
Apoptosis Activation in Human Lung Cancer Cell Lines by a Novel Synthetic Peptide Derived from Conus californicus Venom

 ,
, 
Abstract
:1. Introduction
2. Results
2.1. Effect of s-cal14.1a on Lung Cancer Cells Viability
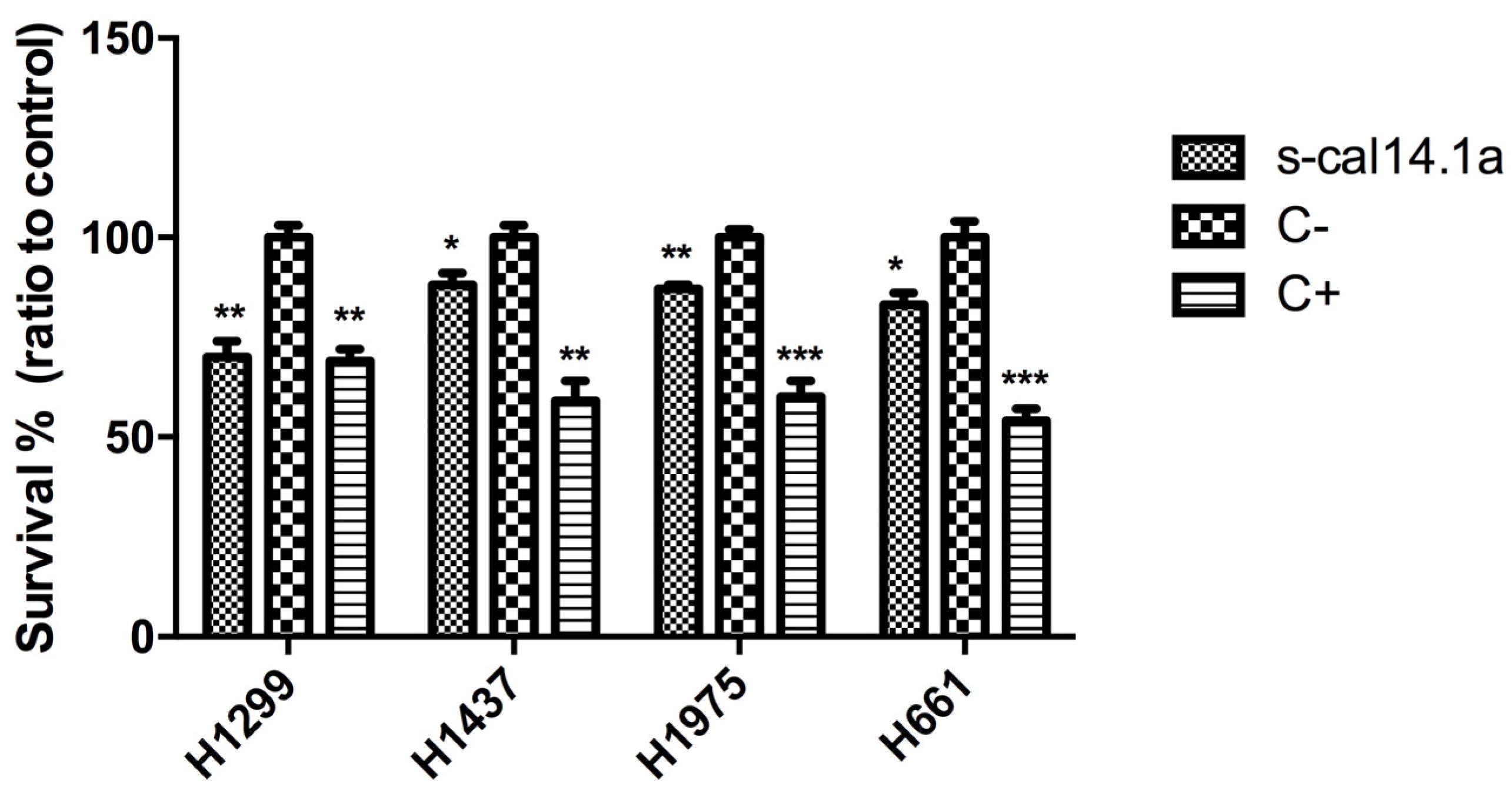
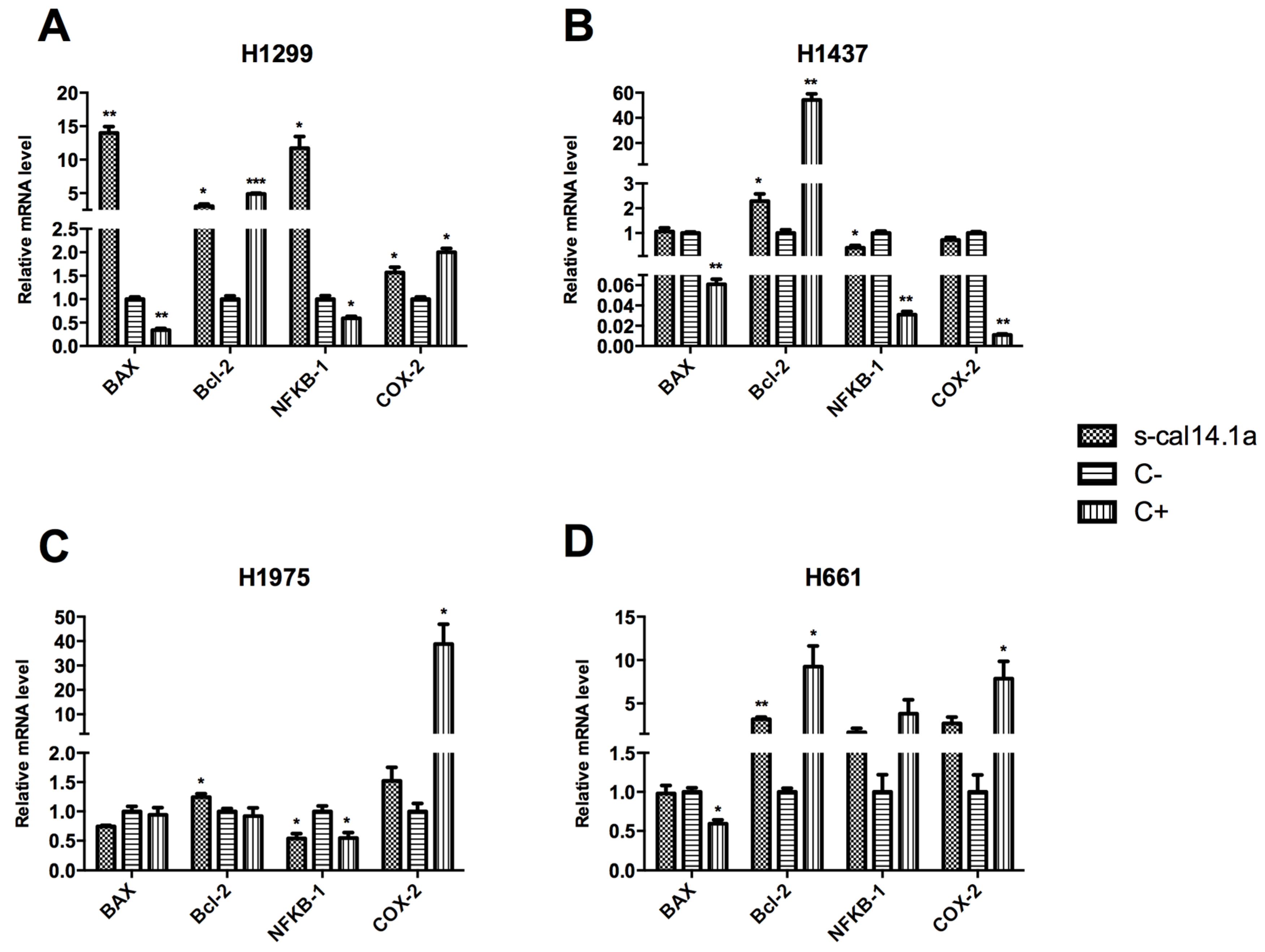
2.2. Expression Analysis of Apoptotic-Related Genes
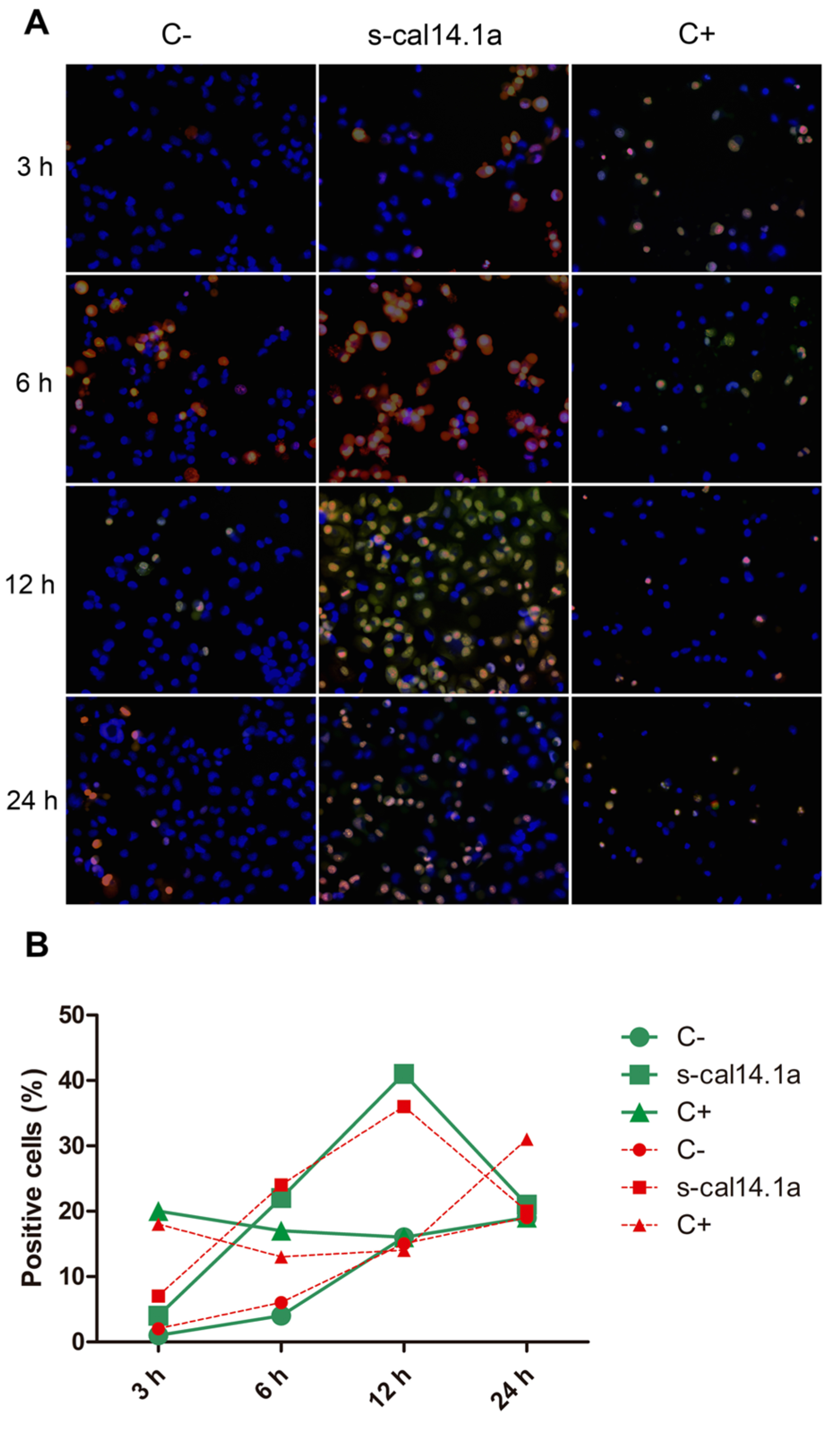
2.3. Activation of Caspase-3 and -7 in Lung Cancer Cell Lines
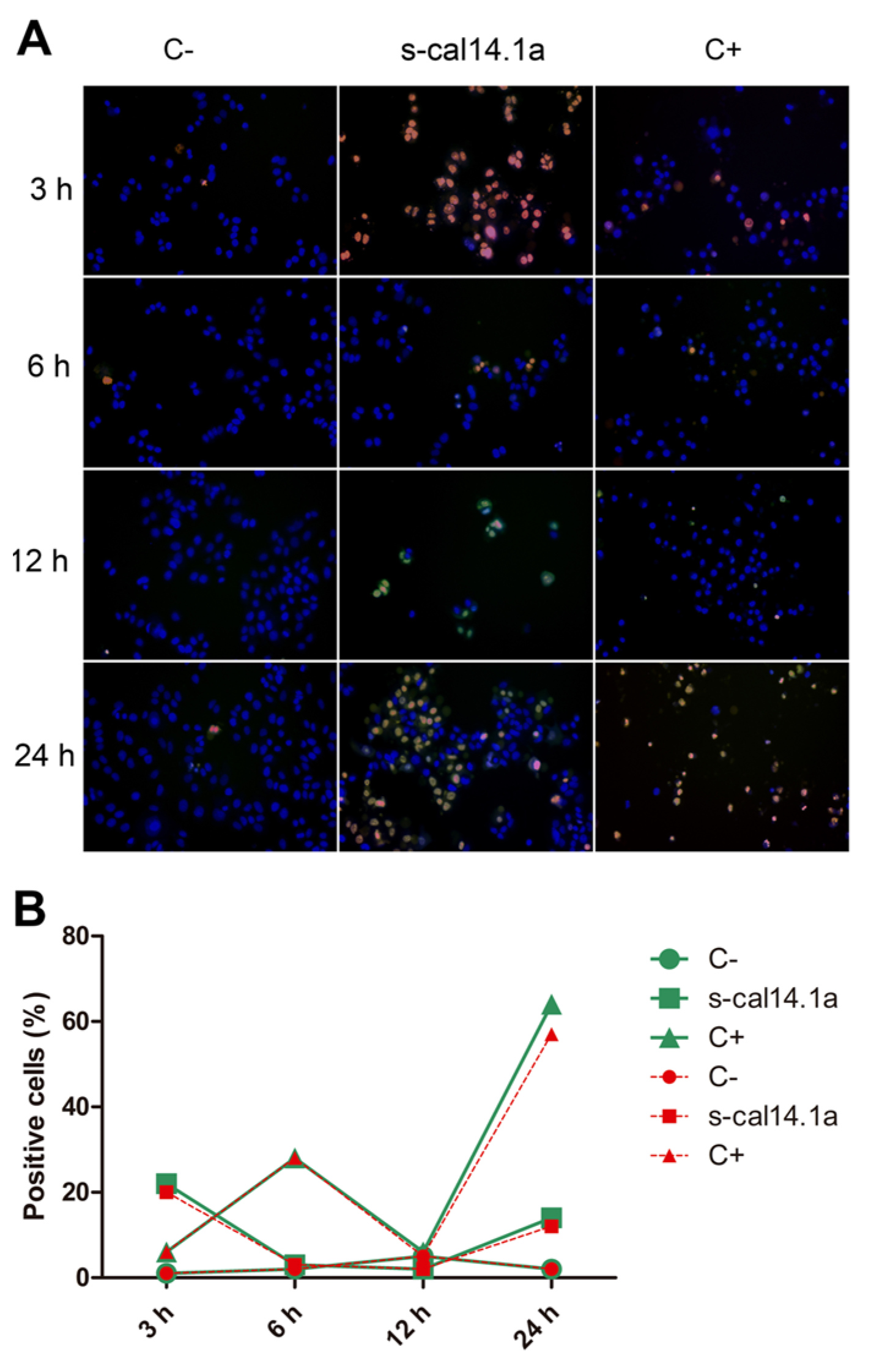

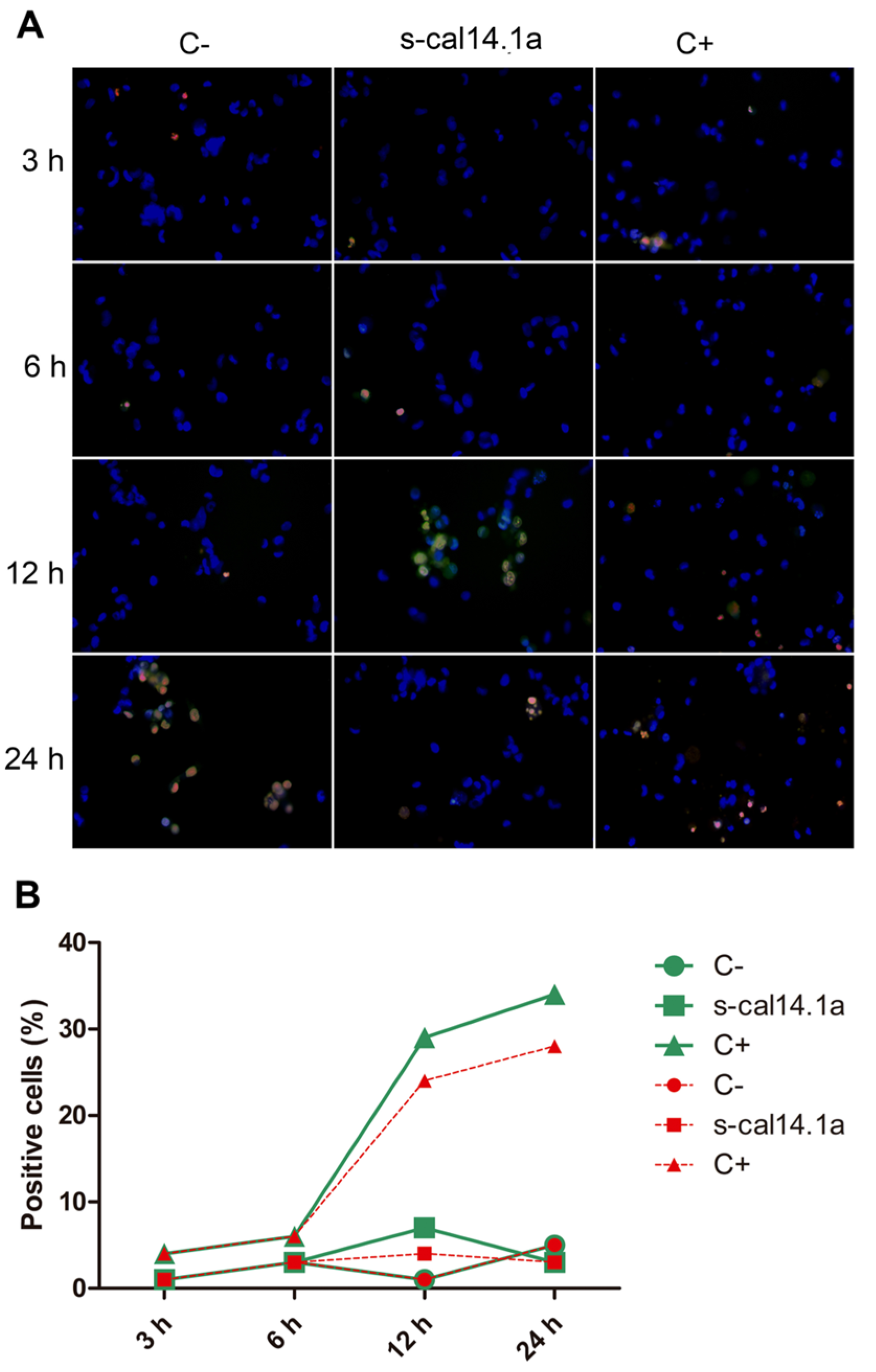
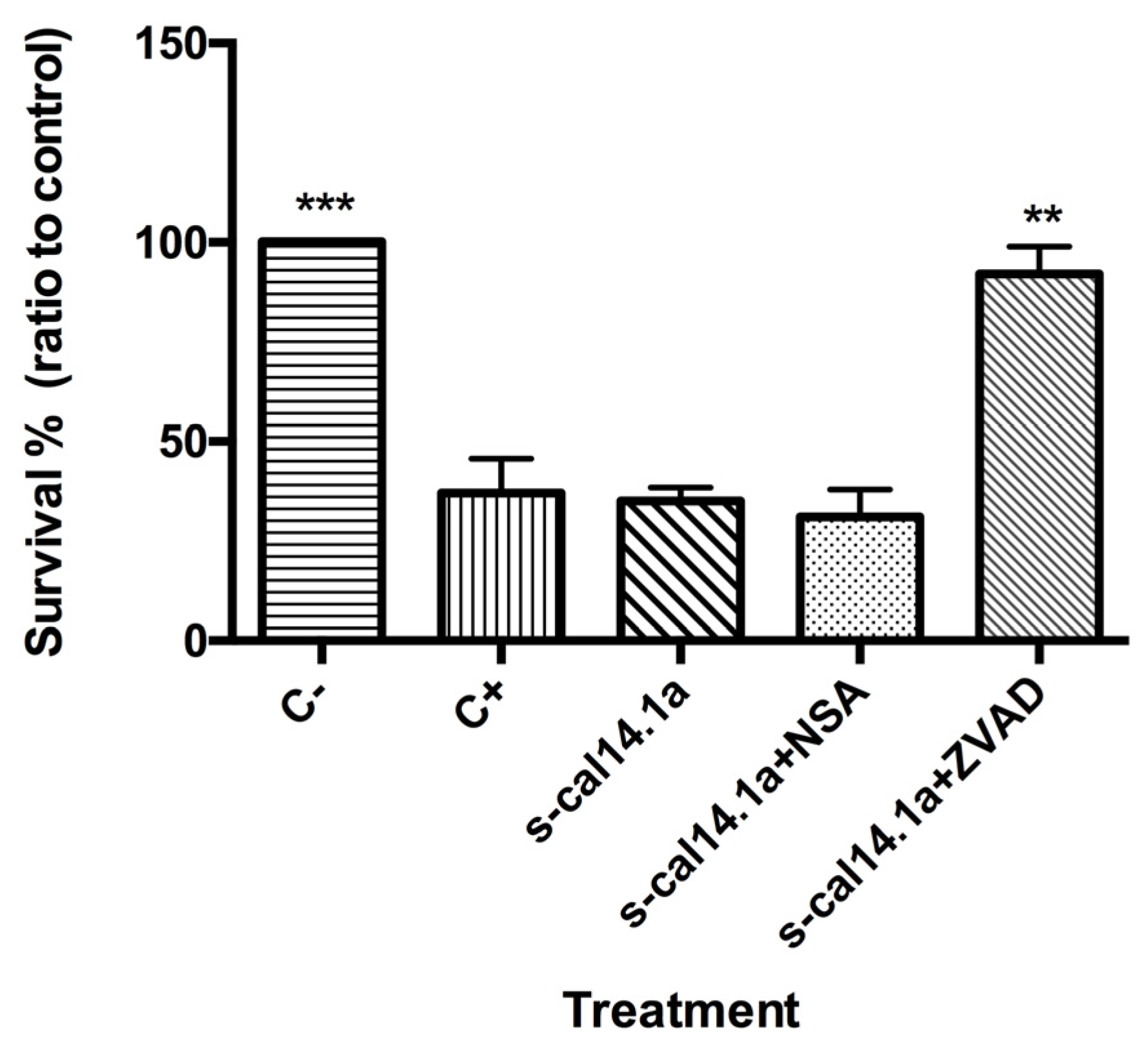
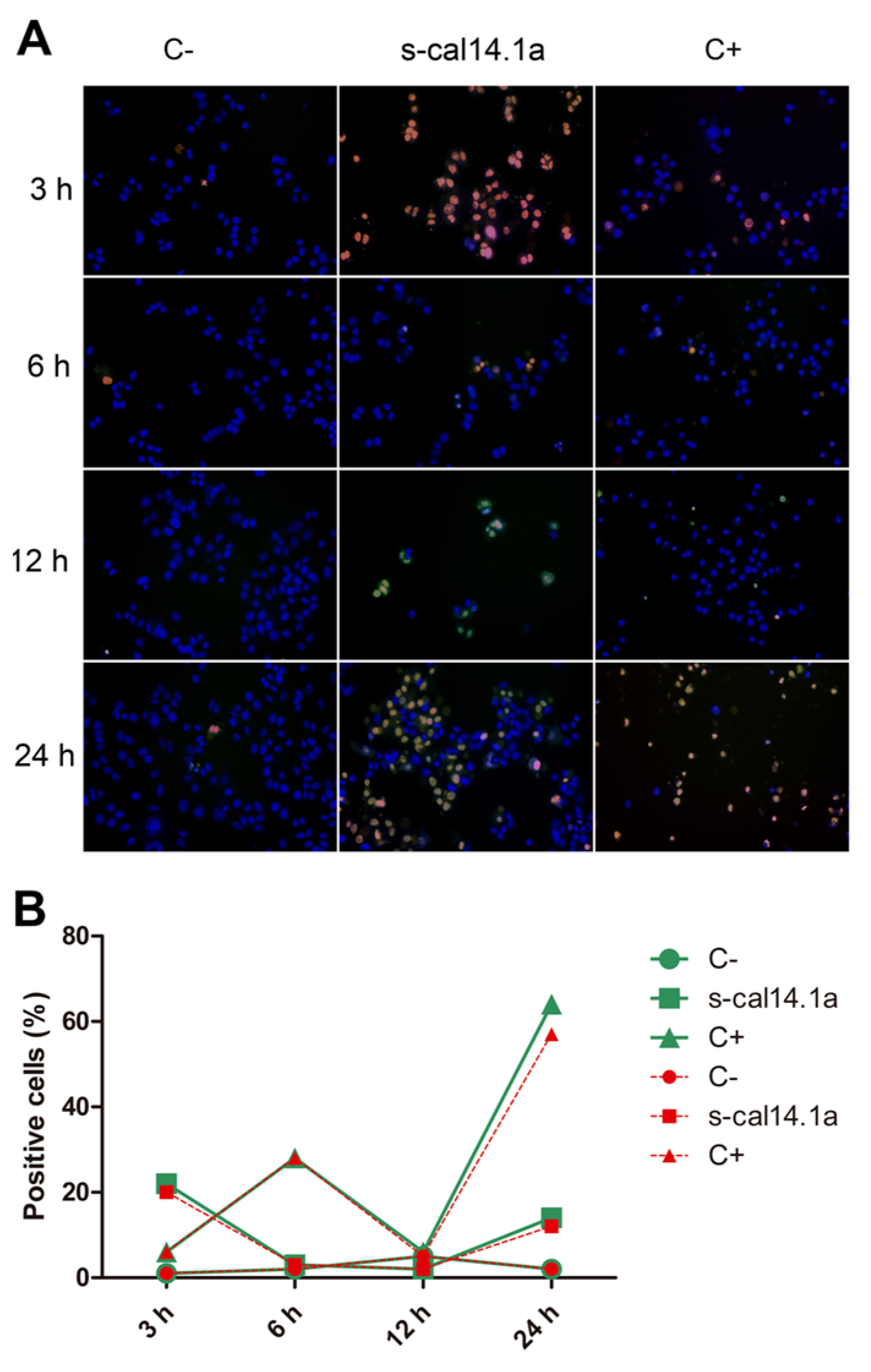
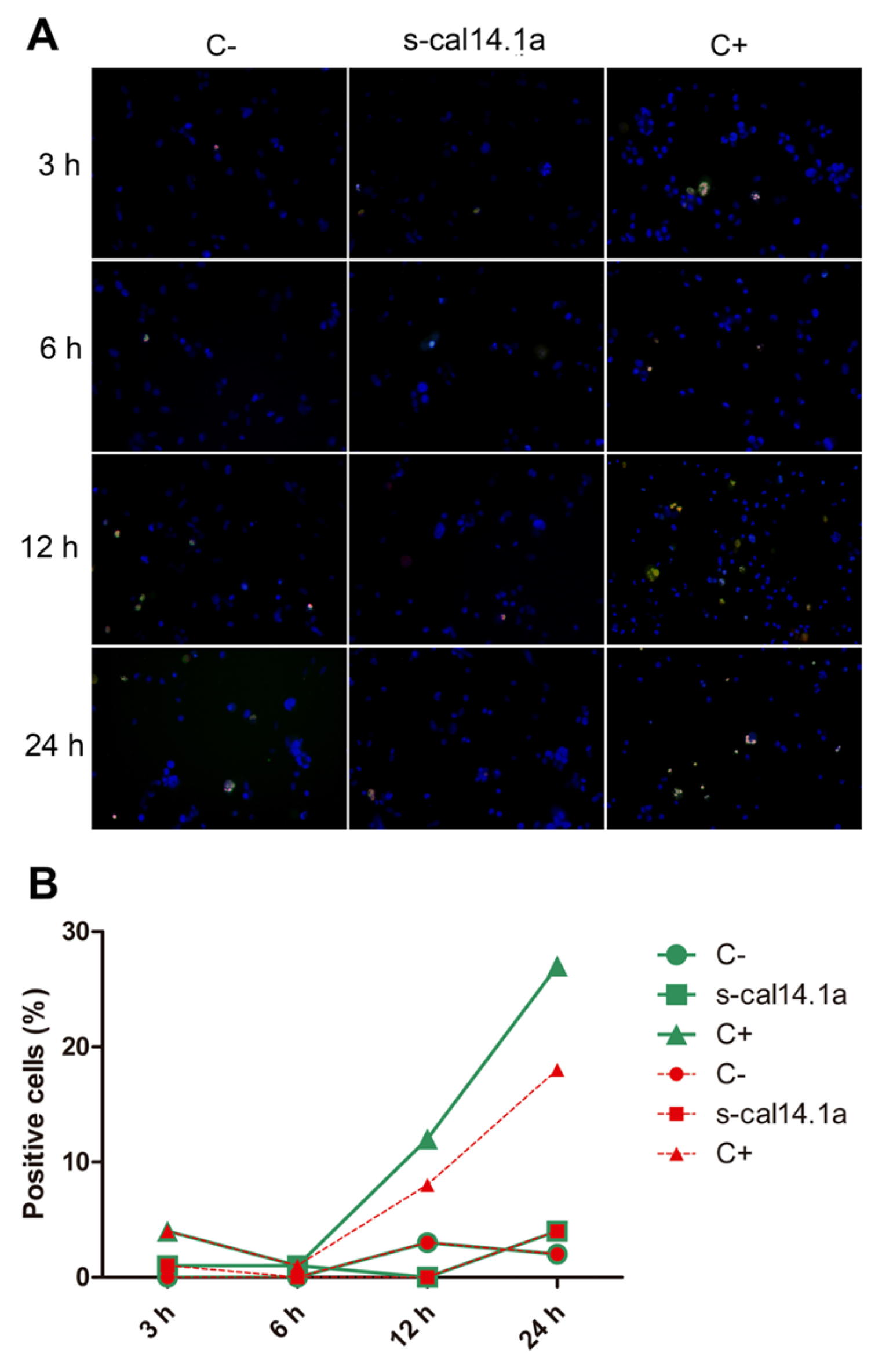
2.4. Apoptosis-Dependent Cell Death Induced by s-cal14.1a on H1437
3. Discussion
3.1. Cytotoxic Effect of s-cal14.1a on Four Lung Cancer Cell Lines
3.2. Apoptosis-Related Genes as Regulators of Cell Death
3.3. Activation of Caspase -3 and -7 and Apoptosis by s-cal14.1a
4. Materials and Methods
4.1. Cancer Cell Lines
4.2. Cell Viability Assay
4.3. RNA Extraction and Quantitative Real-Time PCR (RT-qPCR) Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer | Genbank Accession Number and Reference |
|---|---|---|
| β-actin | F: GCGAGAAGATGACCCAGATC R: CCAGTGGTACGGCCAGAGG | BRWS1 |
| Bcl-2 | F: ATGTGTGTGGAGAGCGTCACC R: TGAGCAGAGTCTTCAGAGACAGCC | BC027258.1 |
| BAX | F: TGGCAGCTGACATGTTTTCTGAC R: TCACCCAACCACCCRGGTCTT | NM_004324.3 |
| NFκB-1 | F: CGCCGCTTAGGAGGGAGA R: AGGTATGGGCCATCTGCTGT | NM_003998.3 |
| COX-2 | F: TGCATTCTTTGCCCCAGCACT R: AAAGGCGCAGTTTACGCTGT | Inoue et al., 2013 [75] |
4.4. Caspase-3 and -7 Activity Assay
4.5. Cell Imaging
4.6. Statistic Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| nAChRs | acetylcholine nicotinic receptors |
| ACh | acetylcholine |
| PI | propidium iodide |
| NSCLC | non-small cell lung cancer |
| EMT | epithelial-to-mesenchymal transition |
| TNF-α/CHX | tumor necrosis factor- α and cycloheximide |
References
- Improgo, M.R.; Tapper, A.R.; Gardner, P.D. Nicotinic acetylcholine receptor-mediated mechanisms in lung cancer. Biochem. Pharmacol. 2011, 82, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Jia, Y.; Zu, S.; Li, R.; Jia, Y.; Zhao, Y.; Xiao, D.; Dang, N.; Wang, Y. Alpha5 nicotinic acetylcholine receptor mediates nicotine-induced HIF-1α and VEGF expression in non-small cell lung cancer. Toxicol. Appl. Pharmacol. 2014, 278, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Reina Improgo, M.; Soll, L.G.; Tapper, A.R.; Gardner, P.D. Nicotinic acetylcholine receptors mediate lung cancer growth. Front. Physiol. 2013, 4, 1–6. [Google Scholar]
- Zhang, H.; Zhang, C.; Wu, D. Activation of insulin-like growth factor 1 receptor regulates the radiation-induced lung cancer cell apoptosis. Immunobiology 2015, 220, 1136–1140. [Google Scholar] [CrossRef] [PubMed]
- Haque, A.; Rahman, M.A.; Fuchs, J.R.; Chen, Z.G.; Khuri, F.R.; Shin, D.M.; Amin, A.R. FLLL12 induces apoptosis in lung cancer cells through a p53/p73-independent but death receptor 5-dependent pathway. Cancer Lett. 2015, 363, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Franklin, J.B.; Rajesh, R.P. A sleep-inducing peptide from the venom of the Indian cone snail Conus araneosus. Toxicon 2015, 103, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Akondi, K.B.; Muttenthaler, M.; Dutertre, S.; Kaas, Q.; Craik, D.J.; Lewis, R.J.; Alewood, P.F. Discovery, synthesis, and structure-activity relationships of conotoxins. Chem. Rev. 2014, 114, 5815–5847. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M. Conus venom peptides: Correlating chemistry and behavior. J. Comp. Physiol. A Sens. Neural Behav. Physiol. 1999, 185, 353–359. [Google Scholar] [CrossRef]
- Wang, C.; Chi, C. Conus peptides—A rich pharmaceutical treasure the biology of cone snails classification and nomenclature of Conus peptides. Acta Biochim. Biophys. Sin. (Shanghai) 2004, 36, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wang, L.; Wu, Y.; Zhu, X.; Feng, Y.; Chen, Z.; Li, Y.; Sun, D.; Ren, Z.; Xu, A. Characterizing the evolution and functions of the M-superfamily conotoxins. Toxicon 2013, 76, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. ConoServer: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Garrett, J.E.; Watkins, M.; Olivera, B.M. Purification and characterization of a novel excitatory peptide from Conus distans venom that defines a novel gene superfamily of conotoxins. Toxicon 2008, 52, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, M.B.; Ortiz, E.; Kaas, Q.; López-Vera, E.; Becerril, B.; Possani, L.D.; De La Cotera, E.P.H. Precursor De13.1 from Conus delessertii defines the novel G gene superfamily. Peptides 2013, 41, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Oliver, L.; Vallette, F.M. The role of caspases in cell death and differentiation. Drug Resist. Updat. 2005, 8, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Korbakis, D.; Scorilas, A. Quantitative expression analysis of the apoptosis-related genes BCL2, BAX and BCL2L12 in gastric adenocarcinoma cells following treatment with the anticancer drugs cisplatin, etoposide and taxol. Tumor Biol. 2012, 33, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kass, G.E.N.; Szegezdi, E.; Joseph, B. The mitochondrial death pathway: A promising therapeutic target in diseases. J. Cell. Mol. Med. 2009, 13, 1004–1033. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G. Mitochondrial control of apoptosis: An introduction. Biochem. Biophys. Res. Commun. 2003, 304, 433–435. [Google Scholar] [CrossRef]
- Ozören, N.; El-Deiry, W.S. Cell surface death receptor signaling in normal and cancer cells. Semin. Cancer Biol. 2003, 13, 135–147. [Google Scholar] [CrossRef]
- Thorburn, A. Death receptor-induced cell killing. Cell. Signal. 2004, 16, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Pore, M.M.; Hiltermann, T.J.; Kruyt, F.A. Targeting apoptosis pathways in lung cancer. Cancer Lett. 2013, 332, 359–368. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Hashemi, M.; Ande, S.R.; Yeganeh, B.; Xiao, W.; Eshraghi, M.; Bus, C.J.; Kadkhoda, K.; Wiechec, E.; Halayko, A.J.; et al. Apoptosis and cancer: Mutations within caspase genes. J. Med. Genet. 2009, 46, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Lin, X.; Wu, N.; Liu, M.; Zheng, Y.; Sheng, J.; Ji, X.; Sun, M. Targeting cellular apoptotic pathway with peptides from marine organisms. Biochim. Biophys. Acta Rev. Cancer 2013, 1836, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Biggs, J.S.; Watkins, M.; Puillandre, N.; Ownby, J.P.; Lopez-Vera, E.; Christensen, S.; Moreno, K.J.; Bernaldez, J.; Licea-Navarro, A.; Corneli, P.S.; et al. Evolution of Conus peptide toxins: Analysis of Conus californicus Reeve, 1844. Mol. Phylogenet. Evol. 2010, 56, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Lluisma, A.O.; Milash, B.A.; Moore, B.; Olivera, B.M.; Bandyopadhyay, P.K. Novel venom peptides from the cone snail Conus pulicarius discovered through next-generation sequencing of its venom duct transcriptome. Mar. Genomics 2012, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Dowell, C.; Olivera, B.M.; Garrett, J.E.; Staheli, S.T.; Watkins, M.; Kuryatov, A.; Yoshikami, D.; Lindstrom, J.M.; McIntosh, J.M. Alpha-conotoxin PIA is selective for alpha6 subunit-containing nicotinic acetylcholine receptors. J. Neurosci. 2003, 23, 8445–8452. [Google Scholar] [PubMed]
- Pucci, L.; Grazioso, G.; Dallanoce, C.; Rizzi, L.; De Micheli, C.; Clementi, F.; Bertrand, S.; Bertrand, D.; Longhi, R.; De Amici, M.; et al. Engineering of α-conotoxin MII-derived peptides with increased selectivity for native α6β2* nicotinic acetylcholine receptors. FASEB J. 2011, 25, 3775–3789. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Li, L.; Wang, W.; Pan, Z.; Zhou, Q.; Wu, Z. Mitochondrial reactive oxygen species mediates nicotine-induced hypoxia-inducible factor-1α expression in human non-small cell lung cancer cells. Biochim. Biophys. Acta 2012, 1822, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.W.; Romano, M.A.; Kudrimoti, M.R.; Randall, M.E.; McGarry, R.C.; Singh, A.K.; Rangnekar, V.M. Nicotinic modulation of therapeutic response in vitro and in vivo. Int. J. Cancer 2012, 131, 2519–2527. [Google Scholar] [CrossRef] [PubMed]
- Malich, G.; Markovic, B.; Winder, C. The sensitivity and specificity of the MTS tetrazolium assay for detecting the in vitro cytotoxicity of 20 chemicals using human cell lines. Toxicology 1997, 124, 179–192. [Google Scholar] [CrossRef]
- Yadav, S.S.; Prasad, C.B.; Prasad, S.B.; Pandey, L.K.; Singh, S.; Pradhan, S.; Narayan, G. Anti-tumor activity of staurosporine in the tumor microenvironment of cervical cancer: An in vitro study. Life Sci. 2015, 133, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Skrzypski, M. Quantitative reverse transcriptase real-time polymerase chain reaction (qRT-PCR) in translational oncology: Lung cancer perspective. Lung Cancer 2008, 59, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Paul-Samojedny, M.; Kokocińska, D.; Samojedny, A.; Mazurek, U.; Partyka, R.; Lorenz, Z.; Wilczok, T. Expression of cell survival/death genes: Bcl-2 and Bax at the rate of colon cancer prognosis. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1741, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Akl, H.; Vervloessem, T.; Kiviluoto, S.; Bittremieux, M.; Parys, J.B.; De Smedt, H.; Bultynck, G. A dual role for the anti-apoptotic Bcl-2 protein in cancer: Mitochondria versus endoplasmic reticulum. Biochim. Biophys. Acta Mol. Cell Res. 2014, 1843, 2240–2252. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.; Qian, Y.; Tang, J.; Liang, Z.; Zhang, M.; Si, J.; Li, X.; Huang, W.; Xu, R.; Wang, K. Ca2+/calmodulin-dependent protein kinase IIγ, a critical mediator of the NF-κB network, is a novel therapeutic target in non-small cell lung cancer. Cancer Lett. 2014, 344, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.G.; Cullen, S.P.; Sheridan, C.; Lüthi, A.U.; Gerner, C.; Martin, S.J. Executioner caspase-3 and caspase-7 are functionally distinct proteases. Proc. Natl. Acad. Sci. USA 2008, 105, 12815–12819. [Google Scholar] [CrossRef] [PubMed]
- Wolbers, F.; Buijtenhuijs, P.; Haanen, C.; Vermes, I. Apoptotic cell death kinetics in vitro depend on the cell types and the inducers used. Apoptosis 2004, 9, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Van Noorden, C.J.F. Editorial: The history of Z-VAD-FMK, a tool for understanding the significance of caspase inhibition. Acta Histochem. 2001, 103, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wang, H.; Wang, Z.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; Wang, X. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Lebbe, E.K. M.; Peigneur, S.; Maiti, M.; Mille, B.G.; Devi, P.; Ravichandran, S.; Lescrinier, E.; Waelkens, E.; D’Souza, L.; Herdewijn, P.; et al. Discovery of a new subclass of α-conotoxins in the venom of Conus australis. Toxicon 2014, 91, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Tang, S.; Pi, C.; Liu, J.; Wang, F.; Wang, L.; Zhou, W.; Xu, A. Discovery of a novel class of conotoxin from Conus litteratus, lt14a, with a unique cysteine pattern. Peptides 2006, 27, 2174–2181. [Google Scholar] [CrossRef] [PubMed]
- Lam, D.C.-L.; Girard, L.; Ramirez, R.; Chau, W.-S.; Suen, W.; Sheridan, S.; Tin, V.P.C.; Chung, L.; Wong, M.P.; Shay, J.W.; et al. Expression of nicotinic acetylcholine receptor subunit genes in non-small-cell lung cancer reveals differences between smokers and nonsmokers. Cancer Res. 2007, 67, 4638–4647. [Google Scholar] [CrossRef] [PubMed]
- Tsurutani, J.; Castillo, S.S.; Brognard, J.; Granville, C.A.; Zhang, C.; Gills, J.J.; Sayyah, J.; Dennis, P.A. Tobacco components stimulate Akt-dependent proliferation and NFkB-dependent survival in lung cancer cells. Carcinogenesis 2005, 26, 1182–1195. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, M.G.; Codignola, A.; Vicentini, L.M.; Clementi, F.; Sher, E. Nicotine stimulates a serotonergic autocrine loop in human small-cell lung carcinoma. Cancer Res. 1993, 53, 5566–5568. [Google Scholar] [PubMed]
- Ho, Y.S.; Lee, C.H.; Wu, C.H. The alpha 9-nicotinic acetylcholine receptor serves as a molecular target for breast cancer therapy. J. Exp. Clin. Med. 2011, 3, 246–251. [Google Scholar] [CrossRef]
- Mei, D.; Lin, Z.; Fu, J.; He, B.; Gao, W.; Ma, L.; Dai, W.; Zhang, H.; Wang, X.; Wang, J.; et al. The use of α-conotoxin ImI to actualize the targeted delivery of paclitaxel micelles to α7 nAChR-overexpressing breast cancer. Biomaterials 2015, 42, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Grando, S.A. Basic and clinical aspects of non-neuronal acetylcholine: Biological and clinical significance of non-canonical ligands of epithelial nicotinic acetylcholine receptors. J. Pharmacol. Sci. 2008, 106, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Warren, G.; Singh, A. Nicotine and lung cancer. J. Carcinog. 2013, 12, 1. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, A.; Nastrucci, C.; Cesario, A.; Russo, P. Nicotine: Specific role in angiogenesis, proliferation and apoptosis. Crit. Rev. Toxicol. 2012, 42, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Nakada, T.; Kiyotani, K.; Iwano, S.; Uno, T.; Yokohira, M.; Yamakawa, K.; Fujieda, M.; Saito, T.; Yamazaki, H.; Imaida, K.; et al. Lung tumorigenesis promoted by anti-apoptotic effects of cotinine, a nicotine metabolite through activation of PI3K/Akt pathway. J. Toxicol. Sci. 2012, 37, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Improgo, M.R. D.; Scofield, M.D.; Tapper, A.R.; Gardner, P.D. The nicotinic acetylcholine receptor CHRNA5/A3/B4 gene cluster: Dual role in nicotine addiction and lung cancer. Prog. Neurobiol. 2010, 92, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-S.; Peng, Y.-J.; Wei, P.-L.; Lee, C.-H.; Su, H.-Y.; Ho, Y.-S.; Lin, S.-Y.; Wu, C.-H.; Chang, Y.-J. The alpha9 nicotinic acetylcholine receptor is the key mediator in nicotine-enhanced cancer metastasis in breast cancer cells. J. Exp. Clin. Med. 2011, 3, 283–292. [Google Scholar] [CrossRef]
- Khan, K.H.; Blanco-Codesido, M.; Molife, L.R. Cancer therapeutics: Targeting the apoptotic pathway. Crit. Rev. Oncol. Hematol. 2014, 90, 200–219. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Chonghaile, T.N.; Letai, A. Mimicking the BH3 domain to kill cancer cells. Oncogene 2008, 27, S149–S157. [Google Scholar] [CrossRef] [PubMed]
- Pellecchia, M.; Reed, J.C. Inhibition of anti-apoptotic Bcl-2 family proteins by natural polyphenols: New avenues for cancer chemoprevention and chemotherapy. Curr. Pharm. Des. 2004, 10, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Blando, J.; Perez, C.J.; Wang, H.; Benavides, F.J.; Kazanietz, M.G. Activation of nuclear factor B (NF-kB) in prostate cancer is mediated by protein kinase C (PKC ). J. Biol. Chem. 2012, 287, 37570–37582. [Google Scholar] [CrossRef] [PubMed]
- Magné, N.; Toillon, R.A.; Bottero, V.; Didelot, C.; Houtte, P. Van; Gérard, J.P.; Peyron, J.F. NF-κB modulation and ionizing radiation: Mechanisms and future directions for cancer treatment. Cancer Lett. 2006, 231, 158–168. [Google Scholar]
- Maeng, H.J.; Lee, W.J.; Jin, Q.R.; Chang, J.E.; Shim, W.S. Upregulation of COX-2 in the lung cancer promotes overexpression of multidrug resistance protein 4 (MRP4) via PGE2-dependent pathway. Eur. J. Pharm. Sci. 2014, 62, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Gehlot, P. Inflammation and cancer: How friendly is the relationship for cancer patients? Curr. Opin. Pharmacol. 2009, 9, 351–369. [Google Scholar] [CrossRef] [PubMed]
- Korbakis, D.; Scorilas, A. Treatment of gastric cancer cells with 5-fluorouracil/leucovorin and irinotecan induces distinct alterations in the mRNA expression of the apoptosis-related genes, including the novel gene BCL2L12. Tumor Biol. 2009, 30, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Almeida, L.O.; Abrahao, A.C.; Rosselli-Murai, L.K.; Giudice, F.S.; Zagni, C.; Leopoldino, A.M.; Squarize, C.H.; Castilho, R.M. NFκB mediates cisplatin resistance through histone modifications in head and neck squamous cell carcinoma (HNSCC). FEBS Open Biol. 2014, 4, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sethi, G. Targeting transcription factor NF-κB to overcome chemoresistance and radioresistance in cancer therapy. Biochim. Biophys. Acta Rev. Cancer 2010, 1805, 167–180. [Google Scholar] [CrossRef] [PubMed]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Pillai, G.R.; Srivastava, A.S.; Hassanein, T.I.; Chauhan, D.P.; Carrier, E. Induction of apoptosis in human lung cancer cells by curcumin. Cancer Lett. 2004, 208, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Earnshaw, W.C.; Martins, L.M.; Kaufmann, S.H. Mammalian caspases: Structure, activation, substrates, and functions during apoptosis. Annu. Rev. Biochem. 1999, 68, 383–424. [Google Scholar] [CrossRef] [PubMed]
- Deveraux, Q.L.; Roy, N.; Stennicke, H.R.; Van Arsdale, T.; Zhou, Q.; Srinivasula, S.M.; Alnemri, E.S.; Salvesen, G.S.; Reed, J.C. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J. 1998, 17, 2215–2223. [Google Scholar] [CrossRef] [PubMed]
- Shioiri, T.; Muroi, M.; Hatao, F.; Nishida, M.; Ogawa, T.; Mimura, Y.; Seto, Y.; Kaminishi, M.; Tanamoto, K.-I. Caspase-3 is activated and rapidly released from human umbilical vein endothelial cells in response to lipopolysaccharide. Biochim. Biophys. Acta 2009, 1792, 1011–1018. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R. Apoptotic pathways: Ten minutes to dead. Cell 2005, 121, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, E.; Sahin, A.A; Chen, J.S.; Krishnamurthy, R.R.; Aggarwal, N.; Brun, A.-M.; Sapino, A.; Zhang, F.; Sharma, D.; Yang, X.-H.; et al. Down-regulation of caspase 3 in breast cancer: A possible mechanism for chemoresistance. Oncogene 2002, 21, 8843–8851. [Google Scholar] [CrossRef] [PubMed]
- Staal, J.; Bekaert, T.; Beyaert, R. Regulation of NF-κB signaling by caspases and MALT1 paracaspase. Cell Res. 2011, 21, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Inoue, T.; Anai, S.; Onishi, S.; Miyake, M.; Tanaka, N.; Hirayama, A.; Fujimoto, K.; Hirao, Y. Inhibition of COX-2 expression by topical diclofenac enhanced radiation sensitivity via enhancement of TRAIL in human prostate adenocarcinoma xenograft model. BMC Urol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oroz-Parra, I.; Navarro, M.; Cervantes-Luevano, K.E.; Álvarez-Delgado, C.; Salvesen, G.; Sanchez-Campos, L.N.; Licea-Navarro, A.F. Apoptosis Activation in Human Lung Cancer Cell Lines by a Novel Synthetic Peptide Derived from Conus californicus Venom. Toxins 2016, 8, 38. https://doi.org/10.3390/toxins8020038
Oroz-Parra I, Navarro M, Cervantes-Luevano KE, Álvarez-Delgado C, Salvesen G, Sanchez-Campos LN, Licea-Navarro AF. Apoptosis Activation in Human Lung Cancer Cell Lines by a Novel Synthetic Peptide Derived from Conus californicus Venom. Toxins. 2016; 8(2):38. https://doi.org/10.3390/toxins8020038
Chicago/Turabian StyleOroz-Parra, Irasema, Mario Navarro, Karla E. Cervantes-Luevano, Carolina Álvarez-Delgado, Guy Salvesen, Liliana N. Sanchez-Campos, and Alexei F. Licea-Navarro. 2016. "Apoptosis Activation in Human Lung Cancer Cell Lines by a Novel Synthetic Peptide Derived from Conus californicus Venom" Toxins 8, no. 2: 38. https://doi.org/10.3390/toxins8020038