
Microfluidic-Based Multi-Organ Platforms for Drug Discovery

Abstract
:
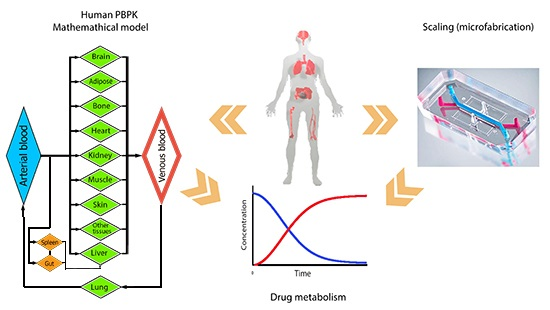{kind=link}
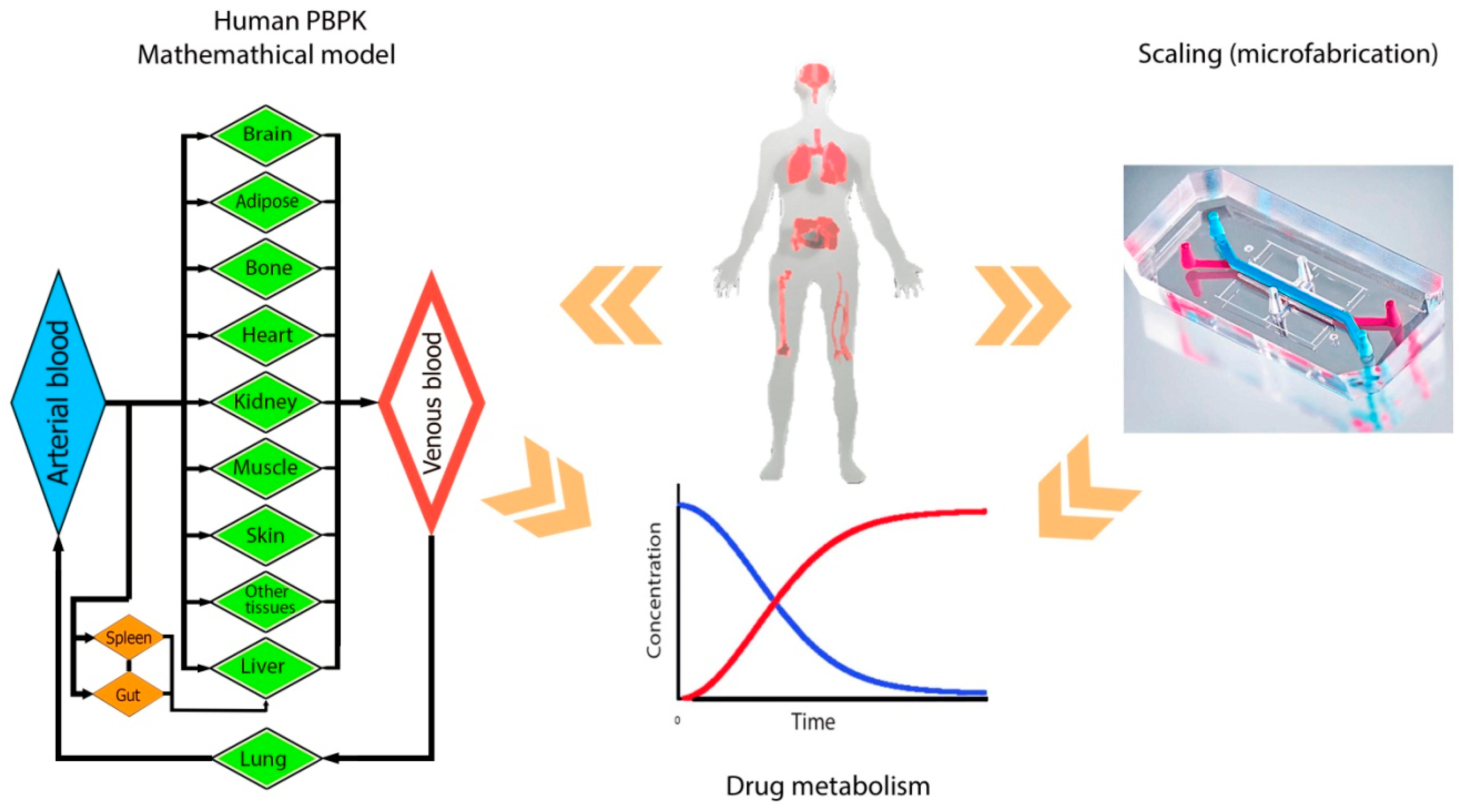{kind=link}
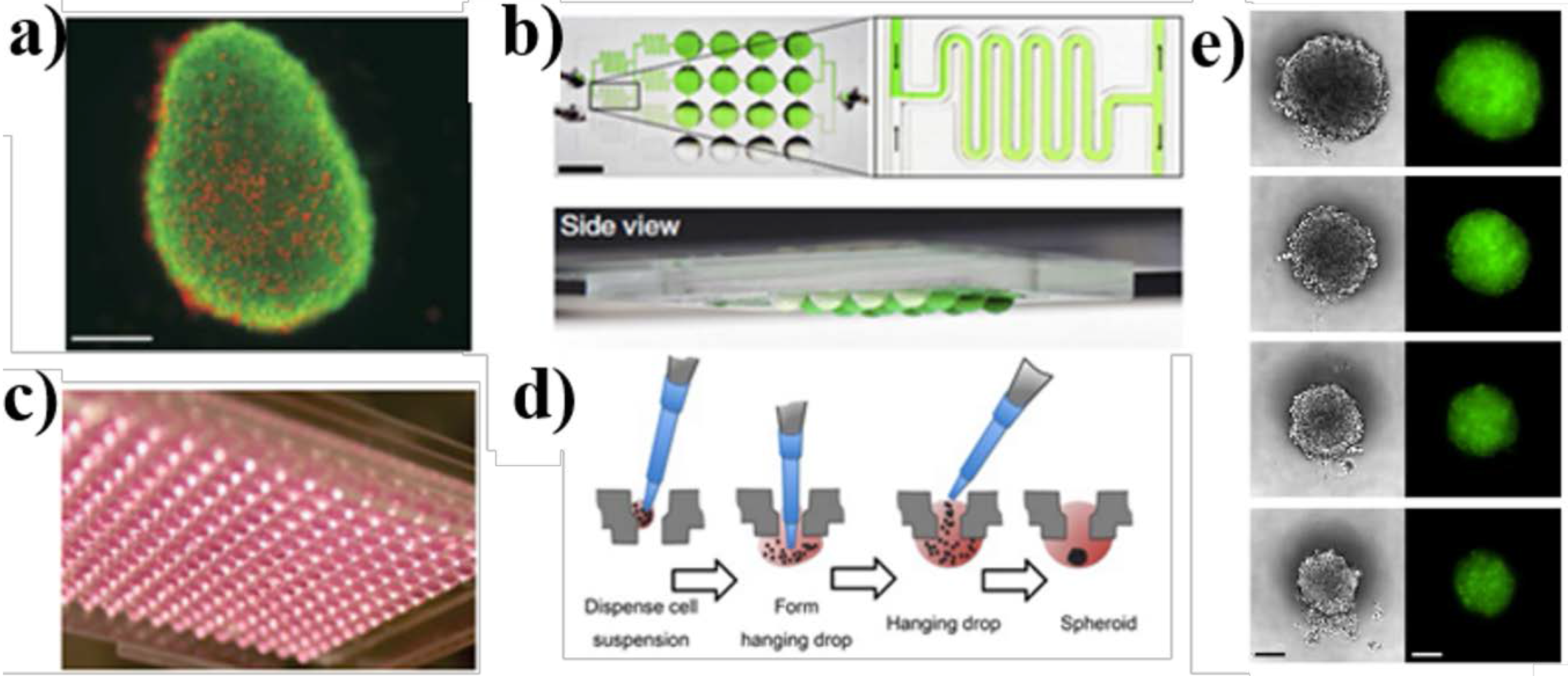{kind=link}
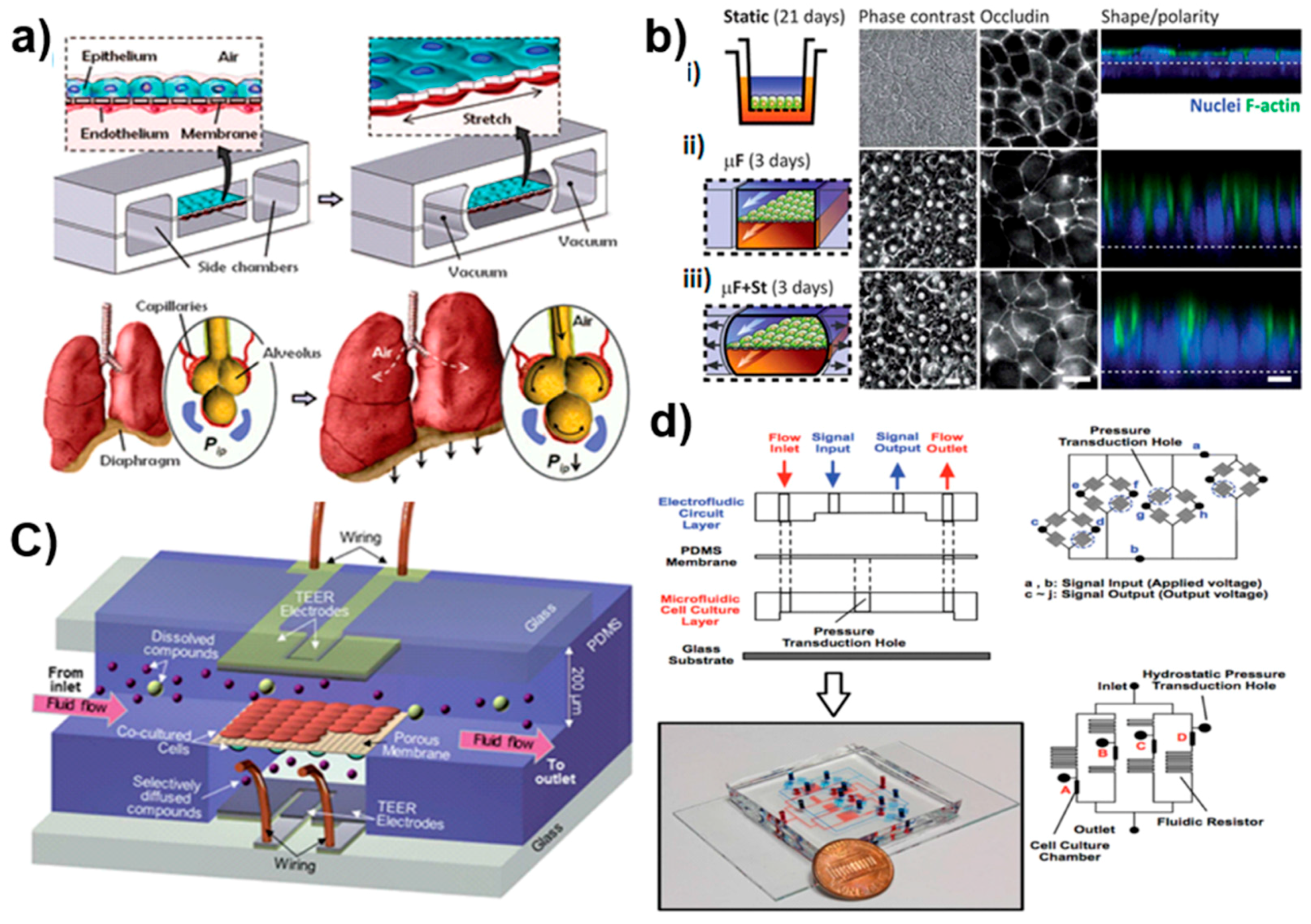{kind=link}
{kind=link}
1. Introduction
2. Drug Testing and Design of BOC
3. Multi-Organs on Chip
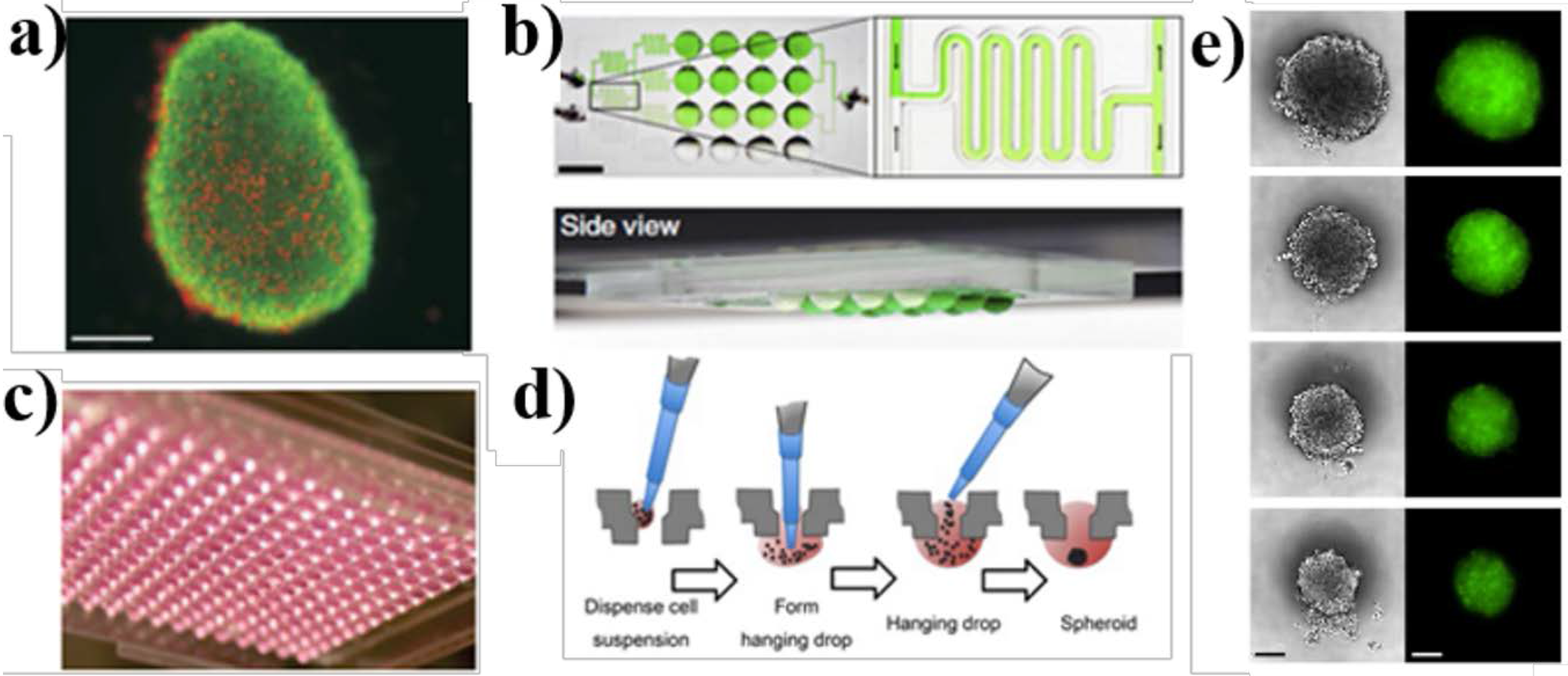
3.1. Organoid and Hanging Drop Spheroid Culture Models
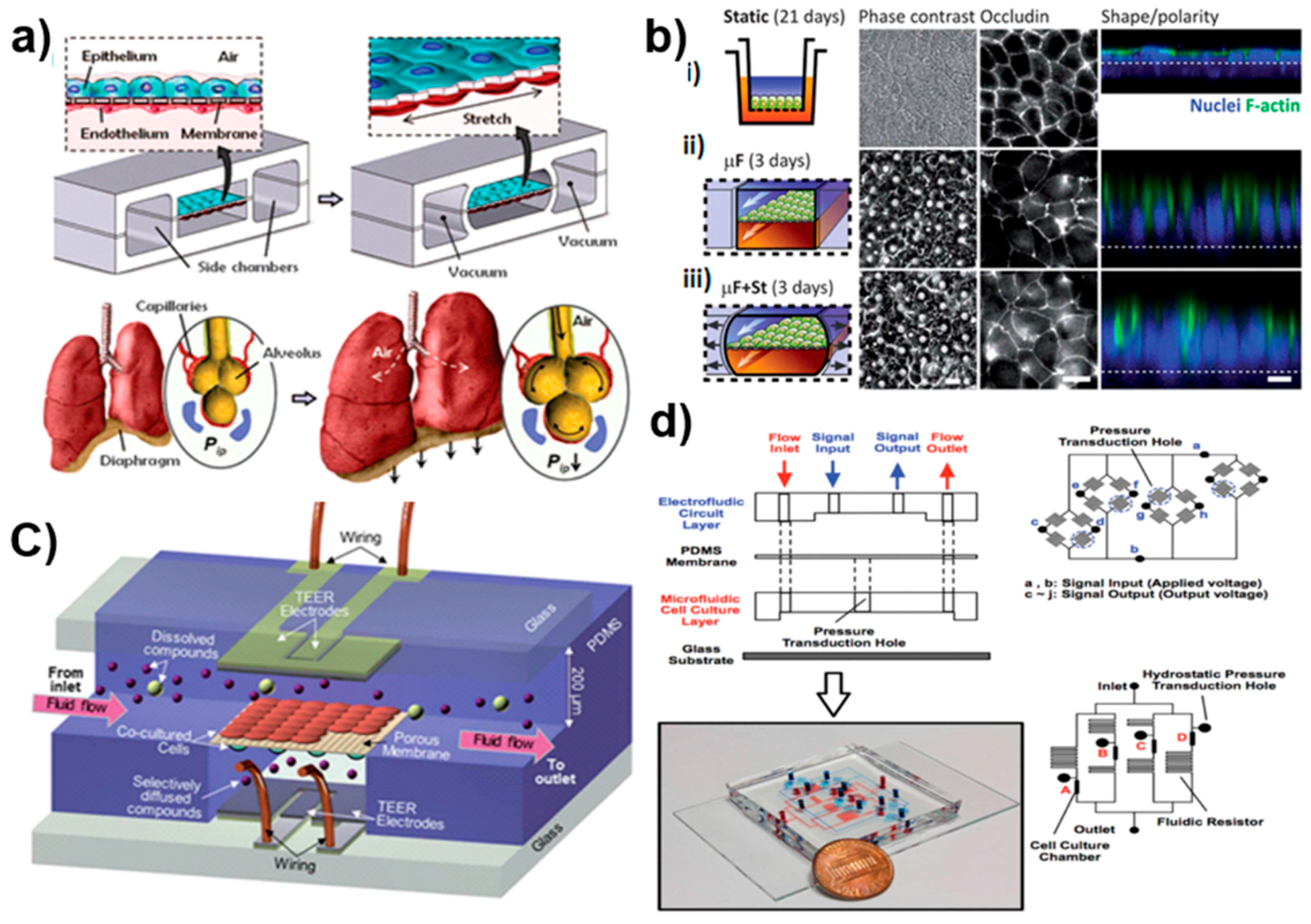
3.2. Microfluidics: A Proficient Framework for Multi-Organ Studies
3.3. Microfluidic-Based BOC Models for Drug Development
3.4. Two Organ Models
3.4.1. Liver-Heart Co-Culture
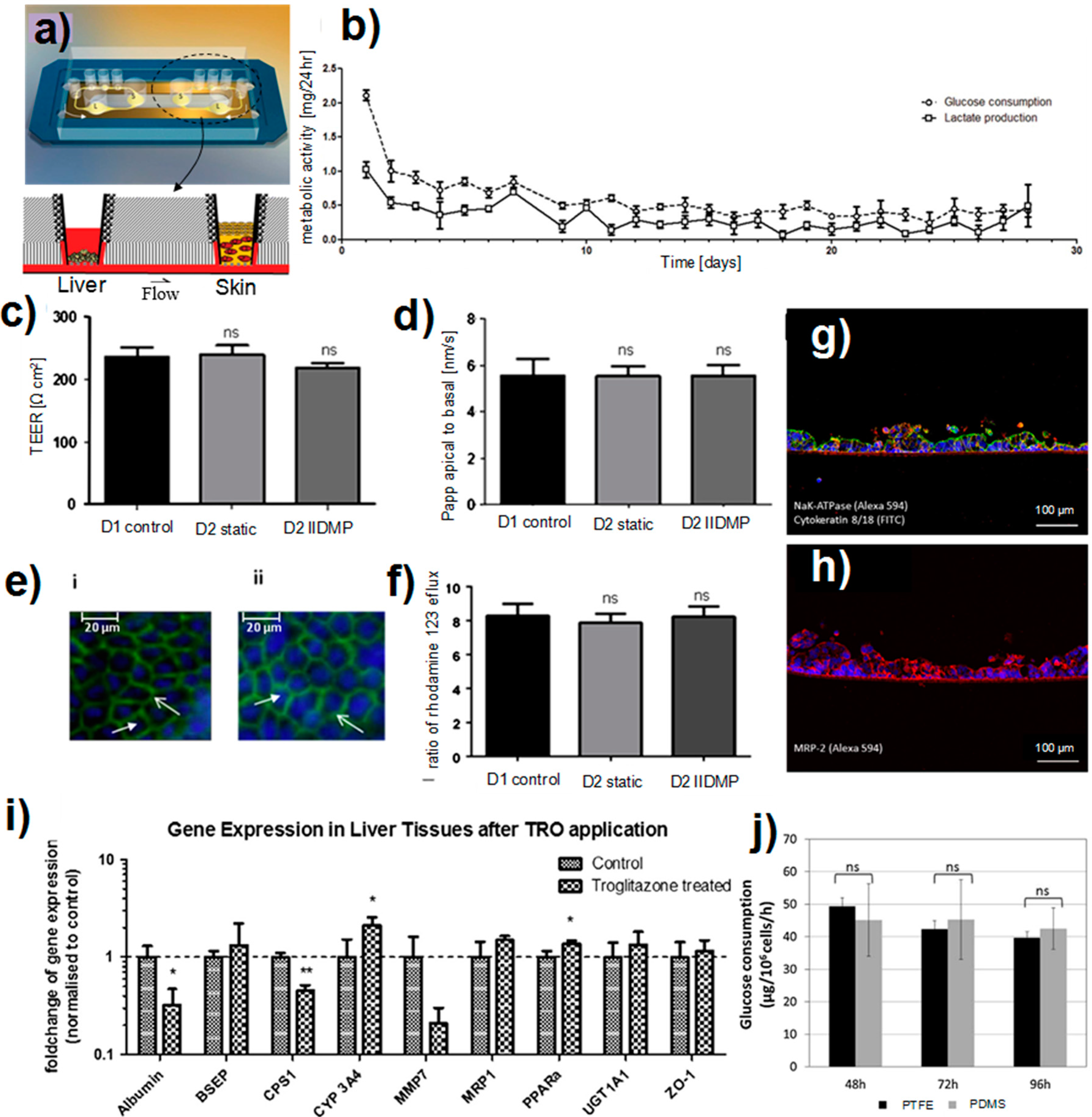
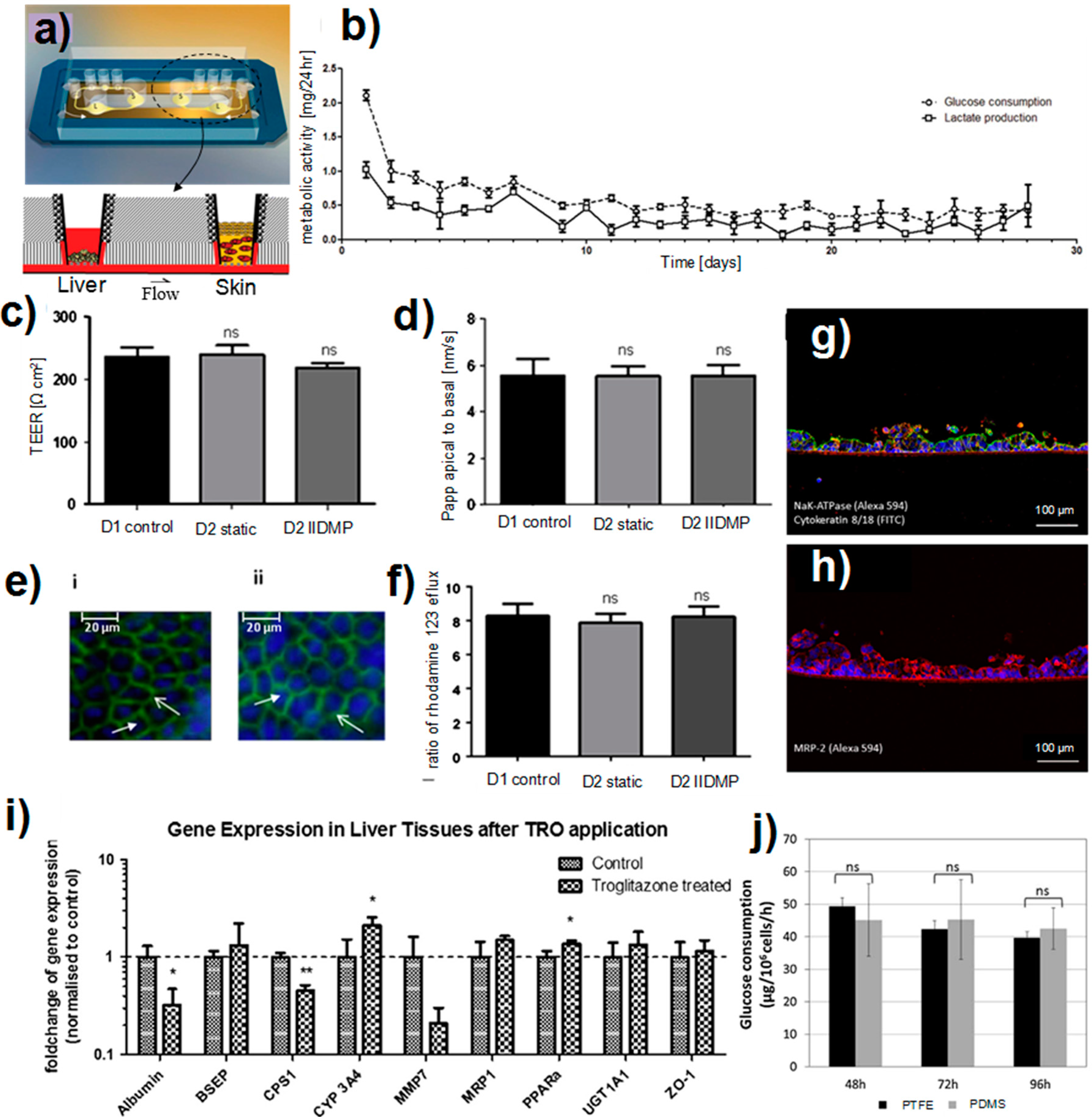
3.4.2. Liver-Skin Co-Culture
3.4.3. Liver-Intestine Co-Culture
3.4.4. Liver-Kidney Co-Culture
3.5. Multi-ORGAN Models
4. Challenges and Future of Multi-Organ Systems
4.1. Engineering Challenges
4.2. Scaling
4.3. Cell Sources: Cancer Cells versus Stem Cells
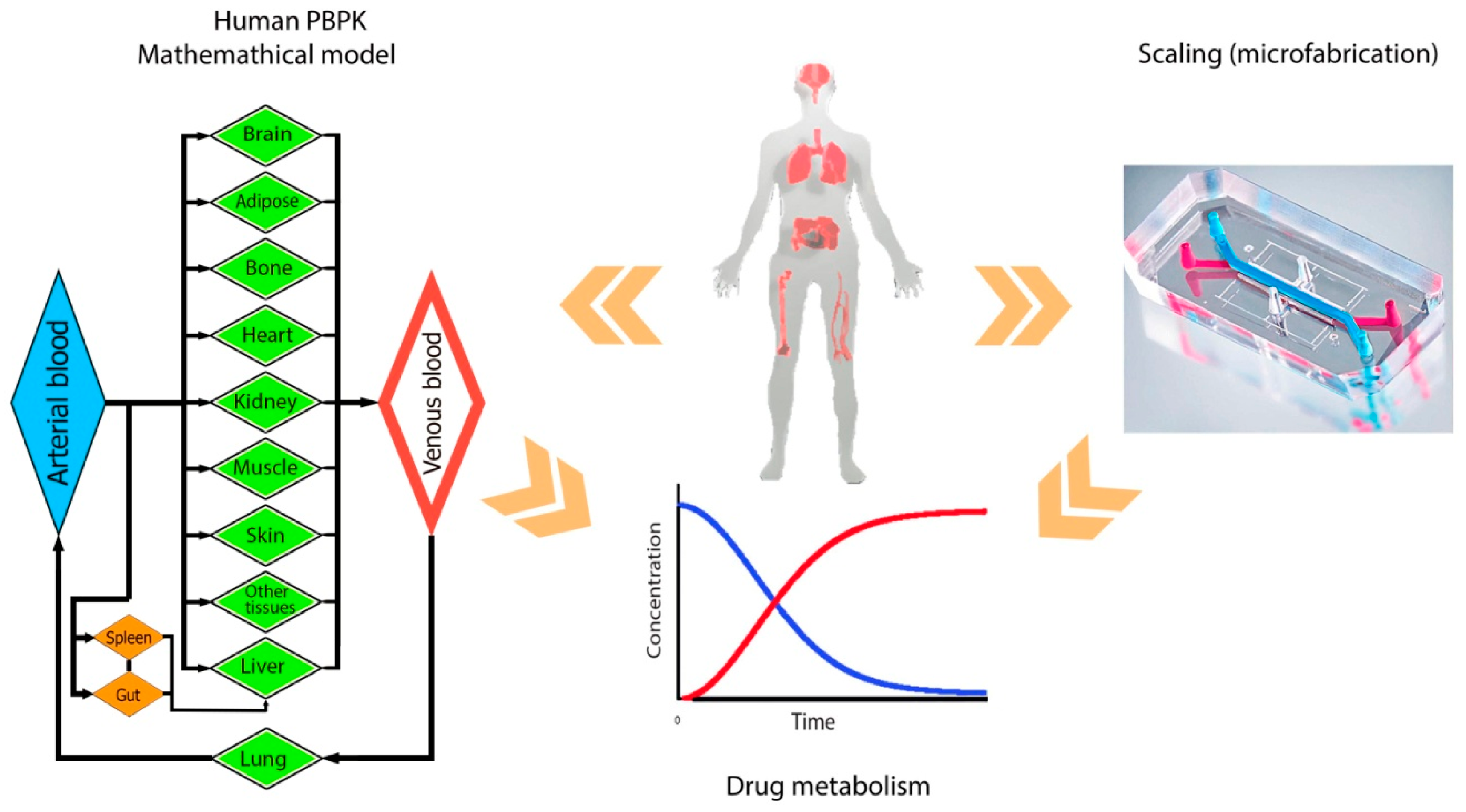
4.4. Computational Bioinformatics Opportunities for Drug Design in Multi-Organ Platforms
4.5. Biosensors for On-Chip Technologies
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Raies, B.A.; Bajic, V.B. In silico toxicology: Computational methods for the prediction of chemical toxicity. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 147–172. [Google Scholar] [CrossRef] [PubMed]
- Madan, A.; Bajaj, S.; Dureja, H. Classification models for safe drug molecules. Comput. Toxicol. 2013, 2, 99–124. [Google Scholar]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [PubMed]
- Mak, I.; Evaniew, N.; Ghert, M. Lost in translation: Animal models and clinical trials in cancer treatment. Am. J. Transl. Res. 2014, 6, 114–118. [Google Scholar] [PubMed]
- O’Connor, A.K.; Roth, B.L. Finding new tricks for old drugs: An efficient route for public-sector drug discovery. Nat. Rev. Drug Discov. 2005, 4, 1005–1014. [Google Scholar]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Tiong, H.Y.; Huang, P.; Xiong, S.; Li, Y.; Vathsala, A.; Zink, D. Drug-induced nephrotoxicity: Clinical impact and preclinical in vitro models. Mol. Pharm. 2014, 11, 1933–1948. [Google Scholar] [CrossRef] [PubMed]
- Astashkina, A.; Mann, B.; Grainger, D.W. A critical evaluation of in vitro cell culture models for high-throughput drug screening and toxicity. Pharmacol. Ther. 2012, 134, 82–106. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, K.L.; Keppler, D.; Hoffmaster, K.A.; Bow, D.A.; Cheng, Y.; Lai, Y.; Palm, J.E.; Stieger, B.; Evers, R. In vitro methods to support transporter evaluation in drug discovery and development. Clin. Pharmacol. Ther. 2013, 94, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Frey, O.; Misun, P.M.; Fluri, D.A.; Hengstler, J.G.; Hierlemann, A. Reconfigurable microfluidic hanging drop network for multi-tissue interaction and analysis. Nat. Commun. 2014, 5, 4250. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; di Carlo, D.; Lee, L.P. Microfluidic self-assembly of tumor spheroids for anticancer drug discover. Biomed. Microdevices 2008, 10, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Kunz-Schughart, L.A.; Freyer, J.P.; Hofstaedter, F.; Ebner, R. The use of 3-D cultures for high-throughput screening: The multicellular spheroid model. J. Biomol. Screen. 2004, 9, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Bricks, T.; Paullier, P.; Legendre, A.; Fleury, M.-J.; Zeller, P.; Merlier, F.; Anton, P.M.; Leclerc, E. Development of a new microfluidic platform integrating co-cultures of intestinal and liver cell lines. Toxicol. In Vitro 2014, 28, 885–895. [Google Scholar] [CrossRef] [PubMed]
- Maschmeyer, I.; Hasenberg, T.; Jaenicke, A.; Lindner, M.; Lorenz, A.K.; Zech, J.; Garbe, L.-A.; Sonntag, F.; Hayden, P.; Ayehunie, S.; et al. Chip-based human liver–intestine and liver–skin co-cultures–A first step toward systemic repeated dose substance testing in vitro. Eur. J. Pharm. Biopharm. 2015, 95, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Jellali, R.; Paullier, P.; Fleury, M.-J.; Leclerc, E. Liver and kidney cells cultures in a new perfluoropolyether biochip. Sens. Actuators B Chem. 2016, 229, 396–407. [Google Scholar] [CrossRef]
- Wikswo, J.P.; Curtis, E.L.; Eagleton, Z.E.; Evans, B.C.; Kole, A.; Hofmeister, L.H.; Matloff, W.J. Scaling and systems biology for integrating multiple organs-on-a-chip. Lab Chip 2013, 13, 3496–3511. [Google Scholar] [CrossRef] [PubMed]
- Materne, E.-M.; Maschmeyer, I.; Lorenz, A.K.; Horland, R.; Schimek, K.M.; Busek, M.; Sonntag, F.; Lauster, R.; Marx, U. The multi-organ chip—A microfluidic platform for long-term multi-tissue coculture. J. Vis. Exp. 2015, 98, 52526. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Fu, F.; Cheng, Y.; Wang, C.; Zhao, Y.; Gu, Z. Organ-on-a-Chip Systems: Microengineering to Biomimic Living Systems. Small 2016, 12, 2253–2282. [Google Scholar] [CrossRef] [PubMed]
- Maschmeyer, I.; Lorenz, A.K.; Schimek, K.; Hasenberg, T.; Ramme, A.P.; Hubner, J.; Lindner, M.; Drewell, C.; Bauer, S.; Thomas, A.; et al. A four-organ-chip for interconnected long-term co-culture of human intestine, liver, skin and kidney equivalents. Lab Chip 2015, 15, 2688–2699. [Google Scholar] [CrossRef] [PubMed]
- Wikswo, J.P. The relevance potential roles microphysiological systems in biology medicine. Exp. Biol. Med. 2014, 239, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Wikswo, J.P.; Porter, A.P. Biology coming full circle: Joining the whole the parts. Exp. Biol. Med. 2015, 240, 3–7. [Google Scholar] [CrossRef] [PubMed]
- An, F.; Qu, Y.; Luo, Y.; Fang, N.; Liu, Y.; Gao, Z.; Zhao, W.; Lin, B. A Laminated Microfluidic Device for Comprehensive Preclinical Testing in the Drug ADME Process. Sci. Rep. 2016, 6, 25022. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Kam, C.; Shuler, M.L. A microfluidic device for a pharmacokinetic-pharmacodynamic (PK-PD) model on a chip. Lab Chip 2010, 10, 446–455. [Google Scholar] [CrossRef] [PubMed]
- Derendorf, H.; Lesko, L.J.; Chaikin, P.; Colburn, W.A.; Lee, P.; Miller, R.; Powell, R.; Rhodes, G.; Stanski, D.; Venitz, J. Pharmacokinetic/Pharmacodynamic Modeling in Drug Research and Development. J. Clin. Pharmacol. 2000, 40, 1399–1418. [Google Scholar] [PubMed]
- McGonigle, P.; Ruggeri, B. Animal models human disease: Challenges in enabling translation. Biochem. Pharmacol. 2014, 87, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Gardner, H.L.; Fenger, J.M.; London, C.A. Dogs as a Model for Cancer. Annu. Rev. Anim. Biosci. 2016, 4, 199–222. [Google Scholar] [CrossRef] [PubMed]
- Ghanem, A.; Shuler, M.L. Combining Cell Culture Analogue Reactor Designs PBPK Models to Probe Mechanisms Naphthalene Toxicity. Biotechnol. Prog. 2000, 16, 334–345. [Google Scholar] [CrossRef] [PubMed]
- Rasool, M.F.; Khalil, F.; Läer, S. A Physiologically based pharmacokinetic drug–disease model to predict carvedilol exposure in adult paediatric heart failure patients by incorporating pathophysiological changes in Hepatic Renal Blood Flows. Clin. Pharmacokinet. 2015, 54, 943–962. [Google Scholar] [CrossRef] [PubMed]
- Esch, M.B.; Sung, J.H.; Shuler, M.L. Promises challenges and future directions of μCCAs. J. Biotechnol. 2010, 148, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Esch, M.B.; King, T.L.; Shuler, M.L. The role body-on-a-chip devices in drug toxicity studies. Annu. Rev. Biomed. Eng. 2011, 13, 55–72. [Google Scholar] [CrossRef] [PubMed]
- Esch, M.B.; Smith, A.S.T.; Prot, J.-M.; Oleaga, C.; Hickman, J.J.; Shuler, M.L. How multi-organ microdevices can help foster drug development. Adv. Drug Deliv. Rev. 2014, 69–70, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Nestorov, I. Whole-body physiologically based pharmacokinetic models. Expert Opin. Drug Metab. Toxicol. 2007, 3, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, M.E.; Evans, M.V.; Wilson, C.A.; Beesley, L.J.; Leon, L.S.; Eklund, C.R.; Croom, E.L.; Pegram, R.A. Development of a human physiologically based pharmacokinetic (PBPK) model for dermal permeability for lindane. Toxicol. Lett. 2016, 245, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Horiuchi, J.; Sato, A. Development a physiologically based pharmacokinetic model organic solvent in rats. Pharmacol. Res. 2000, 42, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Dostalek, M.; Gardner, I.; Gurbaxani, B.M.; Rose, R.H.; Chetty, M. Pharmacokinetics, Pharmacodynamics and Physiologically-Based Pharmacokinetic Modelling of Monoclonal Antibodies. Clin. Pharmacokinet. 2013, 52, 83–124. [Google Scholar] [CrossRef] [PubMed]
- Tsamandouras, N.; Rostami-Hodjegan, A.; Aarons, L. Combining the ‘bottom up’ ‘top down’ approaches in pharmacokinetic modelling: Fitting PBPK models to observed clinical data. Br. J. Clin. Pharmacol. 2015, 79, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Croom, E.L.; Shafer, T.J.; Evans, M.V.; Mundy, W.R.; Eklund, C.R.; Johnstone, A.F.M.; Mack, C.M.; Pegram, R.A. Improving in vitro to in vivo extrapolation by incorporating toxicokinetic measurements: A case study of lindane-induced neurotoxicity. Toxicol. Appl. Pharmacol. 2015, 283, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Shankaran, H.; Adeshina, F.; Teeguarden, J.G. Physiologically-based pharmacokinetic model for Fentanyl in support the development Provisional Advisory Levels. Toxicol. Appl. Pharmacol. 2013, 273, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Kostewicz, E.S.; Aarons, L.; Bergstrand, M.; Bolger, M.B.; Galetin, A.; Hatley, O.; Jamei, M.; Lloyd, R.; Pepin, X.; Rostami-Hodjegan, A.; et al. PBPK models for the prediction of in vivo performance of oral dosage forms. Eur. J. Pharm. Sci. 2014, 57, 300–321. [Google Scholar] [CrossRef] [PubMed]
- Olivier, B.G.; Swat, M.J.; Moné, M.J. Modeling Simulation Tools: From Systems Biology to Systems Medicine in Systems Medicine; Schmitz, U., Wolkenhauer, O., Eds.; Springer: New York, NY, USA, 2016; pp. 441–463. [Google Scholar]
- Yoon, M.; Campbell, J.L.; Andersen, M.E.; Clewell, H.J. Quantitative in vitro to in vivo extrapolation of cell-based toxicity assay results. Crit. Rev. Toxicol. 2012, 42, 633–652. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.S.T.; Long, C.J.; Berry, B.J.; McAleer, C.; Stancescu, M.; Molnar, P.; Miller, P.G.; Esch, M.B.; Prot, J.-M.; Hickman, J.J.; et al. Microphysiological systems and low-cost microfluidic platform with analytics. Stem Cell Res. Ther. 2013, 4, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Shuler, M.L. Modeling Life. Ann. Biomed. Eng. 2012, 40, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Esch, M.B.; Prot, J.-M.; Long, C.J.; Smith, A.; Hickman, J.J.; Shuler, M.L. Microfabricated mammalian organ systems and their integration into models of whole animals and humans. Lab Chip 2013, 13, 1201–1212. [Google Scholar] [CrossRef] [PubMed]
- Van Duinen, V.; Trietsch, S.J.; Joore, J.; Vulto, P.; Hankemeier, T. Microfluidic 3D cell culture: From tools to tissue models. Curr. Opin. Biotechnol. 2015, 35, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Rismani Yazdi, S.; Shadmani, A.; Burgel, S.C.; Misun, P.M.; Hierlemann, A.; Frey, O. Adding the ‘heart’ to hanging drop networks for microphysiological multi-tissue experiments. Lab Chip 2015, 15, 4138–4147. [Google Scholar] [CrossRef] [PubMed]
- Tasaka, M.; Takeuchi, I. Sorting out behaviour disaggregated cells in the absence morphogenesis in Dictyostelium discoideum. Development 1979, 49, 89–102. [Google Scholar]
- Erlichman, C.; Vidgen, D. Cytotoxicity adriamycin in MGH-U1 cells grown as monolayer cultures, spheroids, and xenografts in immune-deprived mice. Cancer Res. 1984, 44, 5369–5375. [Google Scholar] [PubMed]
- Kelm, J.M.; Timmins, N.E.; Brown, C.J.; Fussenegger, M.; Nielsen, L.K. Method for generation of homogeneous multicellular tumor spheroids applicable to a wide variety of cell types. Biotechnol. Bioeng. 2003, 83, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Horman, S.R.; To, J.; Orth, A.P.; Slawny, N.; Cuddihy, M.J.; Caracino, D. High-content analysis of three-dimensional tumor spheroids: Investigating signaling pathways using small hairpin RNA. Nat. Methods 2013, 10. [Google Scholar] [CrossRef]
- Kim, J.Y.; Fluri, D.A.; Kelm, J.M.; Hierlemann, A.; Frey, O. 96-well format-based microfluidic platform for parallel interconnection of multiple multicellular spheroids. J. Lab. Autom. 2015, 20, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Yuhas, J.M.; Li, A.P.; Martinez, A.O.; Ladman, A.J. A simplified method for production and growth of multicellular tumor spheroids. Cancer Res. 1977, 37, 3639–3643. [Google Scholar] [PubMed]
- Haji-Karim, M.; Carisson, J. Proliferation viability in cellular spheroids human origin. Cancer Res. 1978, 38, 1457–1464. [Google Scholar] [PubMed]
- Walsh, A.J.; Cook, R.S.; Sanders, M.E.; Aurisicchio, L.; Ciliberto, G.; Arteaga, C.L.; Skala, M.C. Quantitative optical imaging of primary tumor organoid metabolism predicts drug response in breast cancer. Cancer Res. 2014, 74, 5184–5194. [Google Scholar] [CrossRef] [PubMed]
- Ruppen, J.; Cortes-Dericks, L.; Marconi, E.; Karoubi, G.; Schmid, R.A.; Peng, R.; Marti, T.M.; Guenat, O.T. A microfluidic platform for chemoresistive testing of multicellular pleural cancer spheroids. Lab Chip 2014, 14, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Xinaris, C.; Benedetti, V.; Rizzo, P.; Abbate, M.; Corna, D.; Azzollini, N.; Conti, S.; Unbekandt, M.; Davies, J.A.; Morigi, M.; et al. In vivo maturation of functional renal organoids formed from embryonic cell suspensions. J. Am. Soc. Nephrol. 2012, 23, 1857–1868. [Google Scholar] [CrossRef] [PubMed]
- Drost, J.; Karthaus, W.R.; Gao, D.; Driehuis, E.; Sawyers, C.L.; Chen, Y.; Clevers, H. Organoid culture systems for prostate epithelial and cancer tissue. Nat. Protoc. 2016, 11, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.A.; Tiriac, H.; Clevers, H.; Tuveson, D.A. Modeling pancreatic cancer with organoids. Trends Cancer 2016, 2, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Kuo, T.-T.; Shih, L.-Y. Histologic types of thymoma associated with pure red cell aplasia: A study of five cases including a composite tumor of organoid thymoma associated with an unusual lipofibroadenoma. Int. J. Surg. Pathol. 2001, 9, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Chio, I.I.; Engle, D.D.; Corbo, V.; Jager, M.; Ponz-Sarvise, M.; Tiriac, H.; Spector, M.S.; et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015, 160, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Clevers, H. Growing self-organizing mini-guts from a single intestinal stem cell: Mechanism applications. Science 2013, 340, 1190–1194. [Google Scholar] [CrossRef] [PubMed]
- Huch, M.; Boj, S.F.; Clevers, H. Lgr5+ liver stem cells, hepatic organoids and regenerative medicine. Regen. Med. 2013, 8, 385–387. [Google Scholar] [CrossRef] [PubMed]
- Gessner, R.C.; Hanson, A.D.; Feingold, S.; Cashion, A.T.; Corcimaru, A.; Wu, B.T.; Mullins, C.R.; Aylward, S.R.; Reid, L.M.; Dayton, P.A. Functional ultrasound imaging for assessment of extracellular matrix scaffolds used for liver organoid formation. Biomaterials 2013, 34, 9341–9351. [Google Scholar] [CrossRef] [PubMed]
- Skardal, A.; Devarasetty, M.; Rodman, C.; Atala, A.; Soker, S. Liver-tumor hybrid organoids for modeling tumor growth and drug response in vitro. Ann. Biomed. Eng. 2015, 43, 2361–2373. [Google Scholar] [CrossRef] [PubMed]
- Yamada, M.; Utoh, R.; Ohashi, K.; Tatsumi, K.; Yamato, M.; Okano, T.; Seki, M. Controlled formation of heterotypic hepatic micro-organoids in anisotropic hydrogel microfibers for long-term preservation of liver-specific functions. Biomaterials 2012, 33, 8304–8315. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Matsuura, T.; Nagatsuma, K.; Tanaka, K.; Maehashi, H.; Shimizu, K.; Hataba, Y.; Kato, F.; Kashimori, I.; Tajiri, H. The functional interrelationship between gap junctions and fenestrae in endothelial cells of the liver organoid. J. Membr. Biol. 2007, 217, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Allinen, M.; Beroukhim, R.; Cai, L.; Brennan, C.; Lahti-Domenici, J.; Huang, H.; Porter, D.; Hu, M.; Chin, L.; Richardson, A.; et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 2004, 6, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Gschweng, E.; Oliveira, S.; Kohn, D.B. Hematopoietic stem cells for cancer immunotherapy. Immunol. Rev. 2014, 257, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.J.; Davidenko, N.; Caffarel, M.M.; Cameron, R.E.; Watson, C.J. A multifunctional 3D co-culture system for studies of mammary tissue morphogenesis and stem cell biology. PLoS ONE 2011, 6, e25661. [Google Scholar] [CrossRef] [PubMed]
- Markov, D.A.; Lu, J.Q.; Samson, P.C.; Wikswo, J.P.; McCawley, L.J. Thick-tissue bioreactor as a platform for long-term organotypic culture and drug delivery. Lab Chip 2012, 12, 4560–4568. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Khademhosseini, A.; Langer, R.; Borenstein, J.; Vacanti, J.P. Microscale technologies for tissue engineering and biology. Proc. Natl. Acad. Sci. USA 2006, 103, 2480–2487. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, A.T.; Monteiro-Riviere, N.A.; Walker, G.M. Characterization of microfluidic human epidermal keratinocyte culture. Cytotechnology 2008, 56, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, Q.; Gijs, M.A. In vitro micro-physiological models for translational immunology. Lab Chip 2015, 15, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Carraro, A.; Hsu, W.M.; Kulig, K.M.; Cheung, W.S.; Miller, M.L.; Weinberg, E.J.; Swart, E.F.; Kaazempur-Mofrad, M.; Borenstein, J.T.; Vacanti, J.P.; et al. In vitro analysis of a hepatic device with intrinsic microvascular-based channels. Biomed. Microdevices 2008, 10, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Griep, L.M.; Wolbers, F.; de Wagenaar, B.; ter Braak, P.M.; Weksler, B.B.; Romero, I.A.; Couraud, P.O.; Vermes, I.; van der Meer, A.D.; van den Berg, A. BBB on chip: Microfluidic platform to mechanically and biochemically modulate blood-brain barrier function. Biomed. Microdevices 2013, 15, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Shao, J.; Wu, L.; Wu, J.; Zheng, Y.; Zhao, H.; Jin, Q.; Zhao, J. Integrated microfluidic chip for endothelial cells culture and analysis exposed to a pulsatile and oscillatory shear stress. Lab Chip 2009, 9, 3118–3125. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Leslie, D.C.; Matthews, B.D.; Fraser, J.P.; Jurek, S.; Hamilton, G.A.; Thorneloe, K.S.; McAlexander, M.A.; Ingber, D.E. A human disease model of drug toxicity-induced pulmonary edema in a lung-on-a-chip microdevice. Sci. Transl. Med. 2012, 4, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Torisawa, Y.S.; Hamilton, G.A.; Kim, H.J.; Ingber, D.E. Human gut-on-a-chip inhabited by microbial flora that experiences intestinal peristalsis-like motions and flow. Lab Chip 2012, 12, 2165–2174. [Google Scholar]
- Huh, D.; Torisawa, Y.S.; Hamilton, G.A.; Kim, H.J.; Ingber, D.E. Microengineered physiological biomimicry: Organs-on-chips. Lab Chip 2012, 12, 2156–2164. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Sim, W.Y.; Min, B.H.; Yang, S.S.; Khademhosseini, A.; Kaplan, D.L. Chip-based comparison of the osteogenesis of human bone marrow- and adipose tissue-derived mesenchymal stem cells under mechanical stimulation. PLoS ONE 2012, 7, e46689. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, T.; Ingber, D.E. Mechanical control of tissue and organ development. Development 2010, 137, 1407–1420. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.C.; Shih, H.C.; Wu, J.G.; Weng, T.W.; Wu, C.Y.; Lu, J.C.; Tung, Y.C. Electrofluidic pressure sensor embedded microfluidic device: A study of endothelial cells under hydrostatic pressure and shear stress combinations. Lab Chip 2013, 13, 1743. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, N. Idiosyncratic drug hepatotoxicity. Nat. Rev. Drug Discov. 2005, 4, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Takayama, S.; Ostuni, E.; LeDuc, P.; Naruse, K.; Ingber, D.E.; Whitesides, G.M. Subcellular positioning of small molecules. Nature 2001, 411, 1016. [Google Scholar] [CrossRef] [PubMed]
- Li Jeon, N.; Baskaran, H.; Dertinger, S.K.; Whitesides, G.M.; Van de Water, L.; Toner, M. Neutrophil chemotaxis in linear and complex gradients of interleukin-8 formed in a microfabricated device. Nat. Biotechnol. 2002, 20, 826–830. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.R.; Zeng, W.J.; Li, Y.T.; Zou, W.; Wang, L.; Pei, X.F.; Xie, M.; Huang, W.H. Simultaneous generation of gradients with gradually changed slope in a microfluidic device for quantifying axon response. Anal. Chem. 2013, 85, 7842–7850. [Google Scholar] [CrossRef] [PubMed]
- Cimetta, E.; Cannizzaro, C.; James, R.; Biechele, T.; Moon, R.T.; Elvassore, N.; Vunjak-Novakovic, G. Microfluidic device generating stable concentration gradients for long term cell culture: Application to Wnt3a regulation of beta-catenin signaling. Lab Chip 2010, 10, 3277–3283. [Google Scholar] [CrossRef] [PubMed]
- Radisic, M.; Deen, W.; Langer, R.; Vunjak-Novakovic, G. Mathematical model of oxygen distribution in engineered cardiac tissue with parallel channel array perfused with culture medium containing oxygen carriers. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H1278–H1289. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.J.; Hung, P.J.; Lee, L.P. An artificial liver sinusoid with a microfluidic endothelial-like barrier for primary hepatocyte culture. Biotechnol. Bioeng. 2007, 97, 1340–1346. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.J.; Mehr, A.P.; Hamilton, G.A.; McPartlin, L.A.; Chung, S.; Suh, K.Y.; Ingber, D.E. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol. 2013, 5, 1119–1129. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Yan, J.J.; Shin, Y.; Jeon, J.J.; Won, J.; Jeong, H.E.; Kamm, R.D.; Kim, Y.J.; Chung, S. A versatile assay for monitoring in vivo-like transendothelial migration of neutrophils. Lab Chip 2012, 12, 3861–3865. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.-H.T.; Stapleton, S.C.; Yang, M.T.; Cha, S.S.; Choi, C.K.; Galie, P.A.; Chen, C.S. Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 6712–6717. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Goss, J.A.; Cho, A.; McCain, M.L.; Parker, K.K. Microfluidic heart on a chip for higher throughput pharmacological studies. Lab Chip 2013, 13, 3599–3608. [Google Scholar] [CrossRef] [PubMed]
- Kane, B.J.; Zinner, M.J.; Yarmush, M.L.; Toner, M. Liver-specific functional studies in a microfluidic array of primary mammalian hepatocytes. Anal. Chem. 2006, 78, 4291–4298. [Google Scholar] [CrossRef] [PubMed]
- Esch, M.B.; Sung, J.H.; Yang, J.; Yu, C.; Yu, J.; March, J.C.; Shuler, M.L. On chip porous polymer membranes for integration of gastrointestinal tract epithelium with microfluidic ‘body-on-a-chip’ devices. Biomed. Microdevices 2012, 14, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Booth, R.; Kim, H. Characterization of a microfluidic in vitro model of the blood-brain barrier (µBBB). Lab Chip 2012, 12, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
- Douville, N.J.; Tung, Y.-C.; Li, R.; Wang, J.D.; El-Sayed, M.E.; Takayama, S. Fabrication of two-layered channel system with embedded electrodes to measure resistance across epithelial and endothelial barriers. Anal. Chem. 2010, 82, 2505–2511. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.A.; Pensabene, V.; Markov, D.A.; Allwardt, V.; Neely, M.D.; Shi, M.; Britt, C.M.; Hoilett, O.S.; Yang, Q.; Brewer, B. Recreating blood-brain barrier physiology and structure on chip: A novel neurovascular microfluidic bioreactor. Biomicrofluidics 2015, 9, 054124. [Google Scholar] [CrossRef] [PubMed]
- Prabhakarpandian, B.; Shen, M.-C.; Nichols, J.B.; Mills, I.R.; Sidoryk-Wegrzynowicz, M.; Aschner, M.; Pant, K. SyM-BBB: A microfluidic blood brain barrier model. Lab Chip 2013, 13, 1093–1101. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, A.; Leach, J.; Townsend, S.; Iida, T.; Hogan, B.; Stolz, D.B.; Fry, R.; Samson, L.; Tannenbaum, S.; Griffith, L. A microscale in vitro physiological model of the liver: Predictive screens for drug metabolism and enzyme induction. Curr. Drug Metab. 2005, 6, 569–591. [Google Scholar] [CrossRef] [PubMed]
- Toh, Y.C.; Lim, T.C.; Tai, D.; Xiao, G.; van Noort, D.; Yu, H. A microfluidic 3D hepatocyte chip for drug toxicity testing. Lab Chip 2009, 9, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Novik, E.; Maguire, T.J.; Chao, P.; Cheng, K.C.; Yarmush, M.L. A microfluidic hepatic coculture platform for cell-based drug metabolism studies. Biochem. Pharmacol. 2010, 79, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, E.; Stoppini, L.; Balestrino, M.; Eskes, C.; Griesinger, C.; Sobanski, T.; Whelan, M.; Hartung, T.; Coecke, S. Electrophysiological recording of re-aggregating brain cell cultures on multi-electrode arrays to detect acute neurotoxic effects. NeuroToxicology 2007, 28, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Pan, L.; Cheng, X.; Berdichevsky, Y. Perfused drop microfluidic device for brain slice culture-based drug discovery. Biomed. Microdevices 2016, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, D.; Weng, L.; Wang, L. Early Lung Cancer Diagnosis by Biosensors. Int. J. Mol. Sci. 2013, 14, 15479–15509. [Google Scholar] [CrossRef] [PubMed]
- Altintas, Z.; Tothill, I. Biomarkers and biosensors for the early diagnosis of lung cancer. Sens. Actuators B Chem. 2013, 188, 988–998. [Google Scholar] [CrossRef]
- Wang, J.; Wu, C.; Hu, N.; Zhou, J.; Du, L.; Wang, P. Microfabricated Electrochemical Cell-Based Biosensors for Analysis of Living Cells In Vitro. Biosensor 2012, 2, 127–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Wu, C.; Cai, H.; Hu, N.; Zhou, J.; Wang, P. Cell-Based Biosensors and Their Application in Biomedicine. Chem. Rev. 2014, 114, 6423–6461. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, Y.; Sonkusale, S.R. Microfluidic optoelectronic sensor for salivary diagnostics of stomach cancer. Biosens. Bioelectron. 2015, 67, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Ferrie, A.M.; Wang, C.; Deng, H.; Fang, Y. A label-free optical biosensor with microfluidics identifies an intracellular signalling wave mediated through the β 2-adrenergic receptor. Integr. Biol. 2013, 5, 1253–1261. [Google Scholar] [CrossRef] [PubMed]
- Ges, I.A.; Brindley, R.L.; Currie, K.P.; Baudenbacher, F.J. A microfluidic platform for chemical stimulation and real time analysis of catecholamine secretion from neuroendocrine cells. Lab Chip 2013, 13, 4663–4673. [Google Scholar] [CrossRef] [PubMed]
- Perestrelo, A.R.; Aguas, A.C.; Rainer, A.; Forte, G. Microfluidic Organ/Body-on-a-Chip Devices at the Convergence of Biology and Microengineering. Sensors 2015, 15, 31142–31170. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Yin, T.I.; Reyes, D.; Urban, G.A. Microfluidic chip with integrated electrical cell-impedance sensing for monitoring single cancer cell migration in three-dimensional matrixes. Anal. Chem. 2013, 85, 11068–11076. [Google Scholar] [CrossRef] [PubMed]
- Lee, L.M.; Liu, A.P. The Application of Micropipette Aspiration in Molecular Mechanics of Single Cells. J. Nanotechnol. Eng. Med. 2014, 5, 0408011–0408016. [Google Scholar] [CrossRef] [PubMed]
- Giobbe, G.G.; Michielin, F.; Luni, C.; Giulitti, S.; Martewicz, S.; Dupont, S.; Floreani, A.; Elvassore, N. Functional differentiation of human pluripotent stem cells on a chip. Nat. Methods 2015, 12, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Vasiliauskas, R.; Liu, D.; Cito, S.; Zhang, H.; Shahbazi, M.A.; Sikanen, T.; Mazutis, L.; Santos, H.A. Simple Microfluidic Approach to Fabricate Monodisperse Hollow Microparticles for Multidrug Delivery. ACS Appl. Mater. Interfaces 2015, 7, 14822–14832. [Google Scholar] [CrossRef] [PubMed]
- Kilian, K.A.; Bugarija, B.; Lahn, B.T.; Mrksich, M. Geometric cues for directing the differentiation of mesenchymal stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 4872–4877. [Google Scholar] [CrossRef] [PubMed]
- Burdick, J.A.; Vunjak-Novakovic, G. Engineered microenvironments for controlled stem cell differentiation. Tissue Eng. A 2008, 15, 205–219. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.F.; Chen, Y.; Marcus, J.S.; Scherer, A.; Quake, S.R.; Taylor, C.R.; Weiner, L.P. A microfluidic processor for gene expression profiling of single human embryonic stem cells. Lab Chip 2008, 8, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Abhyankar, V.V.; Beebe, D.J. Human embryonic stem cells and microfluidics. In Lab-on-Chips for Cellomics; Springer Netherlands: Dordrecht, The Netherlands, 2004; pp. 257–272. [Google Scholar]
- Wan, C.R.; Chung, S.; Kamm, R.D. Differentiation of embryonic stem cells into cardiomyocytes in a compliant microfluidic system. Ann. Biomed. Eng. 2011, 39, 1840–1847. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.W.; Ripplinger, C.M.; van der Meer, P.; Sheehy, S.P.; Domian, I.; Chien, K.R.; Parker, K.K. Functional differences in engineered myocardium from embryonic stem cell-derived versus neonatal cardiomyocytes. Stem Cell Rep. 2013, 1, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Cimetta, E.; Sirabella, D.; Yeager, K.; Davidson, K.; Simon, J.; Moon, R.T.; Vunjak-Novakovic, G. Microfluidic bioreactor for dynamic regulation of early mesodermal commitment in human pluripotent stem cells. Lab Chip 2013, 13, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Kim, S.K.; Woo, D.H.; Lee, E.J.; Kim, J.H.; Lee, S.H. Differentiation of neural progenitor cells in a microfluidic chip-generated cytokine gradient. Stem Cell 2009, 27, 2646–2654. [Google Scholar] [CrossRef] [PubMed]
- Chung, B.G.; Flanagan, L.A.; Rhee, S.W.; Schwartz, P.H.; Lee, A.P.; Monuki, E.S.; Jeon, N.L. Human neural stem cell growth and differentiation in a gradient-generating microfluidic device. Lab Chip 2005, 5, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Han, S.; Shin, Y.; Ko, E.; Kim, J.; Park, K.I.; Chung, S.; Cho, S.W. A microfluidic array for quantitative analysis of human neural stem cell self-renewal and differentiation in three-dimensional hypoxic microenvironment. Biomaterials 2013, 34, 6607–6614. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.M.; Trappmann, B.; Stapleton, S.C.; Toro, E.; Chen, C.S. Microfluidics embedded within extracellular matrix to define vascular architectures and pattern diffusive gradients. Lab Chip 2013, 13, 3246–3252. [Google Scholar] [CrossRef] [PubMed]
- Moya, M.L.; Hsu, Y.H.; Lee, A.P.; Hughes, C.C.; George, S.C. In vitro perfused human capillary networks. Tissue Eng. C Methods 2013, 19, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Bischel, L.L.; Young, E.W.; Mader, B.R.; Beebe, D.J. Tubeless microfluidic angiogenesis assay with three-dimensional endothelial-lined microvessels. Biomaterials 2013, 34, 1471–1477. [Google Scholar] [CrossRef] [PubMed]
- Jeon, J.S.; Zervantonakis, I.K.; Chung, S.; Kamm, R.D.; Charest, J.L. In vitro model of tumor cell extravasation. PLoS ONE 2013, 8, e56910. [Google Scholar] [CrossRef] [PubMed]
- Snouber, L.C.; Bunescu, A.; Legallais, C.; Brochot, C.; Dumas, M.E.; Elena-Herrmann, B.; Leclerc, E. Metabolomics-on-a-chip of hepatotoxicity induced by anticancer drug flutamide and its active metabolite hydroxyflutamide using HepG2/C3a microfluidic biochips. Toxicol. Sci. 2013, 132, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Chao, P.; Maguire, T.; Novik, E.; Cheng, K.C.; Yarmush, M.L. Evaluation of a microfluidic based cell culture platform with primary human hepatocytes for the prediction of hepatic clearance in human. Biochem. Pharmacol. 2009, 78, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Shayan, G.; Choi, Y.S.; Shusta, E.V.; Shuler, M.L.; Lee, K.H. Murine in vitro model of the blood-brain barrier for evaluating drug transport. Eur. J. Pharm. Sci. 2011, 42, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Tatosian, D.A.; Shuler, M.L. A novel system for evaluation of drug mixtures for potential efficacy in treating multidrug resistant cancers. Biotechnol. Bioeng. 2009, 103, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Faley, S.L.; Copland, M.; Wlodkowic, D.; Kolch, W.; Seale, K.T.; Wikswo, J.P.; Cooper, J.M. Microfluidic single cell arrays to interrogate signalling dynamics of individual, patient-derived hematopoietic stem cells. Lab Chip 2009, 9, 2659–2664. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Gao, Y.; Hao, Y.; Li, E.; Wang, Y.; Zhang, J.; Wang, W.; Gao, Z.; Wang, Q. Application of a microfluidic chip-based 3D co-culture to test drug sensitivity for individualized treatment of lung cancer. Biomaterials 2013, 34, 4109–4117. [Google Scholar] [CrossRef] [PubMed]
- Grosberg, A.; Nesmith, A.P.; Goss, J.A.; Brigham, M.D.; McCain, M.L.; Parker, K.K. Muscle on a chip: In vitro contractility assays for smooth and striated muscle. J. Pharmacol. Toxicol. Methods 2012, 65, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, V.; Doshi, A.; Kurup, G.K.; Ereifej, E.; Vandevord, P.J.; Basu, A.S. A modular approach for the generation, storage, mixing, and detection of droplet libraries for high throughput screening. Lab Chip 2010, 10, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, K.A.; Ryu, K.S.; Goluch, E.D.; Nam, J.-M.; Liu, J.; Thaxton, C.S.; Chiesl, T.N.; Barron, A.E.; Lu, Y.; Mirkin, C.A.; Liu, C. A modular microfluidic architecture for integrated biochemical analysis. Proc. Natl. Acad. Sci. USA 2005, 102, 9745–9750. [Google Scholar] [CrossRef] [PubMed]
- Rhee, M.; Burns, M.A. Microfluidic assembly blocks. Lab Chip 2008, 8, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Loskill, P.; Marcus, S.G.; Mathur, A.; Reese, W.M.; Healy, K.E. μOrgano: A lego®-like plug & play system for modular multi-organ-chips. PLoS ONE 2015, 10, e0139587. [Google Scholar]
- Bertau, M.; Mosekilde, E.; Westerhoff, H.V. Front Matter. In Biosimulation in Drug Development; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2007; pp. 1–28. [Google Scholar]
- Wu, M.-H.; Huang, S.-B.; Lee, G.-B. Microfluidic cell culture systems for drug research. Lab Chip 2010, 10, 939–956. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Feldman, L.; Seckler, A.; Wilson, A. Trends in risks associated with new drug development: Success rates for investigational drugs. Clin. Pharmacol. Ther. 2010, 87, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Jin, H.; Yu, Y. A review of the application of body-on-a-chip for drug test and its latest trend of incorporating barrier tissue. J. Lab. Autom. 2015. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Paules, R.S. Use of transcriptomics in understanding mechanisms of drug-induced toxicity. Pharmacogenomics 2010, 11, 573–585. [Google Scholar] [CrossRef] [PubMed]
- Bowes, J.; Brown, A.J.; Hamon, J.; Jarolimek, W.; Sridhar, A.; Waldron, G.; Whitebread, S. Reducing safety-related drug attrition: The use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 2012, 11, 909–922. [Google Scholar] [CrossRef] [PubMed]
- Björnmalm, M.; Yan, Y.; Caruso, F. Engineering and evaluating drug delivery particles in microfluidic devices. J. Controll. Release 2014, 190, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Vunjak-Novakovic, G.; Bhatia, S.; Chen, C.; Hirschi, K. HeLiVa platform: Integrated heart-liver-vascular systems for drug testing in human health and disease. Stem Cell Res. Ther. 2013, 4, S8. [Google Scholar] [CrossRef] [PubMed]
- Theodoridis, K.; Tudorache, I.; Calistru, A.; Cebotari, S.; Meyer, T.; Sarikouch, S.; Bara, C.; Brehm, R.; Haverich, A.; Hilfiker, A. Successful matrix guided tissue regeneration of decellularized pulmonary heart valve allografts in elderly sheep. Biomaterials 2015, 52, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Loskill, P.; Hong, S.; Lee, J.Y.; Marcus, S.G.; Dumont, L.; Conklin, B.R.; Willenbring, H.; Lee, L.P.; Healy, K.E. Human induced pluripotent stem cell-based microphysiological tissue models of myocardium and liver for drug development. Stem Cell Res. Ther. 2013, 4, S14. [Google Scholar] [CrossRef] [PubMed]
- Küchler, S.; Strüver, K.; Friess, W. Reconstructed skin models as emerging tools for drug absorption studies. Expert Opin. Drug Metab. Toxicol. 2013, 9, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Bagherifard, S.; Tamayol, A.; Mostafalu, P.; Akbari, M.; Comotto, M.; Annabi, N.; Ghaderi, M.; Sonkusale, S.; Dokmeci, M.R.; Khademhosseini, A. Dermal Patch with Integrated Flexible Heater for on Demand Drug Delivery. Adv. Healthc. Mater. 2016, 5, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Khetani, S.R.; Berger, D.R.; Ballinger, K.R.; Davidson, M.D.; Lin, C.; Ware, B.R. Microengineered liver tissues for drug testing. J. Lab. Autom. 2015, 20, 216–250. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Zhao, L.; Zhou, E.-M.; Xu, J.; Shen, S.; Wang, J. On-Chip Construction of Liver Lobule-like Microtissue and Its Application for Adverse Drug Reaction Assay. Anal. Chem. 2016, 16, 2609–2617. [Google Scholar] [CrossRef] [PubMed]
- Wagner, I.; Materne, E.-M.; Brincker, S.; Süssbier, U.; Fradrich, C.; Busek, M.; Sonntag, F.; Sakharov, D.A.; Trushkin, E.V.; Tonevitsky, A.G.; et al. A dynamic multi-organ-chip for long-term cultivation and substance testing proven by 3D human liver and skin tissue co-culture. Lab Chip 2013, 13, 3538–3547. [Google Scholar] [CrossRef] [PubMed]
- Welling, P.G. Influence of food and diet on gastrointestinal drug absorption: A review. J. Pharmacokinet. BioPharm. 1977, 5, 291–334. [Google Scholar] [CrossRef] [PubMed]
- Macheras, P.; Argyrakis, P. Gastrointestinal drug absorption: Is it time to consider heterogeneity as well as homogeneity? Pharm. Res. 1997, 14, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Bevernage, J.; Brouwers, J.; Brewster, M.E.; Augustijns, P. Evaluation of gastrointestinal drug supersaturation and precipitation: Strategies and issues. Int. J. Pharm. 2013, 453, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Ensign, L.M.; Cone, R.; Hanes, J. Oral drug delivery with polymeric nanoparticles: The gastrointestinal mucus barriers. Adv. Drug Deliv. Rev. 2012, 64, 557–570. [Google Scholar] [CrossRef] [PubMed]
- Gibaldi, M.; Boyes, R.; Feldman, S. Influence of first-pass effect on availability of drugs on oral administration. J. Pharm. Sci. 1971, 60, 1338–1340. [Google Scholar] [CrossRef] [PubMed]
- Bricks, T.; Hamon, J.; Fleury, M.J.; Jellali, R.; Merlier, F.; Herpe, Y.E.; Seyer, A.; Regimbeau, J.-M.; Bois, F.; Leclerc, E. Investigation of omeprazole and phenacetin first-pass metabolism in humans using a microscale bioreactor and pharmacokinetic models. BioPharm. Drug Dispos. 2015, 36, 275–293. [Google Scholar] [CrossRef] [PubMed]
- Clissold, S.P. Paracetamol and phenacetin. Drugs 1986, 32, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Tassaneeyakul, W.; Tassaneeyakul, W.; Vannaprasaht, S.; Yamazoe, Y. Formation of omeprazole sulphone but not 5-hydroxyomeprazole is inhibited by grapefruit juice. Br. J. Clin. Pharmacol. 2000, 49, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Shintu, L.; Baudoin, R.G.; Navratil, V.; Prot, J.-M.; Pontoizeau, C.M.; Defernez, M.; Blaise, B.J.; Domange, C.L.; Péry, A.R.; Toulhoat, P. Metabolomics-on-a-chip and predictive systems toxicology in microfluidic bioartificial organs. Anal. Chem. 2012, 84, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Ataç, B.; Wagner, I.; Horland, R.; Lauster, R.; Marx, U.; Tonevitsky, A.G.; Azar, R.P.; Lindner, G. Skin and hair on-a-chip: In vitro skin models versus ex vivo tissue maintenance with dynamic perfusion. Lab Chip 2013, 13, 3555–3561. [Google Scholar] [CrossRef] [PubMed]
- Materne, E.M.; Ramme, A.P.; Terrasso, A.P.; Serra, M.; Alves, P.M.; Brito, C.; Sakharov, D.A.; Tonevitsky, A.G.; Lauster, R.; Marx, U. A multi-organ chip co-culture of neurospheres and liver equivalents for long-term substance testing. J. Biotechnol. 2015, 205, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Riahi, R.; Shaegh, S.A.M.; Ghaderi, M.; Zhang, Y.S.; Shin, S.R.; Aleman, J.; Massa, S.; Kim, D.; Dokmeci, M.R.; Khademhosseini, A. Automated microfluidic platform of bead-based electrochemical immunosensor integrated with bioreactor for continual monitoring of cell secreted biomarkers. Sci. Rep. 2016, 6, 24598. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Y.S.; Bakht, S.M.; Aleman, J.; Shin, S.R.; Yue, K.; Sica, M.; Ribas, J.; Duchamp, M.; Ju, J.; et al. Elastomeric free-form blood vessels for interconnecting organs on chip systems. Lab Chip 2016, 16, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.; Sun, Y.; Simmons, C.A. (Micro) managing the mechanical microenvironment. Integr. Biol. 2011, 3, 959–971. [Google Scholar] [CrossRef] [PubMed]
- Douville, N.J.; Zamankhan, P.; Tung, Y.-C.; Li, R.; Vaughan, B.L.; Tai, C.-F.; White, J.; Christensen, P.J.; Grotberg, J.B.; Takayama, S. Combination of fluid and solid mechanical stresses contribute to cell death and detachment in a microfluidic alveolar model. Lab Chip 2011, 11, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Annabi, N.; Tamayol, A.; Uquillas, J.A.; Akbari, M.; Bertassoni, L.E.; Cha, C.; Camci-Unal, G.; Dokmeci, M.R.; Peppas, N.A.; Khademhosseini, A. 25th anniversary article: Rational design and applications of hydrogels in regenerative medicine. Adv. Mater. 2014, 26, 85–124. [Google Scholar] [CrossRef] [PubMed]
- Hook, A.L.; Anderson, D.G.; Langer, R.; Williams, P.; Davies, M.C.; Alexander, M.R. High throughput methods applied in biomaterial development and discovery. Biomaterials 2010, 31, 187–198. [Google Scholar] [CrossRef] [PubMed]
- Nikkhah, M.; Edalat, F.; Manoucheri, S.; Khademhosseini, A. Engineering microscale topographies to control the cell–substrate interface. Biomaterials 2012, 33, 5230–5246. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.; Labuz, J.M.; Leung, B.M.; Inoue, M.; Chun, T.-H.; Takayama, S. On being the right size: Scaling effects in designing a human-on-a-chip. Integr. Biol 2013, 5, 1149–1161. [Google Scholar] [CrossRef] [PubMed]
- Grosberg, A.; Alford, P.W.; McCain, M.L.; Parker, K.K. Ensembles of engineered cardiac tissues for physiological and pharmacological study: Heart on a chip. Lab Chip 2011, 11, 4165–4173. [Google Scholar] [CrossRef] [PubMed]
- van der Meer, A.D.; van den Berg, A. Organs-on-chips: Breaking the in vitro impasse. Integr. Biol. 2012, 4, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Bonnier, F.; Keating, M.; Wrobel, T.P.; Majzner, K.; Baranska, M.; Garcia-Munoz, A.; Blanco, A.; Byrne, H.J. Cell viability assessment using the Alamar blue assay: A comparison of 2D and 3D cell culture models. Toxicol. In Vitro 2015, 29, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Weaver, V.M.; Petersen, O.W.; Larabell, C.A.; Dedhar, S.; Briand, P.; Lupu, R.; Bissell, M.J. Reciprocal interactions between β1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: A different perspective in epithelial biology. Proc. Natl. Acad. Sci. USA 1998, 95, 14821–14826. [Google Scholar] [CrossRef] [PubMed]
- Schütte, J.; Stelzle, M. Organ-Like Cell Cultures in Microfluidic Systems. In Encyclopedia of Microfluidics and Nanofluidics; Li, D.Q., Ed.; Springer-Verlag New York: New York, NY, USA, 2015; pp. 2614–2621. [Google Scholar]
- Schütte, J.; Hagmeyer, B.; Holzner, F.; Kubon, M.; Werner, S.; Freudigmann, C.; Benz, K.; Böttger, J.; Gebhardt, R.; Becker, H. “Artificial micro organs”—A microfluidic device for dielectrophoretic assembly of liver sinusoids. Biomed. Microdevices 2011, 13, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.-T.; Lin, R.-Z.; Chang, W.-Y.; Chang, H.-Y.; Liu, C.-H. Rapid heterogeneous liver-cell on-chip patterning via the enhanced field-induced dielectrophoresis trap. Lab Chip 2006, 6, 724–734. [Google Scholar] [CrossRef] [PubMed]
- Wikswo, J.P.; Block, F.E.; Cliffel, D.E.; Goodwin, C.R.; Marasco, C.C.; Markov, D.A.; McLean, D.L.; McLean, J.A.; McKenzie, J.R.; Reiserer, R.S.; et al. Engineering challenges for instrumenting and controlling integrated organ-on-chip systems. IEEE Trans. Biomed. Eng. 2013, 60, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Berntenis, N.; Roth, A.; Ebeling, M. Data mining reveals a network of early-response genes as a consensus signature of drug-induced in vitro and in vivo toxicity. Pharm. J. 2014, 14, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Berthier, E.; Young, E.W.; Beebe, D. Engineers are from PDMS-land, Biologists are from Polystyrenia. Lab Chip 2012, 12, 1224–1237. [Google Scholar] [CrossRef] [PubMed]
- Eddington, D.T.; Puccinelli, J.P.; Beebe, D.J. Thermal aging and reduced hydrophobic recovery of polydimethylsiloxane. Sens. Actuators B Chem. 2006, 114, 170–172. [Google Scholar] [CrossRef]
- Toepke, M.W.; Beebe, D.J. PDMS absorption of small molecules and consequences in microfluidic applications. Lab Chip 2006, 6, 1484–1486. [Google Scholar] [CrossRef] [PubMed]
- Ren, K.N.; Zhou, J.H.; Wu, H.K. Materials for Microfluidic Chip Fabrication. Acc. Chem. Res. 2013, 46, 2396–2406. [Google Scholar] [CrossRef] [PubMed]
- Borysiak, M.D.; Bielawski, K.S.; Sniadecki, N.J.; Jenkel, C.F.; Vogt, B.D.; Posner, J.D. Simple replica micromolding of biocompatible styrenic elastomers. Lab Chip 2013, 13, 2773–2784. [Google Scholar] [CrossRef] [PubMed]
- Borysiak, M.D.; Yuferova, E.; Posner, J.D. Simple, Low-Cost Styrene-Ethylene/Butylene-Styrene Microdevices for Electrokinetic Applications. Anal. Chem. 2013, 85, 11700–11704. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.H.; Shuler, M.L. A micro cell culture analog (µCCA) with 3-D hydrogel culture of multiple cell lines to assess metabolism-dependent cytotoxicity of anti-cancer drugs. Lab Chip 2009, 9, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Lindstedt, S.L.; Schaeffer, P. Use of allometry in predicting anatomical and physiological parameters of mammals. Lab. Anim. 2002, 36, 1–19. [Google Scholar] [CrossRef] [PubMed]
- West, G.B.; Brown, J.H. The origin of allometric scaling laws in biology from genomes to ecosystems: Towards a quantitative unifying theory of biological structure and organization. J. Exp. Biol. 2005, 208, 1575–1592. [Google Scholar] [CrossRef] [PubMed]
- Ucciferri, N.; Sbrana, T.; Ahluwalia, A. Allometric scaling and cell ratios in multi-organ in vitro models of human metabolism. Front. Bioeng. Biotechnol. 2014, 2, 74. [Google Scholar] [CrossRef] [PubMed]
- Hyder, F.; Rothman, D.L.; Bennett, M.R. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc. Natl. Acad. Sci. USA 2013, 110, 3549–3554. [Google Scholar] [CrossRef] [PubMed]
- Balaban, R.S. Allometry of brain metabolism. Proc. Natl. Acad. Sci. USA 2013, 110, 3216–3217. [Google Scholar] [CrossRef] [PubMed]
- Sbrana, T.; Ahluwalia, A. Engineering Quasi-Vivo® in vitro organ models. Adv. Exp. Med. Biol. 2012, 745, 138–153. [Google Scholar] [PubMed]
- West, G.B.; Woodruff, W.H.; Brown, J.H. Allometric scaling of metabolic rate from molecules and mitochondria to cells and mammals. Proc. Natl. Acad. Sci. USA 2002, 99, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Toussaint, O.; Weemaels, G.; Debacq-Chainiaux, F.; Scharffetter-Kochanek, K.; Wlaschek, M. Artefactual effects of oxygen on cell culture models of cellular senescence and stem cell biology. J. Cell. Physiol. 2011, 226, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Van Noort, D.; Park, S.; Nguyen, N.-T. Towards Human on a Chip: Recent Progress and Future Perspective. Micro Nanosyst. 2014, 6, 215–231. [Google Scholar]
- Bale, S.S.; Moore, L.; Yarmush, M.; Jindal, R. Emerging in vitro liver technologies for drug metabolism and inter-organ interactions. Tissue Eng. B Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Stokes, C.; Cirit, M.; Lauffenburger, D. Physiome-on-a-Chip: The Challenge of “Scaling” in Design, Operation, and Translation of Microphysiological Systems. CPT Pharm. Syst. Pharmacol. 2015, 4, 559–562. [Google Scholar] [CrossRef] [PubMed]
- Selimović, Š.; Dokmeciab, M.R.; Khademhosseini, A. Scaling laws: From human to human-on-a-chip. Lab Chip 2014, 14, 264–267. [Google Scholar] [CrossRef]
- Beißner, N.; Lorenz, T.; Reichl, S. Organ on Chip, in Microsystems for Pharmatechnology; Springer: Berlin, Germany, 2016; pp. 299–339. [Google Scholar]
- Williamson, A.; Singh, S.; Fernekorn, U.; Schober, A. The future of the patient-specific Body-on-a-chip. Lab Chip 2013, 13, 3471–3480. [Google Scholar] [CrossRef] [PubMed]
- Zwi-Dantsis, L.; Huber, I.; Habib, M.; Winterstern, A.; Gepstein, A.; Arbel, G.; Gepstein, L. Derivation and cardiomyocyte differentiation of induced pluripotent stem cells from heart failure patients. Eur. Heart J. 2013, 34, 1575–1586. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Inamura, M.; Kawabata, K.; Katayama, K.; Higuchi, M.; Tashiro, K.; Nonaka, A.; Sakurai, F.; Hayakawa, T.; Furue, M.K. Efficient generation of functional hepatocytes from human embryonic stem cells and induced pluripotent stem cells by HNF4α transduction. Mol. Ther. 2012, 20, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Teo, A.K.; Windmueller, R.; Johansson, B.B.; Dirice, E.; Njolstad, P.R.; Tjora, E.; Raeder, H.; Kulkarni, R.N. Derivation of human induced pluripotent stem cells from patients with maturity onset diabetes of the young. J. Biol. Chem. 2013, 288, 5353–5356. [Google Scholar] [CrossRef] [PubMed]
- Keung, A.J.; Asuri, P.; Kumar, S.; Schaffer, D.V. Soft microenvironments promote the early neurogenic differentiation but not self-renewal of human pluripotent stem cells. Integr. Biol. 2012, 4, 1049–1058. [Google Scholar] [CrossRef] [PubMed]
- Belair, D.G.; Whisler, J.A.; Valdez, J.; Velazquez, J.; Molenda, J.A.; Vickerman, V.; Lewis, R.; Daigh, C.; Hansen, T.D.; Mann, D.A. Human vascular tissue models formed from human induced pluripotent stem cell derived endothelial cells. Stem Cell Rev. Rep. 2015, 11, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Mathur, A.; Loskill, P.; Shao, K.; Huebsch, N.; Hong, S.; Marcus, S.G.; Marks, N.; Mandegar, M.; Conklin, B.R.; Lee, L.P. Human iPSC-based cardiac microphysiological system for drug screening applications. Sci. Rep. 2015, 5, 8883. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Li, J.; Xie, L.; Bourne, P.E. Drug discovery using chemical systems biology: Identification of the protein-ligand binding network to explain the side effects of CETP inhibitors. PLoS Comput. Biol. 2009, 5, e1000387. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Wang, J.; Bourne, P.E. In silico elucidation of the molecular mechanism defining the adverse effect of selective estrogen receptor modulators. PLoS Comput. Biol. 2007, 3, e217. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Roth, B.L.; Armbruster, B.N.; Ernsberger, P.; Irwin, J.J.; Shoichet, B.K. Relating protein pharmacology by ligand chemistry. Nat. Biotechnol. 2007, 25, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, T. Managing the drug discovery/development interface. Drug Discov. Today 1997, 2, 436–444. [Google Scholar] [CrossRef]
- Zimmermann, G.R.; Lehar, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, M.; Campillos, M.; González, P.; Jensen, L.J.; Bork, P. Large-scale prediction of drug–target relationships. FEBS Lett. 2008, 582, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.; Casini, A.; Heine, A.; Kuhn, D.; Supuran, C.T.; Scozzafava, A.; Klebe, G. Unexpected nanomolar inhibition of carbonic anhydrase by COX-2-selective celecoxib: New pharmacological opportunities due to related binding site recognition. J. Med. Chem. 2004, 47, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Campillos, M.; Kuhn, M.; Gavin, A.-C.; Jensen, L.J.; Bork, P. Drug target identification using side-effect similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Young, D.W.; Jenkins, J.L.; Serrano, M.; Mikhailov, D.; Clemons, P.A.; Davies, J.W. Chemogenomic data analysis: Prediction of small-molecule targets and the advent of biological fingerprints. Comb. Chem. High Throughput Screen. 2007, 10, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, E. Chemogenomics: Drug discovery’s panacea? Mol. Biosyst. 2006, 2, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Mestres, J. Computational chemogenomics approaches to systematic knowledge-based drug discovery. Curr. Opin. Drug Discov. Dev. 2004, 7, 304–313. [Google Scholar]
- Rognan, D. Chemogenomic approaches to rational drug design. Br. J. Pharmacol. 2007, 152, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Savchuk, N.P.; Balakin, K.V.; Tkachenko, S.E. Exploring the chemogenomic knowledge space with annotated chemical libraries. Curr. Opin. Chem. Biol. 2004, 8, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.T.; Deshpande, T.; Butte, A.J. Exploiting drug–disease relationships for computational drug repositioning. Brief. Bioinform. 2011, 12, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.I.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B. Predicting new molecular targets for known drugs. Nature 2009, 462, 175. [Google Scholar] [CrossRef] [PubMed]
- Noeske, T.; Sasse, B.C.; Stark, H.; Parsons, C.G.; Weil, T.; Schneider, G. Predicting Compound Selectivity by Self-Organizing Maps: Cross-Activities of Metabotropic Glutamate Receptor Antagonists. ChemMedChem 2006, 1, 1066–1068. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.A.; Gudivada, R.C.; Jegga, A.G.; Neumann, E.K.; Aronow, B.J. Inferring novel disease indications for known drugs by semantically linking drug action and disease mechanism relationships. BMC Bioinform. 2009, 10, S4. [Google Scholar] [CrossRef] [PubMed]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.-P.; Subramanian, A.; Ross, K.N. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriero, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA 2010, 107, 14621–14626. [Google Scholar] [CrossRef] [PubMed]
- Frey, B.J.; Dueck, D. Clustering by passing messages between data points. Science 2007, 315, 972–976. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Mestres, J.; Testa, B. In silico pharmacology for drug discovery: Methods for virtual ligand screening and profiling. Br. J. Pharmacol. 2007, 152, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Agarwal, P. Human disease-drug network based on genomic expression profiles. PLoS ONE 2009, 4, e6536. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Agarwal, P. A pathway-based view of human diseases and disease relationships. PLoS ONE 2009, 4, e4346. [Google Scholar] [CrossRef] [PubMed]
- Suthram, S.; Dudley, J.T.; Chiang, A.P.; Chen, R.; Hastie, T.J.; Butte, A.J. Network-based elucidation of human disease similarities reveals common functional modules enriched for pluripotent drug targets. PLoS Comput. Biol. 2010, 6, e1000662. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.T.; Schadt, E.; Sirota, M.; Butte, A.J.; Ashley, E. Drug discovery in a multidimensional world: Systems, patterns, and networks. J. Cardiovasc. Transl. Res. 2010, 3, 438–447. [Google Scholar] [CrossRef] [PubMed]
- Bodenreider, O. The unified medical language system (UMLS): Integrating biomedical terminology. Nucleic Acids Res. 2004, 32, D267–D270. [Google Scholar] [CrossRef] [PubMed]
- Gerhold, D.; Lu, M.; Xu, J.; Austin, C.; Caskey, C.T.; Rushmore, T. Monitoring expression of genes involved in drug metabolism and toxicology using DNA microarrays. Physiol. Genom. 2001, 5, 161–170. [Google Scholar]
- Thomas, R.S.; Rank, D.R.; Penn, S.G.; Zastrow, G.M.; Hayes, K.R.; Pande, K.; Glover, E.; Silander, T.; Craven, M.W.; Reddy, J.K. Identification of toxicologically predictive gene sets using cDNA microarrays. Mol. Pharmacol. 2001, 60, 1189–1194. [Google Scholar] [PubMed]
- Beger, R.D.; Sun, J.; Schnackenberg, L.K. Metabolomics approaches for discovering biomarkers of drug-induced hepatotoxicity and nephrotoxicity. Toxicol. Appl. Pharmacol. 2010, 243, 154–166. [Google Scholar] [CrossRef] [PubMed]
- McKinney, J.D.; Richard, A.; Waller, C.; Newman, M.C.; Gerberick, F. The practice of structure activity relationships (SAR) in toxicology. Toxicol. Sci. 2000, 56, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Li, A.P. Accurate prediction of human drug toxicity: A major challenge in drug development. Chem. Biol. Interact. 2004, 150, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Thukral, S.K.; Nordone, P.J.; Hu, R.; Sullivan, L.; Galambos, E.; Fitzpatrick, V.D.; Healy, L.; Bass, M.B.; Cosenza, M.E.; Afshari, C.A. Prediction of nephrotoxicant action and identification of candidate toxicity-related biomarkers. Toxicol. Pathol. 2005, 33, 343–355. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Heinloth, A.N.; Zeng, Z.-B.; Paules, R.S.; Bushel, P.R. Genes related to apoptosis predict necrosis of the liver as a phenotype observed in rats exposed to a compendium of hepatotoxicants. BMC Genom. 2008, 9, 288. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, K.; Larsen, P.; Linninger, A.A. Assessing chronic liver toxicity based on relative gene expression data. J. Theor. Biol. 2008, 254, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Low, Y.; Uehara, T.; Minowa, Y.; Yamada, H.; Ohno, Y.; Urushidani, T.; Sedykh, A.; Muratov, E.; Kuz’min, V.; Fourches, D.; et al. Predicting drug-induced hepatotoxicity using QSAR and toxicogenomics approaches. Chem. Res. Toxicol. 2011, 24, 1251–1262. [Google Scholar] [CrossRef] [PubMed]
- Minowa, Y.; Kondo, C.; Uehara, T.; Morikawa, Y.; Okuno, Y.; Nakatsu, N.; Ono, A.; Maruyama, T.; Kato, I.; Yamate, J.; et al. Toxicogenomic multigene biomarker for predicting the future onset of proximal tubular injury in rats. Toxicology 2012, 297, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Valerio, L.G. In silico toxicology for the pharmaceutical sciences. Toxicol. Appl. Pharmacol. 2009, 241, 356–370. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H. From QSAR to QSIIR: Searching for enhanced computational toxicology models. Comput. Toxicol. 2013, 2, 53–65. [Google Scholar]
- Benigni, R.; Battistelli, C.L.; Bossa, C.; Colafranceschi, M.; Tcheremenskaia, O. Mutagenicity, carcinogenicity, and other end points. Comput. Toxicol. 2013, 2, 67–98. [Google Scholar]
- Toropov, A.A.; Toropova, A.P.; Raska, I.; Leszczynska, D.; Leszczynski, J. Comprehension of drug toxicity: Software and databases. Comput. Biol. Med. 2014, 45, 20–25. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.; Hughes, M.; Garrow, A.; White, A. The value of in silico chemistry in the safety assessment of chemicals in the consumer goods and pharmaceutical industries. Drug Discov. Today 2012, 17, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Venkatapathy, R.; Wang, N.C.Y. Developmental toxicity prediction. Comput. Toxicol. 2013, 2, 305–340. [Google Scholar]
- Jack, J.; Wambaugh, J.; Shah, I. Systems Toxicology from Genes to Organs. Comput. Toxicol. 2013, 930, 375–397. [Google Scholar]
- Cannata, N.; Corradini, F.; Merelli, E.; Tesei, L. Agent-Based Models of Cellular Systems. Comput. Toxicol. 2013, 930, 399–426. [Google Scholar]
- Milan, C.; Schifanella, O.; Roncaglioni, A.; Benfenati, E. Comparison and possible use of in silico tools for carcinogenicity within REACH legislation. J. Environ. Sci. Health C 2011, 29, 300–323. [Google Scholar] [CrossRef] [PubMed]
- Cronin, M.; Wilson, A. In silico tools for toxicity prediction. In New Horizons in Predictive Toxicology: Current Status Application; Royal Society of Chemistry: Cambridge, UK, 2011. [Google Scholar]
- Worth, A.P.; Lapenna, S.; Serafimova, R. QSAR and metabolic assessment tools in the assessment of genotoxicity. Comput. Toxicol. 2013, 2, 125–162. [Google Scholar]
- Guha, R. On exploring structure–activity relationships. Methods Mol. Biol. 2013, 993, 81–94. [Google Scholar] [PubMed]
- Bowles, M.; Shigeta, R. Statistical models for predicting liver toxicity from genomic data. Syst. Biomed. 2013, 1, 144–149. [Google Scholar] [CrossRef]
- Lee, W.M. Drug-induced hepatotoxicity. N. Engl. J. Med. 2003, 349, 474–485. [Google Scholar] [CrossRef] [PubMed]
- An, Y.R.; Kim, J.Y.; Kim, Y.S. Construction of a predictive model for evaluating multiple organ toxicity. Mol. Cell. Toxicol. 2016, 12, 1–6. [Google Scholar] [CrossRef]
- Kim, J.; Shin, M. An integrative model of multi-organ drug-induced toxicity prediction using gene-expression data. BMC Bioinform. 2014, 15, S2. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kumar, S.; Ali, M.; Anand, P.; Agrawal, V.V.; John, R.; Maji, S.; Malhotra, B.D. Microfluidic-integrated biosensors: Prospects for point-of-care diagnostics. Biotechnol. J. 2013, 8, 1267–1279. [Google Scholar] [CrossRef] [PubMed]
- Noh, J.; Kim, H.C.; Chung, T.D. Biosensors in Microfluidic Chips. Microfluidics 2011, 117–152. [Google Scholar]
- Zhao, Y.; Stratton, Z.S.; Guo, F.; Lapsley, M.I.; Chan, C.Y.; Lin, S.-C.S.; Huang, T.J. Optofluidic imaging: Now and beyond. Lab Chip 2013, 13, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.-I.; Desmulliez, M.P. Lab-on-a-chip based immunosensor principles and technologies for the detection of cardiac biomarkers: A review. Lab Chip 2011, 11, 569–595. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Kim, H.-J.; Park, J.-H.; Jeong, D.H.; Lee, S.-K. Effects of surface density and size of gold nanoparticles in a fiber-optic localized surface plasmon resonance sensor and its application to peptide detection. Meas. Sci. Technol. 2010, 21, 085805. [Google Scholar] [CrossRef]
- Englebienne, P.; Hoonacker, A.V.; Verhas, M. Surface plasmon resonance: Principles, methods and applications in biomedical sciences. J. Spectrosc. 2003, 17, 255–273. [Google Scholar] [CrossRef]
- Lee, H.; Xu, L.; Koh, D.; Nyayapathi, N.; Oh, K.W. Various on-chip sensors with microfluidics for biological applications. Sensors 2014, 14, 17008–17036. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, L.; Jurasekova, Z.; Domingo, C.; Perez-Mendez, M.; Leyton, P.; Campos-Vallette, M.; Garcia-Ramos, J.; Sanchez-Cortes, S. Importance of metal–adsorbate interactions for the surface-enhanced Raman scattering of molecules adsorbed on plasmonic nanoparticles. Plasmonics 2007, 2, 147–156. [Google Scholar] [CrossRef]
- Arenas, J.F.; Soto, J.; Tocón, I.L.; Fernández, D.J.; Otero, J.C.; Marcos, J.I. The role of charge-transfer states of the metal-adsorbate complex in surface-enhanced Raman scattering. J. Chem. Phys. 2002, 116, 7207–7216. [Google Scholar] [CrossRef]
- Heck, K.N.; Janesko, B.G.; Scuseria, G.E.; Halas, N.J.; Wong, M.S. Observing metal-catalyzed chemical reactions in situ using surface-enhanced Raman spectroscopy on Pd−Au nanoshells. J. Am. Chem. Soc. 2008, 130, 16592–16600. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Herrmann, C.; Kömpe, K.; Haase, M.; Schlücker, S. Synthesis of bifunctional Au/Pt/Au core/shell nanoraspberries for in situ SERS monitoring of platinum-catalyzed reactions. J. Am. Chem. Soc. 2011, 133, 19302–19305. [Google Scholar] [CrossRef] [PubMed]
- Joseph, V.; Engelbrekt, C.; Zhang, J.; Gernert, U.; Ulstrup, J.; Kneipp, J. Characterizing the Kinetics of Nanoparticle-Catalyzed Reactions by Surface-Enhanced Raman Scattering. Angew. Chem. Int. Ed. 2012, 51, 7592–7596. [Google Scholar] [CrossRef] [PubMed]
- Leopold, N.; Haberkorn, M.; Laurell, T.; Nilsson, J.; Baena, J.R.; Frank, J.; Lendl, B. On-line monitoring of airborne chemistry in levitated nanodroplets: In situ synthesis and application of SERS-active Ag-sols for trace analysis by FT-raman spectroscopy. Anal. Chem. 2003, 75, 2166–2171. [Google Scholar] [CrossRef] [PubMed]
- Kneipp, J.; Kneipp, H.; Kneipp, K. SERS—A single-molecule and nanoscale tool for bioanalytics. Chem. Soc. Rev. 2008, 37, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- McLeod, E.; Luo, W.; Mudanyali, O.; Greenbaum, A.; Ozcan, A. Toward giga-pixel nanoscopy on a chip: A computational wide-field look at the nano-scale without the use of lenses. Lab Chip 2013, 13, 2028–2035. [Google Scholar] [CrossRef] [PubMed]
- Keller, P.J.; Schmidt, A.D.; Santella, A.; Khairy, K.; Bao, Z.; Wittbrodt, J.; Stelzer, E.H. Fast, high-contrast imaging of animal development with scanned light sheet-based structured-illumination microscopy. Nat. Methods 2010, 7, 637–642. [Google Scholar] [CrossRef] [PubMed]
- Betzig, E.; Patterson, G.H.; Sougrat, R.; Lindwasser, O.W.; Olenych, S.; Bonifacino, J.S.; Davidson, M.W.; Lippincott-Schwartz, J.; Hess, H.F. Imaging intracellular fluorescent proteins at nanometer resolution. Science 2006, 313, 1642–1645. [Google Scholar] [CrossRef] [PubMed]
- Roth, W.K.; Weber, M.; Seifried, E. Feasibility and efficacy of routine PCR screening of blood donations for hepatitis C virus, hepatitis B virus, and HIV-1 in a blood-bank setting. Lancet 1999, 353, 359–363. [Google Scholar] [CrossRef]
- Matula, P.; Kumar, A.; Wörz, I.; Erfle, H.; Bartenschlager, R.; Eils, R.; Rohr, K. Single-cell-based image analysis of high-throughput cell array screens for quantification of viral infection. Cytometry A 2009, 75, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Tung, Y.-C.; Huang, N.-T.; Oh, B.-R.; Patra, B.; Pan, C.-C.; Qiu, T.; Chu, P.K.; Zhang, W.; Kurabayashi, K. Optofluidic detection for cellular phenotyping. Lab Chip 2012, 12, 3552–3565. [Google Scholar] [CrossRef] [PubMed]
- Pang, L.; Chen, H.M.; Freeman, L.M.; Fainman, Y. Optofluidic devices and applications in photonics, sensing and imaging. Lab Chip 2012, 12, 3543–3551. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zheng, G.; Lee, L.M. Optical imaging techniques in microfluidics and their applications. Lab Chip 2012, 12, 3566–3575. [Google Scholar] [CrossRef] [PubMed]
- Mudanyali, O.; Tseng, D.; Oh, C.; Isikman, S.O.; Sencan, I.; Bishara, W.; Oztoprak, C.; Seo, S.; Khademhosseini, B.; Ozcan, A. Compact, light-weight and cost-effective microscope based on lensless incoherent holography for telemedicine applications. Lab Chip 2010, 10, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Pushkarsky, I.; Liu, Y.; Weaver, W.; Su, T.-W.; Mudanyali, O.; Ozcan, A.; Di Carlo, D. Automated single-cell motility analysis on a chip using lensfree microscopy. Sci. Rep. 2014, 4, 4717. [Google Scholar] [PubMed]
- Isikman, S.O.; Bishara, W.; Sikora, U.; Yaglidere, O.; Yeah, J.; Ozcan, A. Field-portable lensfree tomographic microscope. Lab Chip 2011, 11, 2222–2230. [Google Scholar] [CrossRef] [PubMed]
- Mudanyali, O.; McLeod, E.; Luo, W.; Greenbaum, A.; Coskun, A.F.; Hennequin, Y.; Allier, C.P.; Ozcan, A. Wide-field optical detection of nanoparticles using on-chip microscopy and self-assembled nanolenses. Nat. Photonics 2013, 7, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Schonbrun, E.; Abate, A.R.; Steinvurzel, P.E.; Weitz, D.A.; Crozier, K.B. High-throughput fluorescence detection using an integrated zone-plate array. Lab Chip 2010, 10, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Arpali, S.A.; Arpali, C.; Coskun, A.F.; Chiang, H.-H.; Ozcan, A. High-throughput screening of large volumes of whole blood using structured illumination and fluorescent on-chip imaging. Lab Chip 2012, 12, 4968–4971. [Google Scholar] [CrossRef] [PubMed]
- Coskun, A.F.; Sencan, I.; Su, T.-W.; Ozcan, A. Lensless wide-field fluorescent imaging on a chip using compressive decoding of sparse objects. Opt. Express 2010, 18, 10510–10523. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.A.; Ferrari, F.; Zhang, Y.; Nabavinia, M.; Mohammad, N.B.; Ryan, J.; Pourmand, A.; Laukaitis, E.; Sadeghian, R.; Nadhman, B.; et al. A microfluidic optical platform for real-time monitoring of pH and oxygen in microfluidic bioreactors and organ-on-chip devices. Biomicrofluidics 2016, 10, 044111. [Google Scholar] [CrossRef]
- Goral, V.N.; Zaytseva, N.V.; Baeumner, A.J. Electrochemical microfluidic biosensor for the detection of nucleic acid sequences. Lab Chip 2006, 6, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Boehm, D.A.; Gottlieb, P.A.; Hua, S.Z. On-chip microfluidic biosensor for bacterial detection and identification. Sens. Actuators B Chem. 2007, 126, 508–514. [Google Scholar] [CrossRef]
- Wang, J.; Chatrathi, M.P.; Tian, B.; Polsky, R. Microfabricated electrophoresis chips for simultaneous bioassays of glucose, uric acid, ascorbic acid, and acetaminophen. Anal. Chem. 2000, 72, 2514–2518. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Electrochemical glucose biosensors. Chem. Rev. 2008, 108, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Boo, H.; Kim, Y.; Han, J.-H.; Kim, H.C.; Chung, T.D. pH-sensitive solid-state electrode based on electrodeposited nanoporous platinum. Anal. Chem. 2005, 77, 7695–7701. [Google Scholar] [CrossRef] [PubMed]
- Henry, O.Y.; Fragoso, A.; Beni, V.; Laboria, N.; Sánchez, J.L.A.; Latta, D.; Von Germar, F.; Drese, K.; Katakis, I.; O’Sullivan, C.K. Design and testing of a packaged microfluidic cell for the multiplexed electrochemical detection of cancer markers. Electrophoresis 2009, 30, 3398–3405. [Google Scholar] [CrossRef] [PubMed]
- Länge, K.; Rapp, B.E.; Rapp, M. Surface acoustic wave biosensors: A review. Anal. Bioanal. Chem. 2008, 391, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; Stubbs, D.D.; Cairney, J.; Hunt, W.D. Rapid detection of bacterial spores using a quartz crystal microbalance (QCM) immunoassay. IEEE Sens. J. 2005, 5, 737–743. [Google Scholar]
- Cooper, M.A.; Singleton, V.T. A survey of the 2001, to 2005, quartz crystal microbalance biosensor literature: Applications of acoustic physics to the analysis of biomolecular interactions. J. Mol. Recognit. 2007, 20, 54–184. [Google Scholar] [CrossRef] [PubMed]
- Ergezen, E.; Appel, M.; Shah, P.; Kresh, J.; Lec, R.; Wootton, D. Real-time monitoring of adhesion and aggregation of platelets using thickness shear mode (TSM) sensor. Biosens. Bioelectron. 2007, 23, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Huang, I.-Y.; Lee, M.-C. Development of a FPW allergy biosensor for human IgE detection by MEMS and cystamine-based SAM technologies. Sens. Actuators B Chem. 2008, 132, 340–348. [Google Scholar] [CrossRef]
- Rocha-Gaso, M.-I.; March-Iborra, C.; Montoya-Baides, Á.; Arnau-Vives, A. Surface generated acoustic wave biosensors for the detection of pathogens: A review. Sensors 2009, 9, 5740–5769. [Google Scholar] [CrossRef] [PubMed]
- Arntz, Y.; Seelig, J.D.; Lang, H.; Zhang, J.; Hunziker, P.; Ramseyer, J.; Meyer, E.; Hegner, M.; Gerber, C. Label-free protein assay based on a nanomechanical cantilever array. Nanotechnology 2002, 14, 86. [Google Scholar] [CrossRef]
- Savran, C.A.; Knudsen, S.M.; Ellington, A.D.; Manalis, S.R. Micromechanical detection of proteins using aptamer-based receptor molecules. Anal. Chem. 2004, 76, 3194–3198. [Google Scholar] [CrossRef] [PubMed]
- Backmann, N.; Zahnd, C.; Huber, F.; Bietsch, A.; Plückthun, A.; Lang, H.-P.; Güntherodt, H.-J.; Hegner, M.; Gerber, C. A label-free immunosensor array using single-chain antibody fragments. Proc. Natl. Acad. Sci. USA 2005, 102, 14587–14592. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rezaei Kolahchi, A.; Khadem Mohtaram, N.; Pezeshgi Modarres, H.; Mohammadi, M.H.; Geraili, A.; Jafari, P.; Akbari, M.; Sanati-Nezhad, A. Microfluidic-Based Multi-Organ Platforms for Drug Discovery. Micromachines 2016, 7, 162. https://doi.org/10.3390/mi7090162
Rezaei Kolahchi A, Khadem Mohtaram N, Pezeshgi Modarres H, Mohammadi MH, Geraili A, Jafari P, Akbari M, Sanati-Nezhad A. Microfluidic-Based Multi-Organ Platforms for Drug Discovery. Micromachines. 2016; 7(9):162. https://doi.org/10.3390/mi7090162
Chicago/Turabian StyleRezaei Kolahchi, Ahmad, Nima Khadem Mohtaram, Hassan Pezeshgi Modarres, Mohammad Hossein Mohammadi, Armin Geraili, Parya Jafari, Mohsen Akbari, and Amir Sanati-Nezhad. 2016. "Microfluidic-Based Multi-Organ Platforms for Drug Discovery" Micromachines 7, no. 9: 162. https://doi.org/10.3390/mi7090162