
Pertussis Toxin Exploits Host Cell Signaling Pathways Induced by Meningitis-Causing E. coli K1-RS218 and Enhances Adherence of Monocytic THP-1 Cells to Human Cerebral Endothelial Cells

Abstract
:1. Introduction
2. Results
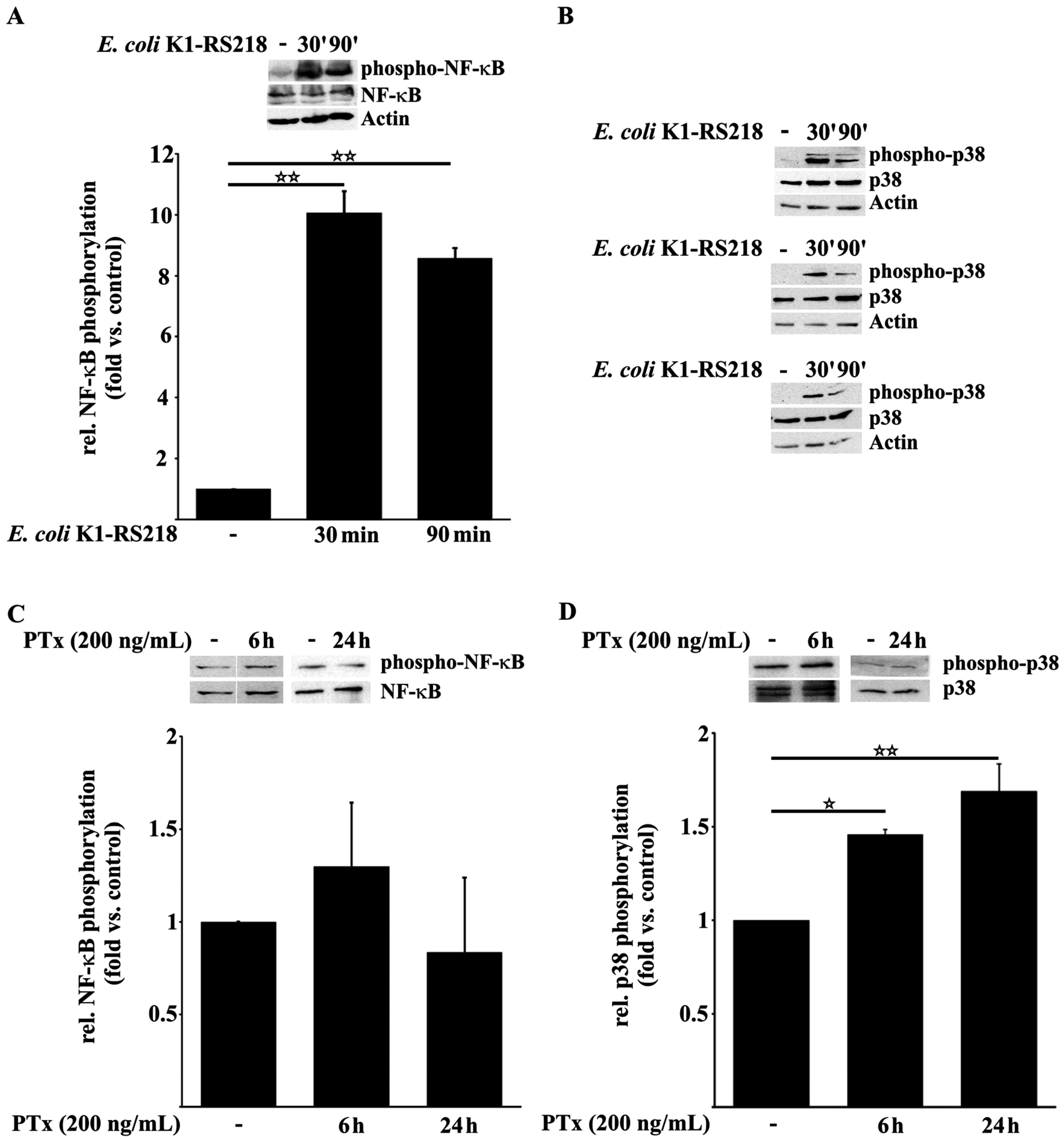
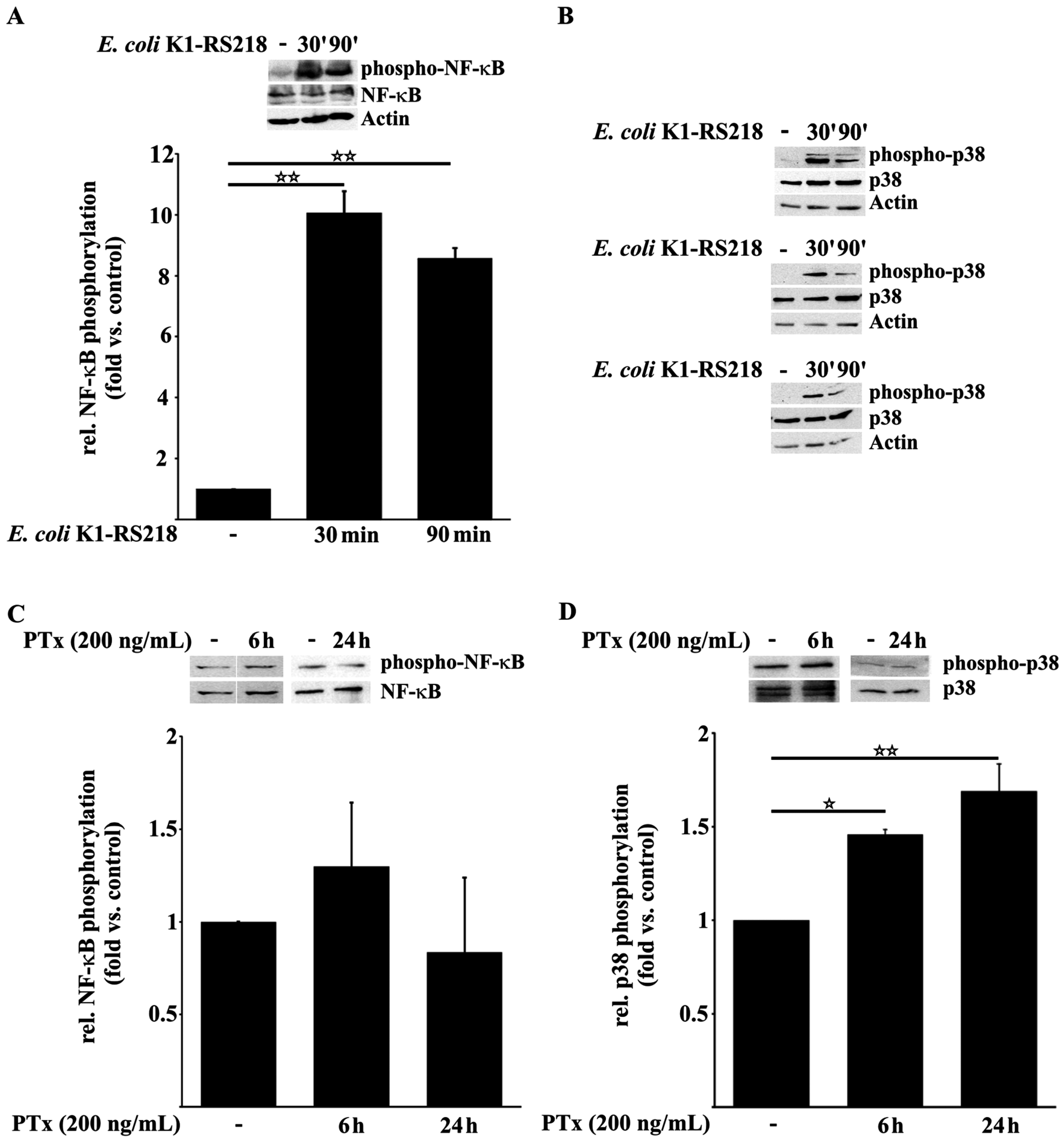
2.1. PTx Enhances p38 but Not NF-κB Phosphorylation
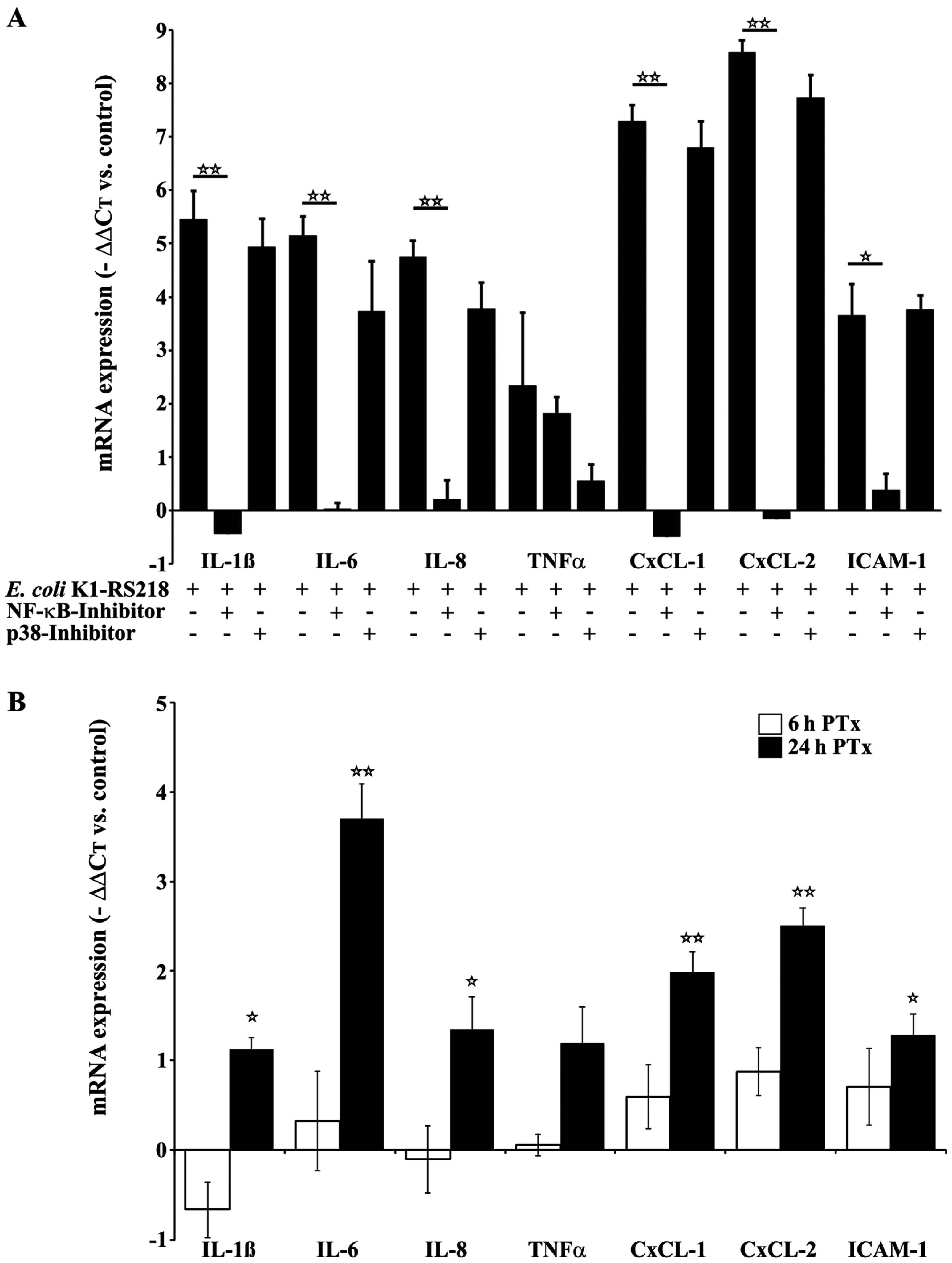
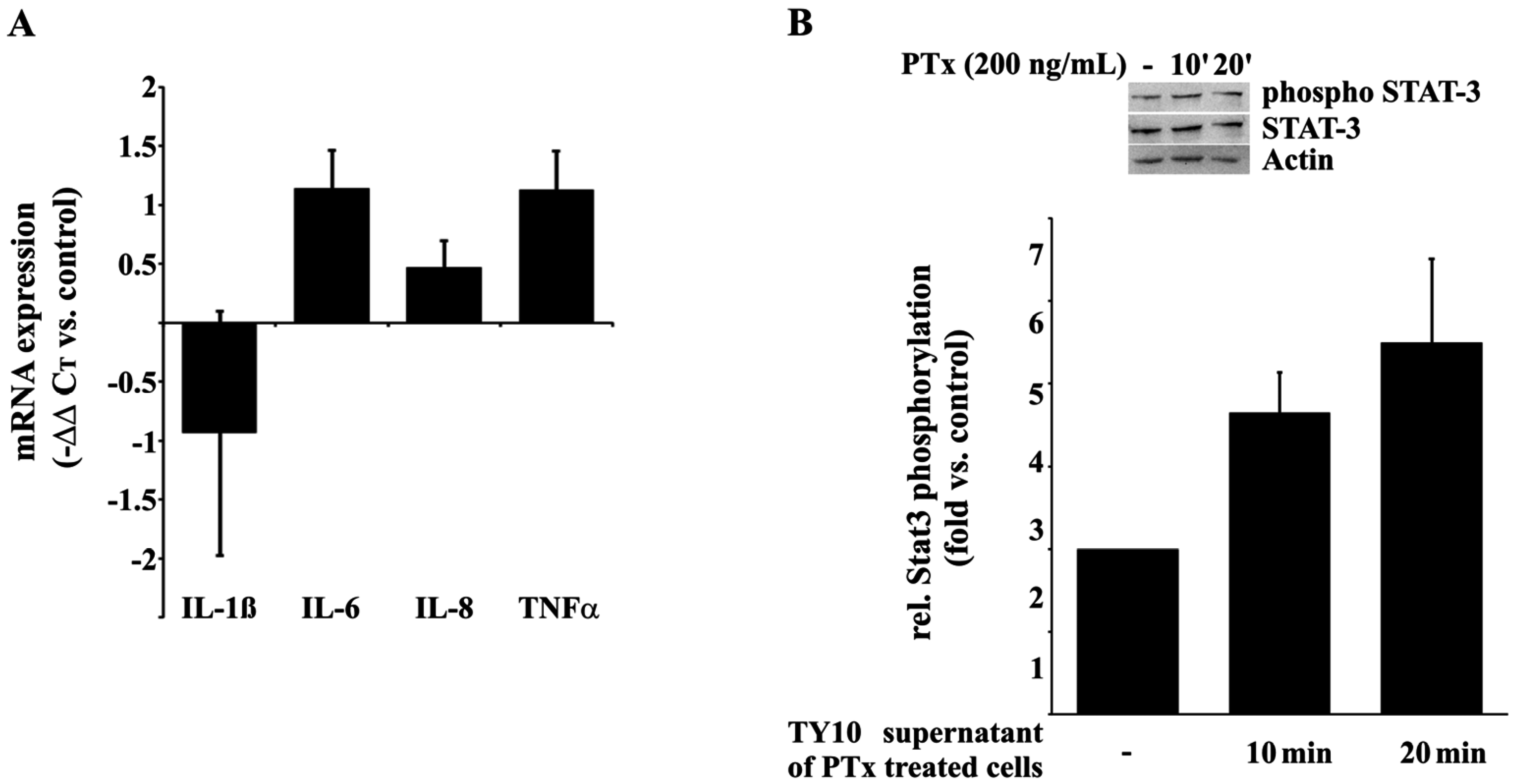
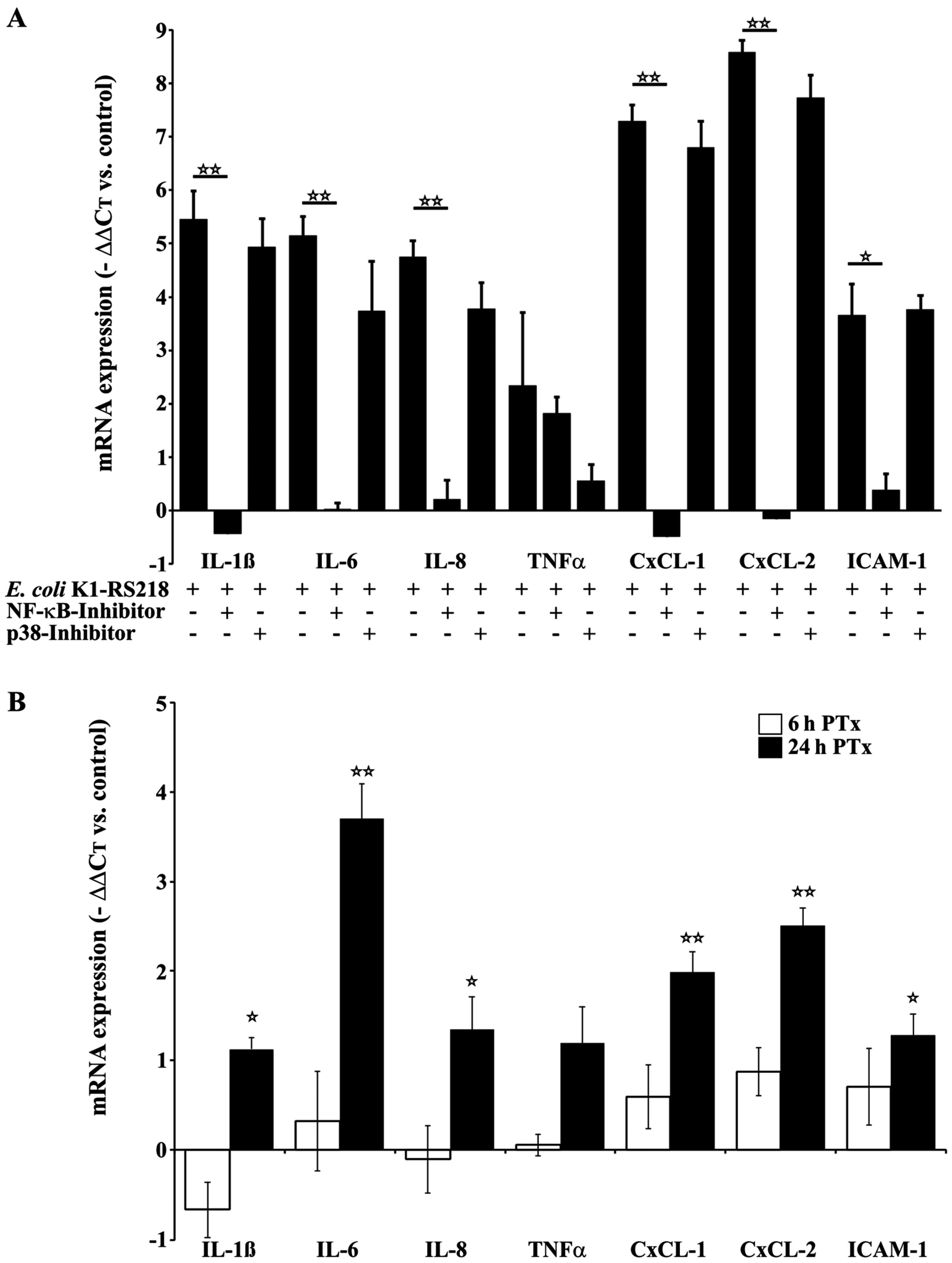
2.2. PTx and E. coli K1-RS218 Enhance mRNA Expression of IL-1β, IL-6, IL-8, (TNFα), CxCL-1, CxCL-2 and ICAM-1
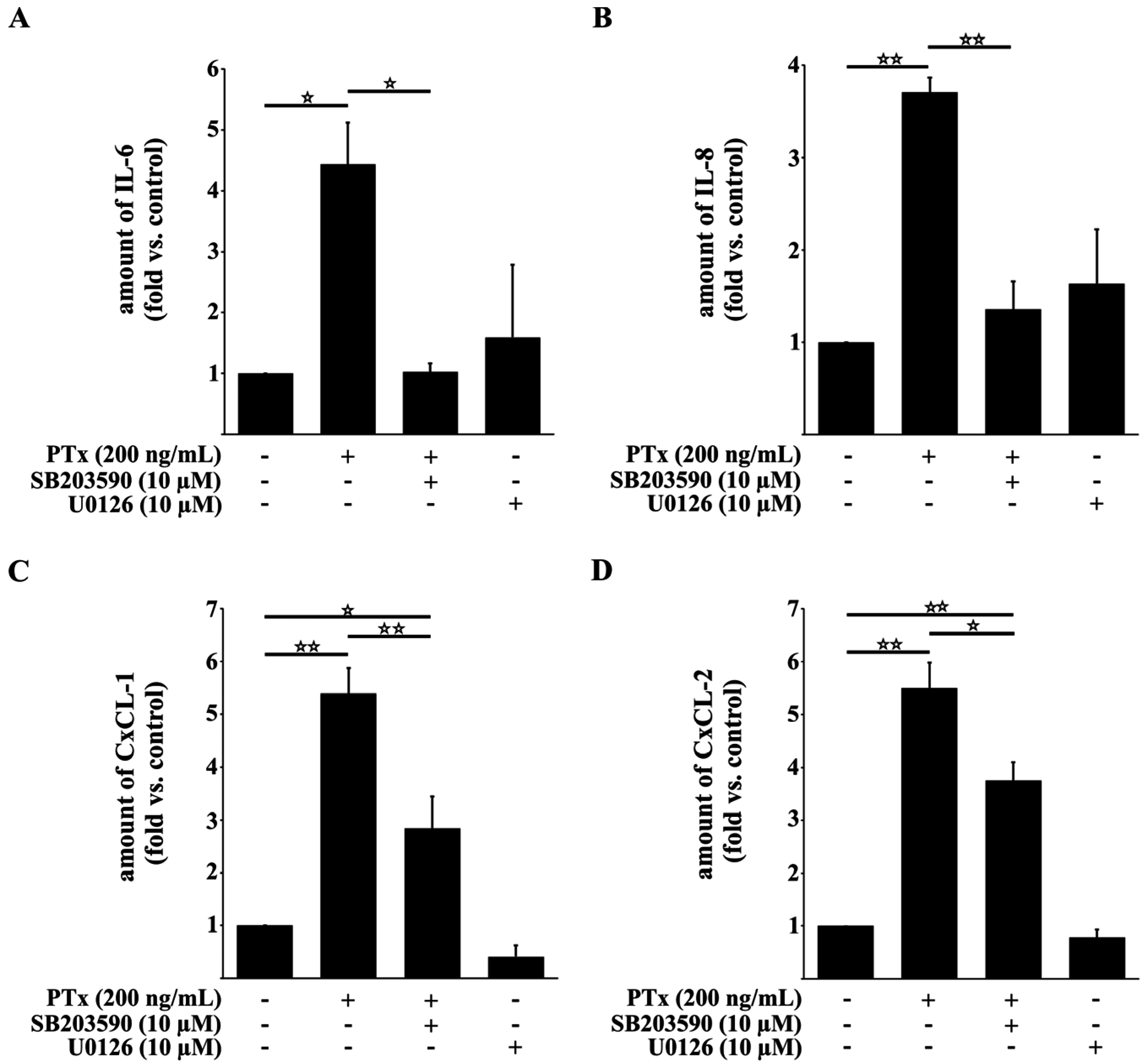
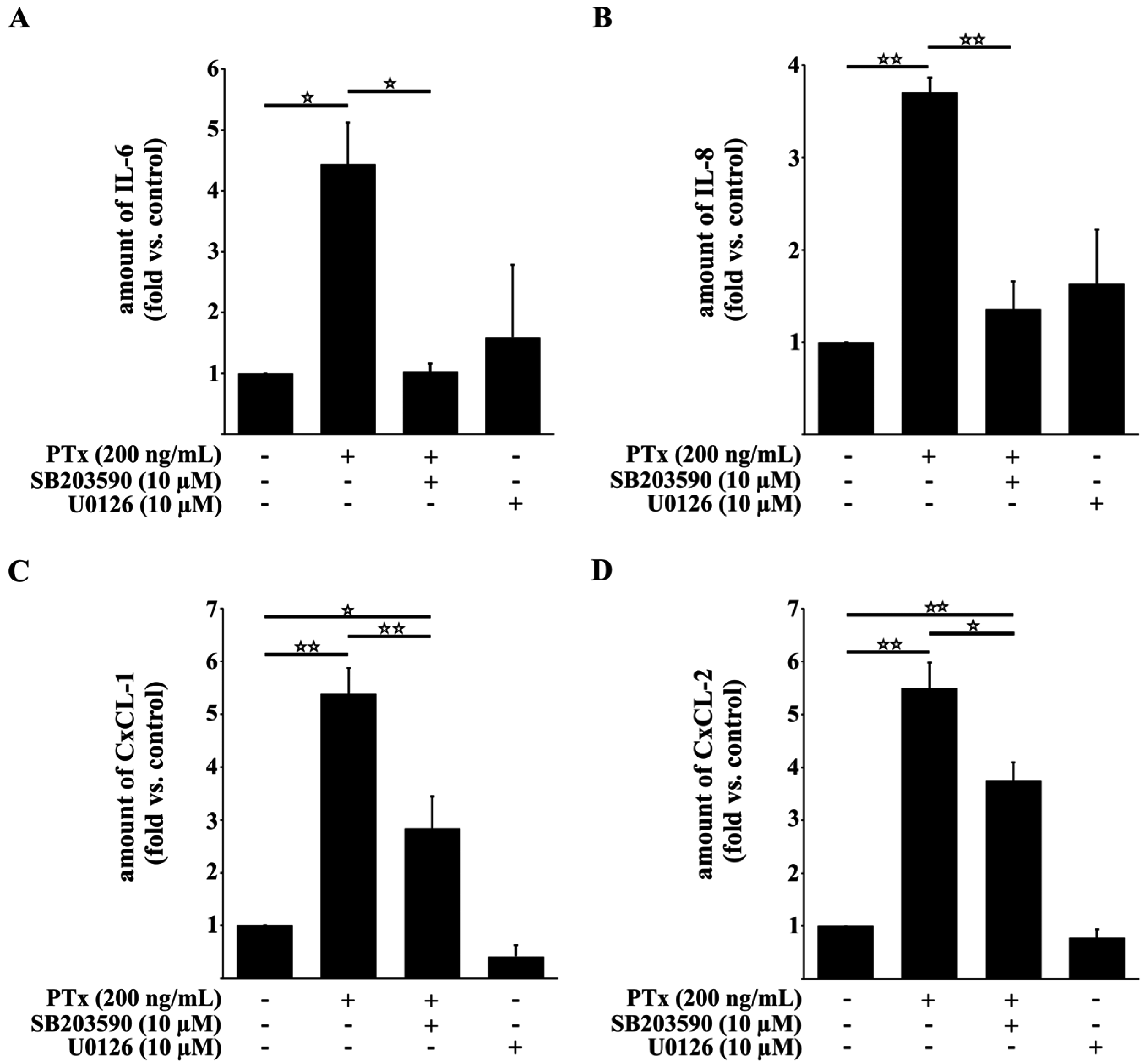
2.3. PTx Increases IL-6, IL-8, CxCL-1, CxCL-2 and ICAM-1 Protein Levels
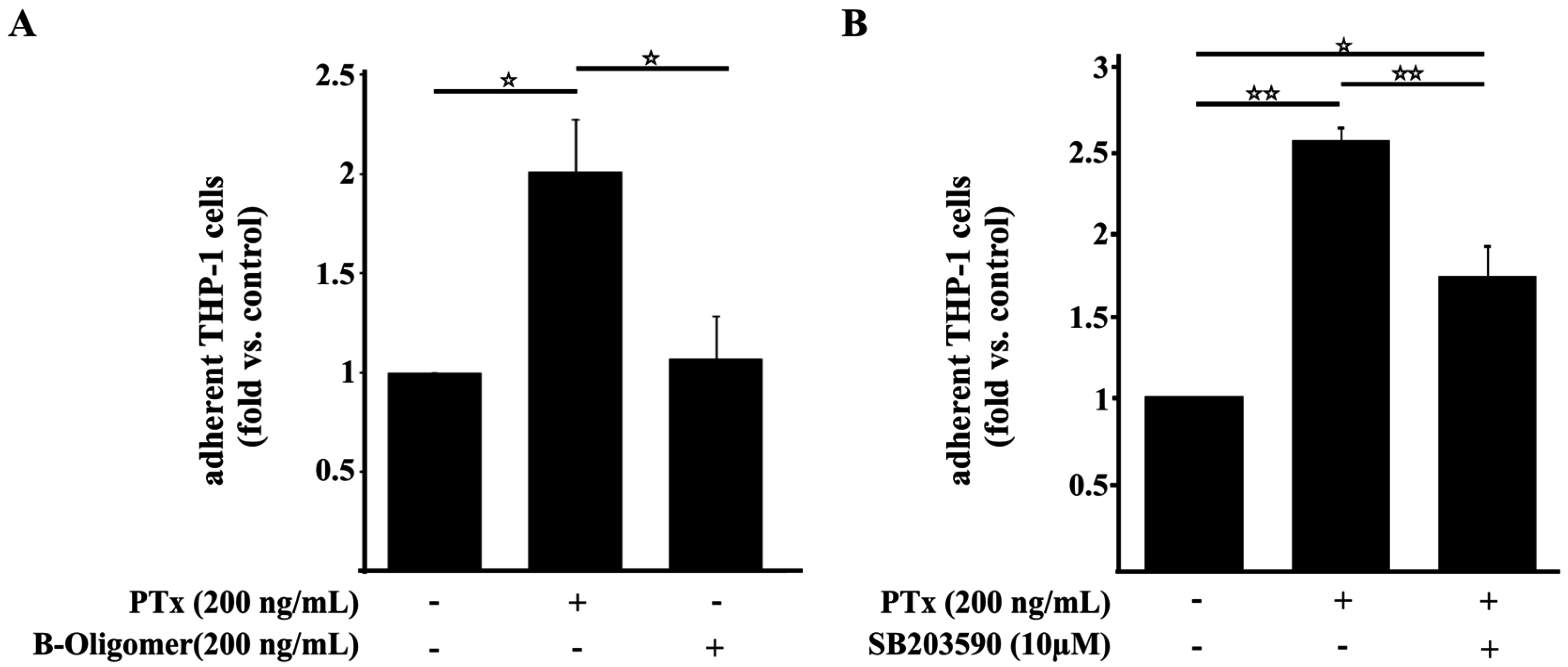
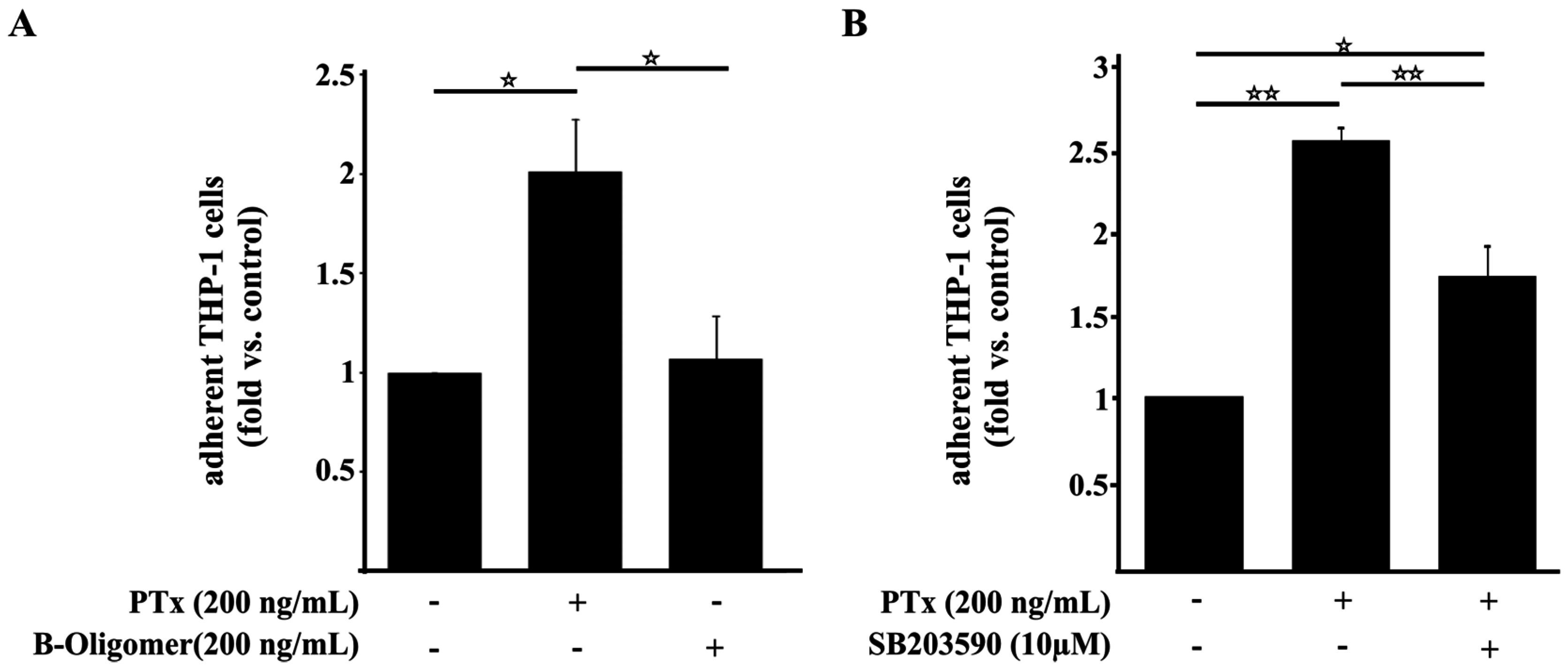
2.4. PTx Enhances the Adherence of Monocytic THP-1 Cells to Cerebral Endothelial TY10 Cells Involving the Activation of the p38 Pathway
2.5. PTx-Treated TY10 Cells Induce the Expression of Pro-Inflammatory Cytokines and the Activation of STAT3 in THP-1 Cells
3. Discussion
4. Materials and Methods
4.1. Chemicals, Antibodies and Bacterial Strains
4.2. Cell Culture
4.3. Western Blotting
4.4. qRT-PCR
4.5. Enzyme-Linked Immunosorbent Assay (ELISA)
4.6. Adherence Assay
4.7. Activation of THP-1 Cells
4.8. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| BBB | Blood-brain barrier |
| CNS | central nervous system |
| ELISA | enzyme-linked immunosorbent assay |
| Erk1/2 | extracellular signal-regulated kinase |
| FBS | fetal bovine serum |
| HBMEC | human brain-derived microvascular endothelial cell |
| HPRT | hypoxanthine phosphoribosyltransferase |
| ICAM-1 | intercellular adhesion molecule 1 |
| IL | interleukin |
| JNK | c-jun N-terminal kinase |
| PBS | phosphate-buffered saline |
| PMA | phorbol 12-myristate 13-acetate |
| PTx | pertussis toxin |
| MAPK | mitogen-activated protein kinase |
| NF-κB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NMEC | neonatal menigitis-causing E. coli |
| qRT-PCR | quantitative real time polymerase chain reaction |
| RIPA | radioimmunoprecipitation |
| SDS-PAGE | sodium dodecyl sulfate polyacrylamide gel electrophoresis |
| STAT3 | signal transducer and activator of transcription 3 |
| TNFα | tumor necrosis factor α |
References
- Pittman, M. Pertussis toxin: The cause of the harmful effects and prolonged immunity of whooping cough. A hypothesis. Rev. Infect. Dis. 1979, 1, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Hewlett, E.L.; Burns, D.L.; Cotter, P.A.; Harvill, E.T.; Merkel, T.J.; Quinn, C.P.; Stibitz, S.E. Pertussis pathogenesis—What we know and what we don’t know. J. Infect. Dis. 2014, 209, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Melvin, J.A.; Scheller, E.V.; Miller, J.F.; Cotter, P.A. Bordetella pertussis pathogenesis: Current and future challenges. Nat. Rev. Microbiol. 2014, 12, 274–288. [Google Scholar] [CrossRef] [PubMed]
- Kilgore, P.E.; Salim, A.M.; Zervos, M.J.; Schmitt, H.J. Pertussis: Microbiology, disease, treatment, and prevention. Clin. Microbiol. Rev. 2016, 29, 449–486. [Google Scholar] [CrossRef] [PubMed]
- Carbonetti, N.H. Bordetella pertussis: New concepts in pathogenesis and treatment. Curr. Opin. Infect. Dis. 2016, 29, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.W.; Wagner, E.K.; Laber, J.R.; Goodfield, L.L.; Smallridge, W.E.; Harvill, E.T.; Papin, J.F.; Wolf, R.F.; Padlan, E.A.; Bristol, A.; et al. A cocktail of humanized anti-pertussis toxin antibodies limits disease in murine and baboon models of whooping cough. Sci. Transl. Med. 2015, 7. [Google Scholar] [CrossRef] [PubMed]
- Vittucci, A.C.; Vennarucci, V.S.; Grandin, A.; Russo, C.; Lancella, L.; Tozzi, A.E.; Bartuli, A.; Villani, A. Pertussis in infants: An underestimated disease. BMC Infect. Dis. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Warfel, J.M.; Zimmermann, L.I.; Merkel, T.J. Acellular pertussis vaccines protect against disease but fail to prevent infection and transmission in a nonhuman primate model. Proc. Natl. Acad. Sci. USA 2014, 111, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Althouse, B.M.; Scarpino, S.V. Asymptomatic transmission and the resurgence of Bordetella pertussis. BMC Med. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Sealey, K.L.; Belcher, T.; Preston, A. Bordetella pertussis epidemiology and evolution in the light of pertussis resurgence. Infect. Genet. Evol. 2016, 40, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Geier, D.A.; Geier, M.R. An evaluation of serious neurological disorders following immunization: A comparison of whole-cell pertussis and acellular pertussis vaccines. Brain Dev. 2004, 26, 296–300. [Google Scholar] [CrossRef]
- Grant, C.C.; McKay, E.J.; Simpson, A.; Buckley, D. Pertussis encephalopathy with high cerebrospinal fluid antibody titers to pertussis toxin and filamentous hemagglutinin. Pediatrics 1998, 102, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Heininger, U.; Kleemann, W.J.; Cherry, J.D. Sudden infant death syndrome study group. A controlled study of the relationship between Bordetella pertussis infections and sudden unexpected deaths among German infants. Pediatrics 2004, 114, e9–e15. [Google Scholar] [CrossRef] [PubMed]
- Brückener, K.E.; el Baya, A.; Galla, H.J.; Schmidt, M.A. Permeabilization in a cerebral endothelial barrier model by pertussis toxin involves the PKC effector pathway and is abolished by elevated levels of cAMP. J. Cell Sci. 2003, 116, 1837–1846. [Google Scholar] [CrossRef] [PubMed]
- Seidel, G.; Böcker, K.; Schulte, J.; Wewer, C.; Greune, L.; Humberg, V.; Schmidt, M.A. Pertussis toxin permeabilization enhances the traversal of Escherichia coli K1, macrophages, and monocytes in a cerebral endothelial barrier model in vitro. Int. J. Med. Microbiol. 2011, 301, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Kügler, S.; Böcker, K.; Heusipp, G.; Greune, L.; Kim, K.S.; Schmidt, M.A. Pertussis toxin transiently affects barrier integrity, organelle organization, and transmigration of monocytes in a human brain microvascular endothelial cell barrier model. Cell. Microbiol. 2007, 9, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Karassek, S.; Starost, L.; Solbach, J.; Greune, L.; Sano, Y.; Kanda, T.; Kim, K.S.; Schmidt, M.A. Pertussis toxin exploits specific host cell signaling pathways for promoting invasion and translocation of E. coli K1-RS218 in human brain-derived microvascular endothelial cells. J. Biol. Chem. 2015, 41, 2835–2843. [Google Scholar]
- Rubin, K.; Glazer, S. The potential role of subclinical Bordetella pertussis colonization in the etiology of multiple sclerosis. Immunobiology 2016, 221, 512–515. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Nogimori, K.; Murai, S.; Yajima, M.; Ito, K.; Katada, T.; Ui, M.; Ishii, S. Subunit structure of islet-activating protein, pertussis toxin, in conformity with the A-B model. Biochemistry 1982, 21, 5516–5522. [Google Scholar] [CrossRef] [PubMed]
- Locht, C.; Coutte, L.; Mielcarek, N. The ins and outs of pertussis toxin. FEBS J. 2011, 278, 4668–4682. [Google Scholar] [CrossRef] [PubMed]
- Tamura, M.; Nogimori, K.; Yajima, M.; Ase, K.; Ui, M. A role of the B-oligomer moiety of islet-activating protein, pertussis toxin, in development of the biological effects on intact cells. J. Biol. Chem. 1983, 258, 6756–6761. [Google Scholar] [PubMed]
- Katada, T.; Ui, M. ADP ribosylation of the specific membrane protein of C6 cells by islet-activating protein associated with modification of adenylate cyclase activity. J. Biol. Chem. 1982, 257, 7210–7216. [Google Scholar] [PubMed]
- El Bayâ, A.; Linnemann, R.; von Olleschik-Elbheim, L.; Schmidt, M.A. Endocytosis and retrograde transport of pertussis toxin to the Golgi complex as a prerequisite for cellular intoxication. Eur. J. Cell Biol. 1997, 73, 40–48. [Google Scholar] [PubMed]
- El Bayâ, A.; Brückener, K.; Schmidt, M.A. Nonrestricted differential intoxication of cells by pertussis toxin. Infect. Immun. 1999, 67, 433–435. [Google Scholar] [PubMed]
- Robbins, J.B.; McCracken, G.H., Jr.; Gotschlich, E.C.; Ørskov, F.; Ørskov, I.; Hanson, L.A. Escherichia coli K1 capsular polysaccharide associated with neonatal meningitis. N. Engl. J. Med. 1974, 290, 1216–1220. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S. Human meningitis-associated Escherichia coli. EcoSal Plus 2016. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S. Strategy of Escherichia coli for crossing the blood-brain barrier. J. Infect. Dis. 2002, 186 (Suppl. 2), S220–S224. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.S. Mechanisms of microbial traversal of the blood-brain barrier. Nat. Rev. Microbiol. 2008, 6, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Dando, S.J.; Mackay-Sim, A.; Norton, R.; Currie, B.J.; St John, J.A.; Ekberg, J.A.; Batzloff, M.; Ulett, G.C.; Beacham, I.R. Pathogens penetrating the central nervous system: Infection pathways and the cellular and molecular mechanisms of invasion. Clin. Microbiol. Rev. 2014, 27, 691–726. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.A.; Shin, S.; Chung, J.W.; Kim, K.J.; Elliott, S.; Wang, Y.; Kim, K.S. Outer membrane protein A and cytotoxic necrotizing factor-1 use diverse signaling mechanisms for Escherichia coli K1 invasion of human brain microvascular endothelial cells. Microb. Pathog. 2003, 35, 35–42. [Google Scholar] [CrossRef]
- Khan, N.A.; Kim, Y.; Shin, S.; Kim, K.S. FimH-mediated Escherichia coli K1 invasion of human brain microvascular endothelial cells. Cell. Microbiol. 2007, 9, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, G.; Sehr, P.; Wilm, M.; Selzer, J.; Mann, M.; Aktories, K. Gln 63 of Rho is deamidated by Escherichia coli cytotoxic necrotizing factor-1. Nature 1997, 387, 725–729. [Google Scholar] [PubMed]
- Maruvada, R.; Kim, K.S. IbeA and OmpA of Escherichia coli K1 exploit Rac1 activation for invasion of human brain microvascular endothelial cells. Infect. Immun. 2012, 80, 2035–2041. [Google Scholar] [CrossRef] [PubMed]
- Stins, M.F.; Badger, J.; Kim, K.S. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb. Pathog. 2001, 30, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.K.; Selvaraj, S.K.; Prasadarao, N.V. Inhibition of apoptosis by Escherichia coli K1 is accompanied by increased expression of BclXL and blockade of mitochondrial cytochrome c release in macrophages. Infect. Immun. 2004, 72, 6012–6022. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.K.; Shimada, H.; Prasadarao, N.V. Entry and intracellular replication of Escherichia coli K1 in macrophages require expression of outer membrane protein A. Infect. Immun. 2003, 71, 5951–5961. [Google Scholar] [CrossRef] [PubMed]
- Rot, A.; Hub, E.; Middleton, J.; Pons, F.; Rabeck, C.; Thierer, K.; Wintle, J.; Wolff, B.; Zsak, M.; Dukor, P. Some aspects of IL-8 pathophysiology. III: Chemokine interaction with endothelial cells. J. Leukoc. Biol. 1996, 59, 39–44. [Google Scholar] [PubMed]
- Middleton, J.; Patterson, A.M.; Gardner, L.; Schmutz, C.; Ashton, B.A. Leukocyte extravasation: Chemokine transport and presentation by the endothelium. Blood 2002, 100, 3853–3860. [Google Scholar] [CrossRef] [PubMed]
- Zer, C.; Sachs, G.; Shin, J.M. Identification of genomic targets downstream of p38 mitogen-activated protein kinase pathway mediating tumor necrosis factor-alpha signaling. Physiol. Genom. 2007, 31, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Rossomando, A.J.; Payne, D.M.; Weber, M.J.; Sturgill, T.W. Evidence that pp42, a major tyrosine kinase target protein, is a mitogen-activated serine/threonine protein kinase. Proc. Natl. Acad. Sci. USA 1989, 86, 6940–6943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Shimizu, F.; Abe, M.; Maeda, T.; Kashiwamura, Y.; Ohtsuki, S.; Terasaki, T.; Obinata, M.; Kajiwara, K.; Fujii, M.; et al. Establishment of a new conditionally immortalized human brain microvascular endothelial cell line retaining an in vivo blood-brain barrier function. J. Cell Physiol. 2010, 225, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Sano, Y.; Kashiwamura, Y.; Abe, M.; Dieu, L.H.; Huwyler, J.; Shimizu, F.; Haruki, H.; Maeda, T.; Saito, K.; Tasaki, A.; et al. Stable human brain microvascular endothelial cell line retaining its barrier-specific nature independent of the passage number. Clin. Exp. Neuroimmunol. 2013, 4, 92–103. [Google Scholar] [CrossRef]
- Rouault, C.; Pellegrinelli, V.; Schilch, R.; Cotillard, A.; Poitou, C.; Tordjman, J.; Sell, H.; Clément, K.; Lacasa, D. Roles of chemokine ligand-2 (CXCL2) and neutrophils in influencing endothelial cell function and inflammation of human adipose tissue. Endocrinology 2013, 154, 1069–1079. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Chesnutt, B.C.; Smith, D.F.; Raffler, N.A.; Smith, M.L.; White, E.J.; Ley, K. Induction of LFA-1-dependent neutrophil rolling on ICAM-1 by engagement of E-selectin. Microcirculation 2006, 13, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Lang, R. Tuning of macrophage responses by Stat3-inducing cytokines: Molecular mechanisms and consequences in infection. Immunobiology 2005, 210, 63–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Peng, L.; Gai, Z.; Zhang, L.; Jong, A.; Cao, H.; Huang, S.-H. Pathogenic triad in bacterial meningitis: Pathogen invasion, NF-kB activation, and leucocyte transmigration that occur at the blood-brain barrier. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Krachler, A.M.; Woolery, A.R.; Orth, K. Manipulation of kinase signaling by bacterial pathogens. J. Cell Biol. 2011, 195, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Burke, S.J.; Lu, D.; Sparer, T.E.; Masi, T.; Goff, M.R.; Karlstad, M.D.; Collier, J.J. NF-kappaB and STAT1 control CXCL1 and CXCL2 gene transcription. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E131–E149. [Google Scholar] [CrossRef] [PubMed]
- Williams, L.M.; Sarma, U.; Willets, K.; Smallie, T.; Brennan, F.; Foxwell, B.M. Expression of constitutively active STAT3 can replicate the cytokine-suppressive activity of interleukin-10 in human primary macrophages. J. Biol. Chem. 2007, 282, 6965–6975. [Google Scholar] [CrossRef] [PubMed]
- Matsukawa, A.; Kudo, S.; Maeda, T.; Numata, K.; Watanabe, H.; Takeda, K.; Akira, S.; Ito, T. Stat3 in resident macrophages as a repressor protein of inflammatory response. J. Immunol. 2005, 175, 3354–3359. [Google Scholar] [CrossRef] [PubMed]
- Ronchi, F.; Basso, C.; Preite, S.; Reboldi, A.; Baumjohann, D.; Perlini, L.; Lanzavecchia, A.; Sallusto, F. Experimental priming of encephalitogenic Th1/Th17 cells requires pertussis-toxin-driven IL-1β production by myeloid cells. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Cuenda, A.; Rouse, J.; Doza, Y.N.; Meier, R.; Cohen, P.; Gallagher, T.F.; Young, P.R.; Lee, J.C. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 1995, 364, 229–233. [Google Scholar] [PubMed]
- Pierce, J.W.; Schoenleber, R.; Jesmok, G.; Best, J.; Moore, S.A.; Collins, T.; Gerritsen, M.E. Novel inhibitors of cytokine-induced IkBα phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J. Biol. Chem. 1997, 272, 21096–21103. [Google Scholar] [CrossRef] [PubMed]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef] [PubMed]
- Silver, R.P.; Aaronson, W.; Sutton, A.; Schneerson, R. Comparative analysis of plasmids and some metabolic characteristics of Escherichia coli K1 from diseased and healthy individuals. Infect. Immun. 1980, 29, 200–206. [Google Scholar] [PubMed]
- Tsuchiya, S.; Yamabe, M.; Yamaguchi, Y.; Kobayashi, Y.; Konno, T.; Tada, K. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int. J. Cancer 1980, 26, 171–176. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | F: Forward R: Reverse | Sequence (5′ -> 3′) |
|---|---|---|
| HPRT | F | GCCAGACTTTGTTGGATTTG |
| HPRT | R | CTCATCTTAGGCTTTGTATTTTG |
| IL-1β | F | TACCTGTCCTGCGTGTTGAA |
| IL-1β | R | TCTTTGGGTAATTTTTGGGATCT |
| IL-6 | F | GTCCTTCGGGCTCCTTGT |
| IL-6 | R | CAGCACAGCAGAGACAGGAC |
| IL-8 | F | GAGCACTCCATAAGGCACAAA |
| IL-8 | R | ATGGTTCCTTCCGGTGGT |
| TNFα | F | GTCCAGGCTTGTCCTGCTAC |
| TNFα | R | AGTCCTGAGGCCTGTGTTTG |
| CxCL-1 | F | GCTGAACAGTGACAAATCCAAC |
| CxCL-1 | R | CTTCAGGAACAGCCACCAGT |
| CxCL-2 | F | CCCATGGTTAAGAAAATCATCG |
| CxCL-2 | R | CTTCAGGAACAGCCACCAAT |
| ICAM-1 | F | CCTTCCTCACCGTGTACTGG |
| ICAM-1 | R | AGCGTAGGGTAAGGTTCTTGC |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Starost, L.J.; Karassek, S.; Sano, Y.; Kanda, T.; Kim, K.S.; Dobrindt, U.; Rüter, C.; Schmidt, M.A. Pertussis Toxin Exploits Host Cell Signaling Pathways Induced by Meningitis-Causing E. coli K1-RS218 and Enhances Adherence of Monocytic THP-1 Cells to Human Cerebral Endothelial Cells. Toxins 2016, 8, 291. https://doi.org/10.3390/toxins8100291
Starost LJ, Karassek S, Sano Y, Kanda T, Kim KS, Dobrindt U, Rüter C, Schmidt MA. Pertussis Toxin Exploits Host Cell Signaling Pathways Induced by Meningitis-Causing E. coli K1-RS218 and Enhances Adherence of Monocytic THP-1 Cells to Human Cerebral Endothelial Cells. Toxins. 2016; 8(10):291. https://doi.org/10.3390/toxins8100291
Chicago/Turabian StyleStarost, Laura Julia, Sascha Karassek, Yasuteru Sano, Takashi Kanda, Kwang Sik Kim, Ulrich Dobrindt, Christian Rüter, and Marcus Alexander Schmidt. 2016. "Pertussis Toxin Exploits Host Cell Signaling Pathways Induced by Meningitis-Causing E. coli K1-RS218 and Enhances Adherence of Monocytic THP-1 Cells to Human Cerebral Endothelial Cells" Toxins 8, no. 10: 291. https://doi.org/10.3390/toxins8100291