Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy

1
Division of Cancer Sciences, University of Manchester, Manchester M13 9NT, UK
2
Department of Medical Oncology, The Christie NHS Foundation Trust, Wilmslow Road, Manchester M20 4BX, UK
*
Author to whom correspondence should be addressed.
Cancers 2018, 10(1), 17; https://doi.org/10.3390/cancers10010017
Submission received: 14 December 2017
/
Revised: 8 January 2018
/
Accepted: 10 January 2018
/
Published: 12 January 2018
(This article belongs to the Special Issue Latest Development in Pancreatic Cancer)
Abstract
:Pancreatic ductal adenocarcinoma (PDAC) continues to be a disease with poor outcomes and short-lived treatment responses. New information is emerging from genome sequencing identifying potential subgroups based on somatic and germline mutations. A variety of different mutations and mutational signatures have been identified; the driver mutation in around 93% of PDAC is KRAS, with other recorded alterations being SMAD4 and CDKN2A. Mutations in the deoxyribonucleic acid (DNA) damage repair pathway have also been investigated in PDAC and multiple clinical trials are ongoing with DNA-damaging agents. Rare mutations in BRAF and microsatellite instability (MSI) have been reported in about 1–3% of patients with PDAC, and agents used in other cancers to target these have also shown some promise. Immunotherapy is a developing field, but has failed to demonstrate benefits in PDAC to date. While many trials have failed to improve outcomes in this deadly disease, there is optimism that by developing a better understanding of the translational aspects of this cancer, future informed therapeutic strategies may prove more successful.
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the deadliest cancers [1] with a five-year overall survival (OS) for all stages of around 8% in the United States (US) [2] and 3% in the United Kingdom (UK) [3]. Even in patients who had potentially curative surgery followed by adjuvant chemotherapy with gemcitabine and capecitabine, the five-year OS was still only 28.8% in the recently reported phase III randomised ESPAC-4 trial [4]. The Phase III ACCORD [5] and MPACT [6] combination chemotherapy trials in patients with advanced PDAC have been the only studies which reported clinically meaningful significant extensions in median OS in the recent decade. Currently, the combination of 5-fluorouracil, oxaliplatin, irinotecan and leucovorin (FOLFIRINOX) from the ACCORD trial has resulted in the longest-reported OS for patients with metastatic PDAC; median OS was 11.1 months compared to 6.8 months with single-agent gemcitabine [5]. Unfortunately, multiple other clinical trials with either chemotherapy combinations or novel agents have failed to demonstrate a significant OS improvement [7,8,9,10]. Due to poor prognosis and very little improvement in survival, PDAC is a major cause of cancer death and it is estimated that it will become the 2nd leading cause of cancer-related death in the US by 2030 [11], being 3rd [2] and 5th [3] currently in the US and UK, respectively.
It has been reported that around 5–10% of pancreatic cancers arise in the presence of a family history of this diagnosis [12]. Hereditary breast and ovarian cancer (HBOC) [13], Peutz-Jeghers syndrome (PJS), hereditary non-polyposis colorectal carcinoma (HNPCC) [14], familial adenomatous polyposis (FAP) [15], familial atypical multiple mole melanoma (FAMMM) [16] and hereditary pancreatitis [17] have been linked to an increased risk of PDAC, although the numbers are relatively small.
The largest study reporting germline mutations in patients with PDAC was recently published by Shindo et al. [18]; germline mutations were identified in 3.9% of patients with PDAC. In their cohort of 854 patients, the most prevalent mutations were breast cancer 2 (BRCA2), ataxia telangiectasia mutated (ATM), breast cancer 1 (BRCA1), partner and localiser of BRCA2 (PALB2), mutL homolog 1 (MLH1), cyclin-dependent kinase inhibitor 2A (CDKN2A) and tumour protein p53 (TP53) [18].
This review aims to interrogate the novel mutations and signatures identified in PDAC and assess their potential clinical significance for the treatment of patients.
2. Genomic Studies in PDAC
Several recent large-scale studies have revolutionised our understanding of PDAC biology and the genome-level drivers of its development. The rate of single nucleotide variants is low at 2.64 mutations per Mb, well below that of cancers such as lung and melanoma that are driven by strong mutagens [19,20].
By analysing the specific trinucleotide context in which single nucleotide variants occur, patterns associated with unique mutagenic processes can be identified [20]. In PDAC, four dominant mutational signatures have been identified—1B, 2, 3, 6. These are associated with age, apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like (APOBEC) family of cytidine deaminases, BRCA and mismatch repair (MMR) mutations, respectively [21]. By far the most prevalent of these are the ageing signature and APOBEC signature, both seen in almost all cases of PDAC. In contrast, the BRCA and MMR signatures are found in isolated cases, both of which have therapeutic relevance [19,22].
Signatures of large structural variation can also be used to classify PDAC. Waddell et al. [19] defined four subclasses based on structural variants (SV) patterns; unstable, stable, locally rearranged and scattered. Importantly, these subtypes have been shown to be independent of cellularity, a known-confounder in PDAC classification. Of therapeutic relevance, the locally rearranged and scattered subtypes demonstrated frequent amplification of known oncogenes, including rare amplifications of potentially druggable kinases such as Fibroblast Growth Factor receptor 1 (FGFR1), B-Raf proto-oncogene, serine/threonine kinase (BRAF), cyclin-dependent kinase 6 (CDK6) and MET proto-oncogene receptor tyrosine kinase (MET). As discussed later, the unstable SV signature was also linked to the presence of homologous recombination deficiency which is another potential therapeutic target [23].
Driver gene identification by integrated analysis of copy number changes, structural variants and single nucleotide variants has shown only a relatively small set of core-mutated genes in PDAC, with a long tail of other more infrequently mutated genes. Interestingly, when clustered into pathways, a clearer picture emerges with 10 main pathways commonly targeted. Commonly targeted gene-sets include, cell cycle, deoxyribonucleic acid (DNA) repair, transforming growth factor beta (TGF beta), NOTCH, wingless-type MMTV integration site (WNT), chromatin, SWItch/sucrose non-fermentable (swi/snf), KRAS proto-oncogene, GTPase (KRAS), mitogen-activated protein kinase (MAPK), roundabout guidance receptor-slit guidance ligand (ROBO-SLIT) (axonal guidance) and ribonucleic acid (RNA) processing [24]. Though specific gene mutations are infrequent, many represent potential novel therapeutic targets [25,26].
The timing of mutation development has implications for the utility of targeted drug therapy. Targeting mutations that occur early in disease development is preferential, as they are present in all clones and metastatic sites of the disease [27]. In addition, mutations that occur in pre-malignant disease may make excellent targets for arresting disease before invasion and metastatic spread. Pancreatic ductal adenocarcinoma develops from premalignant changes—pancreatic in situ neoplasia 1–3, in a well-recognised, step-wise transition accumulating specific mutations at each stage [28,29,30]. The exact timing of these mutations has recently been challenged [31]. It appears that in some cases, mutations of TP53, SMAD family member 4 (SMAD4) and CDKN2A may occur in a single event. This evolutionary model of cancer described as a “punctuated equilibrium”, has been suggested to underlie the aggressive presentation of PDAC [31]. This early and clonal accumulation of mutations has recently been verified by sequencing of PDAC primaries and multiple metastases in a small cohort of patients demonstrating little genetic variation between tumours from the same patient, independent of metastatic or primary location of the tissue [32].
Though sequencing of the PDAC genome has identified recurrent mutations in multiple pathways the exact function of many of these remains elusive. There has therefore been a focus on sub-grouping PDAC using a more multi-platform approach, including mRNA, microRNA and proteomic studies. Bailey et al. [24] published data on 456 resected PDACs, including whole genome sequencing, deep exome sequencing and transcriptomics. They identified 4 gene-expression based subtypes: squamous, pancreatic progenitor, immunogenic and aberrantly differentiated endocrine exocrine (ADEX) [24]. However, subsequent integrated genomic, transcriptomic and proteomic profiling of 150 resected PDAC specimens, found similar mutations, but identified that the proposed ADEX and immunogenic subtypes from Bailey et al. correlated with low-purity samples in their cohort, that could in turn suggest that the gene expression profiles were derived from non-neoplastic cell contamination [33]. Thus, it appears that at the RNA level, PDAC can be divided broadly into two categories, basal-like/squamous and pancreatic progenitor. Intriguingly, TP53 mutations and an overall increase in copy-number change numbers were seen in the Basal-like sub type, whilst GATA binding protein 6 (GATA6) amplification and GNAS complex locus (GNAS) mutations were more prevalent in the classical sub-type (as described by Bailey et al. [24] and Moffitt et al. [34]) [33]. Given this finding, a more thorough analysis of the recently identified structural variant sub-types with the revised mRNA classification will be informative.
3. The Role of Different Mutations in PDAC
3.1. KRAS Mutations in PDAC
The majority of PDACs are known to harbour mutations in KRAS with a prevalence of around 90–95% in most studies [35]. In the most recent and comprehensive study by the cancer genome atlas research network, KRAS mutations were identified in 93% of patients with PDAC. In the KRAS wild-type tumours, mutations in other RAS pathway genes were identified in 60% of cases, demonstrating the central importance of this pathway to PDAC development [33].
Due to the vast majority of PDACs harbouring KRAS mutations, multiple clinical trials have tried to target this aberration with novel treatments, but unfortunately all have failed to demonstrate clinically meaningful benefits in OS [36]. Targeting downstream components in the pathway showed promise in phase 1 trials of mitogen-activated protein kinase (MEK) inhibitors, although subsequent trials reported no benefit of MEK inhibition either in monotherapy or in combination with chemotherapy [37,38]. In addition, targeting KRAS activation more directly with a farnesyl transferase inhibitor showed no benefit in a large phase III trial [8].
To date, there is no novel agent that has managed to target mutated KRAS in PDAC, though given its centrality, it remains a key target.
3.2. DNA Damage Repair Mutations in PDAC
The seminal study of mutational patterns in cancer by Alexandrov et al. [20] showed a unique mutational pattern, signature 3, to be associated with BRCA1/2 mutations and homologous recombination deficiency in breast, ovarian and pancreatic cancers. This DNA damage repair (DDR) signature was also seen in PDACs in the study by Waddell et al. [19]. In this study, the signature was associated with the unstable structural variant subtype of PDACs, and both somatic and germline mutations of BRCA1, BRCA2 and PALB2.
Platinum agents have been shown to be more effective in tumours with deficient DDR, potentially due to their ability to cause DNA strand crosslinking and induce double-strand breaks, which together with BRCA1/2 mutations, will not be effectively repaired [39]. To date, there hasn’t been a randomised trial confirming this in pancreatic cancers, although data are promising from retrospective cohorts [40,41].
Homologous recombination (HR) deficiency has also been shown to sensitise cancers to poly (ADP-ribose) polymerase (PARP) inhibition in ovarian cancer [42], and, more recently, also in early phase trials in PDAC [43]. Original trials with PARP inhibitors targeted patients with germline mutations of BRCA genes [43]. Of late, the focus has shifted to understanding the role of somatic DDR-pathway mutations and homologous recombination deficiency as a signal for PARP inhibition efficacy. In particular, biomarkers of this phenotype are eagerly sought to potentially widen the group of patients that could be treated with these agents [23].
A study by Riaz et al. [44] has shown that in a pan-cancer analysis, bi-allelic somatic and germline alterations in multiple DNA repair genes occur across many cancer types, and are associated with genomic features consistent with a deficiency in HR. Interestingly only 45% of bi-allelic mutation cases were in cancers traditionally thought of as hereditary HR deficiency cancer types. This provides a wider population of patients with cancer who may exhibit deficiency in HR characteristics, and thus may allow reclassification of targetable HR deficiency in these tumours.
3.3. ATM Mutations in PDAC
In one of the earliest PDAC whole genome sequencing studies, Biankin et al. [35] reported that ATM aberrations were present in 8% of their samples of 99 resected early PDAC cases. In another study by the same group, ATM was also associated with tumours with unstable genome or the BRCA mutational signature [19]. Perkhofer et al. have also shown that ATM deficiency leads to chromosomal instability in PDAC mouse models [47]. As ATM is involved in cellular response to replication stress, and double-strand breaks [48], it is thought that mutations in this gene could also be linked with aberrant DDR or HR deficiency and sensitivity to PARP inhibitors [49].
The role of ATM in homologous recombination repair and signature 3 was recently questioned in patients with breast cancer as Polak et al. [50] published their work showing that germline pathogenic variants in ATM were not associated with a high level of signature 3. This raises a question of whether novel agents targeting DDR and HR deficiency would also work in ATM-mutated tumours. There is currently an ongoing trial in patients with PDAC, that defines BRCAness as HR deficient but germline BRCA proficient (tumours with somatic BRCA mutation, Fanconi anemia gene, ATM or BRCA1/BRCA2-containing complex, subunit 5 (RAD51) mutations) and is investigating the efficacy of treating these patients with the PARP inhibitor olaparib [51].
Defects in ATM could also be compensated through the activity of ataxia telangiectasia And rad3-related protein (ATR), thus indicating potential synthetic lethality interactions between these pathways, as ATM mutated tumours would be vulnerable to ATR inhibition [52]. This was also shown in PDAC cell lines [47] and multiple ongoing early phase clinical trials are investigating the ATM mutation as a potential biomarker in trials with ATR inhibitors for this reason [53,54].
Mutations in ATM have also been linked with a more aggressive form of PDAC. Kim et al. reported that in their cohort of 396 resected PDACs, ATM loss correlated with more vascular invasion (63.3%) and metastatic lymph nodes (92.2%) compared to tumours without ATM loss [55]. They also reported decreased OS in patients with ATM loss, but only in those who also had normal TP53 expression. Russell et al. reported that ATM loss in PDAC also correlated with poorer prognosis and less differentiated tumour phenotype in mice and men [56]. Drosos et al. has also demonstrated that ATM deficiency correlated with KRAS mutations and promoted highly metastatic PDAC [57]. They also showed that ATM loss accelerated KRAS-induced carcinogenesis and ATM loss increased genomic instability and thus cell lines were more sensitive to DNA damage-inducing agents like radiotherapy [57].
A recent paper by Ayars et al. [58] also showed that ATM-deficient PDAC cells were exquisitely sensitised to radiation, thus illustrating that ATM-mutated PDAC could be more sensitive to radiotherapy, and may thus provide more personalised treatment options for these patients in the future.
3.4. TP53 Mutations in PDAC
Mutations in TP53 have been shown to occur in 50–75% [59] of PDAC and in a more recent analysis in around 73% [33]. Morton et al. have shown that after the initiating activating mutation in the KRAS gene, the TP53 mutation drives the rapid progression of KRAS-induced premalignant lesions to PDAC. They also reported that mutation rather than genetic loss of p53 specifically promotes PDAC metastasis. Weissmueller et al. [60] have shown that promotion of metastasis in mutant p53 cells is induced through platelet-derived growth factor receptor b (PDGFRb), and this in turn could be a prognostic marker and possible target for clinical trials.
Significant multidrug resistance-associated protein 1 (MRP-1) and B-cell lymphoma 2 (Bcl-2) overexpression due to TP53 mutations has been linked to gemcitabine resistance in PDAC cell lines [61].
Early clinical trials targeting TP53 in other cancers have shown exciting results. The most promising agents include WEE1 G2 checkpoint kinase (Wee1) inhibitors and APR-246—a small molecule that reactivates mutant TP53—both in combination with chemotherapy [62,63]. Currently there are a number of clinical trials ongoing evaluating these agents in p53-mutated gastric, ovarian, lung and other cancers [64,65,66]. To date, there are no clinical trials of these agents specifically targeting PDAC.
3.5. SMAD4 Mutations in PDAC
Mutation and dysregulation of SMAD4 has been identified in around 30–64% [19,67] of PDACs, either by whole genome sequencing (WGS) or immunohistochemistry. Although this is common in both resected and advanced PDACs, the role of this SMAD4-dysregulation is still unknown.
Many previous studies have reported that SMAD4 mutations, and loss, might be associated with worse outcomes and patterns of recurrence after PDAC resections [68,69,70,71]. A meta-analysis by Shugang et al. [72] reported that loss of SMAD4 in patients with PDAC was associated with poorer OS and was a negative prognostic factor. In contrast, a study by Ormanns et al. [67] showed that at least in advanced PDACs, SMAD4 loss had no impact on OS, and that its presence actually resulted in increases in progression-free survival (PFS) in patients treated with gemcitabine-based chemotherapy.
Besides the synthetic lethality seen in HR-deficient cancers with PARP inhibitors, collateral lethality was shown to play a role in SMAD4-mutated pancreatic cancers. Dey et al. [73] reported that in SMAD4-mutated PDAC, the loss of the neighbouring housekeeping gene malic enzyme 2 (ME2) in these tumours, created a cancer-specific metabolic vulnerability to Malic Enzyme 3 (ME3) inhibition. They reported that depleting ME3 selectively killed ME2-null PDAC cells, and so hypothesised potential new specific targets for novel inhibitors in SMAD4-mutated tumours. Currently, there are no such inhibitors on the market, and further research is needed in the collateral lethality approach in PDAC.
3.6. CDKN2A Mutations in PDAC
Cell cycle progression through the G1-S phase checkpoint is driven by phosphorylation of the retinoblastoma protein (Rb) by CDK4/6, allowing release of the key transcription factor E2F1. The protein p16, one of the products of the tumour suppressor gene CDKN2A, acts to inhibit CDK4/6-cyclin-D complex activation, thus preventing the G1-S phase transition. Familial atypical multiple mole melanoma may be caused by a mutation in CDKN2A, and in around 40% of cases the second most commonly observed malignancy in families with the CDKN2A mutation is PDAC [74].
Varying numbers of CDKN2A mutations have been reported in patients with PDAC. Older polymerase chain reaction (PCR)-based studies have reported mutational rates between 5–12% [12,75]. In an Italian cohort study of mainly advanced (67.9% stage III–IV) PDACs, CDKN2A mutations were detected in only 5.7% of patients [75]. In another study by Salo-Mullen et al. [12], PCR-based testing in 17 patients identified 2 mutations (11.8%) in CDKN2A. In WGS studies, mutations in CDKN2A have been found in 35% (11 structural variants and 24 mutations) of resected PDAC samples [19]. Thus, it appears that deeper sequencing and more comprehensive analysis of mutation types identified significantly more aberrations in CDKN2A in these tumours.
The predictive and prognostic role of CDKN2A mutations in PDAC is largely unknown. A single study has shown that immunohistochemically detected p16 loss correlated with lymphatic invasion and postoperative widespread metastases, together with TP53 and SMAD4 loss [76]. A meta-analysis [72] which evaluated the effects of SMAD4 loss on the outcomes of patients with PDAC did not address the specific role of CDKN2A loss.
Targeted therapy clinical trials in patients with a variety of cancers and mutations in CDKN2A are currently ongoing. One study includes ilorasertib [77], an inhibitor of Aurora kinases, vascular endothelial growth factor (VEGF) and platelet-derived growth factor receptor (PDGFRs) and another is investigating the effects of AZD1775, a Wee1 inhibitor [65] in a variety of cancers. Single case reports have also shown good response to palbociclib, an inhibitor of cyclin-dependent kinases 4 and 6 (CDK4/6) in a patient with CDKN2A-mutated breast cancer [26] and uterine leiomyosarcoma [78].
Chou et al. [79] have reported that dysregulation of the p16-cyclinD-CDK4/6-RB pathway in PDAC correlates with sensitivity to CDK4/6 inhibitors. Increased total RB or phospho-RB was predictive of response to CDK inhibitors, irrespective of p16 expression or pathway mutations in patient-derived xenograft and cell line models. Therefore, RB protein expression represents a potential new biomarker in PDAC for clinical trials with CDK4/6 inhibitors.
3.7. BRAF Mutations in PDAC
In a whole exome sequencing study by Witkiewicz et al. [82], it was shown that mutations in BRAF V600 were present in 3% of mainly early stage pancreatic cancers (different histological subtypes) and were mutually exclusive with KRAS mutations. The same number of BRAF mutations in PDAC were also reported in the most recent study from the cancer genome atlas research network [33].
In patients with BRAF V600-mutated advanced melanoma, treatment with the BRAF kinase inhibitor vemurafenib is now the standard of care first or second-line treatment [83]. In a phase II ‘basket’ study of 122 patients with different BRAF V600-mutated cancers treated with vemurafenib [71], some positive survival results were reported, including two patients with pancreatic cancer (one of whom had stable disease for seven months and the other progressed and died within one month). Results of ongoing trials for BRAF V600-mutated solid tumours are awaited.
3.8. Microsatellite Instability in PDAC
The extent of PDAC risk in hereditary non-polyposis colorectal cancer or Lynch syndrome is still debated, although small numbers of mismatch repair (MMR) mutations have been detected in PDAC [12,19]. Humphris et al. [84] recently reported that MMR deficiency was found in 1% of their cohort of 385 resected pancreatic cancers samples, and all of these MMR-deficient tumours had different somatic inactivations of MutL homolog 1 (MLH1) and MutS protein homolog 2 (MSH2), without any germline mutations.
Microsatellite instability as the hypermutable phenotype caused by MMR deficiency has been recently shown to predict response to Programmed death-ligand 1 (PDL-1) blockade in solid tumours [22], and has been licenced in the US for any MSI-high cancer [85]. Although MSI is rare in PDAC, there have been promising results in studies of novel treatments with immunotherapy in solid tumours (including PDAC) [22,86], and thus mutational testing for this deficiency in PDAC could be considered.
4. Mutation Burden in PDAC
Next generation sequencing assays and panels are being more widely used by many centres [87,88,89], and with these come new challenges of understanding the results in the context of the tumour types. Many assays are now measuring mutational burden in cancers in the hope that novel treatments with immunotherapy could be indicated for mutational burden high groups [90].
In the previously mentioned work by Humphris et al. [84] they reported that out of the 385 resected PDACs, five were hypermutated, containing ≥12 somatic mutations/Mb and another 15 were classified as having a high mutational burden with ∼4–12 mutations/Mb. This classification of mutational burden comes from colorectal cancer studies, where the threshold for hypermutation was 12 mutations/Mb [91]. The 5 hypermutated tumours were all MSI high, and thus could potentially benefit from treatment with immunotherapy, based on the study by Le et al. where mismatch-repair deficiency predicted response of solid tumours to PD-1 blockade [22]. The additional 15 samples in the high mutational burden group showed no evidence of MMR deficiency, although did show evidence of HR deficiency in 8/15 samples (as a contributor to the mutational burden). The potential effect of immunotherapy in this high but not hypermutated group is unknown.
The immunogenic subtype of PDAC from the Bailey et al. data [24] also showed upregulation of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and PD-1, hypothesising that this subgroup could be treated with novel immunotherapies. Unfortunately, early phase clinical trials with immunotherapy in all patients with PDAC have failed to show response both with CTLA-4 inhibitors and anti-PD-1 therapy [92,93,94]. To the authors’ knowledge, no current clinical trials have specifically looked at targeting the immunogenic subtype of PDAC.
Recently published hypermutation analysis on >81,000 tumours from the Foundation One assay have reported interesting results for PDAC [90]. Similar to previous studies, they reported MSI in pancreatic cancers. However, they also showed that in a proportion of pancreatic cancers, hypermutations could be attributed to tobacco smoke and alkylating agent mutational signatures. Interestingly, alkylating agents are not used in the treatment of pancreatic cancer, thus the presence of this signature is hard to explain.
Multiple combination immunotherapy clinical trials are currently ongoing in patients with PDAC, looking at combining immunotherapy with either chemotherapy, targeted therapy, another checkpoint inhibitor or radiation (Table 1), and thus the hope of a breakthrough for immunotherapy in treating non-MSI pancreatic cancers continues.
5. Other Rare Alterations in PDAC
Additional alterations in the WNT signalling pathway like ring finger protein 43 (RNF43) have been found in 5–10% of patients with PDAC [19,24]. In a recent study by Steinhart et al. [112], it has been shown that mutations in RNF43 resulted in vulnerability of pancreatic cancer cell lines to WNT inhibition, and thus could be a target for these agents. Currently there is a phase I clinical trial on-going evaluating WNT inhibitors in patients with malignancies dependent on WNT ligands and includes patients with RNF43-mutated pancreatic cancer [113].
Oncogene amplification is a common mechanism of deregulation in multiple cancer types and is frequently used as a predictive biomarker for targeted therapies [114]. In PDAC, amplification of oncogenes is relatively uncommon in comparison to other gastrointestinal (GI) malignancies, however the locally rearranged and scattered SV subsets of PDACs are enriched for these [19]. In particular, amplifications of Erb-B2 Receptor Tyrosine Kinase 2 (ERBB2), CDK6 and PIK3CA are seen in rare cases, all of which may represent potential drug targeting opportunities [114,115,116].
6. The Feasibility and Challenges of Clinical Molecular Profiling in PDAC
As molecular profiling of all cancers is getting more accessible around the world and different assays are performed in major cancer centres, the actionability of these alterations becomes more and more important. Currently there are no standard-of-care targeted therapies in patients with PDAC, although multiple trials are ongoing.
In their recent paper, Lowery et al. [117] reported on the feasibility of genomic profiling of PDAC in a real-time clinical setting. In their prospective next generation sequencing (NGS) based assay analysis of tumour tissue and matched normal DNA from 336 patients with PDACs, they reported a median of 45 days between sample collection and the clinical team receiving genomic results. There is no approved targeted therapy in pancreatic cancer, but they did find that 5.5% of patients harboured somatic alterations that are US Food and Drug Administration (FDA)-approved treatment biomarkers in other cancers. These alterations included BRCA1/2 mutations, ERBB2 amplifications, CDK4 amplifications, BRAF V600E mutations, ROS proto-oncogene 1 (ROS-1) and Anaplastic Lymphoma Receptor Tyrosine Kinase (ALK1) involving fusion events. Unfortunately, only three patients (1%) went on to have targeted treatment based on these results; no responses were observed. In addition, six patients with previously known germline BRCA mutations received treatment with gemcitabine and cisplatin in combination with a PARP inhibitor, and in total 14 patients with germline-mutated BRCA had prolonged responses to platinum-based chemotherapy [117].
Similarly, another study by Johns et al. [118] reported that in their cohort of 392 patients with PDAC, 5.1% had somatic mutations that were deemed actionable, but only 1.78% were actioned. Of the 7 actioned, 5 received genetic counselling and 2 received personalised treatment.
Thus, current experience highlights that although actionable alterations in PDAC are rare, it is feasible and may be beneficial in some cases to perform tumour genome analysis (potentially targetable mutations in PDAC are illustrated in Figure 1). Whilst the role for such tests in stratifying patients to receive targeted treatments is currently minimal, there is certainly a role for testing for those changes which might have an impact on genetic counselling of the patient and the family.
7. Discussion
This review aimed to interrogate the current reported data on specific mutations and mutational signatures in patients with PDAC.
Although the majority of PDACs harbour mutations in KRAS, to date there has not been a clinical trial showing efficacy of targeting this mutation in patients with PDAC. Tumours that were previously thought to be KRAS wild-type have now been also shown to have mutations in other parts of the RAS pathway indicating that this 5–7% of PDACs are still driven by the same pathway.
The DDR pathway and its potential implications for treatment with platinum-containing chemotherapy regimens and novel targeted therapies such as PARP inhibitors has been most-extensively researched to date. However, prospective clinical evidence on the benefits of platinum agents is currently lacking in PDAC, and is mainly borrowed from other tumour types like ovarian and breast cancers harbouring BRCA mutations. Thus, clinical trials on the efficacy of both platinum agents and other DDR-targeting novel agents in this disease subgroup are needed and multiple are on-going.
The role of SMAD4 and CDKN2A mutations in PDAC is still largely unknown, although there is some evidence that their presence might predict poorer prognosis in patients harbouring these mutations. The concept of novel collateral lethality in SMAD4-mutated PDACs has been reported, and clinical trials investigating this phenomenon are eagerly awaited. In addition, novel concepts of measuring RB as a biomarker for CDK4/6 inhibitors and targeting BRAF in PDAC are in the early stages of investigation and may give hope for this subgroup of patients.
The use of immunotherapy as a therapeutic option has also been researched in patients with PDAC, with disappointing results in the original single agent trials. Although MSI is rare in PDAC, treatment with the PD-L1 inhibitor pembrolizumab has recently shown promising results in different tumour types harbouring this deficiency, and thus may be an option for a subset of patients with PDAC. In addition, the emerging practice of tumour burden measurement may also help to identify a further subgroup of patients with PDAC who may benefit from this therapeutic approach.
However, to date there are no effective targeted therapies utilised in standard clinical practice for the treatment of patients with PDAC, and so guidelines nor recommendations for regular PDAC profiling for mutations cannot be made. However, novel study designs and initiatives evaluating their clinical relevance as part of a clinical trial are warranted.
At least two therapeutic development platforms have been established for PDAC: "PRECISION-Promise" in the US and “PRECISION-Panc” in the UK, with the vision being to deliver drug discovery and personalised medicine to this cohort of patients [119]. It is hoped that through coordinated effective preclinical and clinical research, the outcomes for patients with this dismal disease can be substantially enhanced.
8. Conclusions
Over the last few years, the genetics of PDAC have been revealed through large scale sequencing studies. These studies have clarified oncogenic drivers and their prevalence in PDAC. However, whilst precision medicine has revolutionised the care of many malignancies, no targeted agent has yet been successful in improving outcomes in PDAC. Innovative clinical trials like PRECISION-Panc will be crucial in addressing this deficit.
Acknowledgments
Rille Pihlak is funded by the Collins Clinical Research fellowship and by Pancreatic Cancer UK.
Conflicts of Interest
The authors declare no conflict of interest.
References
- De Angelis, R.; Sant, M.; Coleman, M.P.; Francisci, S.; Baili, P.; Pierannunzio, D.; Trama, A.; Visser, O.; Brenner, H.; Ardanaz, E.; et al. Cancer survival in europe 1999–2007 by country and age: Results of EUROCARE-5—a population-based study. Lancet Oncol. 2014, 15, 23–34. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA: Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Cancer Research UK, Cancer Statistics, Pancreatic Cancer Statistics. Available online: http://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/pancreatic-cancer (accessed on 30 November 2017).
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouche, O.; Guimbaud, R.; Becouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardiere, C.; et al. Folfirinox versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [PubMed]
- Goel, G.; Sun, W. Novel approaches in the management of pancreatic ductal adenocarcinoma: Potential promises for the future. J. Hematol. Oncol. 2015, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; van de Velde, H.; Karasek, P.; Oettle, H.; Vervenne, W.L.; Szawlowski, A.; Schoffski, P.; Post, S.; Verslype, C.; Neumann, H.; et al. Phase III trial of gemcitabine plus tipifarnib compared with gemcitabine plus placebo in advanced pancreatic cancer. J. Clin. 2004, 22, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Azevedo, S.; Okusaka, T.; Van Laethem, J.L.; Lipton, L.R.; Riess, H.; Szczylik, C.; Moore, M.J.; Peeters, M.; Bodoky, G.; et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: The gamma trial. Ann. Oncol. 2015, 26, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Middleton, G.; Palmer, D.H.; Greenhalf, W.; Ghaneh, P.; Jackson, R.; Cox, T.; Evans, A.; Shaw, V.E.; Wadsley, J.; Valle, J.W.; et al. Vandetanib plus gemcitabine versus placebo plus gemcitabine in locally advanced or metastatic pancreatic carcinoma (ViP): A prospective, randomised, double-blind, multicentre phase 2 trial. Lancet Oncol. 2017, 18, 486–499. [Google Scholar] [CrossRef]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [PubMed]
- Salo-Mullen, E.E.; O’Reilly, E.M.; Kelsen, D.P.; Ashraf, A.M.; Lowery, M.A.; Yu, K.H.; Reidy, D.L.; Epstein, A.S.; Lincoln, A.; Saldia, A.; et al. Identification of germline genetic mutations in patients with pancreatic cancer. Cancer 2015, 121, 4382–4388. [Google Scholar] [CrossRef] [PubMed]
- Lal, G.; Liu, G.; Schmocker, B.; Kaurah, P.; Ozcelik, H.; Narod, S.A.; Redston, M.; Gallinger, S. Inherited predisposition to pancreatic adenocarcinoma: Role of family history and germ-line p16, BRCA1, and BRCA2 mutations. Cancer Res. 2000, 60, 409–416. [Google Scholar] [PubMed]
- Cowgill, S.M.; Muscarella, P. The genetics of pancreatic cancer. Am. J. Surg. 2003, 186, 279–286. [Google Scholar] [CrossRef]
- Gupta, C.; Mazzara, P.F. High-grade pancreatic intraepithelial neoplasia in a patient with familial adenomatous polyposis. Arch. Pathol. Lab. Med. 2005, 129, 1398–1400. [Google Scholar] [PubMed]
- Vasen, H.F.; Gruis, N.A.; Frants, R.R.; van Der Velden, P.A.; Hille, E.T.; Bergman, W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int. J. Cancer 2000, 87, 809–811. [Google Scholar] [CrossRef]
- Lowenfels, A.B.; Maisonneuve, P.; DiMagno, E.P.; Elitsur, Y.; Gates, L.K., Jr.; Perrault, J.; Whitcomb, D.C. Hereditary pancreatitis and the risk of pancreatic cancer. International hereditary pancreatitis study group. J. Nat. Cancer Inst. 1997, 89, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Shindo, K.; Yu, J.; Suenaga, M.; Fesharakizadeh, S.; Cho, C.; Macgregor-Das, A.; Siddiqui, A.; Witmer, P.D.; Tamura, K.; Song, T.J.; et al. Deleterious germline mutations in patients with apparently sporadic pancreatic adenocarcinoma. J. Clin. Oncol. 2017, 35, 3382–3390. [Google Scholar] [CrossRef] [PubMed]
- Waddell, N.; Pajic, M.; Patch, A.-M.; Chang, D.K.; Kassahn, K.S.; Bailey, P.; Johns, A.L.; Miller, D.; Nones, K.; Quek, K.; et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015, 518, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.J.R.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.-L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Catalogue of Somatic Mutations in Cancer. Signatures of Mutational Processes in Human Cancer. Available online: http://cancer.sanger.ac.uk/cosmic/signatures (accessed on 30 November 2017).
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch-repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.K.; Pothuri, B. Homologous recombination deficiency (hrd) testing in ovarian cancer clinical practice: A review of the literature. Gynecol. Oncol. Res. Pract. 2017, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Ke, Z.; Harmston, N.; Ho, S.Y.; Frois, A.O.; Alam, J.; Jeyaraj, D.A.; Pendharkar, V.; Ghosh, K.; Virshup, I.H.; et al. Wnt addiction of genetically defined cancers reversed by porcn inhibition. Oncogene 2016, 35, 2197–2207. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Adams, R.P.; Swain, S.M. Does CDKN2A loss predict palbociclib benefit? Curr. Oncol. 2015, 22, e498–e501. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the evolution of non–small-cell lung cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Wilentz, R.E.; Geradts, J.; Maynard, R.; Offerhaus, G.J.; Kang, M.; Goggins, M.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: Loss of intranuclear expression. Cancer Res. 1998, 58, 4740–4744. [Google Scholar] [PubMed]
- Wilentz, R.E.; Iacobuzio-Donahue, C.A.; Argani, P.; McCarthy, D.M.; Parsons, J.L.; Yeo, C.J.; Kern, S.E.; Hruban, R.H. Loss of expression of DPC4 in pancreatic intraepithelial neoplasia: Evidence that DPC4 inactivation occurs late in neoplastic progression. Cancer Res. 2000, 60, 2002–2006. [Google Scholar] [PubMed]
- Moskaluk, C.A.; Hruban, R.H.; Kern, S.E. P16 and K-ras gene mutations in the intraductal precursors of human pancreatic adenocarcinoma. Cancer Res. 1997, 57, 2140–2143. [Google Scholar] [PubMed]
- Notta, F.; Chan-Seng-Yue, M.; Lemire, M.; Li, Y.; Wilson, G.W.; Connor, A.A.; Denroche, R.E.; Liang, S.B.; Brown, A.M.; Kim, J.C.; et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016, 538, 378–382. [Google Scholar] [CrossRef] [PubMed]
- Makohon-Moore, A.P.; Zhang, M.; Reiter, J.G.; Bozic, I.; Allen, B.; Kundu, D.; Chatterjee, K.; Wong, F.; Jiao, Y.; Kohutek, Z.A.; et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat. Genet. 2017, 49, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Raphael, B.J.; Hruban, R.H.; Aguirre, A.J.; Moffitt, R.A.; Yeh, J.J.; Stewart, C.; Robertson, A.G.; Cherniack, A.D.; Gupta, M.; Getz, G.; et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017, 32, 185–203.e113. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, R.A.; Marayati, R.; Flate, E.L.; Volmar, K.E.; Loeza, S.G.; Hoadley, K.A.; Rashid, N.U.; Williams, L.A.; Eaton, S.C.; Chung, A.H.; et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat. Genet. 2015, 47, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Biankin, A.V.; Waddell, N.; Kassahn, K.S.; Gingras, M.C.; Muthuswamy, L.B.; Johns, A.L.; Miller, D.K.; Wilson, P.J.; Patch, A.M.; Wu, J.; et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012, 491, 399–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bournet, B.; Buscail, C.; Muscari, F.; Cordelier, P.; Buscail, L. Targeting kras for diagnosis, prognosis, and treatment of pancreatic cancer: Hopes and realities. Eur. J. Cancer 2016, 54, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Bodoky, G.; Timcheva, C.; Spigel, D.R.; La Stella, P.J.; Ciuleanu, T.E.; Pover, G.; Tebbutt, N.C. A phase II open-label randomized study to assess the efficacy and safety of selumetinib (AZD6244 [ARRY-142886]) versus capecitabine in patients with advanced or metastatic pancreatic cancer who have failed first-line gemcitabine therapy. Investig. New Drugs 2012, 30, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Lorusso, P.M.; Adjei, A.A.; Varterasian, M.; Gadgeel, S.; Reid, J.; Mitchell, D.Y.; Hanson, L.; DeLuca, P.; Bruzek, L.; Piens, J.; et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J. Clin. Oncol. 2005, 23, 5281–5293. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Kelsen, D.P.; Stadler, Z.K.; Yu, K.H.; Janjigian, Y.Y.; Ludwig, E.; D’Adamo, D.R.; Salo-Mullen, E.; Robson, M.E.; Allen, P.J.; et al. An emerging entity: Pancreatic adenocarcinoma associated with a known brca mutation: Clinical descriptors, treatment implications, and future directions. Oncologist 2011, 16, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Golan, T.; Kanji, Z.S.; Epelbaum, R.; Devaud, N.; Dagan, E.; Holter, S.; Aderka, D.; Paluch-Shimon, S.; Kaufman, B.; Gershoni-Baruch, R.; et al. Overall survival and clinical characteristics of pancreatic cancer in brca mutation carriers. Br. J. Cancer 2014, 111, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Aung, K.L.; Holter, S.; Borgida, A.; Connor, A.; Pintilie, M.; Dhani, N.C.; Hedley, D.W.; Knox, J.J.; Gallinger, S. Overall survival of patients with pancreatic adenocarcinoma and BRCA1 or BRCA2 germline mutation. J. Clin. Oncol. 2016, 34, 4123. [Google Scholar]
- Ledermann, J.; Harter, P.; Gourley, C.; Friedlander, M.; Vergote, I.; Rustin, G.; Scott, C.L.; Meier, W.; Shapira-Frommer, R.; Safra, T.; et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: A preplanned retrospective analysis of outcomes by brca status in a randomised phase 2 trial. Lancet Oncol. 2014, 15, 852–861. [Google Scholar] [CrossRef]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef] [PubMed]
- Riaz, N.; Blecua, P.; Lim, R.S.; Shen, R.; Higginson, D.S.; Weinhold, N.; Norton, L.; Weigelt, B.; Powell, S.N.; Reis-Filho, J.S. Pan-cancer analysis of BI-allelic alterations in homologous recombination DNA repair genes. Nat. Commun. 2017, 8, 857. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute (NCI). A Randomized Phase II Study of Gemcitabine, Cisplatin +/− Veliparib in Patients with Pancreas Adenocarcinoma and a Known BRCA/ PALB2 Mutation (Part I) and a Phase II Single Arm Study of Single-Agent Veliparib in Previously Treated Pancreas Adenocarcinoma (Part II). Available online: https://clinicaltrials.Gov/ct2/show/nct01585805 (accessed on 30 November 2017).
- AstraZeneca. A Phase III, Randomised, Double Blind, Placebo Controlled, Multicentre Study of Maintenance Olaparib Monotherapy in Patients with gBRCA Mutated Metastatic Pancreatic Cancer Whose Disease Has Not Progressed on First Line Platinum Based Chemotherapy. Available online: https://clinicaltrials.Gov/ct2/show/nct02184195 (accessed on 30 November 2017).
- Perkhofer, L.; Schmitt, A.; Romero Carrasco, M.C.; Ihle, M.; Hampp, S.; Ruess, D.A.; Hessmann, E.; Russell, R.; Lechel, A.; Azoitei, N.; et al. ATM deficiency generating genomic instability sensitizes pancreatic ductal adenocarcinoma cells to therapy-induced DNA damage. Cancer Res. 2017, 77, 5576–5590. [Google Scholar] [CrossRef] [PubMed]
- McCabe, N.; Turner, N.C.; Lord, C.J.; Kluzek, K.; Bialkowska, A.; Swift, S.; Giavara, S.; O’Connor, M.J.; Tutt, A.N.; Zdzienicka, M.Z.; et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer Res. 2006, 66, 8109–8115. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Ashworth, A. Brcaness revisited. Nat. Rev. Cancer 2016, 16, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Kim, J.; Braunstein, L.Z.; Karlic, R.; Haradhavala, N.J.; Tiao, G.; Rosebrock, D.; Livitz, D.; Kübler, K.; Mouw, K.W.; et al. A mutational signature reveals alterations underlying deficient homologous recombination repair in breast cancer. Nat. Genet. 2017, 49, 1476. [Google Scholar] [CrossRef] [PubMed]
- MD Anderson Cancer Center. Olaparib for Brcaness Phenotype in Pancreatic Cancer: Phase II Study. Available online: https://clinicaltrials.Gov/ct2/show/nct02677038 (accessed on 30 November 2017).
- Weber, A.M.; Ryan, A.J. Atm and atr as therapeutic targets in cancer. Pharmacol.Ther. 2015, 149, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Royal Marsden NHS Foundation Trust. A Phase I Study to Assess the Tolerability, Safety and Biological Effects of ATR Inhibitor (AZD6738) as a Single Agent and in Combination with Palliative Radiation Therapy in Patients with Solid Tumours. Available online: https://clinicaltrials.Gov/ct2/show/nct02223923 (accessed on 30 November 2017).
- AstraZeneca. A Modular Phase I, Open-Label, Multicentre Study to Assess the Safety, Tolerability, Pharmacokinetics and Preliminary Anti-Tumour Activity of AZD6738 in Combination with Cytotoxic Chemotherapy and/or DNA Damage Repair/Novel Anti-Cancer Agents in Patients with Advanced Solid Malignancies. Available online: https://clinicaltrials.Gov/ct2/show/nct02264678 (accessed on 30 November 2017).
- Kim, H.; Saka, B.; Knight, S.; Borges, M.; Childs, E.; Klein, A.; Wolfgang, C.; Herman, J.; Adsay, V.N.; Hruban, R.H.; et al. Having pancreatic cancer with tumoral loss of ATM and normal TP53 protein expression is associated with a poorer prognosis. Clin. Cancer Res. 2014, 20, 1865–1872. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.; Perkhofer, L.; Liebau, S.; Lin, Q.; Lechel, A.; Feld, F.M.; Hessmann, E.; Gaedcke, J.; Guthle, M.; Zenke, M.; et al. Loss of ATM accelerates pancreatic cancer formation and epithelial-mesenchymal transition. Nat. Commun. 2015, 6, 7677. [Google Scholar] [CrossRef] [PubMed]
- Drosos, Y.; Escobar, D.; Chiang, M.Y.; Roys, K.; Valentine, V.; Valentine, M.B.; Rehg, J.E.; Sahai, V.; Begley, L.A.; Ye, J.; et al. Atm-deficiency increases genomic instability and metastatic potential in a mouse model of pancreatic cancer. Sci. Rep. 2017, 7, 11144. [Google Scholar] [CrossRef] [PubMed]
- Ayars, M.; Eshleman, J.; Goggins, M. Susceptibility of ATM-deficient pancreatic cancer cells to radiation. Cell Cycle 2017, 16, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Weissmueller, S.; Manchado, E.; Saborowski, M.; Morris, J.P.; Wagenblast, E.; Davis, C.A.; Moon, S.H.; Pfister, N.T.; Tschaharganeh, D.F.; Kitzing, T.; et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor β signaling. Cell 2014, 157, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Dhayat, S.A.; Mardin, W.A.; Seggewiss, J.; Strose, A.J.; Matuszcak, C.; Hummel, R.; Senninger, N.; Mees, S.T.; Haier, J. Microrna profiling implies new markers of gemcitabine chemoresistance in mutant p53 pancreatic ductal adenocarcinoma. PLoS ONE 2015, 10, e0143755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oza, A.M.; Weberpals, J.I.; Provencher, D.M.; Grischke, E.-M.; Hall, M.; Uyar, D.; Estevez-Diz, M.D.P.; Marmé, F.; Kuzmin, A.; Rosenberg, P.; et al. An international, biomarker-directed, randomized, phase II trial of AZD1775 plus paclitaxel and carboplatin (p/C) for the treatment of women with platinum-sensitive, TP53-mutant ovarian cancer. J. Clin. Oncol. 2015, 33, 5506. [Google Scholar]
- Gourley, C.; Green, J.; Gabra, H.; Vergote, I.; Basu, B.; Brenton, J.D.; Björklund, U.; Smith, A.M.; Euler, M.V. PISARRO: A EUTROC phase Ib study of APR-246 in combination with carboplatin (C) and pegylated liposomal doxorubicin (PLD) in platinum sensitive relapsed high grade serous ovarian cancer (HGSOC). J. Clin. Oncol. 2016, 34, 5571. [Google Scholar]
- A Phase Ib/2 Study Evaluating the Efficacy of APR-246, a First-In-Class Agent Targeting Mutant p53 in the Treatment of Platinum Resistant Advanced and Metastatic Oesophageal or Gastro-Oesophageal Junction Cancers. Available online: https://clinicaltrials.Gov/show/nct02999893 (accessed on 30 November 2017).
- Phase II, Single-Arm Study of AZD1775 Monotherapy in Relapsed Small Cell Lung Cancer Patients with MYC Family Amplification or CDKN2A Mutation Combined with TP53 Mutation. Available online: https://clinicaltrials.Gov/show/nct02688907 (accessed on 30 November 2017).
- A Multicentre Phase II Study of AZD1775 Plus Chemotherapy in Patients with Platinum-Resistant Epithelial Ovarian, Fallopian Tube, or Primary Peritoneal Cancer. Available online: https://clinicaltrials.Gov/show/nct02272790 (accessed on 30 November 2017).
- Ormanns, S.; Haas, M.; Remold, A.; Kruger, S.; Holdenrieder, S.; Kirchner, T.; Heinemann, V.; Boeck, S. The impact of SMAD4 loss on outcome in patients with advanced pancreatic cancer treated with systemic chemotherapy. Int. J. Mol. Sci. 2017, 18, 1094. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.; Wu, W.; Huang, C.; Cen, G.; Jiang, T.; Cao, J.; Huang, K.; Qiu, Z. SMAD4 and its role in pancreatic cancer. Tumour Boil. 2015, 36, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.H.; Varadhachary, G.R.; Yordy, J.S.; Staerkel, G.A.; Javle, M.M.; Safran, H.; Haque, W.; Hobbs, B.D.; Krishnan, S.; Fleming, J.B.; et al. Phase II trial of cetuximab, gemcitabine, and oxaliplatin followed by chemoradiation with cetuximab for locally advanced (T4) pancreatic adenocarcinoma: Correlation of SMAD4(DPC4) immunostaining with pattern of disease progression. J. Clin. Oncol. 2011, 29, 3037–3043. [Google Scholar] [CrossRef] [PubMed]
- Ottenhof, N.A.; Morsink, F.H.M.; ten Kate, F.; van Noorden, C.J.F.; Offerhaus, G.J.A. Multivariate analysis of immunohistochemical evaluation of protein expression in pancreatic ductal adenocarcinoma reveals prognostic significance for persistent SMAD4 expression only. Cell. Oncol. 2012, 35, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.H.; Kim, H.J.; Hwang, D.W.; Lee, J.H.; Song, K.B.; Jun, E.; Shim, I.K.; Hong, S.M.; Kim, H.J.; Park, K.M.; et al. The DPC4/SMAD4 genetic status determines recurrence patterns and treatment outcomes in resected pancreatic ductal adenocarcinoma: A prospective cohort study. Oncotarget 2017, 8, 17945–17959. [Google Scholar] [PubMed]
- Shugang, X.; Hongfa, Y.; Jianpeng, L.; Xu, Z.; Jingqi, F.; Xiangxiang, L.; Wei, L. Prognostic value of SMAD4 in pancreatic cancer: A meta-analysis. Trans. Oncol. 2016, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Dey, P.; Baddour, J.; Muller, F.; Wu, C.C.; Wang, H.; Liao, W.-T.; Lan, Z.; Chen, A.; Gutschner, T.; Kang, Y.; et al. Genomic deletion of malic enzyme 2 confers collateral lethality in pancreatic cancer. Nature 2017, 542, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Soura, E.; Eliades, P.; Shannon, K.; Stratigos, A.; Tsao, H. Hereditary melanoma: Update on syndromes and management—Genetics of familial atypical multiple mole melanoma syndrome. J. Am. Acad. Der. 2016, 74, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Ghiorzo, P.; Fornarini, G.; Sciallero, S.; Battistuzzi, L.; Belli, F.; Bernard, L.; Bonelli, L.; Borgonovo, G.; Bruno, W.; De Cian, F.; et al. CDKN2A is the main susceptibility gene in italian pancreatic cancer families. J. Med. Genet. 2012, 49, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Oshima, M.; Okano, K.; Muraki, S.; Haba, R.; Maeba, T.; Suzuki, Y.; Yachida, S. Immunohistochemically detected expression of 3 major genes (CDKN2A/p16, TP53, and SMAD4/DPC4) strongly predicts survival in patients with resectable pancreatic cancer. Ann. Surg. 2013, 258, 336–346. [Google Scholar] [CrossRef] [PubMed]
- Phase II Study of Ilorasertib (ABT348) in Patients with CDKN2A Deficient Solid Tumors. Available online: https://clinicaltrials.Gov/show/nct02478320 (accessed on 30 November 2017).
- Elvin, J.A.; Gay, L.M.; Ort, R.; Shuluk, J.; Long, J.; Shelley, L.; Lee, R.; Chalmers, Z.R.; Frampton, G.M.; Ali, S.M.; et al. Clinical benefit in response to palbociclib treatment in refractory uterine leiomyosarcomas with a common CDKN2A alteration. Ooncologist 2017, 22, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Chou, A.; Froio, D.; Nagrial, A.M.; Parkin, A.; Murphy, K.J.; Chin, V.T.; Wohl, D.; Steinmann, A.; Stark, R.; Drury, A.; et al. Tailored first-line and second-line CDK4-targeting treatment combinations in mouse models of pancreatic cancer. Gut 2017. [Google Scholar] [CrossRef] [PubMed]
- An Open-Label Phase Ib Study of Palbociclib (Oral CDK 4/6 Inhibitor) Plus Abraxane (Registered) (Nab-Paclitaxel) in Patients with Metastatic Pancreatic Ductal Adenocarcinoma. Available online: https://clinicaltrials.Gov/show/nct02501902 (accessed on 30 November 2017).
- Phase I Study of the CDK4/6 Inhibitor Palbociclib (PD-0332991) in Combination with the PI3K/mTOR Inhibitor Gedatolisib (PF-05212384) for Patients with Advanced Squamous Cell Lung, Pancreatic, Head & Neck and Other Solid Tumors. Available online: https://clinicaltrials.Gov/show/nct03065062 (accessed on 30 November 2017).
- Witkiewicz, A.K.; McMillan, E.A.; Balaji, U.; Baek, G.; Lin, W.C.; Mansour, J.; Mollaee, M.; Wagner, K.U.; Koduru, P.; Yopp, A.; et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat. Commun. 2015, 6, 6744. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Humphris, J.L.; Patch, A.-M.; Nones, K.; Bailey, P.J.; Johns, A.L.; McKay, S.; Chang, D.K.; Miller, D.K.; Pajic, M.; Kassahn, K.S.; et al. Hypermutation in pancreatic cancer. Gastroenterology 2017, 152, 68–74.e62. [Google Scholar] [CrossRef] [PubMed]
- The US Food and Drug Administration News Release. FDA Approves First Cancer Treatment for Any Solid Tumor with a Specific Genetic Feature. Available online: https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm560167.htm (accessed on 30 November 2017).
- Diaz, L.A.; Marabelle, A.; Delord, J.-P.; Shapira-Frommer, R.; Geva, R.; Peled, N.; Kim, T.W.; Andre, T.; Cutsem, E.V.; Guimbaud, R.; et al. Pembrolizumab therapy for microsatellite instability high (MSI-H) colorectal cancer (CRC) and non-CRC. J. Clin. Oncol. 2017, 35, 3071. [Google Scholar]
- Cheng, D.T.; Mitchell, T.N.; Zehir, A.; Shah, R.H.; Benayed, R.; Syed, A.; Chandramohan, R.; Liu, Z.Y.; Won, H.H.; Scott, S.N.; et al. Memorial sloan kettering-integrated mutation profiling of actionable cancer targets (MSK-impact): A hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J. Mol. Diagn. 2015, 17, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Fox, A.J.; Hiemenz, M.C.; Lieberman, D.B.; Sukhadia, S.; Li, B.; Grubb, J.; Candrea, P.; Ganapathy, K.; Zhao, J.; Roth, D.; et al. Next generation sequencing for the detection of actionable mutations in solid and liquid tumors. J. Vis. Exp. 2016, 52758. [Google Scholar] [CrossRef] [PubMed]
- Eifert, C.; Pantazi, A.; Sun, R.; Xu, J.; Cingolani, P.; Heyer, J.; Russell, M.; Lvova, M.; Ring, J.; Tse, J.Y.; et al. Clinical application of a cancer genomic profiling assay to guide precision medicine decisions. Pers. Med. 2017, 14, 309–325. [Google Scholar] [CrossRef] [PubMed]
- Campbell, B.B.; Light, N.; Fabrizio, D.; Zatzman, M.; Fuligni, F.; de Borja, R.; Davidson, S.; Edwards, M.; Elvin, J.A.; Hodel, K.P.; et al. Comprehensive analysis of hypermutation in human cancer. Cell 2017, 171, 1042–1056. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar]
- Royal, R.E.; Levy, C.; Turner, K.; Mathur, A.; Hughes, M.; Kammula, U.S.; Sherry, R.M.; Topalian, S.L.; Yang, J.C.; Lowy, I.; et al. Phase 2 trial of single agent ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J. Immunother. 2010, 33, 828–833. [Google Scholar] [CrossRef] [PubMed]
- Aglietta, M.; Barone, C.; Sawyer, M.B.; Moore, M.J.; Miller, W.H., Jr.; Bagala, C.; Colombi, F.; Cagnazzo, C.; Gioeni, L.; Wang, E.; et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann. Oncol. 2014, 25, 1750–1755. [Google Scholar] [CrossRef] [PubMed]
- Kunk, P.R.; Bauer, T.W.; Slingluff, C.L.; Rahma, O.E. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J. Immunother. Cancer 2016, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- A Phase Ib/2, Open-Label, Multicenter, Randomized Umbrella Study Evaluating the Efficacy and Safety of Multiple Immunotherapy-Based Treatment Combinations in Patients with Metastatic Pancreatic Ductal Adenocarcinoma (Morpheus-Pancreatic Cancer). Available online: https://clinicaltrials.Gov/show/nct03193190 (accessed on 30 November 2017).
- A Phase 2 Study of Neoadjuvant Chemotherapy with and without Immunotherapy to CA125 (Oregovomab) Followed by Hypofractionated Stereotactic Radiotherapy and Concurrent HIV Protease Inhibitor Nelfinavir in Patients with Locally Advanced Pancreatic Cancer. Available online: https://clinicaltrials.Gov/show/nct01959672 (accessed on 30 November 2017).
- A Phase 2 Study of Cabiralizumab (BMS-986227, FPA008) Administered in Combination with Nivolumab (BMS-936558) with and without Chemotherapy in Patients with Advanced Pancreatic Cancer. Available online: https://clinicaltrials.Gov/show/nct03336216 (accessed on 30 November 2017).
- A Pilot Study of a GVAX Pancreas Vaccine (with Cyclophosphamide) in Combination with a PD-1 Blockade Antibody (Pembrolizumab) and a Macrophage Targeting Agent (CSF1R Inhibitor) for the Treatment of Patients with Borderline Resectable Adenocarcinoma of the Pancreas. Available online: https://clinicaltrials.Gov/show/nct03153410 (accessed on 30 November 2017).
- A Phase Ib Dose-Escalation and Cohort-Expansion Study of the Safety, Tolerability, and Efficacy of a Novel Transforming Growth Factor-β Receptor I Kinase Inhibitor (Galunisertib) Administered in Combination with the Anti-PD-l1 Antibody Durvalumab (MEDI4736) in Recurrent or Refractory Metastatic Pancreatic Cancer. In: Clinicaltrials. Available online: https://clinicaltrials.Gov/show/nct02734160 (accessed on 30 November 2017).
- A Phase 2 Study of GM-CSF Secreting Allogeneic Pancreatic Cancer Vaccine in Combination with PD-1 Blockade Antibody (Pembrolizumab) and Stereotactic Body Radiation Therapy (SBRT) for the Treatment of Patients with Locally Advanced Adenocarcinoma of the Pancreas. Available online: https://clinicaltrials.Gov/show/nct02648282 (accessed on 30 November 2017).
- A Phase 2 Clinical Trial of GVAX Pancreas Vaccine (with Cyclophosphamide) in Combination with Nivolumab and Stereotactic Body Radiation Therapy (SBRT) Followed by Definitive Resection for Patients with Borderline Resectable Pancreatic Adenocarcinoma. Available online: https://clinicaltrials.Gov/show/nct03161379 (accessed on 30 November 2017).
- A Randomized Multicenter Phase Ib/2 Study to Assess the Safety and the Immunological Effect of Chemoradiation Therapy (CRT) in Combination with Pembrolizumab (MK-3475) Compared to CRT Alone in Patients with Resectable or Borderline Resectable Pancreatic Cancer. Available online: https://clinicaltrials.Gov/show/nct02305186 (accessed on 30 November 2017).
- Nivolumab and Ipilimumab and Radiation Therapy in Microsatellite Stable (MSS) and Microsatellite Instability (MSI) High Colorectal and Pancreatic Cancer. Available online: https://clinicaltrials.Gov/show/nct03104439 (accessed on 30 November 2017).
- A Randomized Phase 2 Study of the Safety, Efficacy, and Immune Response of CRS-207, Nivolumab, and Ipilimumab with or without GVAX Pancreas Vaccine (with Cyclophosphamide) in Patients with Previously Treated Metastatic Pancreatic Adenocarcinoma. Available online: https://clinicaltrials.Gov/show/nct03190265 (accessed on 30 November 2017).
- Phase 2 Study of Epacadostat, Pembrolizumab, and CRS-207, with or without Cyclophosphamide and GVAX Pancreas Vaccine in Patients with Metastatic Pancreas Cancer. Available online: https://clinicaltrials.Gov/show/nct03006302 (accessed on 30 November 2017).
- Phase 1/2 Study to Evaluate the Safety and Preliminary Efficacy of Nivolumab Combined with Daratumumab in Participants with Advanced or Metastatic Solid Tumors. Available online: https://clinicaltrials.Gov/show/nct03098550 (accessed on 30 November 2017).
- A Phase 1 Immunotherapy Study of Evofosfamide in Combination with Ipilimumab in Patients with Advanced Solid Malignancies. Available online: https://clinicaltrials.Gov/show/nct03098160 (accessed on 30 November 2017).
- A Phase 1b and 2 Open-Label, Multi-Center Study of MEDI4736 Evaluated in Different Combinations in Patients with Metastatic Pancreatic Ductal Adenocarcinoma. Available online: https://clinicaltrials.Gov/show/nct02583477 (accessed on 30 November 2017).
- A Two-Part, Open-Label Phase 1/2 Study to Evaluate Pharmacodynamic Effects and Safety of Olaptesed Pegol Monotherapy and Safety and Efficacy of Olaptesed Pegol/Pembrolizumab Combination Therapy in Metastatic Colorectal and Pancreatic Cancer. Available online: https://clinicaltrials.Gov/show/nct03168139 (accessed on 30 November 2017).
- Nant Pancreatic Cancer Vaccine: Combination Immunotherapy with High-Affinity Natural Killer (HANK) in Subjects with Pancreatic Cancer Who Have Progressed on or after Standard-of-Care Therapy. Available online: https://clinicaltrials.Gov/show/nct03329248 (accessed on 30 November 2017).
- Immunotherapy and Irreversible Electroporation in the Treatment of Advanced Pancreatic Adenocarcinoma. Available online: https://clinicaltrials.Gov/show/nct03080974 (accessed on 30 November 2017).
- Steinhart, Z.; Pavlovic, Z.; Chandrashekhar, M.; Hart, T.; Wang, X.; Zhang, X.; Robitaille, M.; Brown, K.R.; Jaksani, S.; Overmeer, R.; et al. Genome-wide CRISPR screens reveal a Wnt-FZD5 signaling circuit as a druggable vulnerability of RNF43-mutant pancreatic tumors. Nat. Med. 2017, 23, 60–68. [Google Scholar] [CrossRef] [PubMed]
- A Phase I, Open-Label, Dose Escalation Study of Oral LGK974 in Patients with Malignancies Dependent on Wnt Ligands. Available online: https://clinicaltrials.Gov/show/nct01351103 (accessed on 30 November 2017).
- Ross, J.S.; Slodkowska, E.A.; Symmans, W.F.; Pusztai, L.; Ravdin, P.M.; Hortobagyi, G.N. The HER-2 receptor and breast cancer: Ten years of targeted anti-HER-2 therapy and personalized medicine. Oncologist 2009, 14, 320–368. [Google Scholar] [CrossRef] [PubMed]
- Tadesse, S.; Yu, M.; Kumarasiri, M.; Le, B.T.; Wang, S. Targeting CDK6 in cancer: State of the art and new insights. Cell Cycle 2015, 14, 3220–3230. [Google Scholar] [CrossRef] [PubMed]
- Karakas, B.; Bachman, K.E.; Park, B.H. Mutation of the PIK3CA oncogene in human cancers. Br. J. Cancer 2006, 94, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Lowery, M.A.; Jordan, E.J.; Basturk, O.; Ptashkin, R.N.; Zehir, A.; Berger, M.F.; Leach, T.; Herbst, B.; Askan, G.; Maynard, H.; et al. Real-time genomic profiling of pancreatic ductal adenocarcinoma: Potential actionability and correlation with clinical phenotype. Clin. Cancer Res. 2017, 23, 6094–6100. [Google Scholar] [CrossRef] [PubMed]
- Johns, A.L.; McKay, S.H.; Humphris, J.L.; Pinese, M.; Chantrill, L.A.; Mead, R.S.; Tucker, K.; Andrews, L.; Goodwin, A.; Leonard, C.; et al. Lost in translation: Returning germline genetic results in genome-scale cancer research. Genome Med. 2017, 9, 41. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, S.B.; Chang, D.K.; Bailey, P.; Biankin, A.V. Pancreatic cancer genomes: Implications for clinical management and therapeutic development. Clin. Cancer Res. 2017, 23, 1638–1646. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Prevalence of potentially targetable mutations in PDAC. KRAS—KRAS proto-oncogene, GTPase [33,35]; TP53—tumour protein p53 [59]; SMAD4—SMAD Family Member 4 [19,67]; DDR—DNA damage repair pathway mutations [18,19]; CDKN2A—cyclin-dependent kinase inhibitor 2A [12,75]; RNF43—ring finger protein 43 [19,24]; BRAF B-Raf proto-oncogene, serine/threonine kinase [82]; MSI—microsatellite instability [84]. Dark colour signifies minimal range of mutation reported, while lighter shade signifies maximum range of mutation reported within different references.
Figure 1.
Prevalence of potentially targetable mutations in PDAC. KRAS—KRAS proto-oncogene, GTPase [33,35]; TP53—tumour protein p53 [59]; SMAD4—SMAD Family Member 4 [19,67]; DDR—DNA damage repair pathway mutations [18,19]; CDKN2A—cyclin-dependent kinase inhibitor 2A [12,75]; RNF43—ring finger protein 43 [19,24]; BRAF B-Raf proto-oncogene, serine/threonine kinase [82]; MSI—microsatellite instability [84]. Dark colour signifies minimal range of mutation reported, while lighter shade signifies maximum range of mutation reported within different references.


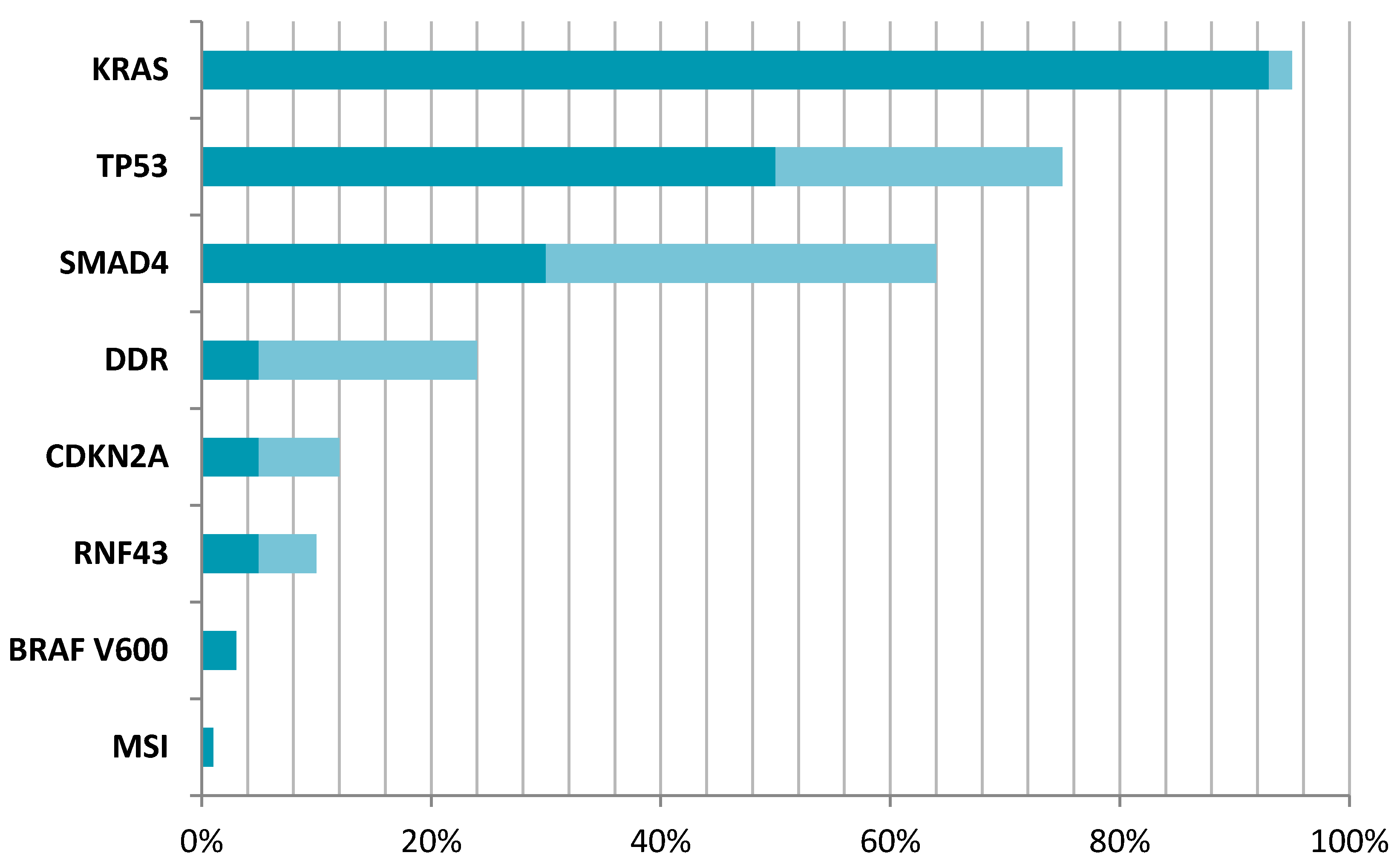{kind=link}
Table 1.
Clinical trials registered on Clinicaltrials.gov that are utilising checkpoint inhibitor-based immunotherapy, and are currently recruiting or due to recruit patients with pancreatic ductal adenocarcinoma.
Table 1.
Clinical trials registered on Clinicaltrials.gov that are utilising checkpoint inhibitor-based immunotherapy, and are currently recruiting or due to recruit patients with pancreatic ductal adenocarcinoma.
| Trial ID | Stage of Disease | Checkpoint Inhibitor | Combined with | Phase | Number of Patients | Countries Involved |
|---|---|---|---|---|---|---|
| NCT03193190 [95] | IV | Atezolizumab | Cobimetinib or PEGPH20 or BL-8040 | Ib–II | 185 | International |
| NCT01959672 [96] | I–III | Oregovomab | As single agent after chemotherapy and prior to Stereotactic Body Radiation Therapy | II | 66 | US |
| NCT03336216 [97] | III–IV | Nivolumab | Cabiralizumab or Cabiralizumab + chemotherapy | II | 160 | US |
| NCT03153410 [98] | II * | Pembrolizumab | Cyclophosphamide + GVAX Pancreas Vaccine + IMC-CS4 | I | 12 | US |
| NCT02734160 [99] | IV | Durvalumab | Galunisertib | Ib | 37 | International |
| NCT02648282 [100] | III | Pembrolizumab | Cyclophosphamide + GVAX Pancreas Vaccine + Stereotactic Body Radiation Therapy | II | 54 | US |
| NCT03161379 [101] | II * | Nivolumab | Cyclophosphamide + GVAX Pancreas Vaccine + Stereotactic Body Radiation Therapy | II | 50 | US |
| NCT02305186 [102] | I–II * | Pembrolizumab | Radiotherapy + capecitabine | Ib–II | 56 | US |
| NCT03104439 [103] | IV | Nivolumab and Ipilimumab | Radiotherapy | II | 80 | US |
| NCT03190265 [104] | IV | Nivolumab and Ipilimumab | Cyclophosphamide + GVAX Pancreas Vaccine + CRS-207 | II | 63 | US |
| NCT03006302 [105] | IV | Pembrolizumab | Cyclophosphamide + Epacadostat + CRS-207 + GVAX Pancreas Vaccine | II | 70 | US |
| NCT03098550 [106] | III–IV | Nivolumab | Daratumumab | I–II | 120 | International |
| NCT03098160 [107] | IV | Ipilimumab | Evofosfamide | I | 69 | US |
| NCT02583477 [108] | IV | Durvalumab | Nab-paclitaxel and gemcitabine or AZD5069 | Ib–II | 19 | US, UK |
| NCT03168139 [109] | IV | Pembrolizumab | Olaptesed pegol | I–II | 20 | Germany |
| NCT03329248 [110] | III–IV | Avelumab | ALT-803 + ETBX-011 + GI-4000 + haNK for infusion + bevacizumab + Capecitabine + Cyclophosphamide + Fluorouracil + Leucovorin + nab-Paclitaxel + lovaza + Oxaliplatin + SBRT | I–II | 80 | US |
| NCT03080974 [111] | III | Nivolumab | Irreversible Electroporation | II | 10 | US |
* Borderline resectable; US, United States; UK, United Kingdom.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Pihlak, R.; Weaver, J.M.J.; Valle, J.W.; McNamara, M.G. Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy. Cancers 2018, 10, 17. https://doi.org/10.3390/cancers10010017
AMA Style
Pihlak R, Weaver JMJ, Valle JW, McNamara MG. Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy. Cancers. 2018; 10(1):17. https://doi.org/10.3390/cancers10010017
Chicago/Turabian StylePihlak, Rille, Jamie M. J. Weaver, Juan W. Valle, and Mairéad G. McNamara. 2018. "Advances in Molecular Profiling and Categorisation of Pancreatic Adenocarcinoma and the Implications for Therapy" Cancers 10, no. 1: 17. https://doi.org/10.3390/cancers10010017
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.