
Triggering of Erythrocyte Death by Triparanol

1
Department of Physiology, University of Tübingen, Gmelinstr. 5, 72076 Tuebingen, Germany
2
Department of Biochemistry, Biophysics and General Pathology, School of Medicine and Surgery, Second University of Naples, Via L. De Crecchio 7, 80138 Naples, Italy
*
Author to whom correspondence should be addressed.
Toxins 2015, 7(8), 3359-3371; https://doi.org/10.3390/toxins7083359
Submission received: 22 July 2015
/
Revised: 11 August 2015
/
Accepted: 12 August 2015
/
Published: 24 August 2015
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The cholesterol synthesis inhibitor Triparanol has been shown to trigger apoptosis in several malignancies. Similar to the apoptosis of nucleated cells, erythrocytes may enter eryptosis, the suicidal death characterized by cell shrinkage and cell membrane scrambling with phosphatidylserine translocation to the erythrocyte surface. Triggers of eryptosis include oxidative stress which may activate erythrocytic Ca2+ permeable unselective cation channels with subsequent Ca2+ entry and increase of cytosolic Ca2+ activity ([Ca2+]i). The present study explored whether and how Triparanol induces eryptosis. To this end, phosphatidylserine exposure at the cell surface was estimated from annexin-V-binding, cell volume from forward scatter, hemolysis from hemoglobin release, [Ca2+]i from Fluo3-fluorescence, and ROS formation from 2’,7’-dichlorodihydrofluorescein diacetate (DCFDA) dependent fluorescence. As a result, a 48 h exposure of human erythrocytes to Triparanol (20 µM) significantly increased DCFDA fluorescence and significantly increased Fluo3-fluorescence. Triparanol (15 µM) significantly increased the percentage of annexin-V-binding cells, and significantly decreased the forward scatter. The effect of Triparanol on annexin-V-binding was significantly blunted, but not abolished by removal of extracellular Ca2+. In conclusion, Triparanol leads to eryptosis, the suicidal erythrocyte death characterized by cell shrinkage and phospholipid scrambling of the erythrocyte cell membrane. Triparanol is at least in part effective by stimulating ROS formation and Ca2+ entry.
1. Introduction
Triparanol, a 3β-hydroxysterol-Δ24-reductase inhibitor and thus inhibitor of cholesterol synthesis [1,2,3,4], has been shown to inhibit proliferation and trigger apoptosis in several malignancies including leukemia, melanoma, chondrosarcoma, and cancer of lung, breast, liver, pancreas, or prostate [5,6,7]. Triparanol may be teratogenic [4,8] and may lead to myotonia, cataract and baldness [9].
Triparanol is partially effective by interference with the Hedgehog pathway [1,5,7,10,11,12]. Triparanol has further been shown to increase cytosolic Ca2+ activity ([Ca2+]i), an effect attributed to Ca2+ release from intracellular stores [9]. Moreover, Triparanol indirectly modifies activation of K+ channels [2] and decreases Na+/K+ ATPase activity [13].
Similar to apoptosis of nucleated cells, erythrocytes may enter eryptosis, the suicidal death of erythrocytes characterized by cell shrinkage [14] and phosphatidylserine translocation from the inner cell membrane leaflet to the cell surface [15]. As erythrocytes lack nuclei and mitochondria, eryptosis lacks several aspects of apoptosis, such as mitochondrial depolarization and altered gene expression [15]. However, similar to apoptotic cells, eryptotic erythrocytes are engulfed by phagocytosing cells and thus rapidly removed from circulating blood [15]. Triggers of eryptosis include oxidative stress, opening of oxidant sensitive cation channels, Ca2+ entry and increase of [Ca2+]i. Eryptosis is further triggered by heat stress [15], ceramide exposure [16], ATP depletion [15], and caspase activation [15,17,18]. Moreover, eryptosis is influenced by casein kinase 1α, Janus-activated kinase JAK3, protein kinase C, p38 kinase, PAK2 kinase, AMP activated kinase AMPK, cGMP-dependent protein kinase, and sorafenib/sunitinib sensitive kinases [15]. Eryptosis is triggered by a variety of xenobiotics [15,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43].
The present study explored whether Triparanol triggers eryptosis. To this end, human erythrocytes from healthy volunteers were treated with Triparanol and phosphatidylserine surface abundance, cell volume, [Ca2+]i and ROS formation determined by flow cytometry.
2. Results and Discussion
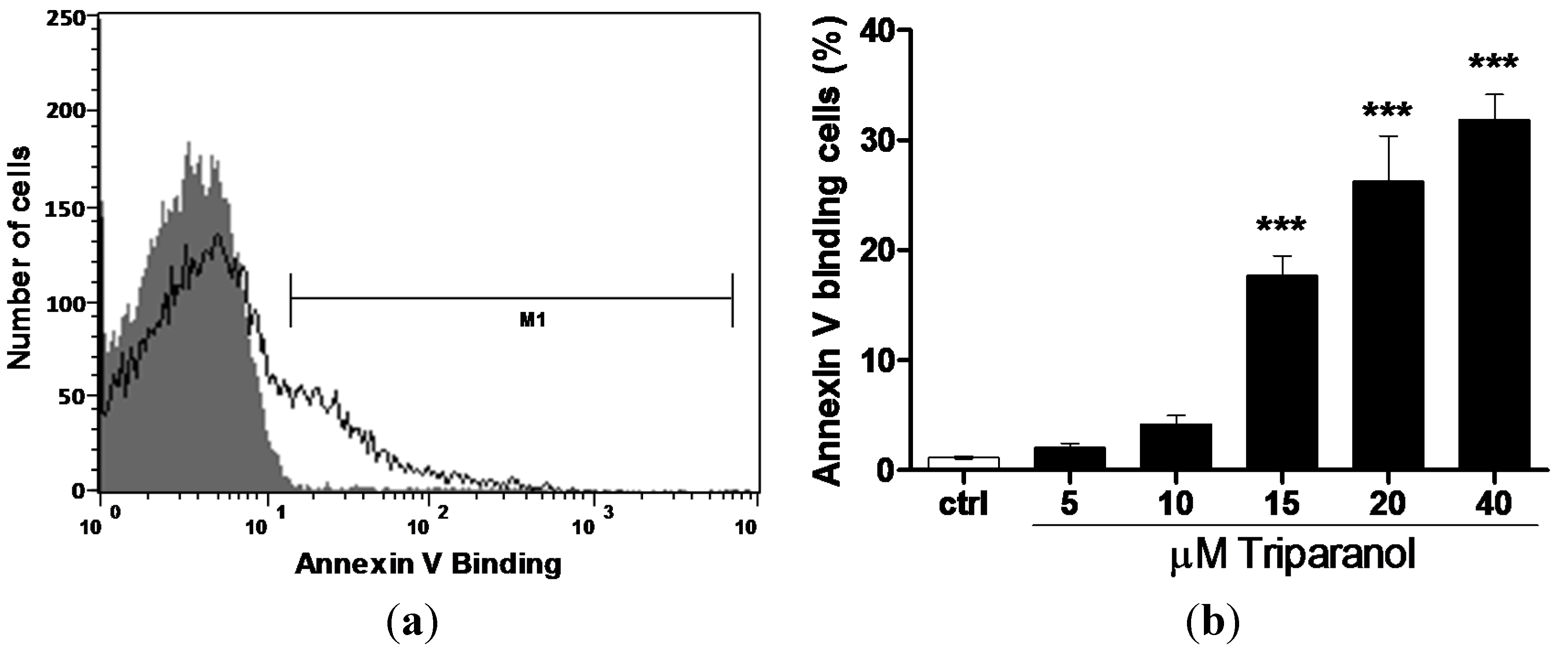
The present study addressed the hypothesis that Triparanol may stimulate eryptosis, the suicidal erythrocyte death characterized by cell shrinkage and cell membrane scrambling with phosphatidylserine translocation to the cell surface. In order to identify phosphatidylserine exposing erythrocytes, phosphatidylserine was quantified utilizing annexin-V-binding, as determined by flow cytometry (BD, Heidelberg, Germany).The erythrocytes were analyzed following incubation for 48 h in Ringer solution without or with Triparanol (5–40 µM). As illustrated in Figure 1, a 48 h exposure to Triparanol was followed by an increase of the percentage phosphatidylserine exposing erythrocytes, an effect reaching statistical significance at 15 µM Triparanol concentration.
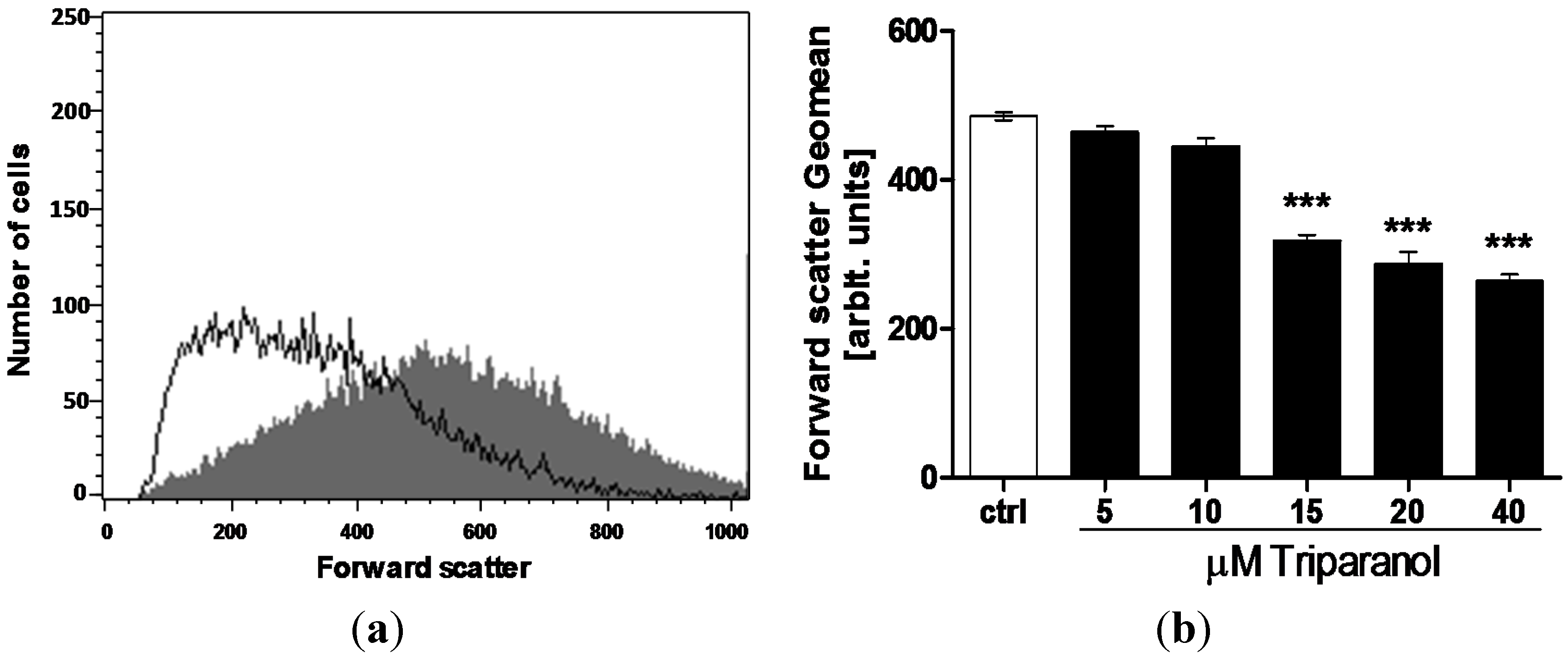
In order to quantify erythrocyte volume, forward scatter was determined utilizing flow cytometry. The measurements were again performed following a 48 h incubation in Ringer solution without or with Triparanol (5–40 µM). As shown in Figure 2, the treatment with Triparanol was followed by a decrease of erythrocyte forward scatter, an effect reaching statistical significance at 15 µM Triparanol concentration.
In order to quantify hemolysis, the hemoglobin concentration in the supernatant was determined by photometry. As a result, following a 48 h incubation the percentage of hemolytic erythrocytes was significantly (p < 0.01) higher following exposure to 20 µM Triparanol (2.52 ± 0.30% n = 8) than in the absence of Triparanol (0.10 ± 0.02% n = 8). The percentage of hemolytic erythrocytes remained, however, one order of magnitude lower than the percentage of annexin-V-binding erythrocytes.
Figure 1.
Effect of Triparanol on phosphatidylserine exposure (a) Original histogram of annexin-V-binding of erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of 20 µM Triparanol; (b) Arithmetic means ± SEM (n = 4) of erythrocyte annexin-V-binding (black bars) following incubation for 48 h to Ringer solution without or with presence of Triparanol (5–40 µM). *** (p < 0.001) indicate significant difference from the absence of Triparanol (ANOVA).
Figure 1.
Effect of Triparanol on phosphatidylserine exposure (a) Original histogram of annexin-V-binding of erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of 20 µM Triparanol; (b) Arithmetic means ± SEM (n = 4) of erythrocyte annexin-V-binding (black bars) following incubation for 48 h to Ringer solution without or with presence of Triparanol (5–40 µM). *** (p < 0.001) indicate significant difference from the absence of Triparanol (ANOVA).

Figure 2.
Effect of Triparanol on erythrocyte forward scatter (a) Original histogram of forward scatter of erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of 20 µM Triparanol; (b) Arithmetic means ± SEM (n = 4) of the erythrocyte forward scatter (FSC) following incubation for 48 h to Ringer solution without (white bar) or with (black bars) Triparanol (5–40 µM). *** (p < 0.001) indicate significant difference from the absence of Triparanol (ANOVA).
Figure 2.
Effect of Triparanol on erythrocyte forward scatter (a) Original histogram of forward scatter of erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of 20 µM Triparanol; (b) Arithmetic means ± SEM (n = 4) of the erythrocyte forward scatter (FSC) following incubation for 48 h to Ringer solution without (white bar) or with (black bars) Triparanol (5–40 µM). *** (p < 0.001) indicate significant difference from the absence of Triparanol (ANOVA).

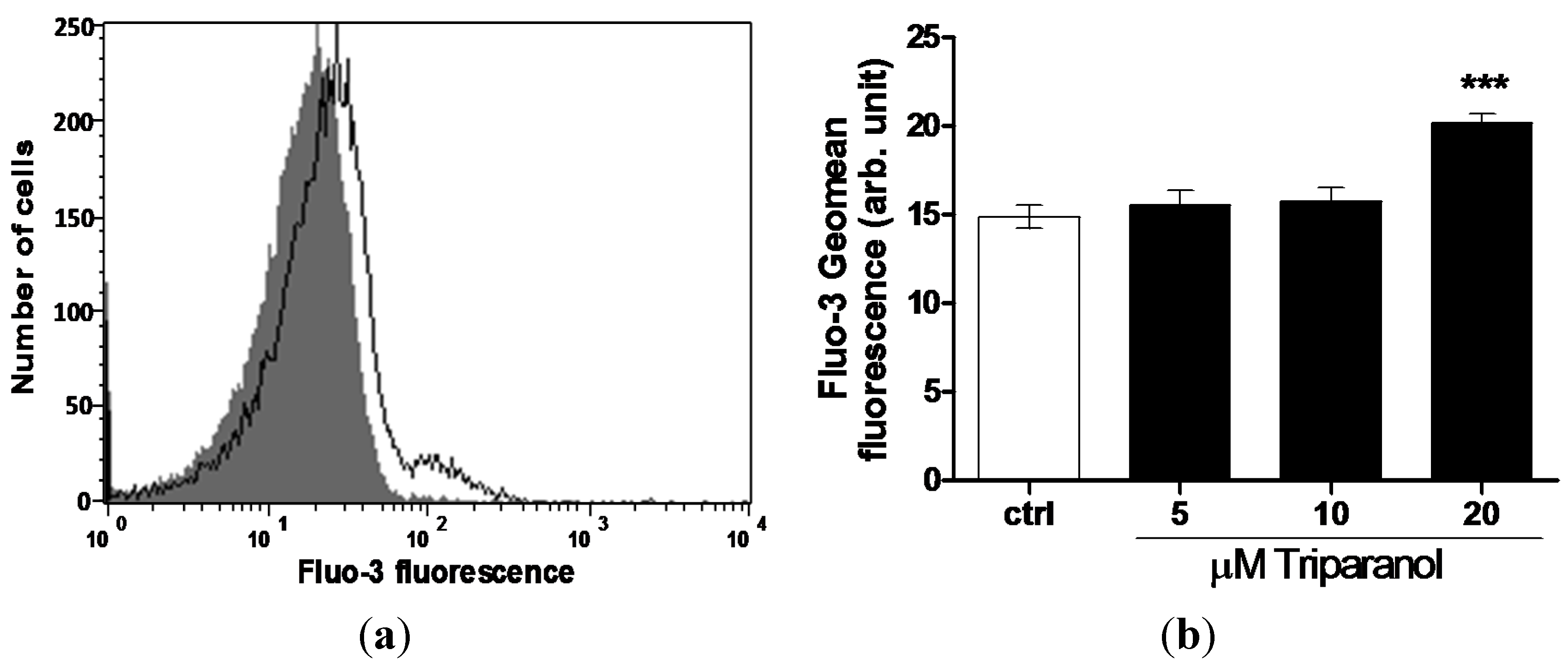
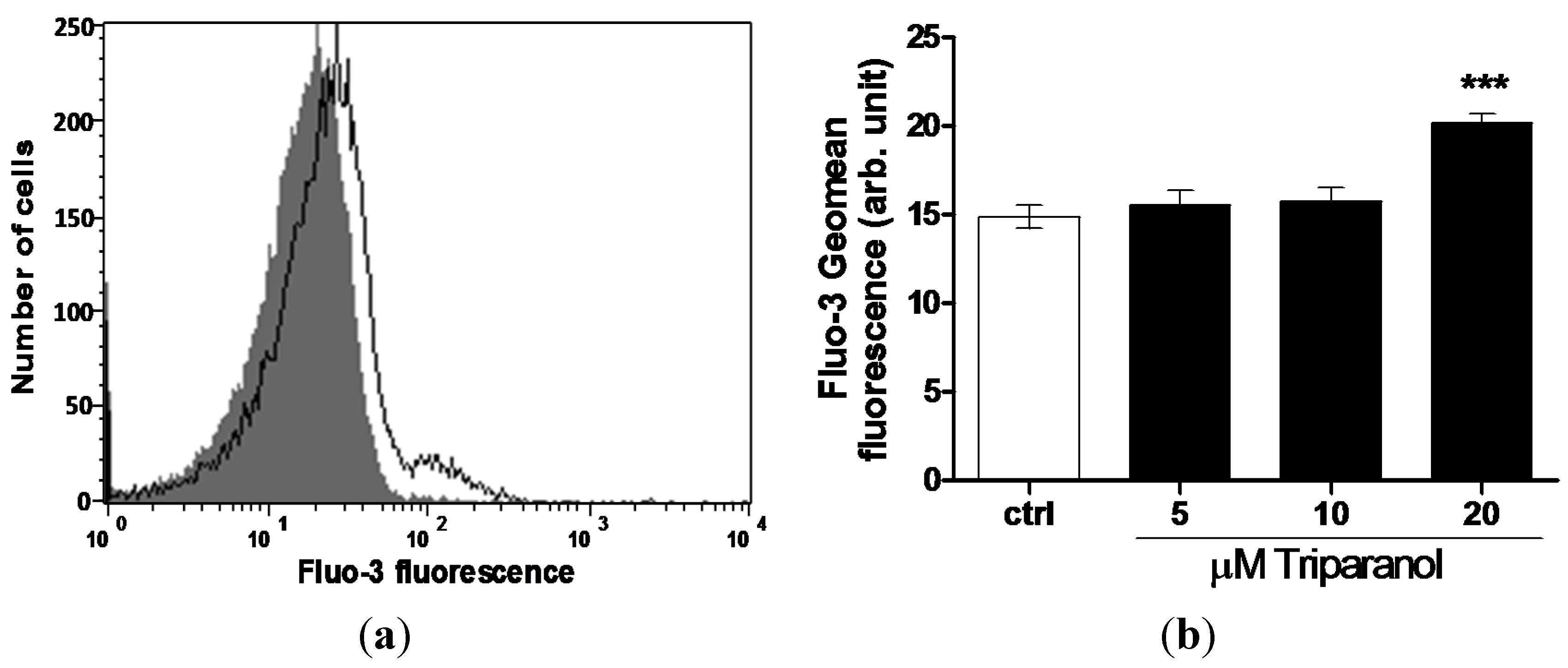
For measurement of cytosolic Ca2+ activity ([Ca2+]i), the erythrocytes were loaded with Fluo3 and Fluo3-fluorescence determined following a 48 h incubation in Ringer solution without or with Triparanol (5–20 µM). As shown in Figure 3, a 48 h exposure to Triparanol increased the Fluo3-fluorescence, an effect reaching statistical significance at 20 µM Triparanol.
Figure 3.
Effect of Triparanol on erythrocyte Ca2+ activity (a) Original histogram of Fluo3-fluorescence in erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of Triparanol (20 µM); (b) Arithmetic means ± SEM (n = 4) of the Fluo3-fluorescence (arbitrary units) in erythrocytes exposed for 48 h to Ringer solution without (white bar) or with (black bars) Triparanol (5–20 µM). *** (p < 0.001) indicate significant difference from the absence of Triparanol (ANOVA).
Figure 3.
Effect of Triparanol on erythrocyte Ca2+ activity (a) Original histogram of Fluo3-fluorescence in erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of Triparanol (20 µM); (b) Arithmetic means ± SEM (n = 4) of the Fluo3-fluorescence (arbitrary units) in erythrocytes exposed for 48 h to Ringer solution without (white bar) or with (black bars) Triparanol (5–20 µM). *** (p < 0.001) indicate significant difference from the absence of Triparanol (ANOVA).
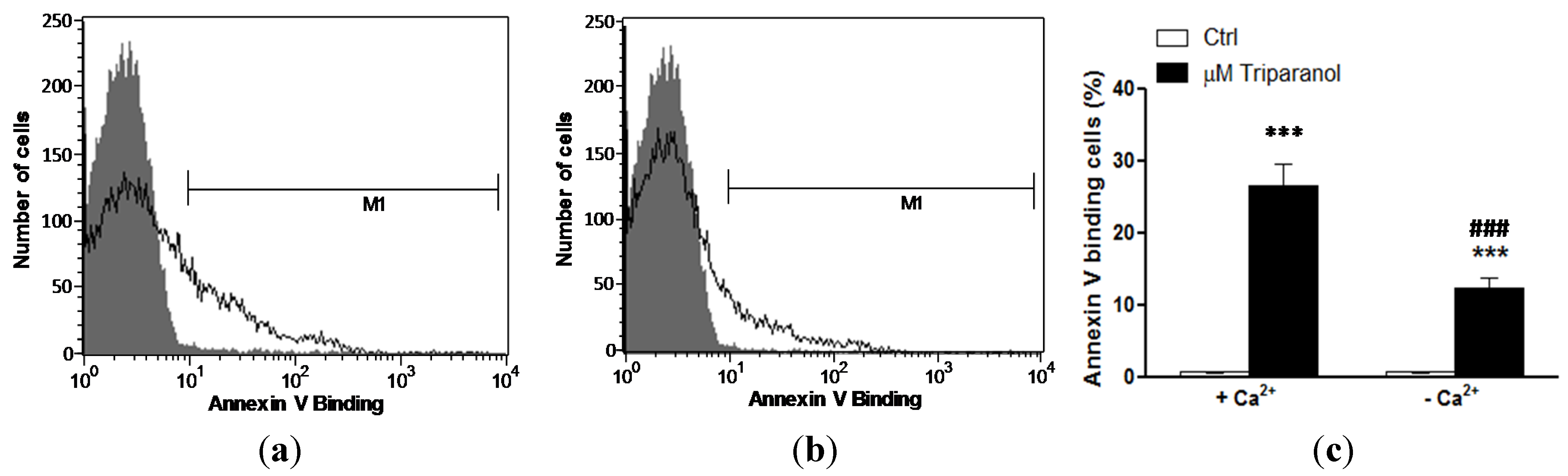
In order to test whether Triparanol-induced translocation of phosphatidylserine required entry of extracellular Ca2+, erythrocytes were incubated for 48 h in the absence or presence of 20 µM Triparanol in the presence or nominal absence of extracellular Ca2+. As displayed in Figure 4, removal of extracellular Ca2+ significantly blunted the effect of Triparanol on annexin-V-binding. However, Triparanol significantly increased the percentage of annexin-V-binding erythrocytes even in the absence of extracellular Ca2+. Accordingly, the effect of Triparanol on cell membrane scrambling was in large part but not fully due to entry of extracellular Ca2+.
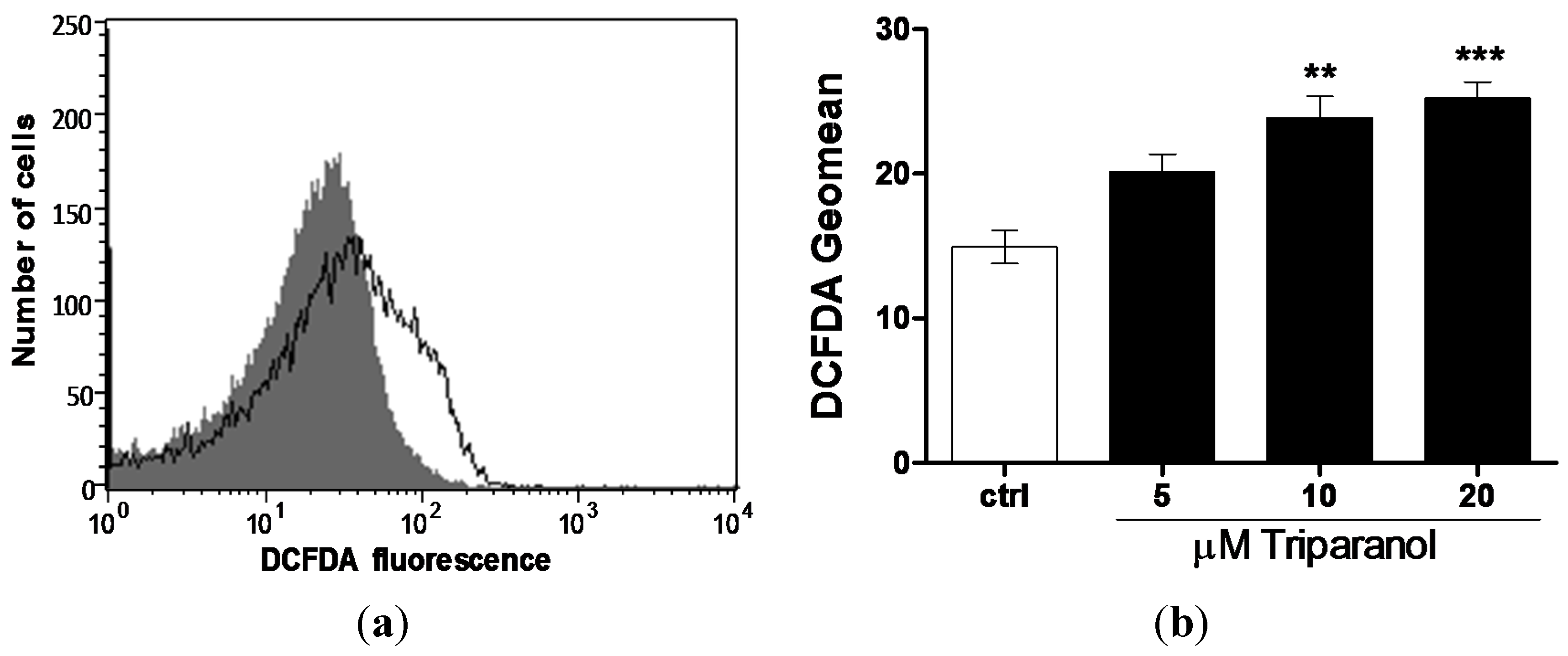
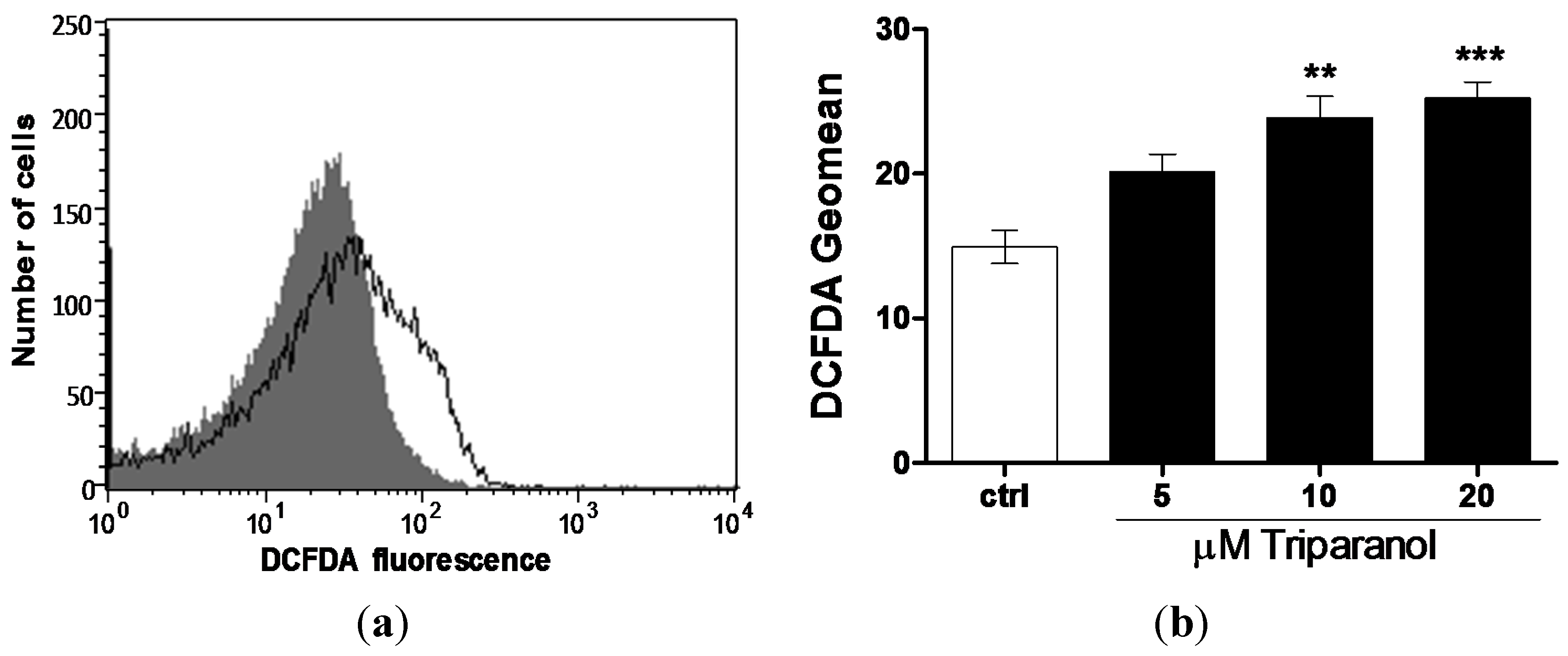
Since Ca2+ entry and subsequent eryptosis could have been triggered by oxidative stress, reactive oxygen species (ROS) was determined utilizing 2’,7’-dichlorodihydrofluorescein diacetate (DCFDA). As illustrated in Figure 5, a 48 h exposure to Triparanol increased the DCFDA fluorescence, an effect reaching statistical significance at 10 µM Triparanol.
The concentration of reduced glutathione (GSH) was significantly higher in untreated (223 ± 13 n = 4) than in Triparanol (20 µM) treated (96 ± 12, n = 4) erythrocytes.
To test whether Triparanol is similarly effective in other enucleated cells, blood platelets were exposed to Triparanol (20 µM) for 30 min. As a result, the percentage annexin-V-binding blood platelets was significantly higher in Triparanol (20 µM) treated (50 ± 13, n = 5) than in untreated (0.5 ± 0.1, n = 5) blood platelets.
The present study reveals a novel effect of Triparanol, i.e. the triggering of suicidal erythrocyte death or eryptosis. Triparanol treatment for 48 h was followed by cell shrinkage and cell membrane scrambling with phosphatidylserine translocation to the erythrocyte surface. The concentrations required for the stimulation of eryptosis were within the range expected following the administration of 200 mg in humans [44].
Figure 4.
Ca2+ sensitivity of Triparanol-induced phosphatidylserine exposure (a) Original histogram of annexin-V-binding of erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of Triparanol (20 µM) in the presence; and absence (b) of extracellular Ca2+; (c) Arithmetic means ± SEM (n = 4) of annexin-V-binding of erythrocytes after a 48 h treatment with Ringer solution without (white bars) or with (black bars) Triparanol (20 µM) in the presence (left bars, +Ca2+) and absence (right bars, −Ca2+) of Ca2+. *** (p < 0.001) indicates significant difference from the absence of Triparanol, ### (p < 0.001) indicate significant difference from the presence of Ca2+ (ANOVA).
Figure 4.
Ca2+ sensitivity of Triparanol-induced phosphatidylserine exposure (a) Original histogram of annexin-V-binding of erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of Triparanol (20 µM) in the presence; and absence (b) of extracellular Ca2+; (c) Arithmetic means ± SEM (n = 4) of annexin-V-binding of erythrocytes after a 48 h treatment with Ringer solution without (white bars) or with (black bars) Triparanol (20 µM) in the presence (left bars, +Ca2+) and absence (right bars, −Ca2+) of Ca2+. *** (p < 0.001) indicates significant difference from the absence of Triparanol, ### (p < 0.001) indicate significant difference from the presence of Ca2+ (ANOVA).
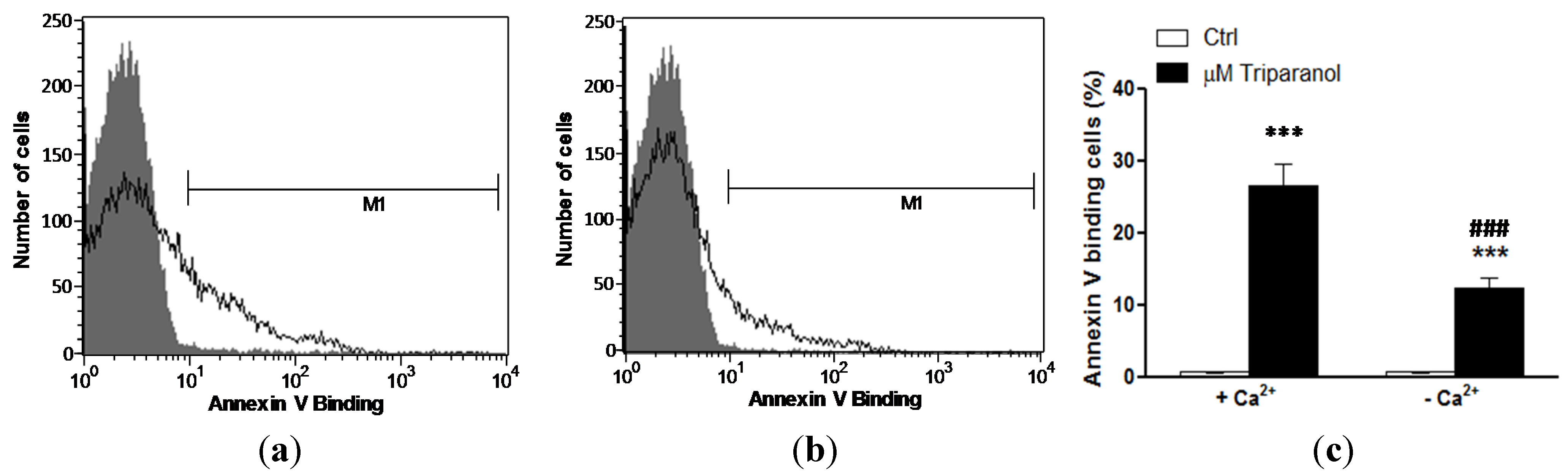
Figure 5.
Effect of Triparanol on erythrocyte ROS formation (a) Original histogram of 2’,7’-dichlorodihydrofluorescein diacetate (DCFDA) fluorescence in erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of Triparanol (20 µM); (b) Arithmetic means ± SEM (n = 4) of the DCFDA fluorescence (arbitrary units) in erythrocytes exposed for 48 h to Ringer solution without (white bar) or with (black bars) Triparanol (5–20 µM). ** (p < 0.01), *** (p < 0.001) indicate significant difference from the absence of Triparanol (ANOVA).
Figure 5.
Effect of Triparanol on erythrocyte ROS formation (a) Original histogram of 2’,7’-dichlorodihydrofluorescein diacetate (DCFDA) fluorescence in erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of Triparanol (20 µM); (b) Arithmetic means ± SEM (n = 4) of the DCFDA fluorescence (arbitrary units) in erythrocytes exposed for 48 h to Ringer solution without (white bar) or with (black bars) Triparanol (5–20 µM). ** (p < 0.01), *** (p < 0.001) indicate significant difference from the absence of Triparanol (ANOVA).
The present observations were made in erythrocytes drawn from healthy individuals. The rate of eryptosis may be higher in clinical conditions with enhanced susceptibility to triggers of eryptosis, such as dehydration [32], hyperphosphatemia [42] chronic kidney disease (CKD) [24,45,46,47], hemolyticuremic syndrome [48], diabetes [49], hepatic failure [50], malignancy [15], sepsis [51], sickle-cell disease [15], beta-thalassemia [15], Hb-C and G6PD-deficiency [15], or Wilsons disease [52]. In those erythrocytes, lower Triparanol concentrations may be required for the triggering of eryptosis.
As Triparanol was added to isolated erythrocytes in vitro, the effect on erythrocytes was presumably due to mechanisms other than inhibition of cholesterol synthesis. The effect of Triparanol on cell membrane scrambling and cell shrinkage was paralleled by an increase of cytosolic Ca2+ activity ([Ca2+]i). Moreover, the effect of Triparanol on cell membrane scrambling was significantly blunted in the absence of extracellular Ca2+. Thus, the effect was in large part due to Ca2+ entry. An increase of [Ca2+]i has previously been shown to trigger cell membrane scrambling by activating an ill-defined scramblase [15]. An increase of [Ca2+]i has further been shown to cause erythrocyte shrinkage by activation of Ca2+ sensitive K+ channels with subsequent K+ exit, cell membrane hyperpolarization, Cl− exit and thus cellular loss of KCl with water [14].
The stimulation of Ca2+ entry with subsequent increase of [Ca2+]i following Triparanol treatment was paralleled by and at least in part due to triggering of oxidative stress, which has previously been shown to activate Ca2+ permeable cation channels with subsequent stimulation of Ca2+ entry and increase of [Ca2+]i [15].
The physiological purpose of eryptosis is the clearance of defective erythrocytes from circulating blood prior to hemolysis [15]. Eryptosis thus serves to prevent release of hemoglobin, which would be filtered in renal glomerula, precipitate in the acidic lumen of renal tubules and thus occlude nephrons [53]. Eryptosis further accomplishes clearance of erythrocytes infected with the malaria pathogen Plasmodium. The pathogen imposes oxidative stress on the host erythrocyte thus leading to opening of Ca2+-permeable erythrocyte cation channels [15,54]. Sickle-cell trait, beta-thalassemia-trait, Hb-C and G6PD-deficiency accelerate eryptosis and subsequent clearance of infected erythrocytes, thus decreasing parasitemia and protecting against a severe course of malaria [15,55,56,57]. Accelerated eryptosis in iron deficiency [58], and following treatment with lead [58], chlorpromazine [59] or NO synthase inhibitors [59] similarly counteracts development of parasitemia. Possibly, Triparanol similarly enhances the susceptibility of plasmodium infected erythrocytes to eryptosis.
Stimulation of eryptosis may, however, lead to anemia, if the erythrocyte loss is not counterbalanced by an equivalent increase of erythropoiesis [15]. Moreover, phosphatdylserine exposing erythrocytes may adhere to the vascular wall [60], stimulate blood clotting and trigger thrombosis [61,62,63], thus impairing microcirculation [16,61,64,65,66,67].
3. Experimental Section
3.1. Erythrocytes, Platelets, Solutions and Chemicals
Fresh Li-Heparin-anticoagulated blood samples were kindly provided by the blood bank of the University of Tübingen and were drawn from healthy individuals. The study is approved by the ethics committee of the University of Tübingen (184/2003 V). The blood was centrifuged at 120 g for 20 min at 21 °C and the platelets and leukocytes-containing supernatant was disposed. Erythrocytes were incubated in vitro at a hematocrit of 0.4% in Ringer solution containing (in mM) 125 NaCl, 5 KCl, 1 MgSO4, 32 N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES; pH 7.4), 5 glucose, 1 CaCl2, at 37 °C for 24 h. Where indicated, erythrocytes were exposed to Triparanol (Sigma Aldrich, Hamburg, Germany) at the indicated concentrations.
In one series of experiments blood platelets were isolated from wild type mice. The mice were anesthetized and blood was drawn from the retroorbital plexus into tubes with 300 µL acid-citrate-dextrose buffer. Platelet rich plasma (PRP) was obtained by centrifugation at 260 g for 5 min. Afterwards PRP was centrifuged at 640 g for 5 min to pellet the platelets. After two washing steps the pellet of washed platelets was resuspended in modified Tyrode-HEPES buffer (pH 7.4, supplemented with 1 mM CaCl2 (Sigma Aldrich, Hamburg, Germany). All animal experiments were conducted according to the German law for the welfare of animals and were approved by local authorities.
3.2. Annexin-V-binding and Forward Scatter
After incubation under the respective experimental condition, 150 µL cell suspension was washed in Ringer solution containing 5 mM CaCl2 and then stained with Annexin-V-FITC (1:200 dilution; ImmunoTools, Friesoythe, Germany) in this solution at 37 °C for 20 min under protection from light. The annexin V abundance at the erythrocyte surface was subsequently determined on a FACS Calibur (BD, Heidelberg, Germany). A dot plot of forward scatter (FSC) vs. side scatter (SSC) was set to linear scale for both parameters. The threshold of forward scatter was set at the default value of “52”.
3.3. Hemolysis
For the determination of hemolysis, the samples were centrifuged (3 min at 1600 rpm, room temperature) after incubation under the respective experimental conditions and the supernatants were harvested. As a measure of hemolysis, the hemoglobin (Hb) concentration of the supernatant was determined photometrically at 405 nm. The absorption of the supernatant of erythrocytes lysed in distilled water was defined as 100% hemolysis.
3.4. Intracellular Ca2+
After incubation, erythrocytes were washed in Ringer solution and then loaded with Fluo-3/AM (Biotium, CA, USA) in Ringer solution containing 5 mM CaCl2 and 5 µM Fluo-3/AM. The cells were incubated at 37 °C for 30 min and washed once in Ringer solution containing 5 mM CaCl2. The Fluo-3/AM-loaded erythrocytes were resuspended in 200 µL Ringer. Then, Ca2+-dependent fluorescence intensity was measured with an excitation wavelength of 488 nm and an emission wavelength of 530 nm on a FACS Calibur.
3.5. Reactive Oxidant Species (ROS)
Oxidative stress was determined utilizing 2’,7’-dichlorodihydrofluorescein diacetate (DCFDA). After incubation, a 100 µL suspension of erythrocytes was washed in Ringer solution and then stained with DCFDA (Sigma, Schnelldorf, Germany) in PBS containing DCFDA at a final concentration of 10 µM. Erythrocytes were incubated at 37 °C for 30 min in the dark and then washed in PBS. The DCFDA-loaded erythrocytes were resuspended in 200 µL Ringer solution, and ROS-dependent fluorescence intensity was measured at an excitation wavelength of 488 nm and an emission wavelength of 530 nm on a FACS Calibur (BD).
3.6. GSH Abundance
Reduced glutathione (GSH) abundance was determined utilizing 5-chloromethylfluorescein diacetate (5-CMFDA). After incubation, a 100 µL suspension of erythrocytes were centrifuged at 1600 rpm for 3 min at 22 °C, supernatant was discarded and cells were then stained with 5-CMFDA (Santa Cruz Biotechnology, Dallas, TX, USA) in PBS containing 5-CMFDA at a final concentration of 1 µM. Erythrocytes were incubated at 37 °C for 45 min in the dark and then washed in PBS. The 5-CMFDA-loaded erythrocytes were resuspended in 200 µL PBS, and 5-CMFDA-dependent fluorescence intensity was measured at an excitation wavelength of 488 nm and an emission wavelength of 530 nm on a FACS Calibur (BD).
3.7. Statistics
Data are expressed as arithmetic means ± SEM. As indicated in the figure legends, statistical analysis was made using ANOVA with Tukey’s test as post-test and t test as appropriate. n denotes the number of different erythrocyte specimens studied. Some variability is observed between erythrocytes drawn from different individuals. Thus, comparisons have always been made between erythrocytes from the same individual.
4. Conclusions
Triparanol triggers eryptosis with cell shrinkage and cell membrane scrambling, an effect paralleled by and in part due to induction of oxidative stress and increase of cytosolic Ca2+ activity.
Acknowledgments
The authors acknowledge the meticulous preparation of the manuscript by Tanja Loch. The study was supported by the Deutsche Forschungsgemeinschaft. The sponsor did not play a role in study design, collection, analysis and interpretation of data, writing of the report and decision to submit the article for publication.
Author Contributions
Arbace Officioso, Kousi Alzoubi and Caterina Manna performed and designed experiments and evaluated the data, Florian Lang drafted and wrote the manuscript. All authors corrected, read and approved the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gofflot, F.; Hars, C.; Illien, F.; Chevy, F.; Wolf, C.; Picard, J.J.; Roux, C. Molecular mechanisms underlying limb anomalies associated with cholesterol deficiency during gestation: Implications of hedgehog signaling. Hum. Mol. Genet. 2003, 12, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Hihara, T.; Taniguchi, T.; Ueda, M.; Yoshinaga, T.; Miyamoto, N.; Sawada, K. Probucol and the cholesterol synthesis inhibitors simvastatin and triparanol regulate Iks channel function differently. Hum. Exp. Toxicol. 2013, 32, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Meyers, W.C.; Hanks, J.B.; Jakoi, L.; Quarfordt, S.; Jones, R.S. Selective biliary secretion of basal and glucagon-inhibited neutral sterol after triparanol administration. Surgery 1980, 88, 156–161. [Google Scholar] [PubMed]
- Roux, C.; Wolf, C.; Mulliez, N.; Gaoua, W.; Cormier, V.; Chevy, F.; Citadelle, D. Role of cholesterol in embryonic development. Am. J. Clin. Nutr. 2000, 71, 1270S–1279S. [Google Scholar] [PubMed]
- Bi, X.; Han, X.; Zhang, F.; He, M.; Zhang, Y.; Zhi, X.Y.; Zhao, H. Triparanol suppresses human tumor growth in vitro and in vivo. Biochem. Biophys. Res. Commun. 2012, 425, 613–618. [Google Scholar] [CrossRef] [PubMed]
- Popjak, G.; Meenan, A.; Parish, E.J.; Nes, W.D. Inhibition of cholesterol synthesis and cell growth by 24(R,S),25-iminolanosterol and triparanol in cultured rat hepatoma cells. J. Biol. Chem. 1989, 264, 6230–6238. [Google Scholar] [PubMed]
- Tiet, T.D.; Hopyan, S.; Nadesan, P.; Gokgoz, N.; Poon, R.; Lin, A.C.; Yan, T.; Andrulis, I.L.; Alman, B.A.; Wunder, J.S. Constitutive hedgehog signaling in chondrosarcoma up-regulates tumor cell proliferation. Am. J. Pathol. 2006, 168, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.P.; Kimura, T.; Yanagihara, T.; Sakoda, S. Calcium increase in mouse skeletal muscles by triparanol: A drug to induce myotonic dystrophy-like clinical manifestations. Neurosci. Lett. 1999, 272, 87–90. [Google Scholar] [CrossRef]
- Ghanbari-Azarnier, R.; Sato, S.; Wei, Q.; Al-Jazrawe, M.; Alman, B.A. Targeting stem cell behavior in desmoid tumors (aggressive fibromatosis) by inhibiting hedgehog signaling. Neoplasia 2013, 15, 712–719. [Google Scholar] [CrossRef] [PubMed]
- Hopyan, S.; Nadesan, P.; Yu, C.; Wunder, J.; Alman, B.A. Dysregulation of hedgehog signalling predisposes to synovial chondromatosis. J. Pathol. 2005, 206, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Wei, Q.; Han, I.; Sato, S.; Ghanbari-Azarnier, R.; Whetstone, H.; Poon, R.; Hu, J.; Zheng, F.; Zhang, P.; et al. Hedgehog and notch signaling regulate self-renewal of undifferentiated pleomorphic sarcomas. Cancer Res. 2012, 72, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, G.R.; Chapman, C.J.; Chipault, J.R.; Pfeiffer, D.R. Lipid composition and (Na++ K+)-ATPase activity in rat lens during triparanol-induced cataract formation. Biochim. Biophys. Acta 1981, 644, 1–12. [Google Scholar] [CrossRef]
- Lang, P.A.; Kaiser, S.; Myssina, S.; Wieder, T.; Lang, F.; Huber, S.M. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol. Cell. Physiol. 2003, 285, C1553–C1560. [Google Scholar] [CrossRef] [PubMed]
- Qadri, S.M.; Mahmud, H.; Lang, E.; Gu, S.; Bobbala, D.; Zelenak, C.; Jilani, K.; Siegfried, A.; Foller, M.; Lang, F. Enhanced suicidal erythrocyte death in mice carrying a loss-of-function mutation of the adenomatous polyposis coli gene. J. Cell. Mol. Med. 2012, 16, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Towhid, S.T.; Mia, S.; Pakladok, T.; Alesutan, I.; Borst, O.; Gawaz, M.; Gulbins, E.; Lang, F. Sphingomyelinase-induced adhesion of eryptotic erythrocytes to endothelial cells. Am. J. Physiol. Cell. Physiol. 2012, 303, C991–C999. [Google Scholar] [CrossRef] [PubMed]
- Lau, I.P.; Chen, H.; Wang, J.; Ong, H.C.; Leung, K.C.; Ho, H.P.; Kong, S.K. In vitro effect of CTAB- and PEG-coated gold nanorods on the induction of eryptosis/erythroptosis in human erythrocytes. Nanotoxicology 2012, 6, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Maellaro, E.; Leoncini, S.; Moretti, D.; Del Bello, B.; Tanganelli, I.; De Felice, C.; Ciccoli, L. Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol. 2013, 50, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Jilani, K.; Lang, F. Carmustine-induced phosphatidylserine translocation in the erythrocyte membrane. Toxins 2013, 5, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Vota, D.M.; Maltaneri, R.E.; Wenker, S.D.; Nesse, A.B.; Vittori, D.C. Differential erythropoietin action upon cells induced to eryptosis by different agents. Cell. Biochem. Biophys. 2013, 65, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Zappulla, D. Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: Adaptations to CO2 increases? J. Cardiometab. Syndr. 2008, 3, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Patulin-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Zoubi, K.A.; Theurer, M.; Lang, F. Effect of dermaseptin on erythrocytes. Basic Clin. Pharmacol. Toxicol. 2013, 113, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Langer, H.; Abed, M.; Voelkl, J.; Lang, F. The uremic toxin acrolein promotes suicidal erythrocyte death. Kidney Blood Press Res. 2013, 37, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaeinia, M.; Cluitmans, J.C.; Toulany, M.; Saki, M.; Koberle, M.; Lang, E.; Dreischer, P.; Biedermann, T.; Duszenko, M.; Lang, F.; et al. Age sensitivity of NFkB abundance and programmed cell death in erythrocytes induced by NFkB inhibitors. Cell. Physiol. Biochem. 2013, 32, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.; Honisch, S.; Abed, M.; Lang, F. Triggering of suicidal erythrocyte death by penta-o-galloyl-beta-d-glucose. Toxins 2014, 6, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Jilani, K.; Enkel, S.; Bissinger, R.; Almilaji, A.; Abed, M.; Lang, F. Fluoxetine induced suicidal erythrocyte death. Toxins 2013, 5, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Jilani, K.; Lang, F. Triggering of suicidal erythrocyte death by celecoxib. Toxins 2013, 5, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Lang, E.; Modicano, P.; Bissinger, R.; Faggio, C.; Abed, M.; Lang, F. Effect of nitazoxanide on erythrocytes. Basic Clin. Pharmacol. Toxicol. 2014, 114, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Oswald, G.; Alzoubi, K.; Abed, M.; Lang, F. Stimulation of suicidal erythrocyte death by ribavirin. Basic Clin. Pharmacol. Toxicol. 2014, 114, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Herrmann, T.; Oswald, G.; Jilani, K.; Lang, F. Induction of suicidal erythrocyte death by novobiocin. Cell. Physiol. Biochem. 2014, 33, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Feger, M.; Alzoubi, K.; Pakladok, T.; Frauenfeld, L.; Geiger, C.; Towhid, S.T.; Lang, F. Sensitization of erythrocytes to suicidal erythrocyte death following water deprivation. Kidney Blood Press Res. 2013, 37, 567–578. [Google Scholar] [PubMed]
- Alzoubi, K.; Calabròa, S.; Bissinger, R.; Abed, M.; Faggio, C.; Lang, F. Stimulation of suicidal erythrocyte death by artesunate. Cell. Physiol. Biochem. 2014, 34, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Bissinger, R.; Lang, F. Mitoxantrone-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2014, 34, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Fischer, S.; Jilani, K.; Lang, F. Stimulation of erythrocyte death by phloretin. Cell. Physiol. Biochem. 2014, 34, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Lupescu, A.; Zelenak, C.; Jilani, K.; Lang, F. Stimulation of eryptosis by cryptotanshinone. Cell. Physiol. Biochem. 2014, 34, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Modicano, P.; Frauenfeld, L.; Lang, E.; Jacobi, J.; Faggio, C.; Lang, F. Estramustine-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, J.; Lang, E.; Bissinger, R.; Frauenfeld, L.; Modicano, P.; Faggio, C.; Abed, M.; Lang, F. Stimulation of erythrocyte cell membrane scrambling by mitotane. Cell. Physiol. Biochem. 2014, 33, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Warsi, J.; Jilani, K.; Lang, F. Stimulation of erythrocyte cell membrane scrambling by gedunin. Cell. Physiol. Biochem. 2014, 33, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Bissinger, R.; Calabro, S.; Faggio, C.; Jilani, K.; Lang, F. Aristolochic acid induced suicidal erythrocyte death. Kidney Blood Press Res. 2014, 39, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Tesoriere, L.; Attanzio, A.; Allegra, M.; Cilla, A.; Gentile, C.; Livrea, M.A. Oxysterol mixture in hypercholesterolemia-relevant proportion causes oxidative stress-dependent eryptosis. Cell. Physiol. Biochem. 2014, 34, 1075–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelkl, J.; Alzoubi, K.; Mamar, A.K.; Ahmed, M.S.; Abed, M.; Lang, F. Stimulation of suicidal erythrocyte death by increased extracellular phosphate concentrations. Kidney Blood Press Res. 2013, 38, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xiang, Y.; Ran, Q.; Deng, X.; Xiao, Y.; Xiang, L.; Li, Z. Involvement of calcium, reactive oxygen species, and ATP in hexavalent chromium-induced damage in red blood cells. Cell. Physiol. Biochem. 2014, 34, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Steinberg, D.; Avigan, J.; Feigelson, E.B. Effects of triparanol (MER-29) on cholesterol biosynthesis and on blood sterol levels in man. J. Clin. Invest. 1961, 40, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Artunc, F.; Alzoubi, K.; Honisch, S.; Baumann, D.; Foller, M.; Lang, F. Suicidal erythrocyte death in end-stage renal disease. J. Mol. Med. 2014, 92, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Polak-Jonkisz, D.; Purzyc, L. Ca2+ influx versus efflux during eryptosis in uremic erythrocytes. Blood Purif. 2012, 34, 209–210. [Google Scholar] [CrossRef] [PubMed]
- Calderon-Salinas, J.V.; Munoz-Reyes, E.G.; Guerrero-Romero, J.F.; Rodriguez-Moran, M.; Bracho-Riquelme, R.L.; Carrera-Gracia, M.A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in type 2 diabetic mellitus patients with chronic kidney disease. Mol. Cell. Biochem. 2011, 357, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Beringer, O.; Nicolay, J.P.; Amon, O.; Kempe, D.S.; Hermle, T.; Attanasio, P.; Akel, A.; Schafer, R.; Friedrich, B.; et al. Suicidal death of erythrocytes in recurrent hemolytic uremic syndrome. J. Mol. Med. 2006, 84, 378–388. [Google Scholar] [CrossRef] [PubMed]
- Nicolay, J.P.; Schneider, J.; Niemoeller, O.M.; Artunc, F.; Portero-Otin, M.; Haik, G., Jr.; Thornalley, P.J.; Schleicher, E.; Wieder, T.; Lang, F. Stimulation of suicidal erythrocyte death by methylglyoxal. Cell. Physiol. Biochem. 2006, 18, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Gatidis, S.; Freise, N.F.; Bock, H.; Kubitz, R.; Lauermann, C.; Orth, H.M.; Klindt, C.; Schuier, M.; Keitel, V.; et al. Conjugated bilirubin triggers anemia by inducing erythrocyte death. Hepatology 2015, 61, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Kempe, D.S.; Akel, A.; Lang, P.A.; Hermle, T.; Biswas, R.; Muresanu, J.; Friedrich, B.; Dreischer, P.; Wolz, C.; Schumacher, U.; et al. Suicidal erythrocyte death in sepsis. J. Mol. Med. 2007, 85, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Schenck, M.; Nicolay, J.P.; Becker, J.U.; Kempe, D.S.; Lupescu, A.; Koka, S.; Eisele, K.; Klarl, B.A.; Rubben, H.; et al. Liver cell death and anemia in wilson disease involve acid sphingomyelinase and ceramide. Nat. Med. 2007, 13, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Harrison, H.E.; Bunting, H.; Ordway, N.K.; Albrink, W.S. The pathogenesis of the renal injury produced in the dog by hemoglobin or methemoglobin. J. Exp. Med. 1947, 86, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K. Membrane transport in the malaria-infected erythrocyte. Physiol. Rev. 2001, 81, 495–537. [Google Scholar] [PubMed]
- Ayi, K.; Giribaldi, G.; Skorokhod, A.; Schwarzer, E.; Prendergast, P.T.; Arese, P. 16α-Bromoepiandrosterone, an antimalarial analogue of the hormone dehydroepiandrosterone, enhances phagocytosis of ring stage parasitized erythrocytes: A novel mechanism for antimalarial activity. Antimicrob. Agents Chemother. 2002, 46, 3180–3184. [Google Scholar] [CrossRef] [PubMed]
- Ayi, K.; Turrini, F.; Piga, A.; Arese, P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: A common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood 2004, 104, 3364–3371. [Google Scholar] [CrossRef] [PubMed]
- Cappadoro, M.; Giribaldi, G.; O’Brien, E.; Turrini, F.; Mannu, F.; Ulliers, D.; Simula, G.; Luzzatto, L.; Arese, P. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood 1998, 92, 2527–2534. [Google Scholar] [PubMed]
- Koka, S.; Huber, S.M.; Boini, K.M.; Lang, C.; Foller, M.; Lang, F. Lead decreases parasitemia and enhances survival of Plasmodium berghei-infected mice. Biochem. Biophys. Res. Commun. 2007, 363, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Koka, S.; Lang, C.; Niemoeller, O.M.; Boini, K.M.; Nicolay, J.P.; Huber, S.M.; Lang, F. Influence of no synthase inhibitor L-NAME on parasitemia and survival of Plasmodium berghei infected mice. Cell. Physiol. Biochem. 2008, 21, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Foller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol. Cell. Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef] [PubMed]
- Andrews, D.A.; Low, P.S. Role of red blood cells in thrombosis. Curr. Opin. Hematol. 1999, 6, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.M.; Bae, O.N.; Lim, K.M.; Noh, J.Y.; Lee, M.Y.; Jung, Y.S.; Chung, J.H. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.G.; Chang, S.H.; Rettig, M.P.; Neely, J.E.; Hillery, C.A.; Smith, B.D.; Low, P.S. Altered erythrocyte endothelial adherence and membrane phospholipid asymmetry in hereditary hydrocytosis. Blood 2003, 101, 4625–4627. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, A.; Di Pietro, N.; Sirolli, V.; Giardinelli, A.; Di Silvestre, S.; Amoroso, L.; Di Tomo, P.; Capani, F.; Consoli, A.; Bonomini, M. Mechanisms of uremic erythrocyte-induced adhesion of human monocytes to cultured endothelial cells. J. Cell. Physiol. 2007, 213, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.L.; Gibson, D.F.; Tait, J.F. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: Flow-cytometric measurement and clinical associations. Blood 1996, 88, 1873–1880. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Officioso, A.; Manna, C.; Alzoubi, K.; Lang, F. Triggering of Erythrocyte Death by Triparanol. Toxins 2015, 7, 3359-3371. https://doi.org/10.3390/toxins7083359
AMA Style
Officioso A, Manna C, Alzoubi K, Lang F. Triggering of Erythrocyte Death by Triparanol. Toxins. 2015; 7(8):3359-3371. https://doi.org/10.3390/toxins7083359
Chicago/Turabian StyleOfficioso, Arbace, Caterina Manna, Kousi Alzoubi, and Florian Lang. 2015. "Triggering of Erythrocyte Death by Triparanol" Toxins 7, no. 8: 3359-3371. https://doi.org/10.3390/toxins7083359