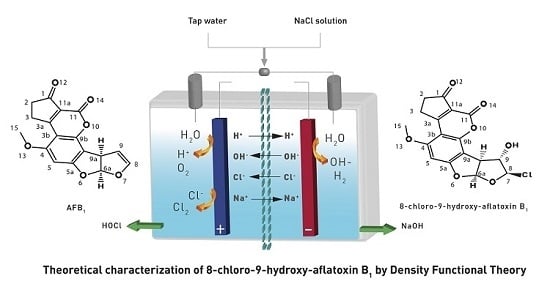
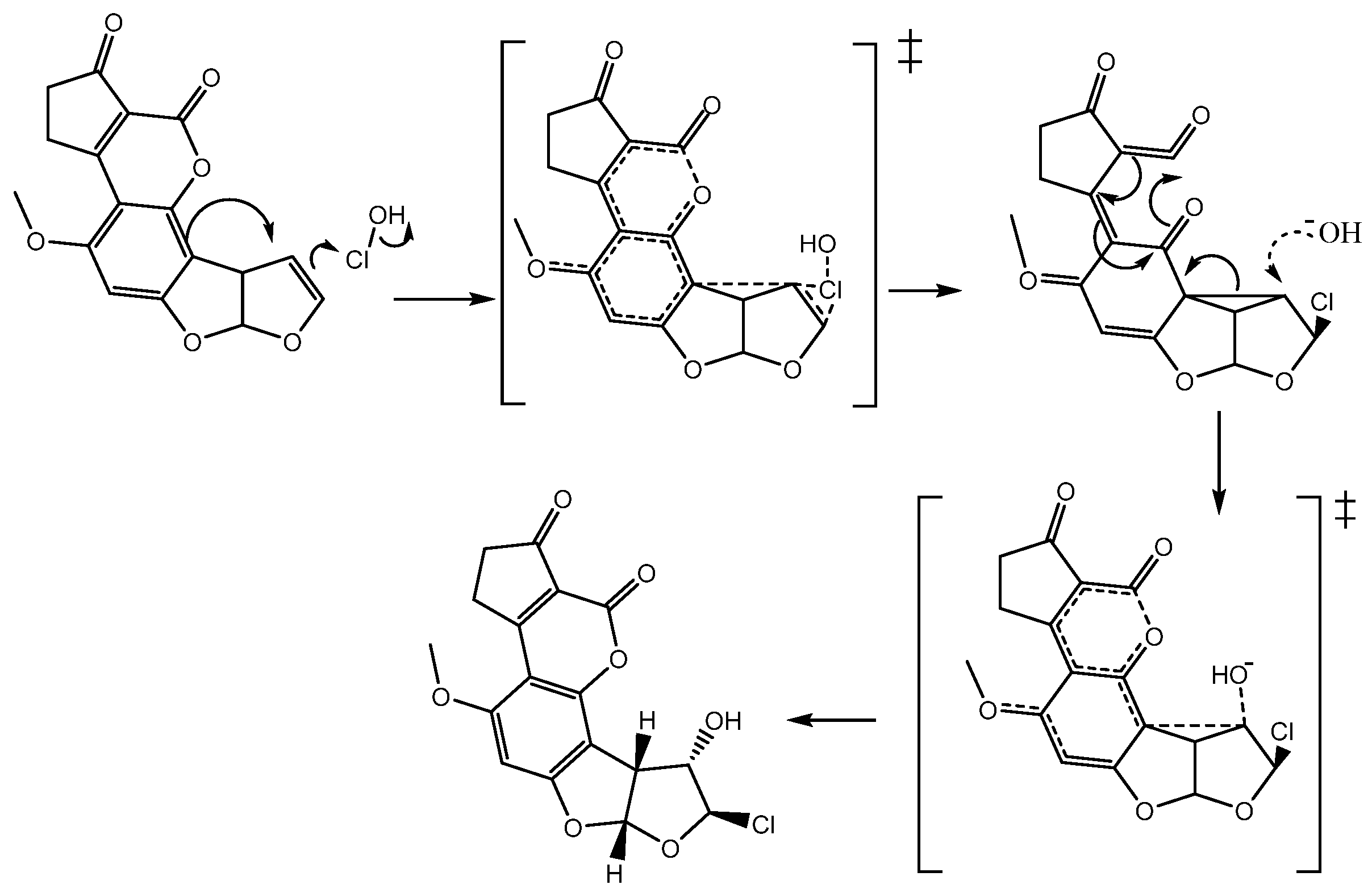
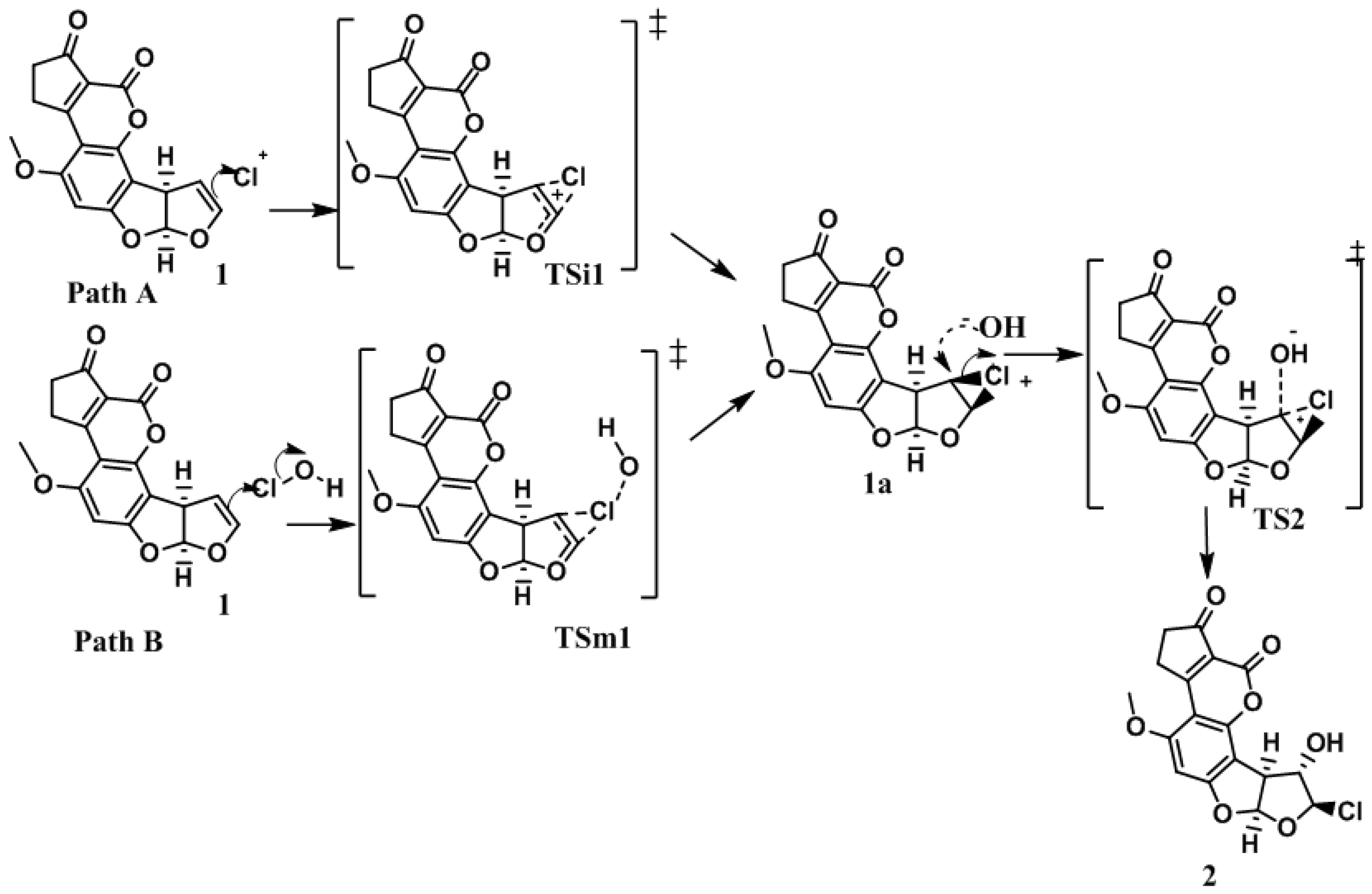
In order to elucidate the formation process of
2, two reaction pathways were considered according to the previously reported typical addition mechanism [
10], one with only ionic species of the HOCl (Path A), and the second, considering the entire HOCl molecule (Path B). These two pathways are shown in
Figure 3. Additionally, it is worth mentioning that both pathways were studied in gas and solution phases.
The reaction begins—in path A and B—with an electrophilic attack by the double bond of
1 to the chlorenium ion (Path A: ionic), or hypochlorous acid (path B: molecular) succeeding the first ionic activated state (
TSi1 for path A) or the molecular activated state (
TSm1 for path B), followed by the production of the chloronium ion reactive intermediate (
1a). In a second step, the hydroxide ion attained a nucleophilic attack to
1a, which produced the second activated state (
TS2), and concluded with the formation of
2 [
11,
12].
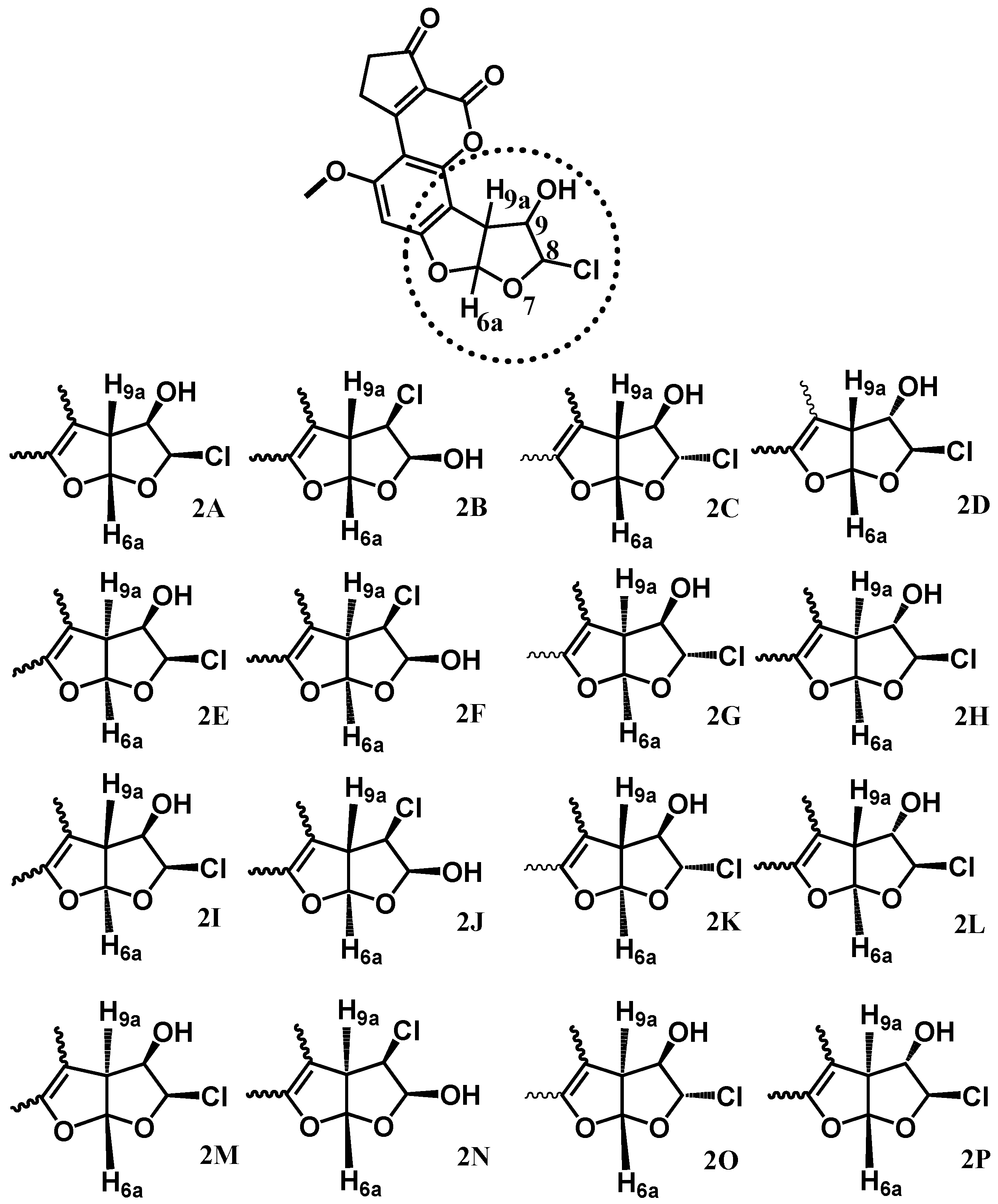
2.2.1. Geometrical Parameters Analysis of the Structures Involved in the Mechanism
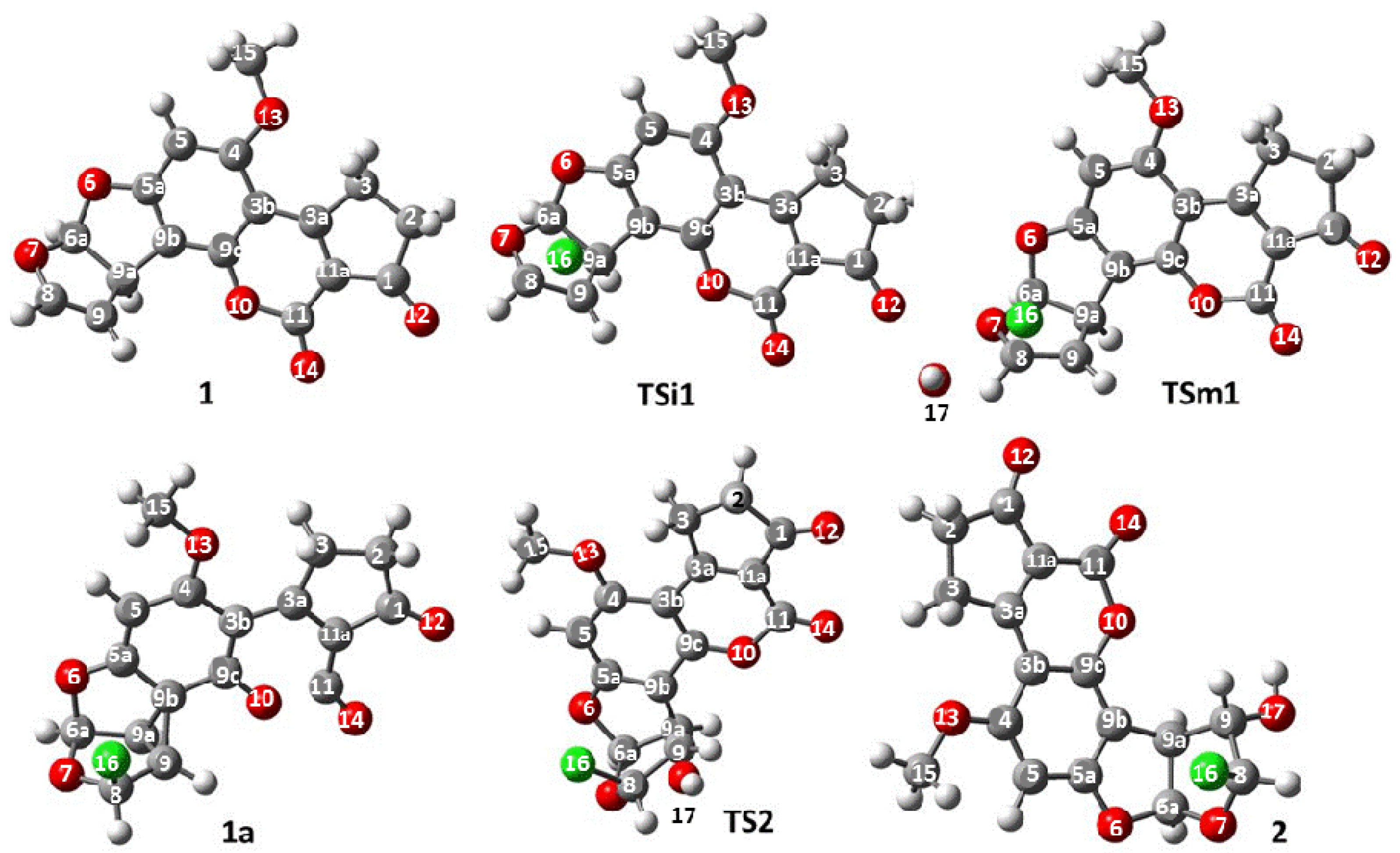
Regarding the active species generation process, the optimized structures are shown in
Figure 4. Selected geometrical parameters of the structures are summarized in
Table 2 and
Table 3. The reaction starts as suggested, with the interaction between the double bond at C
8–C
9 atoms of
1 and the chlorine atom (chlorenium ion or hypochlorous acid) to form the expected chloronium ion. The bond lengths (
Table 2) of
TSi1 show an increase of the distance between C
8–C
9 atoms of
1. Additionally, the distance of C
8–C
9 with the chlorine atom (1.95 and 2.35 Å, respectively) demonstrates a bond formation. However, a minor distance between C
8–Cl atoms was observed, indicating a stronger interaction. Thus, a slight decrease in the distance and angle (
Table 2) between C
9b and C
9 atoms was also observed.
In the same way, a similar behavior in the activated state in path B was found. The bond at C
8–C
9 atoms increased in similar value to that of
TSi1. On the other hand, the distance of C
8–Cl atoms was shorter, while C
9–Cl was longer compared to the same bonds in
TSi1, indicating the preference of the chlorine addition at the C
8 atom. The rest of the bonds in
TSi1 and
TSm1 agree with the experimental values and theoretical calculation for
1. After the addition of the chlorine atom on the double bond, a reactive intermediate is produced. However, the proposed structure of the chloronium ion (
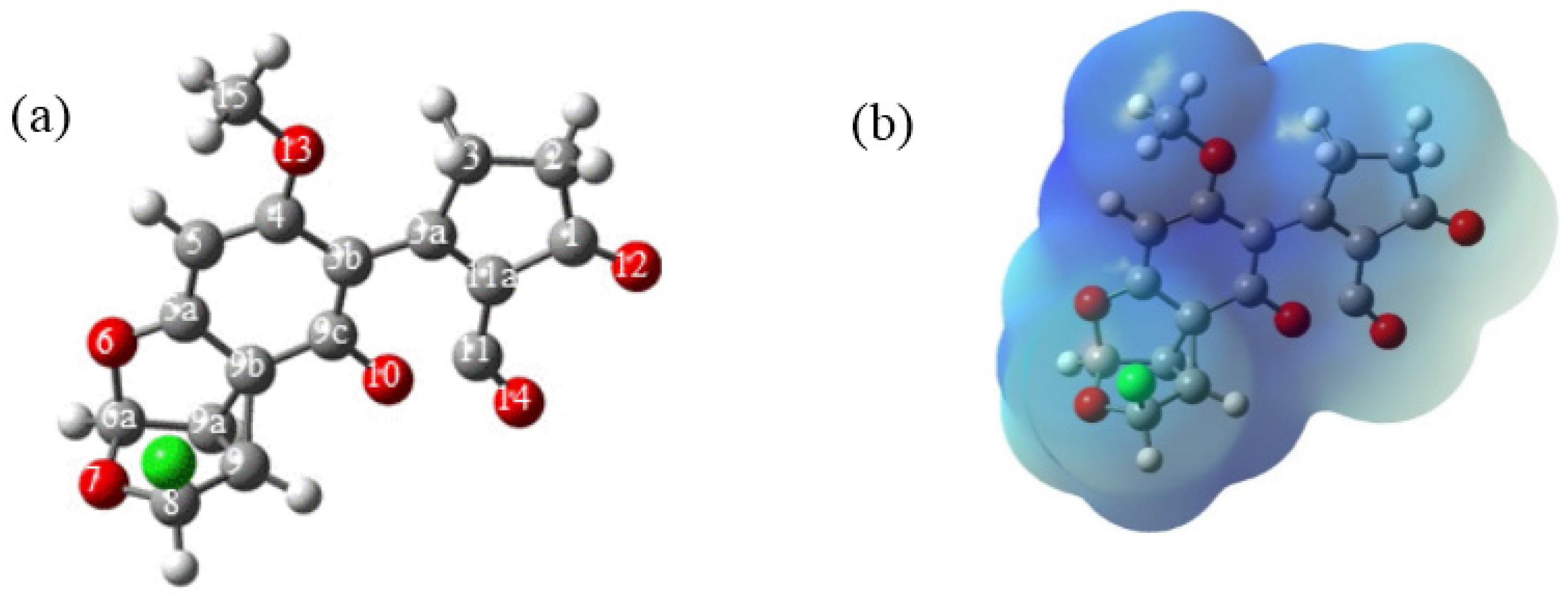
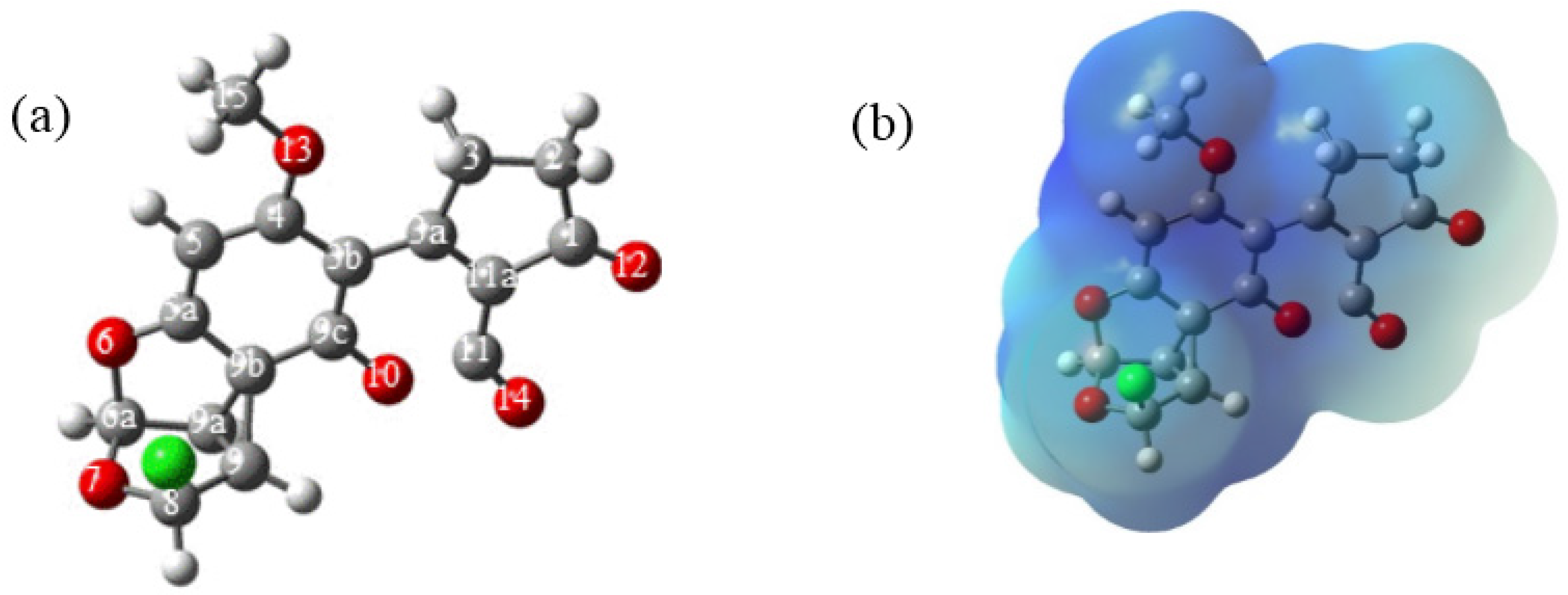
Figure 3) was not obtained; instead, a non-classic carbocation was formed (
Figure 5) [
13].
The change in the structure of the reactive intermediate
1a is a consequence of the interaction of the nearest aromatic system, which opens the chloronium ion and delocalizes the positive charge in the carbons of the benzene ring [
13,
14]. The electrostatic potential molecular surface (
Figure 5b) showed a blue surface denoting the cationic character of
1a; furthermore, the zones of the benzene and lactone rings had the most intense color confirming the electronic delocalization in these moieties. This process produced a cyclopropane moiety between C
9, C
9a and C
9b with a value of 1.47 Å for C
9–C
9a atoms, shorter than in
1, and a value of 1.53 Å for C
9a–C
9b atoms, which is longer in relation to the same bond in
1. Finally, the bond length value for C
9–C
9b atoms corresponds to 1.58 Å. Moreover, the angle for C
9–C
9a–C
9b decreases the value to 63.6°, which is consistent with the expected values for cyclopropane structures.
The next reaction step is the nucleophilic attack of the hydroxide ion at C
9 atom to form
2 via the second activated state (
TS2). In this sense, the closeness of the hydroxide ion to C
9 (2.51 Å) causes the enlargement of the C
9–C
9b bond (from 1.582 in
1a to 1.680 Å); additionally, the angle value at C
9–C
9a–C
9b also increases, signaling the breakage of the bond. The other bond angles and lengths values do not show significant changes. The attack of the hydroxide ion in the cyclopropane ring (instead of other sites with positive charge) is caused by the Bayer’s stress of the cycle and the high stability of the aromatic ring. Additional information presented in
Table 2 and
Table 3 are the geometrical properties of
2. This molecule does not have experimental values of X-ray diffraction reported yet, though the theoretical values for
2 was compared with
1, showing good congruity considering the structural differences.
2.2.2. Natural Charge in the Structures of the Reaction Mechanism
The natural populations of charges provide evidence of the electronic delocalization and the reactive site in the molecule. The charge analysis of the species involved in the reaction mechanism was also studied. The first step consisted of the interaction of the chlorine atom with the double bond to form either:
TSi1 or
TSm1, causing a reduction of the positive charge on C
8 atom. Meanwhile, C
9 atom changed to positive charge as a consequence of partial bonds formed with the chlorine atom. The interaction between C
9b and C
9 atoms was observed by a change in the distance and angle values in
TSi1 and
TSm1, respectively. In addition, the natural charge analysis from
TSm1 (
Table 4) showed that an increment in the negative charge on C
9b atom (−0.202) and the positive charge on C
9 atom (0.108) was greater than that on C
8 atom (0.097), assuming a possible interaction among them.
It is observed that when the C9b atom forms a bond with the C9 atom, the electronic density in C9c decreases. Thereby, O10 shares electronic density from unshared electrons, causing a drop in the negative charge of O10. The resonance effect delocalizes electrons, provoking an increment in the positive value at C4 (from 0.398 in 1 to 0.478 in 1a). The electrodonating effect of the methoxy group is observed in decrement of the negative charge in O13 (from −0.539 to −0.479). Finally, C5a also changes the value from 0.386 to 0.471. Considering the charge values, the atoms in the aromatic ring also change their values more closely to the 1, recovering the original aromatic state.
2.2.3. Thermochemistry of the Reaction
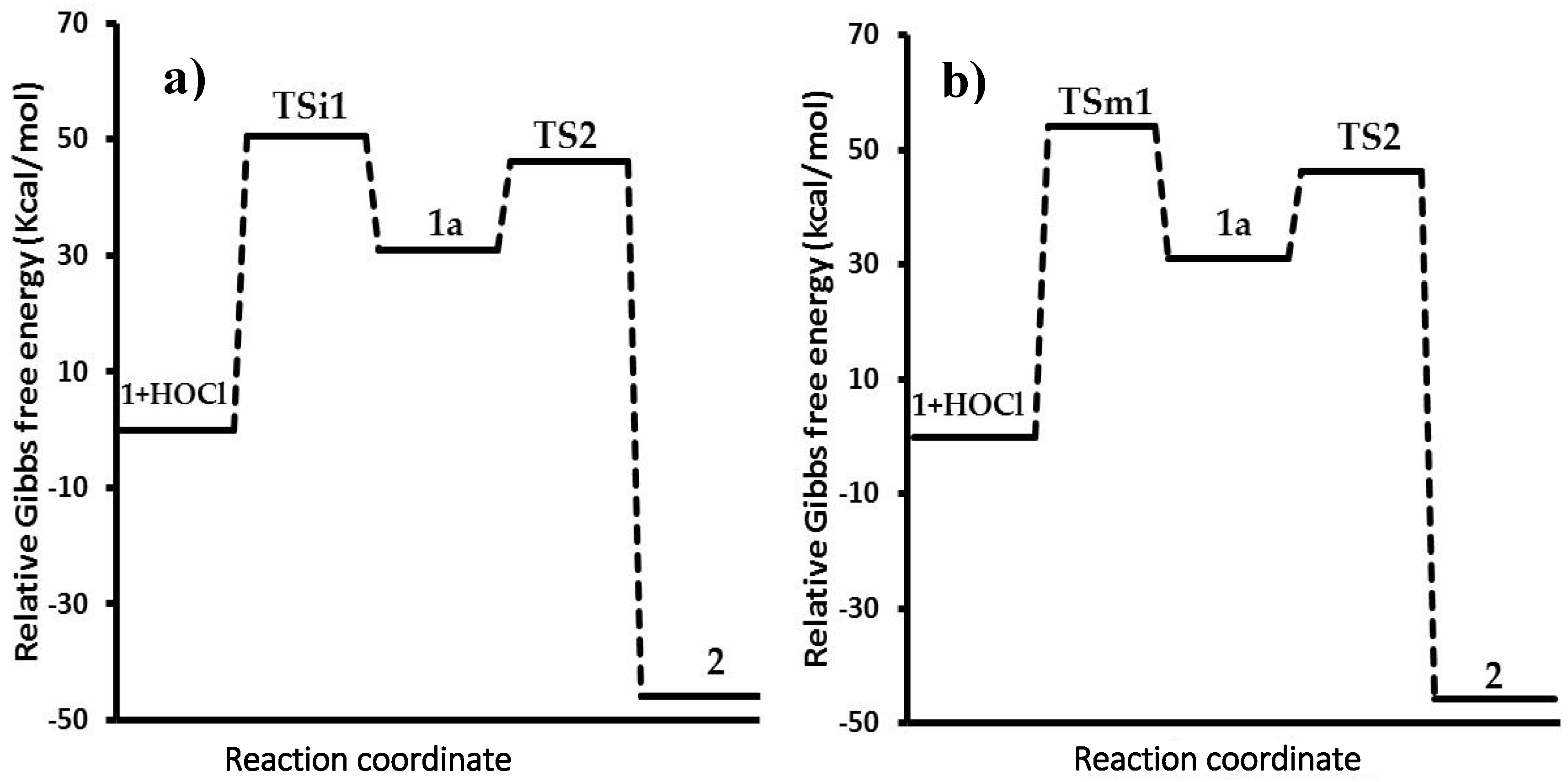
The relative Gibbs free energy profiles for these processes are plotted in
Figure 6. The energy calculations for all species were made in aqueous solution; therefore, two profiles were constructed.
The path A (
Figure 6, profile a) starts with the interaction of the double bond and the chlorine atom via activated state
TSi1 (Path A), which has an energy barrier in gas phase of 173.0 kcal/mol. However, considering the solvent effect, the energy barrier decreases to 50.4 kcal/mol, thus indicating a faster and feasible process. This change is a consequence of the stabilization of the ionic activated state (
TSi1) by a polar solvent such as water [
15,
16,
17], resulting in a faster reaction. The gas phase does not have this effect; consequently, an energy increment was observed. Since
1a is an ionic reactive intermediate, the stability in water solution was greater (30.9 kcal/mol) than in the gas phase (150.0 kcal/mol) by the solvating effect. In the second step, the activated state (
TS2) decreased the charge of the molecule (closely to the neutral charge) provoking a similar energy barrier in gas (62.2 kcal/mol) and solution (46.2 kcal/mol). Finally, the product is considered a polar molecule, which has a hydroxyl group. Thereby, the interaction of this group with water reduces the energy of the product up to −45.8 kcal/mol.
On the other hand, the thermochemical calculations from path B were performed in analogue form, considering the interaction between the double bond of 1 and the hypochlorous acid. In the calculations of gas and solution phases, both values were similar; the relative activation energy of the first activated state (TSm1) was 68.3 kcal/mol, while the energy barrier was 54.1 kcal/mol in solution. The less polarity of TSm1 in comparison with TSi1 allows more stability in gas phase and minor energy; however, partial charges formed were more stable in solution. Similar to path A, the reactive intermediate 1a is produced and reacts with hydroxide ion to form TS2, with the same energy barriers mentioned previously. The results in gas phase showed a great difference in the activation energies (profiles not showed), in contrast with the solution process; for this reason, the process in gas phase is currently under study using other functional and basis sets.
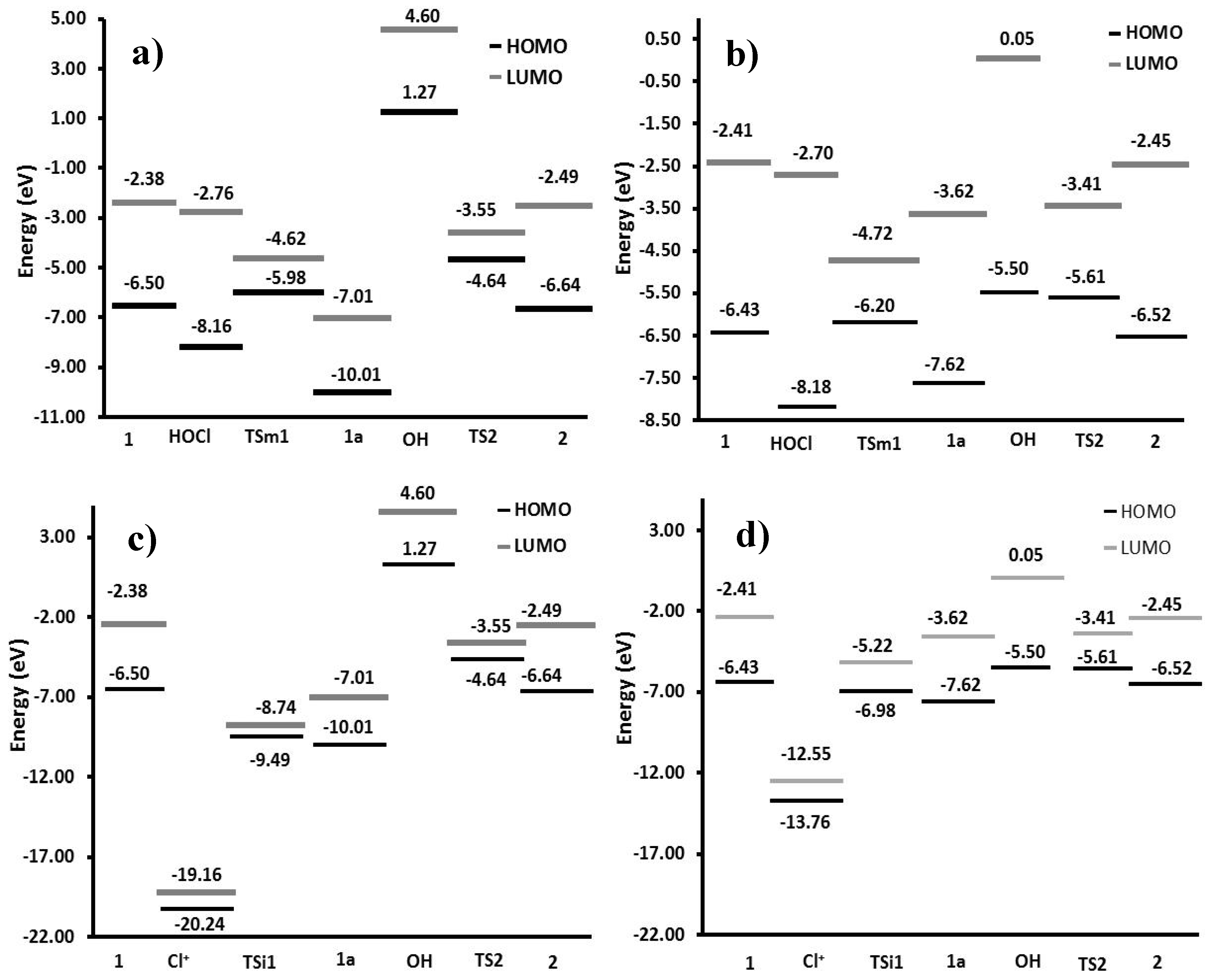
2.2.4. Frontier Orbitals Analysis
In order to describe the reactivity of the species involved in the reaction, a frontier molecular orbital analysis was made (
Figure 7). HOMO–LUMO energy gaps of gas phase and solution were calculated and compared; the most feasible process has the smaller value. Thus, the interaction among the HOMO of
1 with the LUMO of HOCl or Cl
+ in gas phase yielded energy gaps of 3.74 eV and −12.66 eV, respectively. In contrast, values of 3.73 eV and −6.12 eV were found in solution, which showed higher reactivity in aqueous medium. Although the interaction of
1 with the chlorenium ion has a minor value, this specie is unstable (gap value of 1.08 eV) and less probable to exist in the medium; thereby, the study was centered on path B.
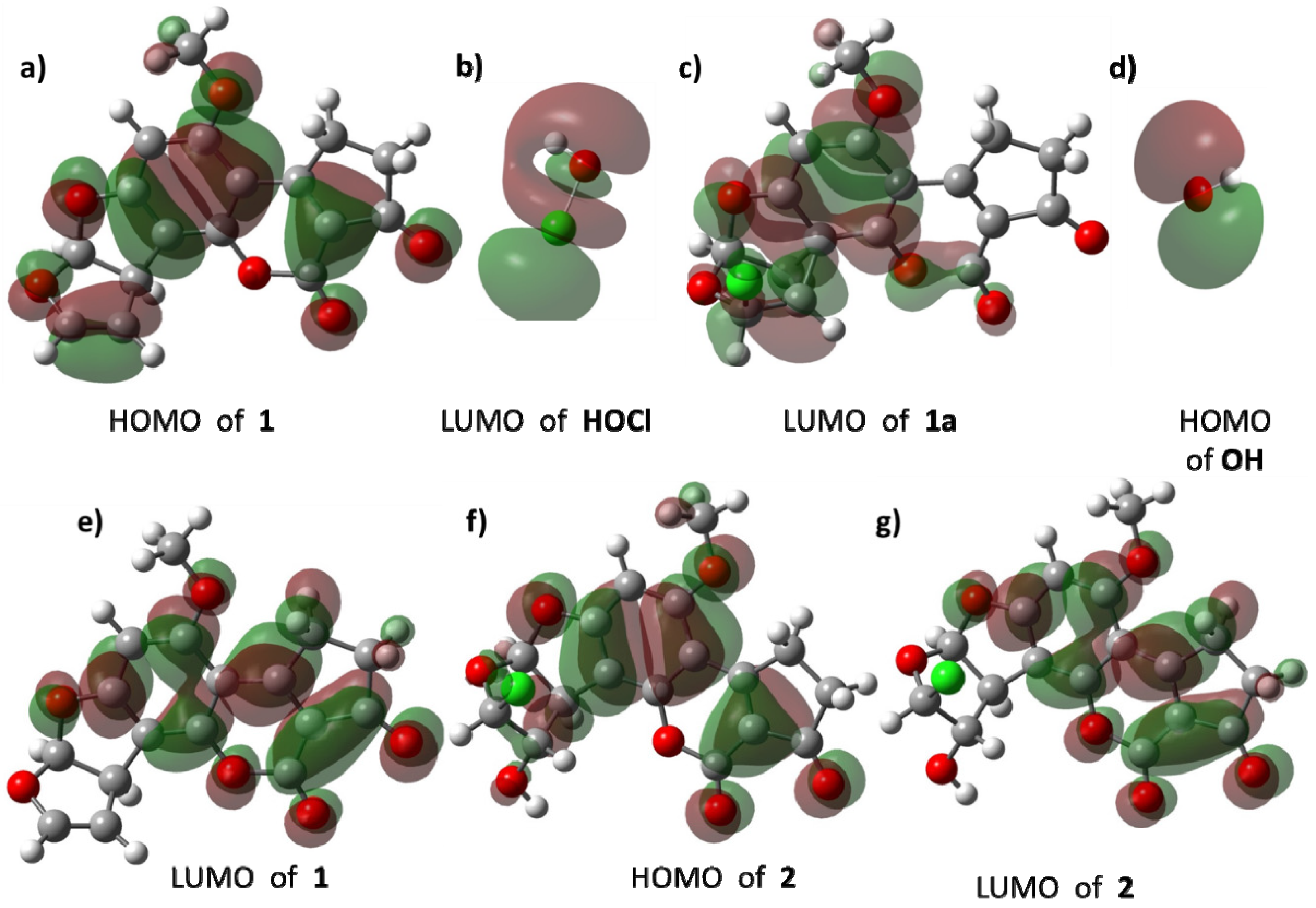
Figure 8 shows the HOMO and LUMO of reactive species. In
1, the HOMO (
Figure 8a) is mainly distributed around the double bond of C
8–C
9 atoms and the aromatic system confirms the electrodonating effect of this moiety towards the LUMO of HOCl (
Figure 8b). The LUMO of
1a (
Figure 8c) is concentrated in the aromatic ring, as a consequence of delocalization of the positive charge, and the cyclopropane motif reacts with the HOMO of the hydroxide ion (
Figure 8d). Regarding product
2, the distribution of its HOMO and LUMO is quite similar to the frontier orbitals of
1.
2.2.5. Bond Order and Reaction Mechanism
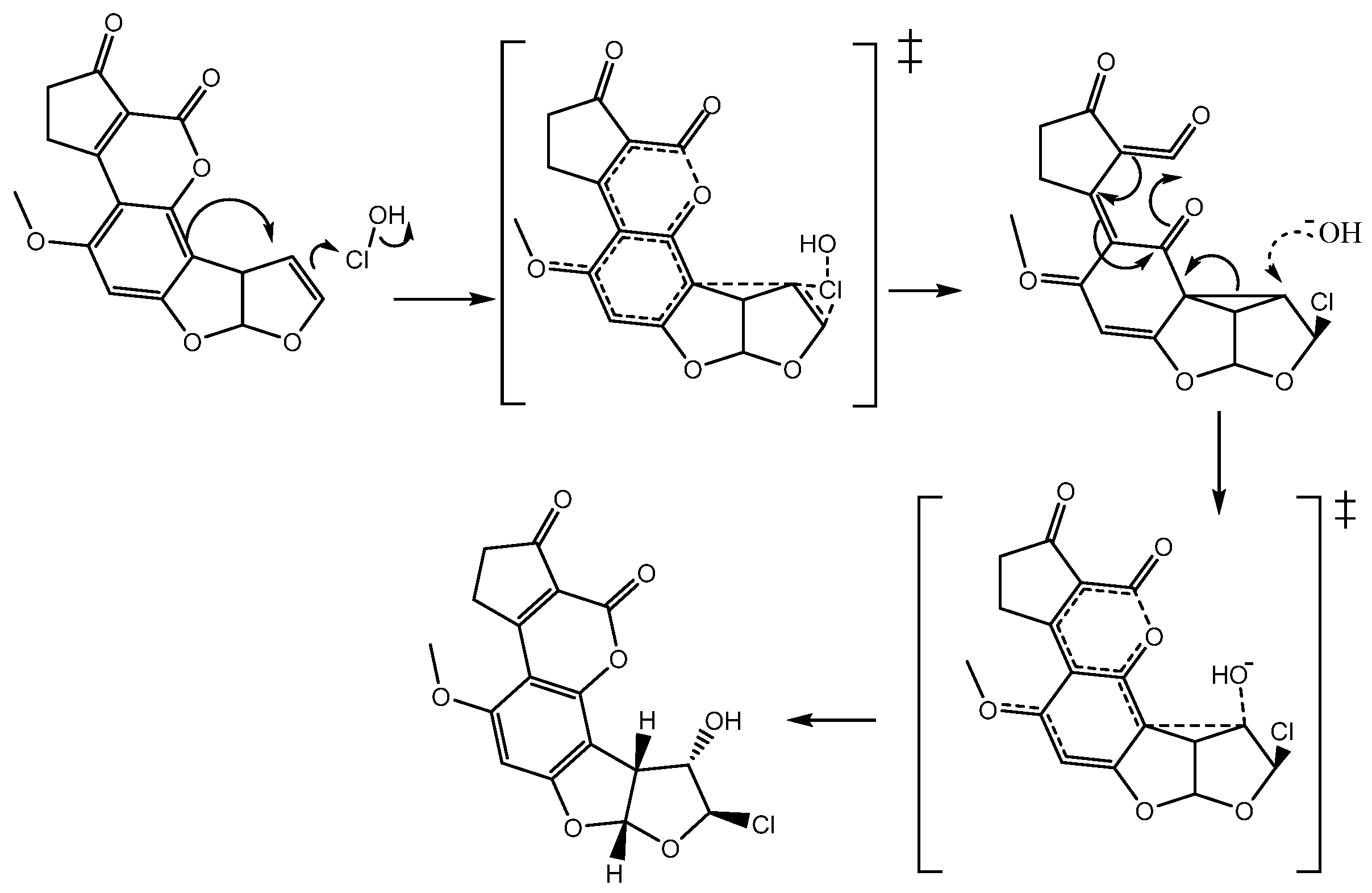
It is noteworthy to mention that, based on theoretical results, it is possible to propose the reaction mechanism with the activated states and intermediates (
Figure 9). In summary, the double bond of
1 makes a nucleophilic attack on the chlorine atom of the hypochlorous acid breaking the Cl-OH bond; however, by assistance of the nearest aromatic system, the chloronium ion is not formed and a non-classic carbocation is obtained [
13].
The bond order values of activated state
TSm1 were compared to that of the reactive
1 (
Table 5). In
TSm1, the C
8–C
9 bond had a bond order of 1.04 in contrast with
1, which had a value of 1.869. In this same sense, the bond order among C
9b and C
9 increased its value in
TSm1, indicating a possible bonding interaction, coinciding with the decrease in the distance and bond angle between these atoms. On the other hand, the partial bond formed within C
8–Cl and C
9–Cl has values of 0.819 and 0.315, respectively, showing a stronger interaction among the C
8 and Cl atoms. The bond orders in the aromatic ring carbon atoms were slightly smaller, and this fact is explained by the increment in bond length (
Table 3).
The same analysis was made for 1a, where it was possible to observe the bonding formation value between C9b–C9 (0.822), a decrement in the bond of C11–O10 and C9b–C9c, and an increment in the bond order among C9c–O10, when compared to 1. All variations are in close agreement with the geometrical parameters formerly described. Finally, it is observed that bond order values of 2 are quite similar to those obtained for 1, only with a significant decrement by the double bond break at C8–C9 atoms.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}