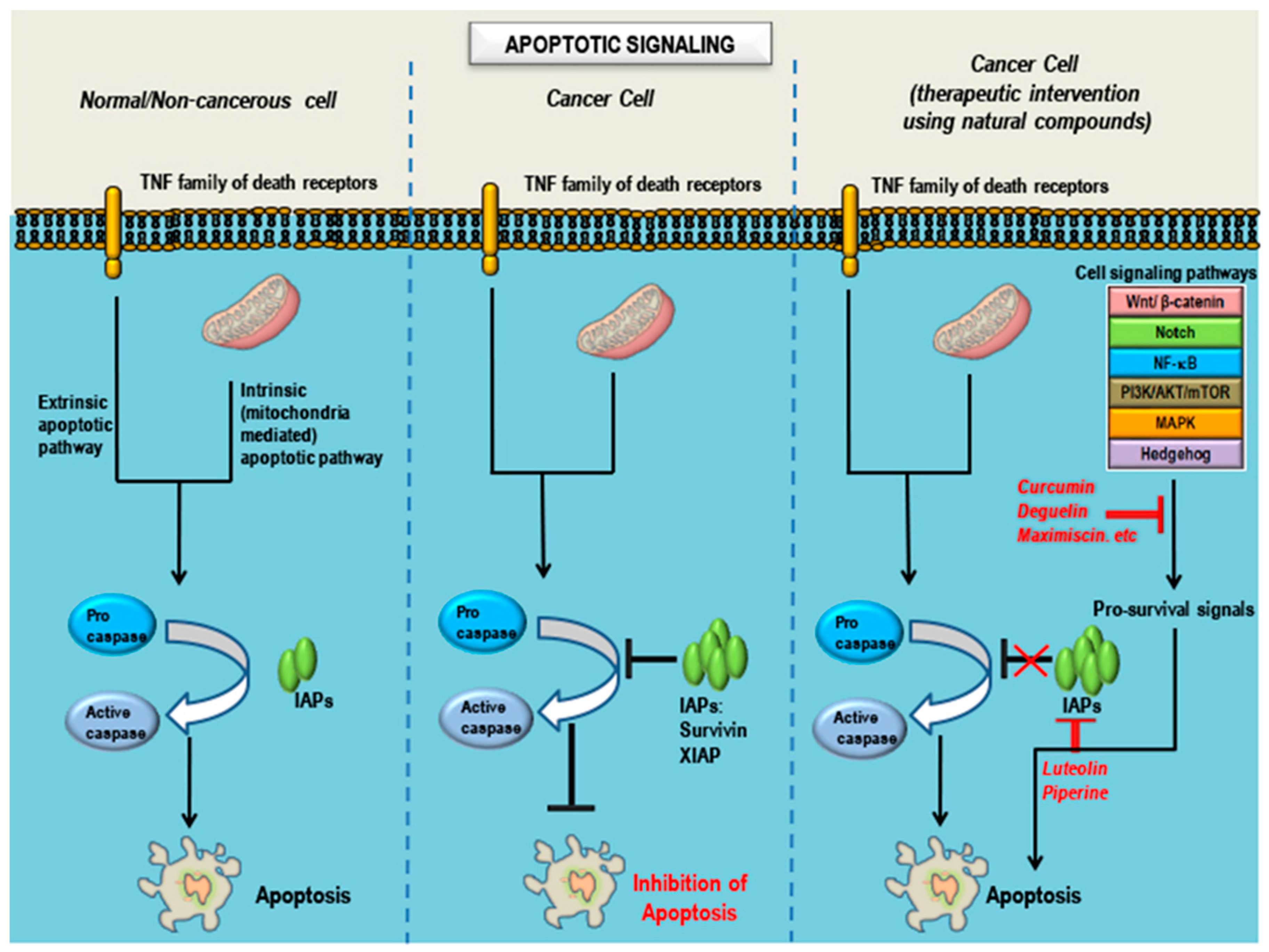{kind=link}
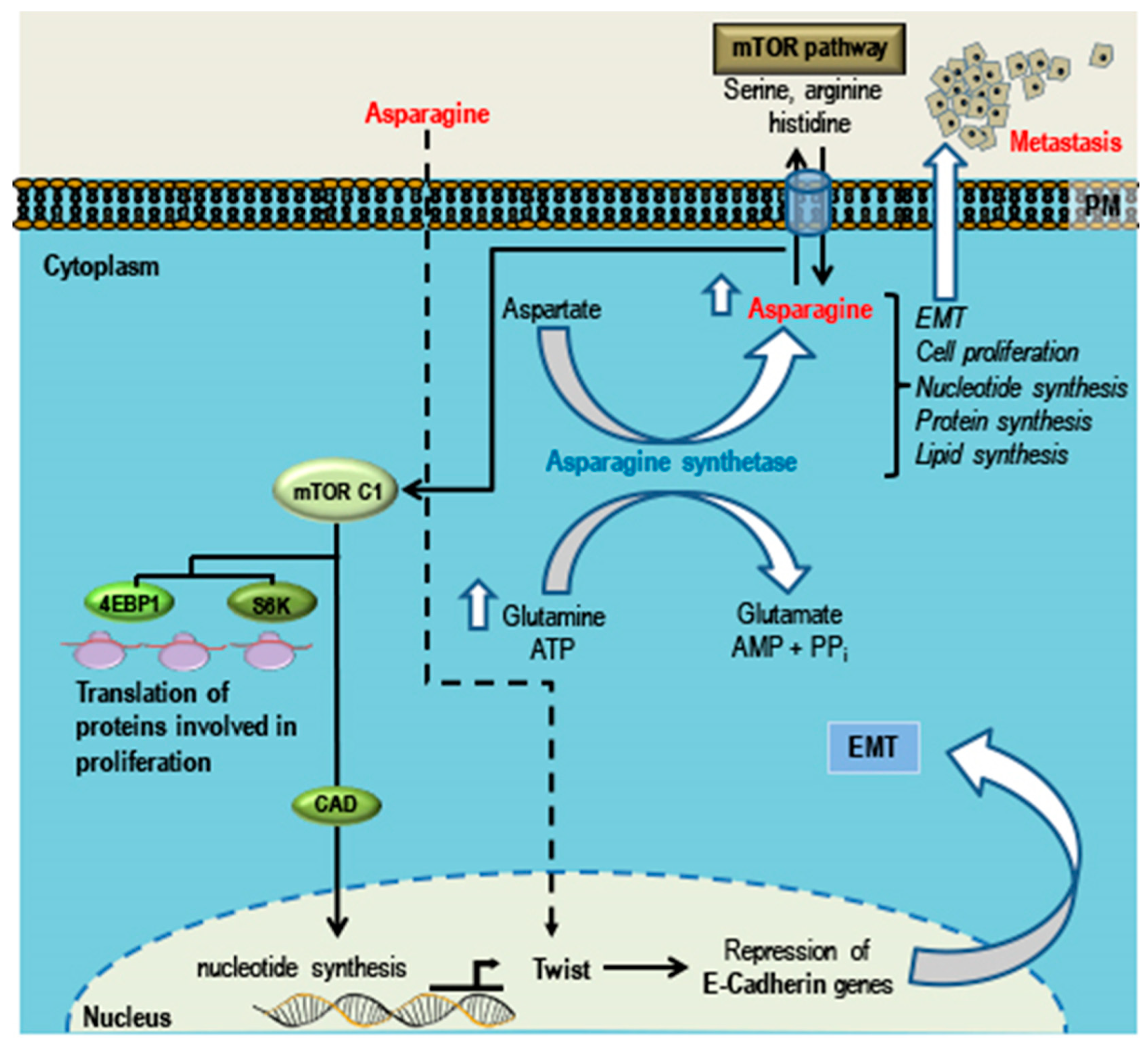{kind=link}
| Subtypes | Characteristics (Based on Gene Ontology and Differential Gene Expression) | Cell Lines |
|---|---|---|
| BL1 (Basal like-1) |
| HCC2157 HCC1599 HCC1937 HCC1143 HCC3153 MDA-MB-468 HCC38 |
| BL2 (Basal like-2) |
| SUM149PT CAL851 HCC70 HCC1806 HDQ-P1 |
| IM (Immunomodulatory) |
| HCC1187 DU4475 |
| M (Mesenchymal like) |
| BT-549 CAL-51 CAL-120 |
| MSL (Mesenchymal Stem Cell-like) |
| Hs578T MDA-MB-157 MDA-MB-436 MDA-MB-231 |
| LAR (Luminal Androgen Receptor) |
| MDA-MB-453 SUM185PE HCC2185 CAL-148 MFM-223 |
| Compound | Chemistry | Source | Conditions Used for | In Vitro | In Vivo | Targets/Markers | Signaling |
|---|---|---|---|---|---|---|---|
| Natural Compounds: Having Anti-Cancer Properties in TNBC and Non-TNBC Cell Lines | |||||||
| (A) Luteolin | Flavonoid (Figure 2A) | Broccoli, Green chilli, Onion leaves, Carrot, Radish, Celery [168] | Hypertension, inflammatory disorders and cancer [156] | MDA-MB-231, LM2-4175, MDA-MB-435, BT-549 | Xenograft | Vimentin, Snail, Slug β-catenin [162] VEGFR2 [161] MMP-2/9 Notch-1, Hes-1, Cyclin D1 | PI3K/Akt, MAPK/ERK1/2, STAT3 [169] Notch [164] |
| (B) Chalcones | Flavonoid (Figure 2B) | Tomatoes, Shallots, Beans, Citrus, Apples | Asthma, gastric ulcer, skin diseases, parasitic infections [170] | MDA-MB-231, BT-549, MDA-MB-468 | MDA-MB 231/4mRL.luc2 (SCID) mice | Cell cycle, NF-κB, p65, p38, Hsp90 [171], Wnt/β-catenin, Bcl2 [172] | Wnt/β-catenin, VEGF/VEGFR2 |
| (C) Piperine | Alkaloid (Figure 2C) | Pepper | Pain, chills, fever, reduces blood cholesterol | MDA-MB-231, MDA-MB-468, T47D, MCF-7 | MDA-MB-468 (NOD/SCID), 4T1-luc mouse TNBC | TRAIL, MMP 2 and 9 [173], survivin, p65 [174], cell cycle components | ERK1/2, p38 MAPK and Akt [175] |
| (D) Deguelin | Flavonoid (rotenoid) (Figure 2D) | Natural insecticides | Insecticide piscicide | BT474, T47D, MDA-MB-231, BT-549, BT20, MCF-7 | MDA-MB-231 (athymic-mouse) | β-catenin, cyclin D1, XIAP, survivin, EGFR and c-Met | EGFR [176] |
| (E) Quercetin (F) Rutin | Flavonoid (Figure 2E) (Figure 2F) | Apples, Onions | Cardio-vascular, common cold, allergy | MDA-MB-157, MDA-MB-231, MDA-MB-468, | C3(1)/SV40Tag transgenic mouse | β-catenin, Foxo3A FASN [177] c-Met, p51, p21 and GADD45 | Wnt/β-catenin [177] PI3K/ERK/MAPK [178] |
| (G) Fisetin | Flavonoid (Figure 2G) | Apples, Onions, Kiwi, Cucumber | Ischemic stroke neuroprotective [179] anticancer | MDA-MB-231, MDA-MB-468, MDA-MB-157, SKBR3, MCF-7 | xenograft | Bid, Bad, Bak, Bax Aurora B kinase | MAPK/ERK1/2 [180] PI3K/Akt/mTOR NF-κB |
| (H) Resveratrol | Phytoalexin (Figure 2H) | Red grapes, Blueberries Raspberries | Hyperlipidemia diabetes, atherosclerosis | MDA-MB-435, MDA-MB-231 | MDA-MB-231 xenograft | GF-1, MMP2, S6 ribosomal protein, MED28, VEGF | EGFR/PI3K/Akt MAPK [181,182] |
| (I) Curcumin | Phytopolyl-phenol (Figure 2I) | Turmeric | Food additive, cosmetics, Neuro-degenerative diseases, arthritis | MDA-MB-231, MDA-MB-468, SUM149 PT, SUM159PT | MDA-MB-231 xenograft | VEGFR2/3, EGFR [183,184], Rac1, NF-kB, Akt [185], p53 | NF-κB [25] EMT |
| (J) Maximiscin | Polyketide-shikimate-NRPS-hybrid metabolite (Figure 2J) | Tolypocladium sp in co-culture with bacteria | Data not available | MDA-MB-468 | MDA-MB-468 xenograft- | p53, Chk-1 and Chk-2 [186] | DNA damage response |
| (K) Cyclopamine | Steroidal jerveratrum alkaloid (Figure 2K) | Corn lily | Hypertension, Cardiac diseases, Psoriasis, Basal cell carcinoma, Teratogenic | MDA-MB-231 (is resistant to cyclopamine due low Smo expression) [187], MDA-MB-435 | Mouse 4T1 | SMO, GLI1 cyclin D1, NF-κB, MMP2 and MMP9 [188] | Hedgehog [189] MAPK/ERK [188] |
| (L) Capsaicin | Alkaloid (Figure 2L) | Chilli pepper | Pain | MDA-MB-231, BT-474, SKBR3 | MDA-MB-231 xenograft | c-Src, FAK and Paxillin MMP2 and MMP9 [190] cyclin D1 | EGFR/HER-2 [191] |
| (M) Genistein | Isoflavanoid (Figure 2M) | Soybeans | Helminthic infection, osteoporosis, cardiovascular diseases, menopause cancer [192] | MDA-MB-231, MDA-MB-468, T47D, MCF-7 | MDA-MB-231 xenograft | MMP-9 p21 VEGF TGF-β [193] Bcl-2, Bax, p53 [194] | Hedgehog [195] PTEN/PI3K/Akt (inhibition of mammosphere formation) [196] EGFR [197] |
| Natural Compound: Having Pro-Carcinogenic Effect in TNBC | |||||||
| (N) Asparagine | Non-essential amino acid (Figure 2N) | Asparagus Potatoes, Legumes, Beef, Egg, Fish | Biosynthetic role | 4T1, MDA-MB-231 | MDA-MB-231 Mouse 4T1 xenograft | Twist E-cadherin [198] | EMT [198] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varghese, E.; Samuel, S.M.; Abotaleb, M.; Cheema, S.; Mamtani, R.; Büsselberg, D. The “Yin and Yang” of Natural Compounds in Anticancer Therapy of Triple-Negative Breast Cancers. Cancers 2018, 10, 346. https://doi.org/10.3390/cancers10100346
Varghese E, Samuel SM, Abotaleb M, Cheema S, Mamtani R, Büsselberg D. The “Yin and Yang” of Natural Compounds in Anticancer Therapy of Triple-Negative Breast Cancers. Cancers. 2018; 10(10):346. https://doi.org/10.3390/cancers10100346
Chicago/Turabian StyleVarghese, Elizabeth, Samson Mathews Samuel, Mariam Abotaleb, Sohaila Cheema, Ravinder Mamtani, and Dietrich Büsselberg. 2018. "The “Yin and Yang” of Natural Compounds in Anticancer Therapy of Triple-Negative Breast Cancers" Cancers 10, no. 10: 346. https://doi.org/10.3390/cancers10100346