
Induction of Suicidal Erythrocyte Death by Nelfinavir

Department of Physiology, University of Tuebingen, Gmelinstr. 5, 72076 Tuebingen, Germany
*
Author to whom correspondence should be addressed.
Toxins 2015, 7(5), 1616-1628; https://doi.org/10.3390/toxins7051616
Submission received: 1 April 2015
/
Revised: 28 April 2015
/
Accepted: 5 May 2015
/
Published: 8 May 2015
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The HIV protease inhibitor, nelfinavir, primarily used for the treatment of HIV infections, has later been shown to be effective in various infectious diseases including malaria. Nelfinavir may trigger mitochondria-independent cell death. Erythrocytes may undergo eryptosis, a mitochondria-independent suicidal cell death characterized by cell shrinkage and phosphatidylserine translocation to the erythrocyte surface. Triggers of eryptosis include oxidative stress and increase of cytosolic Ca2+-activity ([Ca2+]i). During malaria, accelerated death of infected erythrocytes may decrease parasitemia and thus favorably influence the clinical course of the disease. In the present study, phosphatidylserine abundance at the cell surface was estimated from annexin V binding, cell volume from forward scatter, reactive oxidant species (ROS) from 2',7'-dichlorodihydrofluorescein diacetate (DCFDA) fluorescence, and [Ca2+]i from Fluo3-fluorescence. A 48 h treatment of human erythrocytes with nelfinavir significantly increased the percentage of annexin-V-binding cells (≥5µg/mL), significantly decreased forward scatter (≥2.5µg/mL), significantly increased ROS abundance (10 µg/mL), and significantly increased [Ca2+]i (≥5 µg/mL). The up-regulation of annexin-V-binding following nelfinavir treatment was significantly blunted, but not abolished by either addition of the antioxidant N-acetylcysteine (1 mM) or removal of extracellular Ca2+. In conclusion, exposure of erythrocytes to nelfinavir induces oxidative stress and Ca2+ entry, thus leading to suicidal erythrocyte death characterized by erythrocyte shrinkage and erythrocyte membrane scrambling.
1. Introduction
Nelfinavir, a specific HIV protease inhibitor, has originally been developed for the treatment of HIV infections and subsequently been shown to be effective in further infectious diseases including SARS, tuberculosis, and malaria [1,2,3,4,5]. Beyond that, nelfinavir may trigger death of tumor cells and thus counteracts malignancy [2,3,6,7]. Nelfinavir is in part effective by triggering of mitochondria-independent apoptosis [2,3].
In analogy to apoptosis of nucleated cells, erythrocytes could enter eryptosis, a suicidal death characterized by cell shrinkage [8] and translocation of phosphatidylserine from the cell interior to the erythrocyte surface [9]. Cellular mechanisms involved in the stimulation of eryptosis include oxidative stress [9], increased cytosolic Ca2+ activity ([Ca2+]i), ceramide [10], energy depletion [9], and activated caspases [9,11,12]. Moreover, eryptosis may be stimulated by casein kinase 1α, Janus-activated kinase JAK3, protein kinase C, p38 kinase, and PAK2 kinase [9]. Eryptosis is inhibited by AMP activated kinase AMPK, cGMP-dependent protein kinase, sorafenib and sunitinib sensitive kinases [9]. Eryptosis is distinct from programmed erythrocyte necrosis [13], which is triggered by pore-forming bacterial toxins.
Eryptosis is stimulated by diverse xenobiotics [9,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50]. Eryptosis is further triggered during malaria and accelerated eryptosis favourably influences the clinical course of the disease [51].
The present study explored, whether and how nelfinavir stimulates eryptosis. To this end, erythrocytes from healthy volunteers were exposed to nelfinavir and phosphatidylserine abundance at the erythrocyte surface, cell volume, abundance of reactive oxidant species and [Ca2+]i determined utilizing flow cytometry.
2. Results
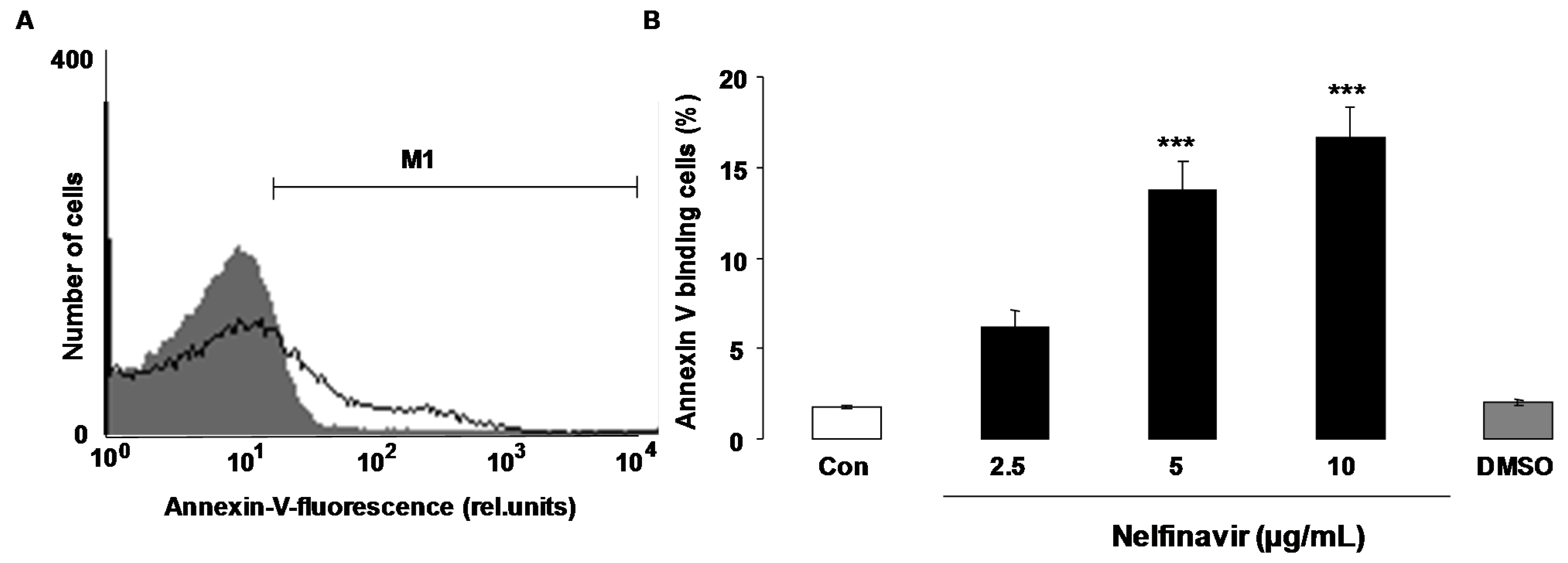
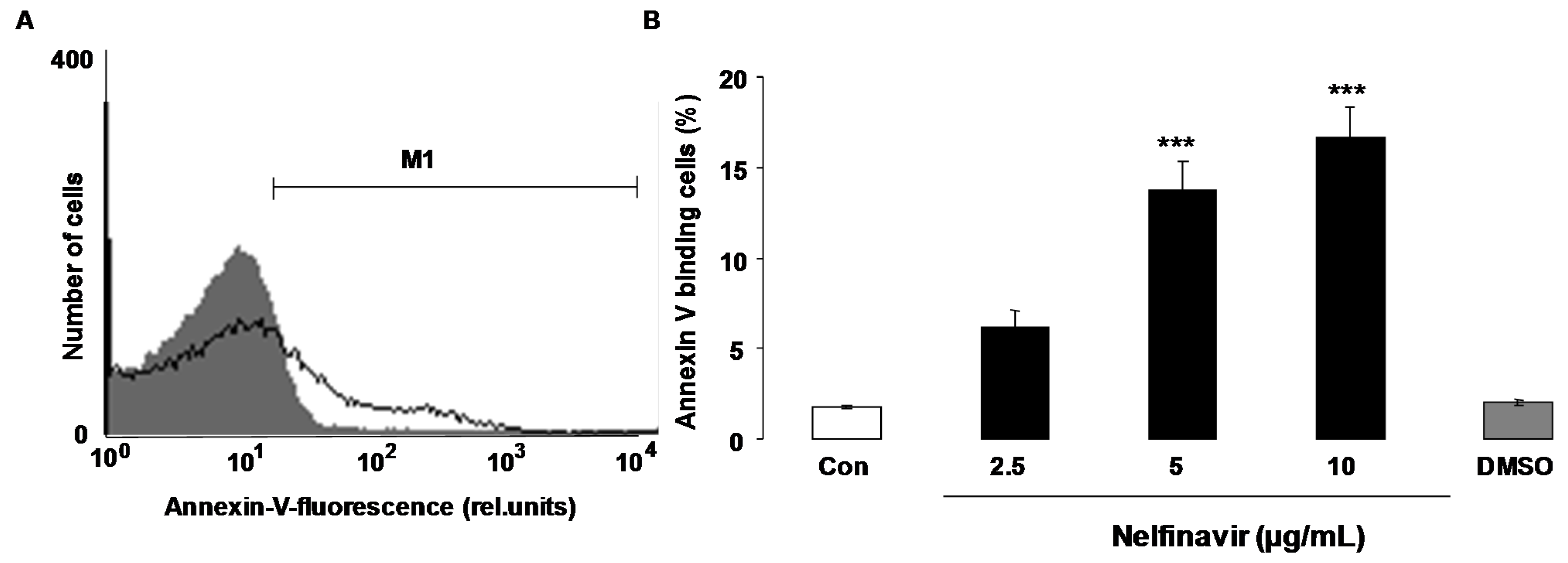
The present study explored whether nelfinavir is capable to trigger eryptosis, the suicidal erythrocyte death. Hallmarks of eryptosis are cell shrinkage and phospholipid scrambling of the cell membrane with phosphatidylserine translocation to the cell surface. In order to quantify phospholipid scrambling of the cell membrane, phosphatidylserine abundance at the cell surface was quantified by determination of phosphatidylserine-binding FITC-labelled annexin-V in flow cytometry. As shown in Figure 1, a 48 h exposure to nelfinavir increased the percentage of annexin-V-binding erythrocytes, an effect reaching statistical significance at 5 μg/mL nelfinavir concentration. Hemoglobin concentration in the supernatant was determined in order to estimate the effect of nelfinavir on hemolysis. According to hemoglobin concentration in the supernatant, a 48 h incubation with 0, 2.5, 5 and 10 μg/mL Nelfinavir resulted in hemolysis of 1.9% ± 0.3%, 3.5% ± 0.3%. 4.0% ± 0.2% and 7.0% ± 1.0% (n = 5), respectively.
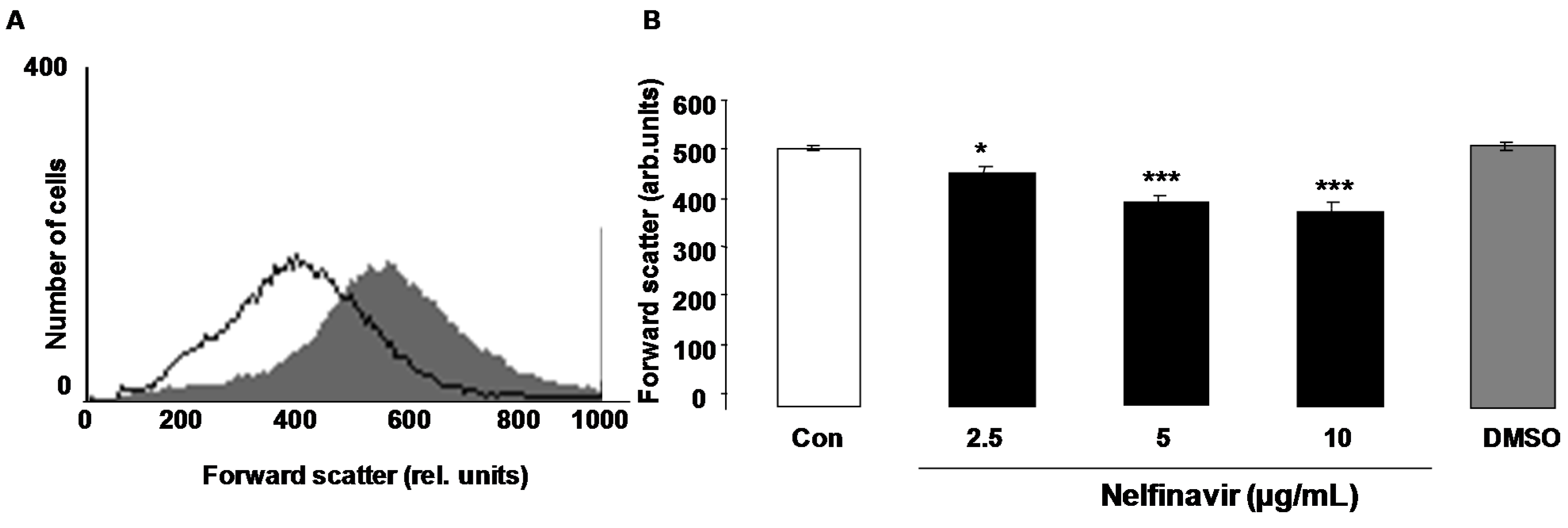
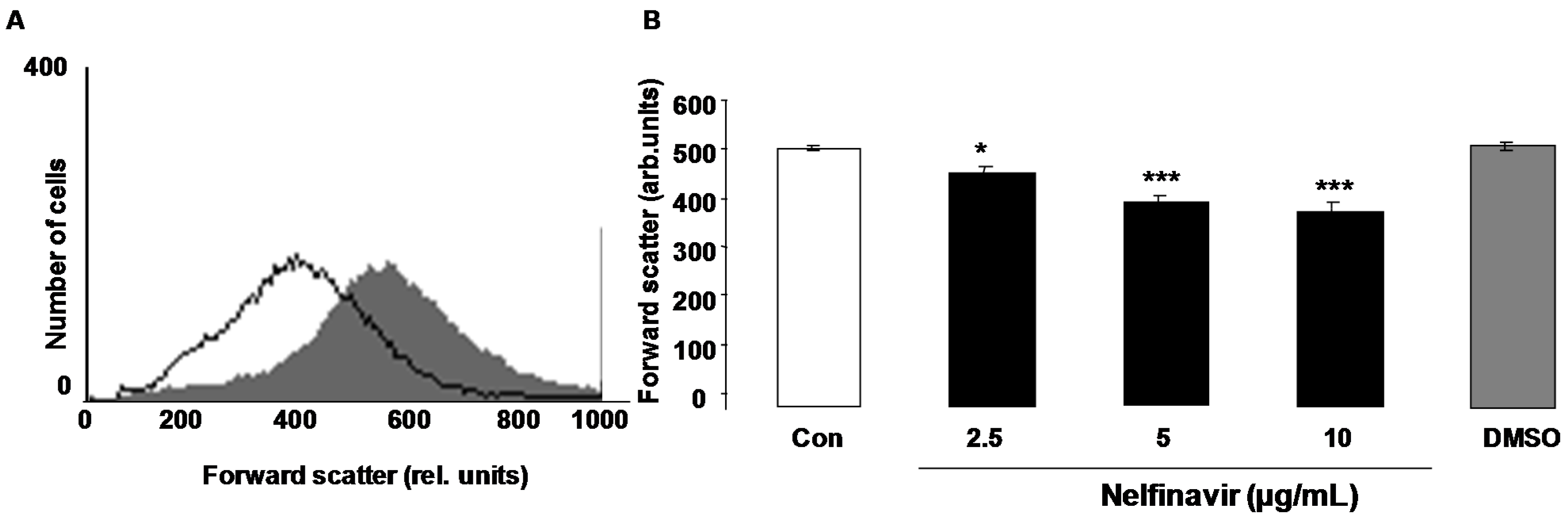
Erythrocyte cell volume was estimated from forward scatter in flow cytometry. As illustrated in Figure 2, a 48 h nelfinavir treatment was followed by a decrease of erythrocyte forward scatter, an effect reaching statistical significance at 2.5 µg/mL nelfinavir concentration.
Figure 1.
Effect of nelfinavir on phosphatidylserine exposure. (A) Original histogram of annexin-V-binding of erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of 10 µg/mL nelfinavir. M1 indicates the annexin-V-fluoresence defining the percentage of annexin-V-binding erythrocytes; (B) Arithmetic means ± SEM of erythrocyte annexin-V-binding (n = 15) following incubation for 48 h to Ringer solution without (white bar) or with (black bars) presence of nelfinavir (2.5–10 µg/mL). For comparison, the effect of the solvent DMSO (1 µL/mL Ringer) is shown (grey bar). *** (p < 0.001) indicates significant difference from the absence of nelfinavir (ANOVA).
Figure 1.
Effect of nelfinavir on phosphatidylserine exposure. (A) Original histogram of annexin-V-binding of erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of 10 µg/mL nelfinavir. M1 indicates the annexin-V-fluoresence defining the percentage of annexin-V-binding erythrocytes; (B) Arithmetic means ± SEM of erythrocyte annexin-V-binding (n = 15) following incubation for 48 h to Ringer solution without (white bar) or with (black bars) presence of nelfinavir (2.5–10 µg/mL). For comparison, the effect of the solvent DMSO (1 µL/mL Ringer) is shown (grey bar). *** (p < 0.001) indicates significant difference from the absence of nelfinavir (ANOVA).
Nelfinavir treatment thus triggered phospholipid scrambling of the erythrocyte membrane and cell shrinkage, the two hallmarks of eryptosis. Additional experiments were performed to shed light on the cellular mechanisms underlying the triggering of eryptosis.
Figure 2.
Effect of nelfinavir on erythrocyte forward scatter. (A) Original histogram of forward scatter of erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of 10 µg/mL nelfinavir; (B) Arithmetic means ± SEM (n = 15) of the erythrocyte forward scatter (FSC) following incubation for 48 h to Ringer solution without (white bar) or with (black bars) nelfinavir (2.5–10 µg/mL). For comparison, the effect of the solvent DMSO (1 µL/mL Ringer) is shown (grey bar). * (p < 0.05), *** (p < 0.001) indicate significant difference from the absence of nelfinavir (ANOVA).
Figure 2.
Effect of nelfinavir on erythrocyte forward scatter. (A) Original histogram of forward scatter of erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of 10 µg/mL nelfinavir; (B) Arithmetic means ± SEM (n = 15) of the erythrocyte forward scatter (FSC) following incubation for 48 h to Ringer solution without (white bar) or with (black bars) nelfinavir (2.5–10 µg/mL). For comparison, the effect of the solvent DMSO (1 µL/mL Ringer) is shown (grey bar). * (p < 0.05), *** (p < 0.001) indicate significant difference from the absence of nelfinavir (ANOVA).
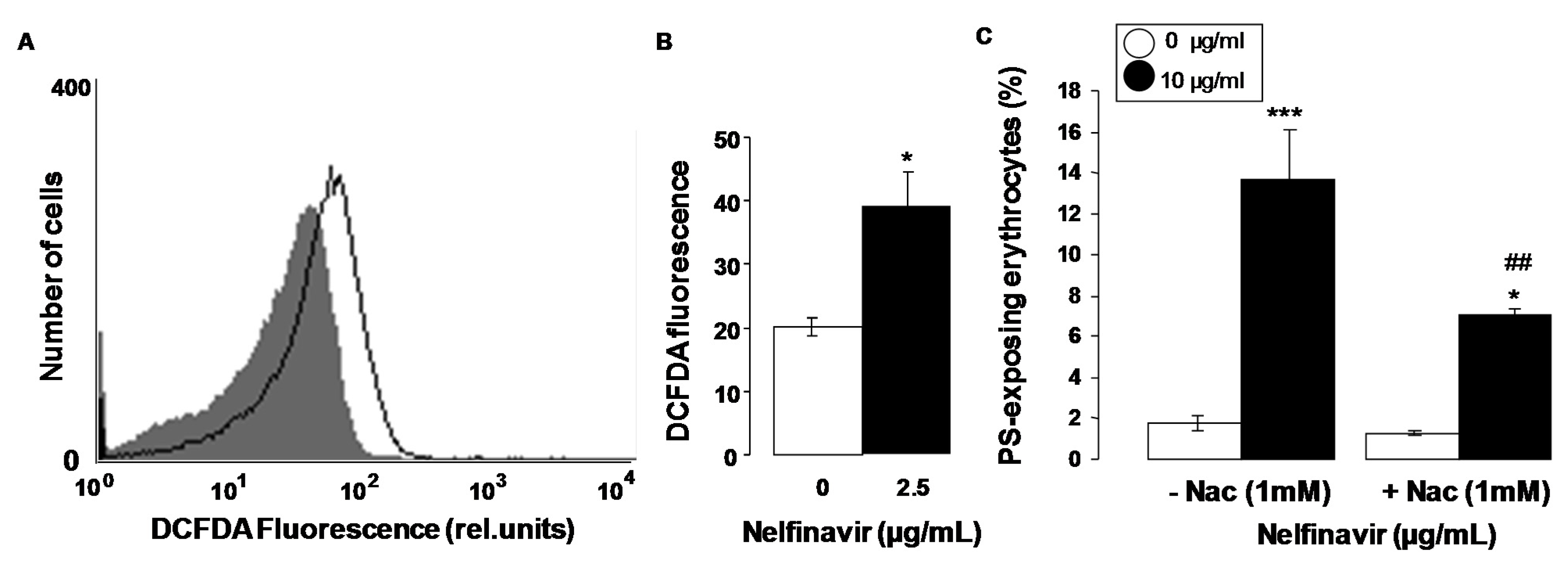
Mechanisms stimulating eryptosis include oxidative stress. Thus, additional experiments explored, whether nelfinavir influences the formation of reactive oxygen species (ROS). To this end, ROS was quantified utilizing 2',7'-dichlorodihydrofluorescein diacetate (DCFDA). As illustrated in Figure 3A,B, a 48 h exposure to nelfinavir (10 µg/mL) was followed by a significant increase of DCFDA fluorescence. Nelfinavir thus induced oxidative stress. An additional series of experiments explored whether nelfinavir-induced translocation of phosphatidylserine to the cell surface required oxidative stress and could thus be abrogated by the reducing substance N-acetylcysteine. To this end, erythrocytes were incubated for 48 h in the absence or presence of 10 µg/mL nelfinavir, both in the absence or presence of N-acetylcysteine (1 mM). As shown in Figure 3C, addition of N-acetylcysteine (1 mM) significantly blunted the effect of nelfinavir on annexin-V-binding, an observation indicating that oxidative stress contributed to the stimulation of cell membrane scrambling by nelfinavir. However, even in the presence of N-acetylcysteine nelfinavir significantly increased the percentage of annexin-V-binding erythrocytes, indicating that eryptosis was in part due to mechanisms other than oxidative stress.
Figure 3.
Effect of nelfinavir on reactive oxygen species. (A) Original histogram of 2',7'-dichlorodihydrofluorescein diacetate (DCFDA) fluorescence in erythrocytes following exposure for 48 h to Ringer solution without (grey shadow) and with (black line) presence of 10 µg/mL nelfinavir; (B) Arithmetic means ± SEM (n = 5) of the erythrocyte DCFDA fluorescence following incubation for 48 h to Ringer solution without (white bar) or with (black bar) presence of 10 µg/mL nelfinavir. * (p<0.05) indicates significant difference from the absence of nelfinavir (t test); (C) Arithmetic means ± SEM (n = 6) of annexin-V-binding of erythrocytes after a 48 h treatment with Ringer solution without (white bars) or with (black bars) 10 µg/mL nelfinavir in the absence (left bars, −Nac) and presence (right bars, +Nac) of the antioxidant N-acetylcysteine (1 mM). * (p < 0.05), *** (p < 0.001) indicate significant difference from the absence of nelfinavir, ## (p < 0.01) indicates significant difference from the respective value in the absence of the antioxidant N-acetylcysteine (1 mM).
Figure 3.
Effect of nelfinavir on reactive oxygen species. (A) Original histogram of 2',7'-dichlorodihydrofluorescein diacetate (DCFDA) fluorescence in erythrocytes following exposure for 48 h to Ringer solution without (grey shadow) and with (black line) presence of 10 µg/mL nelfinavir; (B) Arithmetic means ± SEM (n = 5) of the erythrocyte DCFDA fluorescence following incubation for 48 h to Ringer solution without (white bar) or with (black bar) presence of 10 µg/mL nelfinavir. * (p<0.05) indicates significant difference from the absence of nelfinavir (t test); (C) Arithmetic means ± SEM (n = 6) of annexin-V-binding of erythrocytes after a 48 h treatment with Ringer solution without (white bars) or with (black bars) 10 µg/mL nelfinavir in the absence (left bars, −Nac) and presence (right bars, +Nac) of the antioxidant N-acetylcysteine (1 mM). * (p < 0.05), *** (p < 0.001) indicate significant difference from the absence of nelfinavir, ## (p < 0.01) indicates significant difference from the respective value in the absence of the antioxidant N-acetylcysteine (1 mM).

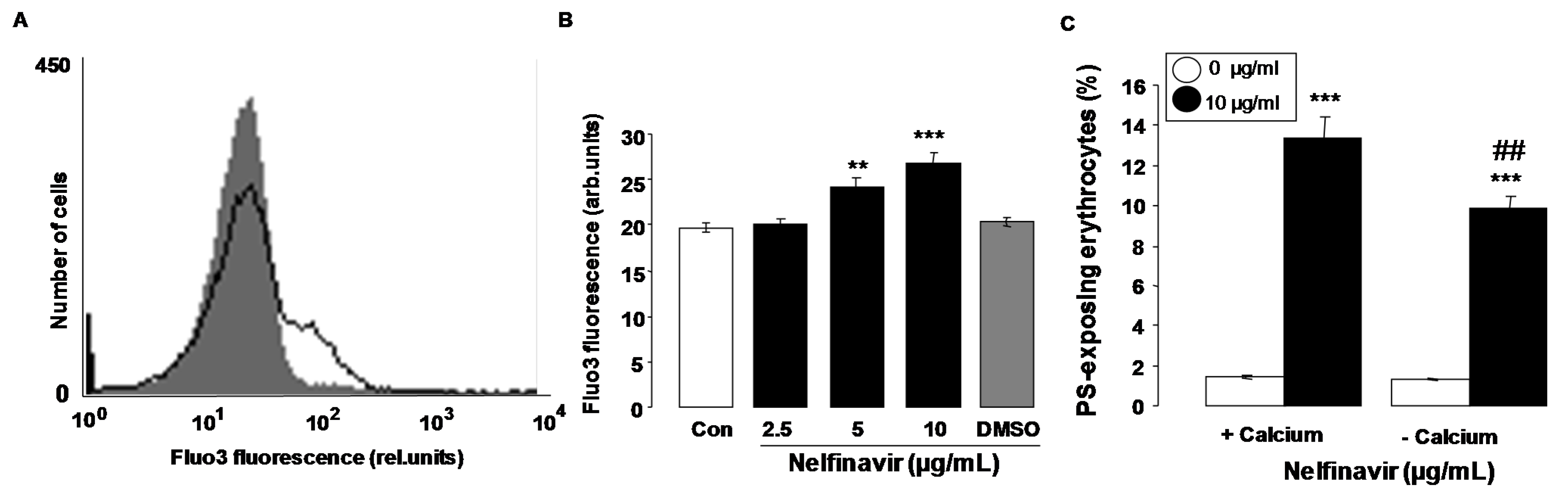
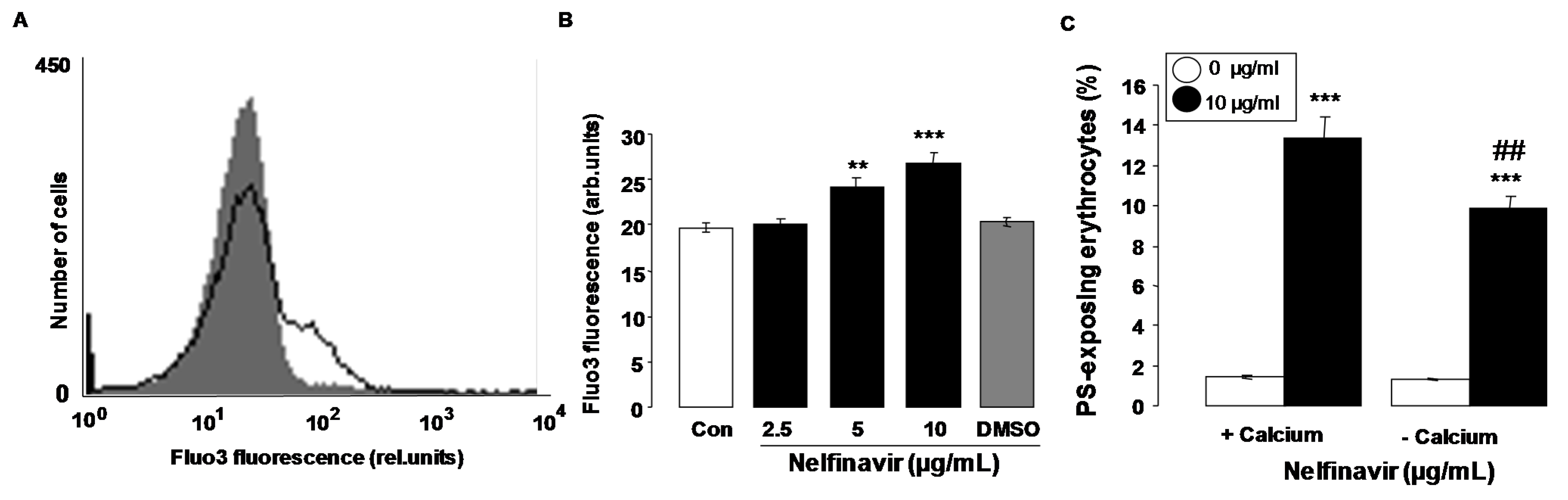
Oxidative stress is known to activate Ca2+ permeable cation channels with subsequent Ca2+ entry. Additional experiments thus explored whether nelfinavir influences cytosolic Ca2+ activity ([Ca2+]i). [Ca2+]i was quantified utilizing Fluo3 fluorescence. As shown in Figure 4A,B, a 48 h exposure to nelfinavir (2.5–10 µg/mL) increased the Fluo3 fluorescence, an effect reaching statistical significance at 5 µg/mL nelfinavir concentration. An additional series of experiments explored whether nelfinavir-induced translocation of phosphatidylserine to the cell surface required entry of extracellular Ca2+. To this end, erythrocytes were incubated for 48 h in the absence or presence of 10 µg/mL nelfinavir, both in the presence or nominal absence of extracellular Ca2+. As shown in Figure 4C, removal of extracellular Ca2+ significantly blunted the effect of nelfinavir on annexin-V-binding, an observation pointing to a role of Ca2+ entry from extracellular space in the stimulation of cell membrane scrambling by nelfinavir. However, even in the absence of extracellular Ca2+, nelfinavir significantly increased the percentage of annexin-V-binding erythrocytes. Thus, eryptosis was in part triggered by mechanisms other than entry of extracellular Ca2+.
Figure 4.
Effect of nelfinavir on erythrocyte Ca2+ activity and Ca2+ dependence of nelfinavir-induced phosphatidylserine exposure (A) Original histogram of Fluo3 fluorescence in erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of nelfinavir (10 µg/mL); (B) Arithmetic means ± SEM (n = 15) of the Fluo3 fluorescence (arbitrary units) in erythrocytes exposed for 48 h to Ringer solution without (white bar) or with (black bars) nelfinavir (2.5–10 µg/mL). For comparison, the effect of the solvent DMSO (1 µL/mL Ringer) is shown (grey bar). ** (p < 0.01), *** (p < 0.001) indicate significant difference from the absence of nelfinavir (ANOVA); (C) Arithmetic means ± SEM (n = 7) of annexin-V-binding of erythrocytes after a 48 h treatment with Ringer solution without (white bars) or with (black bars) 10 µg/mL nelfinavir in the presence (left bars, +Calcium) and absence (right bars, −Calcium) of Ca2+. *** (p < 0.001) indicates significant difference from the absence of nelfinavir, ## (p < 0.01) indicates significant difference from the respective value in the presence of Ca2+.
Figure 4.
Effect of nelfinavir on erythrocyte Ca2+ activity and Ca2+ dependence of nelfinavir-induced phosphatidylserine exposure (A) Original histogram of Fluo3 fluorescence in erythrocytes following exposure for 48 h to Ringer solution without (grey area) and with (black line) presence of nelfinavir (10 µg/mL); (B) Arithmetic means ± SEM (n = 15) of the Fluo3 fluorescence (arbitrary units) in erythrocytes exposed for 48 h to Ringer solution without (white bar) or with (black bars) nelfinavir (2.5–10 µg/mL). For comparison, the effect of the solvent DMSO (1 µL/mL Ringer) is shown (grey bar). ** (p < 0.01), *** (p < 0.001) indicate significant difference from the absence of nelfinavir (ANOVA); (C) Arithmetic means ± SEM (n = 7) of annexin-V-binding of erythrocytes after a 48 h treatment with Ringer solution without (white bars) or with (black bars) 10 µg/mL nelfinavir in the presence (left bars, +Calcium) and absence (right bars, −Calcium) of Ca2+. *** (p < 0.001) indicates significant difference from the absence of nelfinavir, ## (p < 0.01) indicates significant difference from the respective value in the presence of Ca2+.
3. Discussion
The present observations reveal a novel effect of nelfinavir, i.e., the triggering of eryptosis, the suicidal erythrocyte death characterized by cell shrinkage and erythrocyte cell membrane scrambling with phosphatidylserine translocation from the cell interior to the erythrocyte surface. The nelfinavir concentration required for stimulation of erythrocyte cell membrane scrambling (5 µg/mL) was similar to plasma concentrations (10 µg/mL) reported in vivo [52]. It must be kept in mind, though, that 98% of nelfinavir is bound to plasma proteins [53] and that the free nelfinavir concentration may be accordingly lower [52]. How binding to erythrocytes competes with binding to plasma proteins, is, however, not known.
The nelfinavir induced erythrocyte shrinkage was presumably secondary to increase of cytosolic Ca2+ activity ([Ca2+]i), which leads to cell shrinkage by activation of Ca2+ sensitive K+ channels, K+ exit, cell membrane hyperpolarization, Cl− exit and thus cellular loss of KCl with osmotically obliged water [8].
The nelfinavir induced cell membrane scrambling was in part due to stimulation of Ca2+ entry from extracellular space leading to increase of [Ca2+]i, a powerful stimulator of cell membrane scrambling with phosphatidylserine translocation [8]. Removal of extracellular Ca2+ significantly blunted the stimulation of annexin-V-binding following nelfinavir treatment, indicating that Ca2+ entry contributed to the stimulation of nelfinavir induced phosphatidylserine translocation. However, even in the absence of extracellular Ca2+ nelfinavir significantly enhanced the phosphatidylserine abundance at the cell surface. Thus, the effect of nelfinavir on Ca2+ entry contributed to, but did not fully account for the stimulation of phosphatidylserine translocation. Nelfinavir thus triggered cell membrane scrambling in part through mechanisms other than Ca2+ entry.
The effect of nelfinavir on [Ca2+]i was presumably in part the result of oxidative stress, which activates oxidant sensitive Ca2+ permeable erythrocytic cation channels [9]. Beyond that, oxidative stress may trigger eryptosis by further, rather illdefined mechanisms. Nelfinavir has similarly been reported to induce oxidative stress in nucleated cells [2,3,54,55,56,57,58].
Eryptosis serves to clear defective erythrocytes prior to hemolysis with release of hemoglobin, which may undergo glomerular filtration in the kidney and precipitate in the acidic lumen of renal tubules thus occluding affected nephrons [59]. Eryptosis further serves to remove infected erythrocytes during malaria [51]. The malaria pathogen Plasmodium imposes oxidative stress on the host erythrocyte leading to activation of several host cell ion channels including Ca2+-permeable erythrocyte cation channels [9,60]. The subsequent Ca2+ entry triggers cell membrane scrambling, phosphatidylserine translocation, binding to phosphatidylserine receptors at phagocytes, phagocytosis and thus removal of the infected erythrocytes from circulating blood [51]. Eryptotic removal of infected erythrocytes reduces parasitemia and thus favorably influences the clinical course of malaria. Enhanced susceptibility to eryptosis presumably confers protection against a severe course of malaria in several genetic erythrocyte disorders, such as sickle-cell trait, beta-thalassemia-trait, homozygous Hb-C and homozygous G6PD-deficiency [9,61,62,63]. Accelerated eryptosis further contributes to the protective effect against malaria of iron deficiency [64], lead intoxication [64], treatment with chlorpromazine [65] or presence of NO synthase inhibitors [65]. It is tempting to speculate that induction of eryptosis contributes to the antimalarial effect of nelfinavir. As infected erythrocytes are exposed to oxidative stress [51], they are particularly sensitive to triggers of eryptosis and may thus specifically be eleimninated by eryptosis inducing substances. Clearly, additional experimentation is required to confirm or falsify this speculation.
As phosphatidylserine exposing erythrocytes are engulfed by macrophages and thus rapidly cleared from circulating blood, excessive eryptosis may lead to anemia [9]. Moreover, phosphatidylserine exposing erythrocytes may bind to endothelial cells of the vascular wall [66], trigger blood clotting and induce thrombosis [67,68,69]. Phosphatidylserine exposing erythrocytes thus may compromise microcirculation [10,67,70,71,72,73].
4. Experimental Section
4.1. Erythrocytes, Solutions and Chemicals
Fresh Lithium-Heparin-anticoagulated blood samples were kindly provided by the blood bank of the University of Tübingen. The study is approved by the ethics committee of the University of Tübingen (184/2003 V). The blood was centrifuged at 120 g for 20 min at 23 °C and the platelets and leukocytes-containing supernatant was disposed. Erythrocytes were incubated in vitro for 48 h at a hematocrit of 0.4% in Ringer solution containing (in mM) 125 NaCl, 5 KCl, 1 MgSO4, 32 N-2-hydroxyethylpiperazine-N-2-ethanesulfonic acid (HEPES), 5 glucose, and 1 CaCl2; the pH was adjusted to 7.4 and the temperature kept at 37 °C. Where indicated, erythrocytes were exposed to nelfinavir (Sigma Aldrich, Hamburg, Germany), which was dissolved in DMSO (Carl Roth, Karlsruhe, Germany). For comparison, the effect of 1 µL DMSO/mL Ringer was tested.
4.2. Annexin-V-Binding and Forward Scatter
After incubation under the respective experimental condition, a 150 µL cell suspension was washed in Ringer solution containing 5 mM CaCl2 and then stained with Annexin-V-FITC (1:200 dilution; ImmunoTools, Friesoythe, Germany) in this solution at 37 °C for 20 min under protection from light. In the following, the forward scatter (FSC) of the cells was determined, and annexin-V fluorescence intensity was measured with an excitation wavelength of 488 nm and an emission wavelength of 530 nm on a FACS Calibur (BD, Heidelberg, Germany). In some experiments erythrocytes were preincubated in Ca2+ free solution. For determination of annexin-V-binding, addition of Ca2+ was required during the 15 min incubation with FITC-annexin V. Immediately thereafter measurements were done so that the exposure to Ca2+ was too short to trigger significant phosphatidylserine translocation.
4.3. Reactive Oxidant Species (ROS)
Oxidative stress was determined utilizing 2',7'-dichlorodihydrofluorescein diacetate (DCFDA). After incubation, a 150 µL suspension of erythrocytes was washed in Ringer solution and then stained with DCFDA (Sigma, Schnelldorf, Germany) in Ringer solution containing DCFDA at a final concentration of 10 µM. Erythrocytes were incubated at 37 °C for 30 min in the dark and then washed three times in Ringer solution. The DCFDA-loaded erythrocytes were resuspended in 200 µL Ringer solution, and ROS-dependent fluorescence intensity was measured at an excitation wavelength of 488 nm and an emission wavelength of 530 nm on a FACS Calibur (BD).
4.4. Intracellular Ca2+
After incubation, a 150 µL cell suspension was washed in Ringer solution and then loaded with Fluo-3/AM (Biotium, Hayward, CA, USA) in Ringer solution containing 5 mM CaCl2 and 5 µM Fluo-3/AM. The cells were incubated at 37 °C for 30 min and washed twice in Ringer solution containing 5 mM CaCl2. The Fluo-3/AM-loaded erythrocytes were resuspended in 200 µL Ringer. Then, Ca2+-dependent fluorescence intensity was measured with an excitation wavelength of 488 nm and an emission wavelength of 530 nm on a FACS Calibur.
4.5. Hemolysis
For the determination of hemolysis, the samples were centrifuged (3 min at 1600 rpm, room temperature) after incubation under the respective experimental conditions and the supernatants were harvested. As a measure of hemolysis, the hemoglobin (Hb) concentration of the supernatant was determined photometrically at 405 nm. The absorption of the supernatant of erythrocytes lysed in distilled water was defined as 100% hemolysis. Hemolysis is expressed in % of total hemolysis.
4.6. Statistics
Data are expressed as arithmetic means ± SEM. As indicated in the figure legends, statistical analysis was made using ANOVA with Tukey’s test as post-test and t test as appropriate. n denotes the number of different erythrocyte specimens studied. Since different erythrocyte specimens used in distinct experiments are differently susceptible to triggers of eryptosis, the same erythrocyte specimens have been used for control and experimental conditions.
5. Conclusions
Nelfinavir stimulates eryptosis, the suicidal erythrocyte death characterized by erythrocyte cell membrane scrambling and cell shrinkage. The effect is paralleled by, and at least partially due to, oxidative stress and increase of cytosolic Ca2+ activity.
Acknowledgments
The authors acknowledge the meticulous preparation of the manuscript by Tanja Loch. The study was supported by the Deutsche Forschungsgemeinschaft and the Open Access Publishing Fund of Tuebingen University.
Author Contributions
Design of the study, wrote the manuscript: F.L. Production, analysis or interpretation of the results, designed experiments: R.B. and S.W. Approved the submitted manuscript: R.B., S.W. and F.L.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
References
- Baril, J.G.; Lefebvre, E.A.; Lalonde, R.G.; Shafran, S.D.; Conway, B. Nelfinavir and non-nucleoside reverse transcriptase inhibitor-based salvage regimens in heavily HIV pretreated patients. Can. J. Infect. Dis. 2003, 14, 201–205. [Google Scholar] [PubMed]
- Bruning, A.; Gingelmaier, A.; Friese, K.; Mylonas, I. New prospects for nelfinavir in non-HIV-related diseases. Curr. Mol. Pharmacol. 2010, 3, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Bruning, A.; Friese, K.; Burges, A.; Mylonas, I. Tamoxifen enhances the cytotoxic effects of nelfinavir in breast cancer cells. Breast Cancer Res. 2010, 12, R45. [Google Scholar] [CrossRef] [PubMed]
- Olmo, M.; Podzamczer, D. A review of nelfinavir for the treatment of HIV infection. Expert Opin. Drug Metab. Toxicol. 2006, 2, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.M.; Frampton, J.E.; McCormack, P.L.; Siddiqui, M.A.; Cvetkovic, R.S. Nelfinavir: A review of its use in the management of HIV infection. Drugs 2005, 65, 2209–2244. [Google Scholar] [CrossRef] [PubMed]
- Gantt, S.; Casper, C.; Ambinder, R.F. Insights into the broad cellular effects of nelfinavir and the HIV protease inhibitors supporting their role in cancer treatment and prevention. Curr. Opin. Oncol. 2013, 25, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Kast, R.E.; Halatsch, M.E. Matrix metalloproteinase-2 and -9 in glioblastoma: A trio of old drugs-captopril, disulfiram and nelfinavir-are inhibitors with potential as adjunctive treatments in glioblastoma. Arch. Med. Res. 2012, 43, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.A.; Kaiser, S.; Myssina, S.; Wieder, T.; Lang, F.; Huber, S.M. Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol. Cell Physiol. 2003, 285, C1553–C1560. [Google Scholar] [CrossRef] [PubMed]
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly - suicidal erythrocyte death. Int J Biochem Cell Biol 2012, 44, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Towhid, S.T.; Mia, S.; Pakladok, T.; Alesutan, I.; Borst, O.; Gawaz, M.; Gulbins, E.; Lang, F. Sphingomyelinase-induced adhesion of eryptotic erythrocytes to endothelial cells. Am. J. Physiol. Cell Physiol. 2012, 303, C991–C999. [Google Scholar] [CrossRef] [PubMed]
- Lau, I.P.; Chen, H.; Wang, J.; Ong, H.C.; Leung, K.C.; Ho, H.P.; Kong, S.K. In vitro effect of CTAB- and PEG-coated gold nanorods on the induction of eryptosis/erythroptosis in human erythrocytes. Nanotoxicology 2012, 6, 847–856. [Google Scholar] [CrossRef] [PubMed]
- Maellaro, E.; Leoncini, S.; Moretti, D.; Del Bello, B.; Tanganelli, I.; De Felice, C.; Ciccoli, L. Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol. 2013, 50, 489–495. [Google Scholar] [CrossRef] [PubMed]
- LaRocca, T.J.; Stivison, E.A.; Hod, E.A.; Spitalnik, S.L.; Cowan, P.J.; Randis, T.M.; Ratner, A.J. Human-specific bacterial pore-forming toxins induce programmed necrosis in erythrocytes. MBio 2014, 5, e01251-14. [Google Scholar] [CrossRef] [PubMed]
- Bottger, E.; Multhoff, G.; Kun, J.F.; Esen, M. Plasmodium falciparum-infected erythrocytes induce granzyme B by NK cells through expression of host-Hsp70. PLoS One 2012, 7, e33774. [Google Scholar] [CrossRef] [PubMed]
- Firat, U.; Kaya, S.; Cim, A.; Buyukbayram, H.; Gokalp, O.; Dal, M.S.; Tamer, M.N. Increased caspase-3 immunoreactivity of erythrocytes in STZ diabetic rats. Exp. Diabetes Res. 2012, 2012, 316384. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, S.; Chaurasiya, N.D.; Sahu, R.; Walker, L.A.; Tekwani, B.L. Understanding the mechanisms for metabolism-linked hemolytic toxicity of primaquine against glucose 6-phosphate dehydrogenase deficient human erythrocytes: Evaluation of eryptotic pathway. Toxicology 2012, 294, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Cheung, K.L.; Lau, I.P.; Yu, W.S.; Fung, K.P.; Yu, B.; Loo, J.F.; Kong, S.K. Polyphyllin D induces apoptosis in human erythrocytes through Ca2+ rise and membrane permeabilization. Arch. Toxicol. 2012, 86, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaeinia, M.; Cluitmans, J.C.; Akel, A.; Dreischer, P.; Toulany, M.; Koberle, M.; Skabytska, Y.; Saki, M.; Biedermann, T.; Duszenko, M.; et al. The impact of erythrocyte age on eryptosis. Br. J. Haematol. 2012, 157, 606–614. [Google Scholar] [CrossRef] [PubMed]
- Jilani, K.; Qadri, S.M.; Lang, F. Geldanamycin-induced phosphatidylserine translocation in the erythrocyte membrane. Cell. Physiol. Biochem. 2013, 32, 1600–1609. [Google Scholar] [PubMed]
- Polak-Jonkisz, D.; Purzyc, L. Ca influx versus efflux during eryptosis in uremic erythrocytes. Blood Purif. 2012, 34, 209–210. [Google Scholar] [CrossRef] [PubMed]
- Qian, E.W.; Ge, D.T.; Kong, S.K. Salidroside protects human erythrocytes against hydrogen peroxide-induced apoptosis. J. Nat. Prod. 2012, 75, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Vota, D.M.; Maltaneri, R.E.; Wenker, S.D.; Nesse, A.B.; Vittori, D.C. Differential erythropoietin action upon cells induced to eryptosis by different agents. Cell. Biochem. Biophys. 2013, 65, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Weiss, E.; Cytlak, U.M.; Rees, D.C.; Osei, A.; Gibson, J.S. Deoxygenation-induced and Ca2+ dependent phosphatidylserine externalisation in red blood cells from normal individuals and sickle cell patients. Cell Calcium 2012, 51, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Zappulla, D. Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: Adaptations to CO2 increases? J. Cardiometab. Syndr. 2008, 3, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Zbidah, M.; Lupescu, A.; Jilani, K.; Lang, F. Stimulation of suicidal erythrocyte death by fumagillin. Basic Clin. Pharmacol. Toxicol. 2013, 112, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Herrmann, T.; Alzoubi, K.; Pakladok, T.; Lang, F. Tannic Acid induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 1106–1116. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, M.S.; Langer, H.; Abed, M.; Voelkl, J.; Lang, F. The uremic toxin acrolein promotes suicidal erythrocyte death. Kidney Blood Press. Res 2013, 37, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Ghashghaeinia, M.; Cluitmans, J.C.; Toulany, M.; Saki, M.; Koberle, M.; Lang, E.; Dreischer, P.; Biedermann, T.; Duszenko, M.; Lang, F.; et al. Age sensitivity of NFkappaB abundance and programmed cell death in erythrocytes induced by NFkappaB inhibitors. Cell. Physiol. Biochem. 2013, 32, 801–813. [Google Scholar] [CrossRef] [PubMed]
- Alzoubi, K.; Honisch, S.; Abed, M.; Lang, F. Triggering of suicidal erythrocyte death by penta-o-galloyl-beta-d-glucose. Toxins 2014, 6, 54–65. [Google Scholar] [CrossRef]
- Jilani, K.; Lang, F. Carmustine-induced phosphatidylserine translocation in the erythrocyte membrane. Toxins 2013, 5, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Jilani, K.; Enkel, S.; Bissinger, R.; Almilaji, A.; Abed, M.; Lang, F. Fluoxetine induced suicidal erythrocyte death. Toxins 2013, 5, 1230–1243. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Jilani, K.; Lang, F. Triggering of suicidal erythrocyte death by celecoxib. Toxins 2013, 5, 1543–1554. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Jilani, K.; Zbidah, M.; Lang, F. Patulin-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Zoubi, K.A.; Theurer, M.; Lang, F. Effect of dermaseptin on erythrocytes. Basic Clin. Pharmacol. Toxicol. 2013, 113, 347–352. [Google Scholar] [PubMed]
- Arnold, M.; Lang, E.; Modicano, P.; Bissinger, R.; Faggio, C.; Abed, M.; Lang, F. Effect of nitazoxanide on erythrocytes. Basic Clin. Pharmacol. Toxicol. 2014, 114, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Oswald, G.; Alzoubi, K.; Abed, M.; Lang, F. Stimulation of suicidal erythrocyte death by ribavirin. Basic Clin. Pharmacol. Toxicol. 2014, 114, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Malik, A.; Jilani, K.; Lang, F. Triggering of erythrocyte cell membrane scrambling by salinomycin. Basic Clin. Pharmacol. Toxicol. 2014, 115, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, J.; Lang, E.; Bissinger, R.; Frauenfeld, L.; Modicano, P.; Faggio, C.; Abed, M.; Lang, F. Stimulation of erythrocyte cell membrane scrambling by mitotane. Cell. Physiol. Biochem. 2014, 33, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Herrmann, T.; Oswald, G.; Jilani, K.; Lang, F. Induction of suicidal erythrocyte death by novobiocin. Cell. Physiol. Biochem. 2014, 33, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Lupescu, A.; Bissinger, R.; Warsi, J.; Jilani, K.; Lang, F. Stimulation of erythrocyte cell membrane scrambling by gedunin. Cell. Physiol. Biochem. 2014, 33, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Abed, M.; Feger, M.; Alzoubi, K.; Pakladok, T.; Frauenfeld, L.; Geiger, C.; Towhid, S.T.; Lang, F. Sensitization of erythrocytes to suicidal erythrocyte death following water deprivation. Kidney Blood Press. Res. 2013, 37, 567–578. [Google Scholar] [PubMed]
- Alzoubi, K.; Calabro, S.; Bissinger, R.; Abed, M.; Faggio, C.; Lang, F. Stimulation of suicidal erythrocyte death by artesunate. Cell. Physiol. Biochem. 2014, 34, 2232–2244. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Bissinger, R.; Lang, F. Mitoxantrone-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2014, 34, 1756–1767. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Fischer, S.; Jilani, K.; Lang, F. Stimulation of erythrocyte death by phloretin. Cell. Physiol. Biochem. 2014, 34, 2256–2265. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Lupescu, A.; Zelenak, C.; Jilani, K.; Lang, F. Stimulation of eryptosis by cryptotanshinone. Cell. Physiol. Biochem. 2014, 34, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Bissinger, R.; Modicano, P.; Frauenfeld, L.; Lang, E.; Jacobi, J.; Faggio, C.; Lang, F. Estramustine-induced suicidal erythrocyte death. Cell. Physiol. Biochem. 2013, 32, 1426–1436. [Google Scholar] [CrossRef] [PubMed]
- Malik, A.; Bissinger, R.; Calabro, S.; Faggio, C.; Jilani, K.; Lang, F. Aristolochic acid induced suicidal erythrocyte death. Kidney Blood Press. Res. 2014, 39, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Tesoriere, L.; Attanzio, A.; Allegra, M.; Cilla, A.; Gentile, C.; Livrea, M.A. Oxysterol mixture in hypercholesterolemia-relevant proportion causes oxidative stress-dependent eryptosis. Cell. Physiol. Biochem. 2014, 34, 1075–1089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voelkl, J.; Alzoubi, K.; Mamar, A.K.; Ahmed, M.S.; Abed, M.; Lang, F. Stimulation of suicidal erythrocyte death by increased extracellular phosphate concentrations. Kidney Blood Press. Res. 2013, 38, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Xiang, Y.; Ran, Q.; Deng, X.; Xiao, Y.; Xiang, L.; Li, Z. Involvement of calcium, reactive oxygen species, and ATP in hexavalent chromium-induced damage in red blood cells. Cell. Physiol. Biochem. 2014, 34, 1780–1791. [Google Scholar] [CrossRef] [PubMed]
- Foller, M.; Bobbala, D.; Koka, S.; Huber, S.M.; Gulbins, E.; Lang, F. Suicide for survival—Death of infected erythrocytes as a host mechanism to survive malaria. Cell. Physiol. Biochem. 2009, 24, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Sugioka, N.; Haraya, K.; Maeda, Y.; Fukushima, K.; Takada, K. Pharmacokinetics of human immunodeficiency virus protease inhibitor, nelfinavir, in poloxamer 407-induced hyperlipidemic model rats. Biol. Pharm. Bull. 2009, 32, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.L.; Brundage, R.C.; Bushman, L.; Kakuda, T.N.; Remmel, R.P.; Fletcher, C.V. Indinavir plasma protein binding in HIV-1-infected adults. AIDS 2000, 14, 2293–2297. [Google Scholar] [CrossRef] [PubMed]
- Ben-Romano, R.; Rudich, A.; Etzion, S.; Potashnik, R.; Kagan, E.; Greenbaum, U.; Bashan, N. Nelfinavir induces adipocyte insulin resistance through the induction of oxidative stress: Differential protective effect of antioxidant agents. Antivir. Ther. 2006, 11, 1051–1060. [Google Scholar] [PubMed]
- Chandra, S.; Mondal, D.; Agrawal, K.C. HIV-1 protease inhibitor induced oxidative stress suppresses glucose stimulated insulin release: Protection with thymoquinone. Exp. Biol. Med. 2009, 234, 442–453. [Google Scholar] [CrossRef]
- Kumar, P.; Lodge, R.; Trudel, N.; Ouellet, M.; Ouellette, M.; Tremblay, M.J. Nelfinavir, an HIV-1 protease inhibitor, induces oxidative stress-mediated, caspase-independent apoptosis in Leishmania amastigotes. PLoS Negl. Trop. Dis. 2010, 4, e642. [Google Scholar] [CrossRef] [PubMed]
- Kushchayeva, Y.; Jensen, K.; Recupero, A.; Costello, J.; Patel, A.; Klubo-Gwiezdzinska, J.; Boyle, L.; Burman, K.; Vasko, V. The HIV protease inhibitor nelfinavir down-regulates RET signaling and induces apoptosis in medullary thyroid cancer cells. J. Clin. Endocrinol. Metab. 2014, 99, E734–E745. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.; Kost, B.; Renner-Muller, I.; Wolf, E.; Mylonas, I.; Bruning, A. Efavirenz causes oxidative stress, endoplasmic reticulum stress, and autophagy in endothelial cells. Cardiovasc. Toxicol. 2015, in press. [Google Scholar]
- Harrison, H.E.; Bunting, H.; Ordway, N.K.; Albrink, W.S. The pathogenesis of the renal injury produced in the dog by hemoglobin or methemoglobin. J. Exp. Med. 1947, 86, 339–356. [Google Scholar] [CrossRef] [PubMed]
- Kirk, K. Membrane transport in the malaria-infected erythrocyte. Physiol. Rev. 2001, 81, 495–537. [Google Scholar] [PubMed]
- Ayi, K.; Giribaldi, G.; Skorokhod, A.; Schwarzer, E.; Prendergast, P.T.; Arese, P. 16alpha-bromoepiandrosterone, an antimalarial analogue of the hormone dehydroepiandrosterone, enhances phagocytosis of ring stage parasitized erythrocytes: A novel mechanism for antimalarial activity. Antimicrob. Agents Chemother. 2002, 46, 3180–3184. [Google Scholar] [CrossRef] [PubMed]
- Ayi, K.; Turrini, F.; Piga, A.; Arese, P. Enhanced phagocytosis of ring-parasitized mutant erythrocytes: A common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood 2004, 104, 3364–3371. [Google Scholar] [CrossRef] [PubMed]
- Cappadoro, M.; Giribaldi, G.; O’Brien, E.; Turrini, F.; Mannu, F.; Ulliers, D.; Simula, G.; Luzzatto, L.; Arese, P. Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood 1998, 92, 2527–2534. [Google Scholar] [PubMed]
- Koka, S.; Huber, S.M.; Boini, K.M.; Lang, C.; Foller, M.; Lang, F. Lead decreases parasitemia and enhances survival of Plasmodium berghei-infected mice. Biochem. Biophys. Res. Commun. 2007, 363, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Koka, S.; Lang, C.; Niemoeller, O.M.; Boini, K.M.; Nicolay, J.P.; Huber, S.M.; Lang, F. Influence of NO synthase inhibitor L-NAME on parasitemia and survival of Plasmodium berghei infected mice. Cell. Physiol. Biochem. 2008, 21, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Borst, O.; Abed, M.; Alesutan, I.; Towhid, S.T.; Qadri, S.M.; Foller, M.; Gawaz, M.; Lang, F. Dynamic adhesion of eryptotic erythrocytes to endothelial cells via CXCL16/SR-PSOX. Am. J. Physiol. Cell Physiol. 2012, 302, C644–C651. [Google Scholar] [CrossRef] [PubMed]
- Andrews, D.A.; Low, P.S. Role of red blood cells in thrombosis. Curr. Opin. Hematol. 1999, 6, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.M.; Bae, O.N.; Lim, K.M.; Noh, J.Y.; Lee, M.Y.; Jung, Y.S.; Chung, J.H. Lysophosphatidic acid induces thrombogenic activity through phosphatidylserine exposure and procoagulant microvesicle generation in human erythrocytes. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Zwaal, R.F.; Comfurius, P.; Bevers, E.M. Surface exposure of phosphatidylserine in pathological cells. Cell. Mol. Life Sci. 2005, 62, 971–988. [Google Scholar] [CrossRef] [PubMed]
- Closse, C.; Dachary-Prigent, J.; Boisseau, M.R. Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 1999, 107, 300–302. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.G.; Chang, S.H.; Rettig, M.P.; Neely, J.E.; Hillery, C.A.; Smith, B.D.; Low, P.S. Altered erythrocyte endothelial adherence and membrane phospholipid asymmetry in hereditary hydrocytosis. Blood 2003, 101, 4625–4627. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, A.; di Pietro, N.; Sirolli, V.; Giardinelli, A.; di Silvestre, S.; Amoroso, L.; di Tomo, P.; Capani, F.; Consoli, A.; Bonomini, M. Mechanisms of uremic erythrocyte-induced adhesion of human monocytes to cultured endothelial cells. J. Cell. Physiol. 2007, 213, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Wood, B.L.; Gibson, D.F.; Tait, J.F. Increased erythrocyte phosphatidylserine exposure in sickle cell disease: Flow-cytometric measurement and clinical associations. Blood 1996, 88, 1873–1880. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bissinger, R.; Waibel, S.; Lang, F. Induction of Suicidal Erythrocyte Death by Nelfinavir. Toxins 2015, 7, 1616-1628. https://doi.org/10.3390/toxins7051616
AMA Style
Bissinger R, Waibel S, Lang F. Induction of Suicidal Erythrocyte Death by Nelfinavir. Toxins. 2015; 7(5):1616-1628. https://doi.org/10.3390/toxins7051616
Chicago/Turabian StyleBissinger, Rosi, Sabrina Waibel, and Florian Lang. 2015. "Induction of Suicidal Erythrocyte Death by Nelfinavir" Toxins 7, no. 5: 1616-1628. https://doi.org/10.3390/toxins7051616