
Snake and Spider Toxins Induce a Rapid Recovery of Function of Botulinum Neurotoxin Paralysed Neuromuscular Junction

 ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. α -Latrotoxin or β–Bungarotoxin Injection Accelerates the Recovery from BoNTs-Induced Paralysis
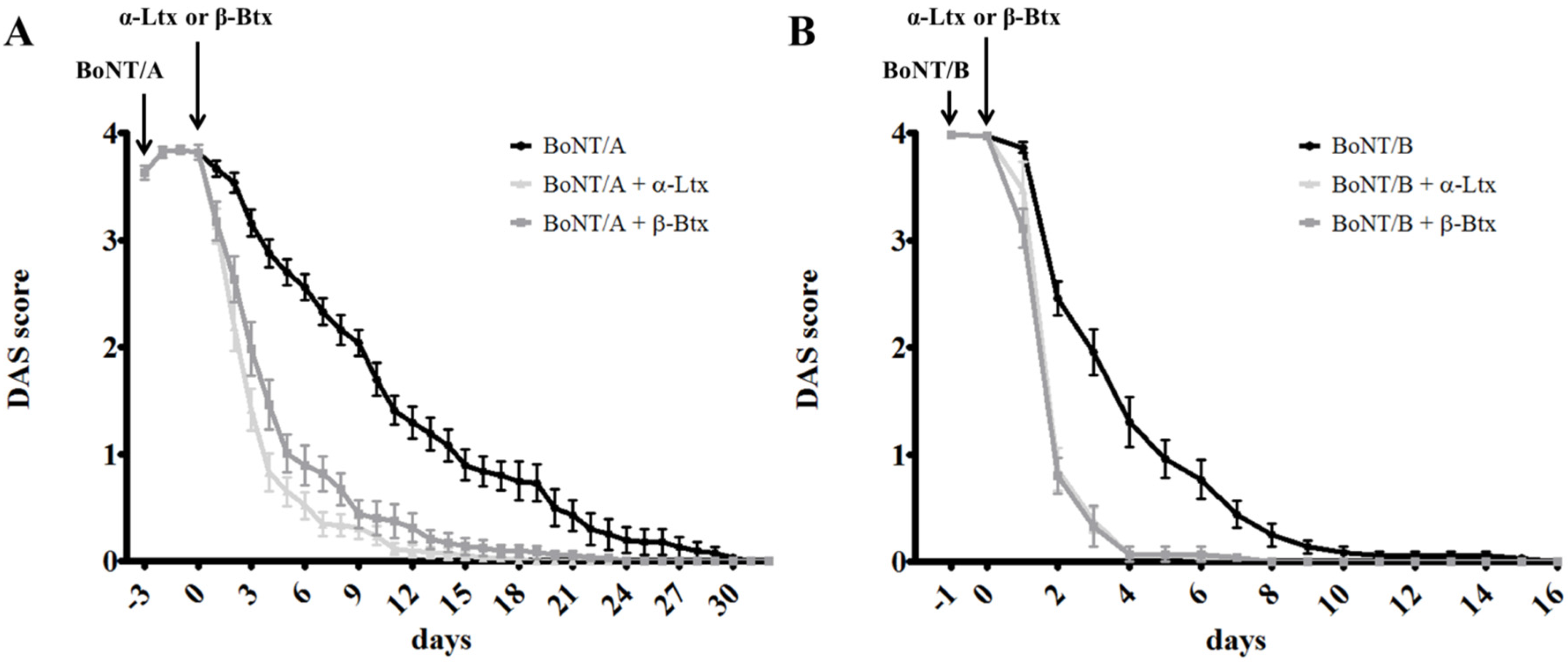
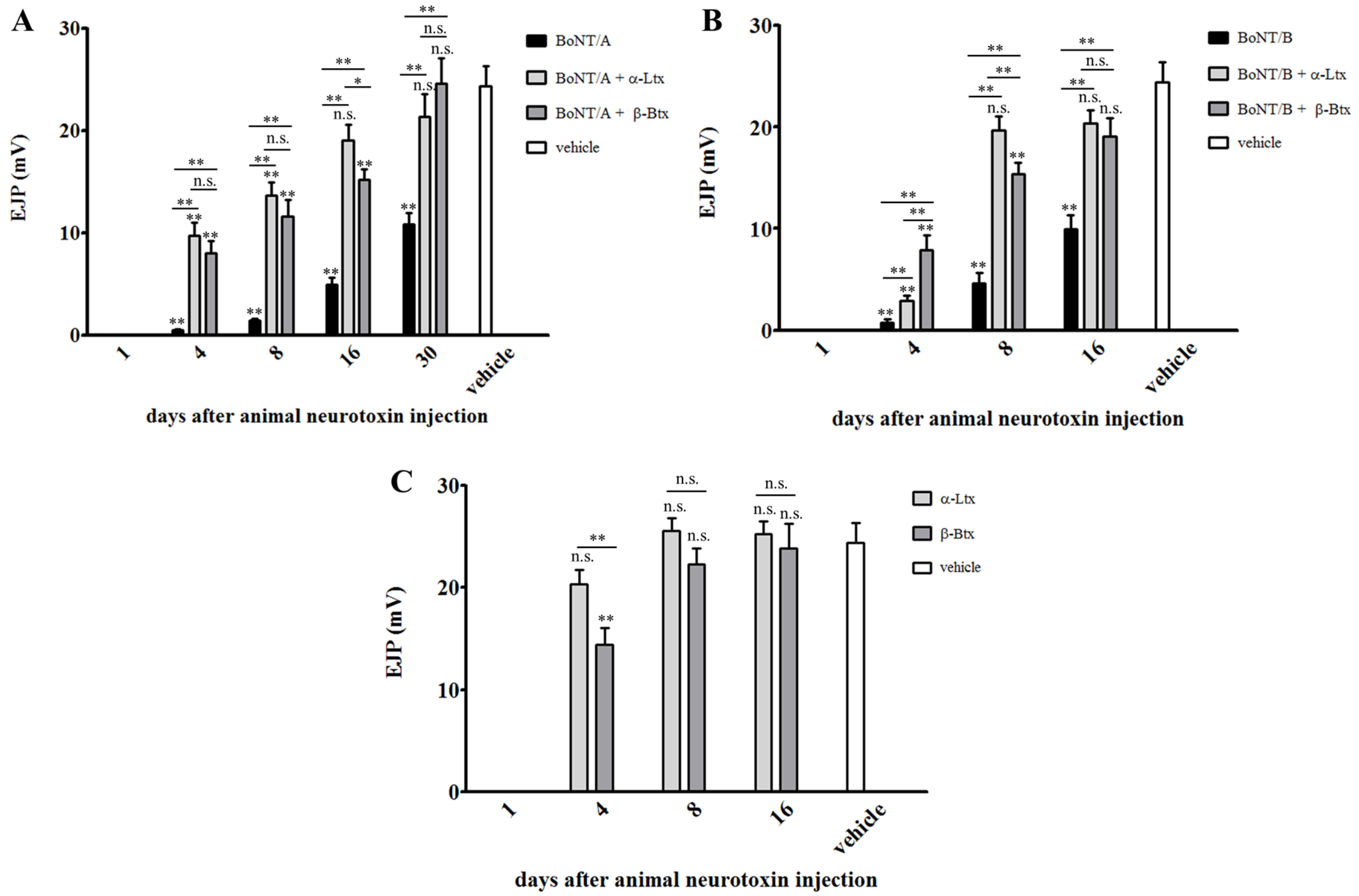
2.2. Synaptic Activity of BoNT-Paralyzed Muscles is Restored Earlier Following α-Latrotoxin or β-Bungarotoxin Injection
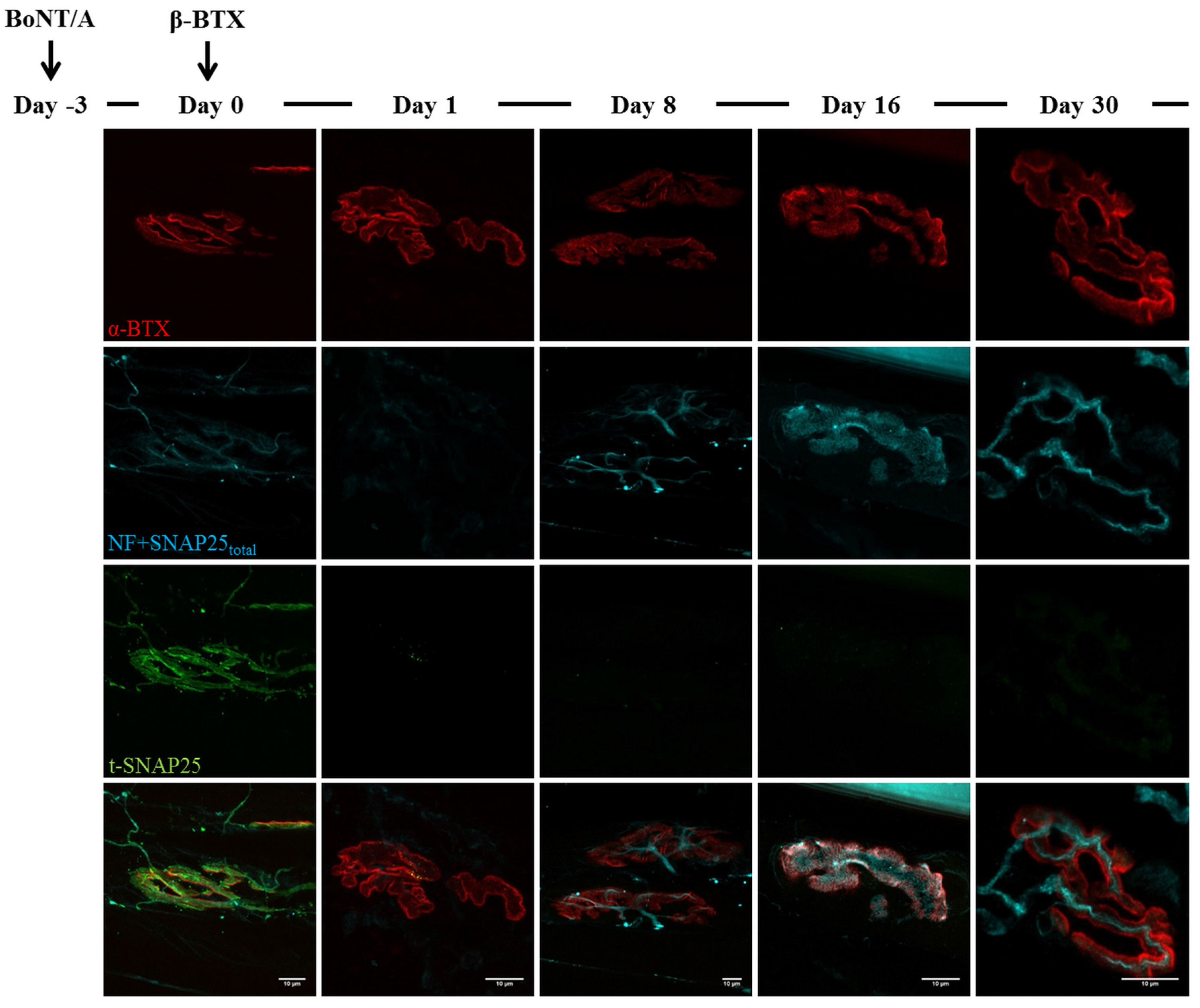
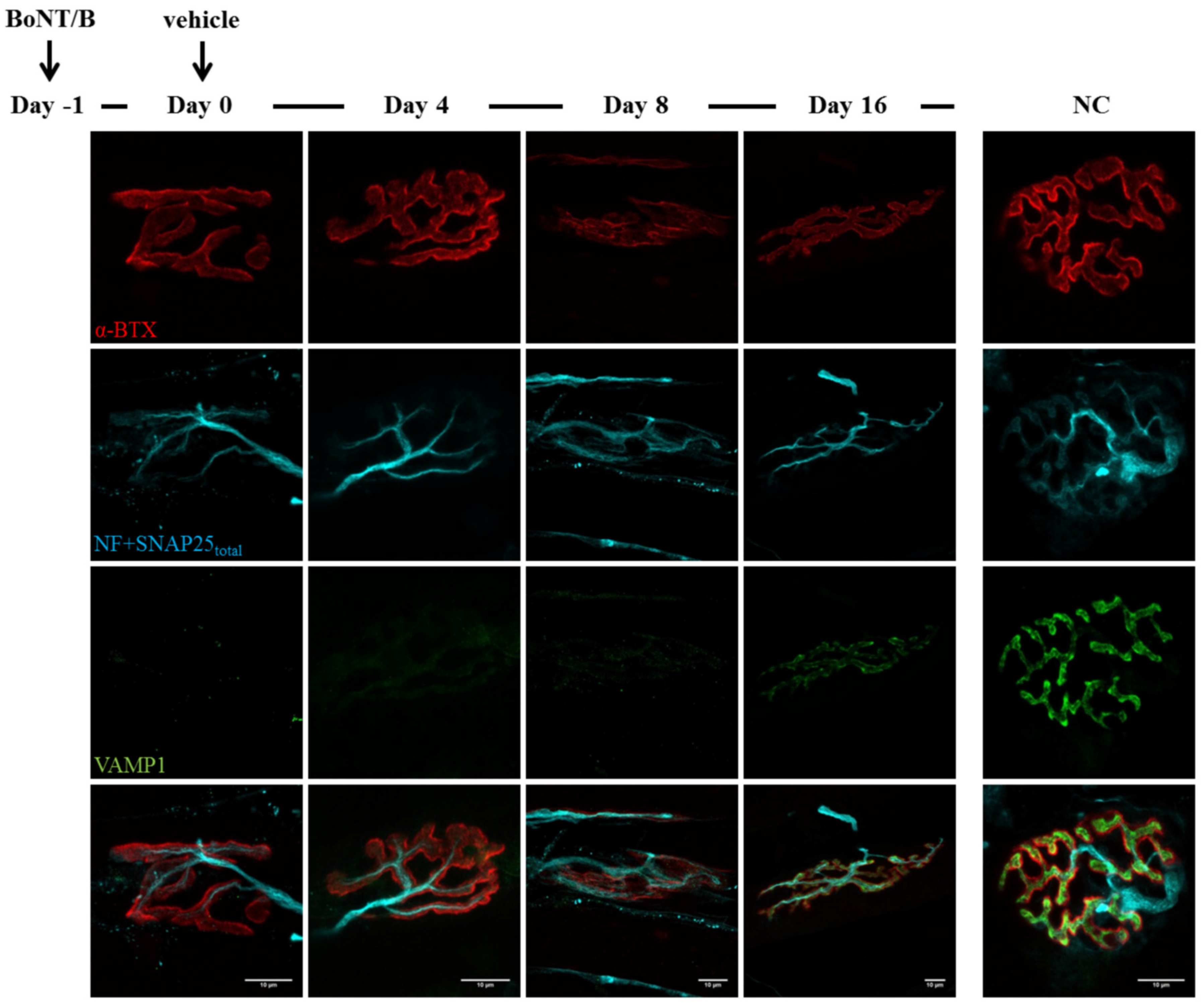
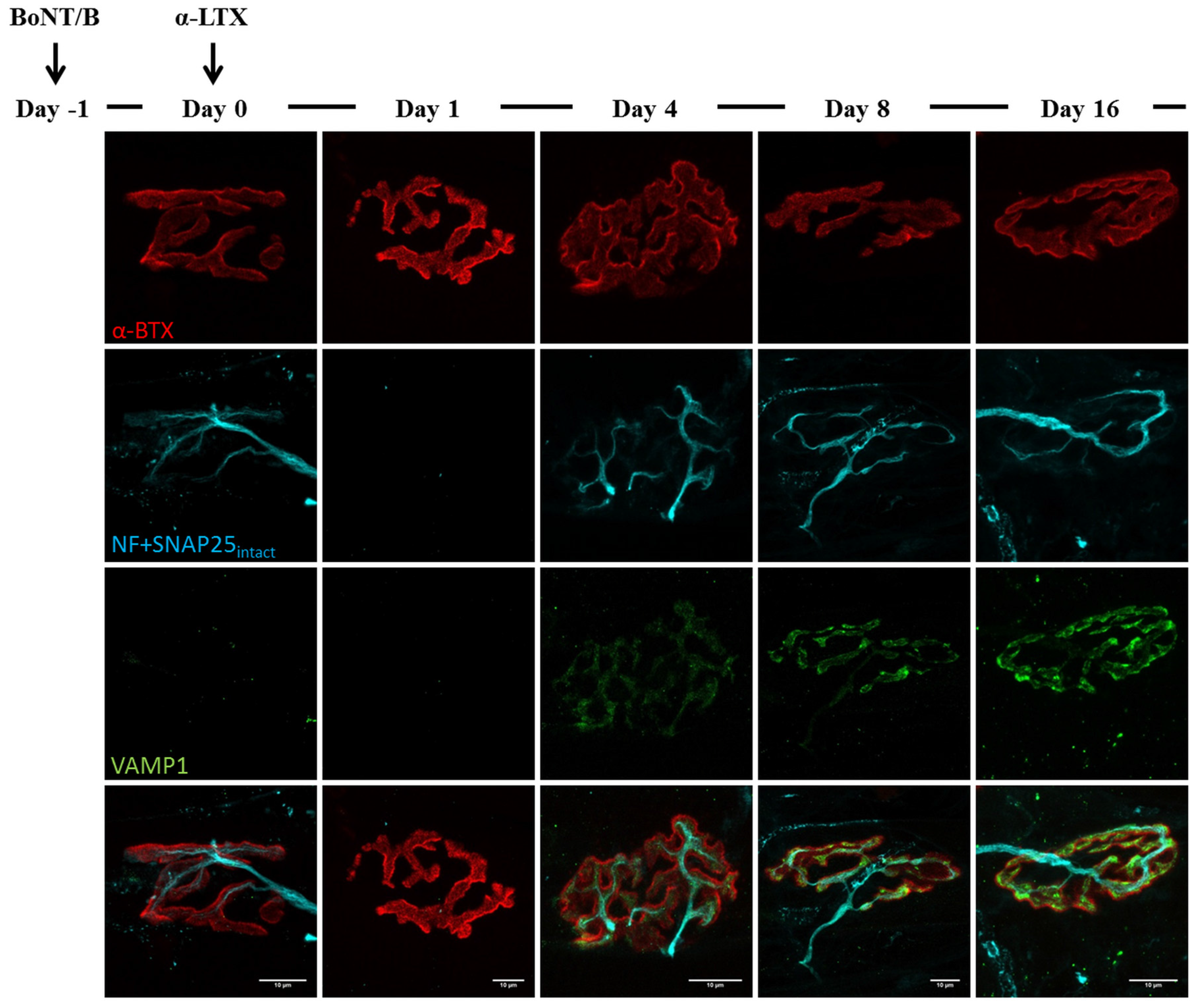
2.3. Fluorescence Microscopy Analysis of SNAP25 and VAMP1 Turn-Over at Single- or Double-Poisoned NMJs
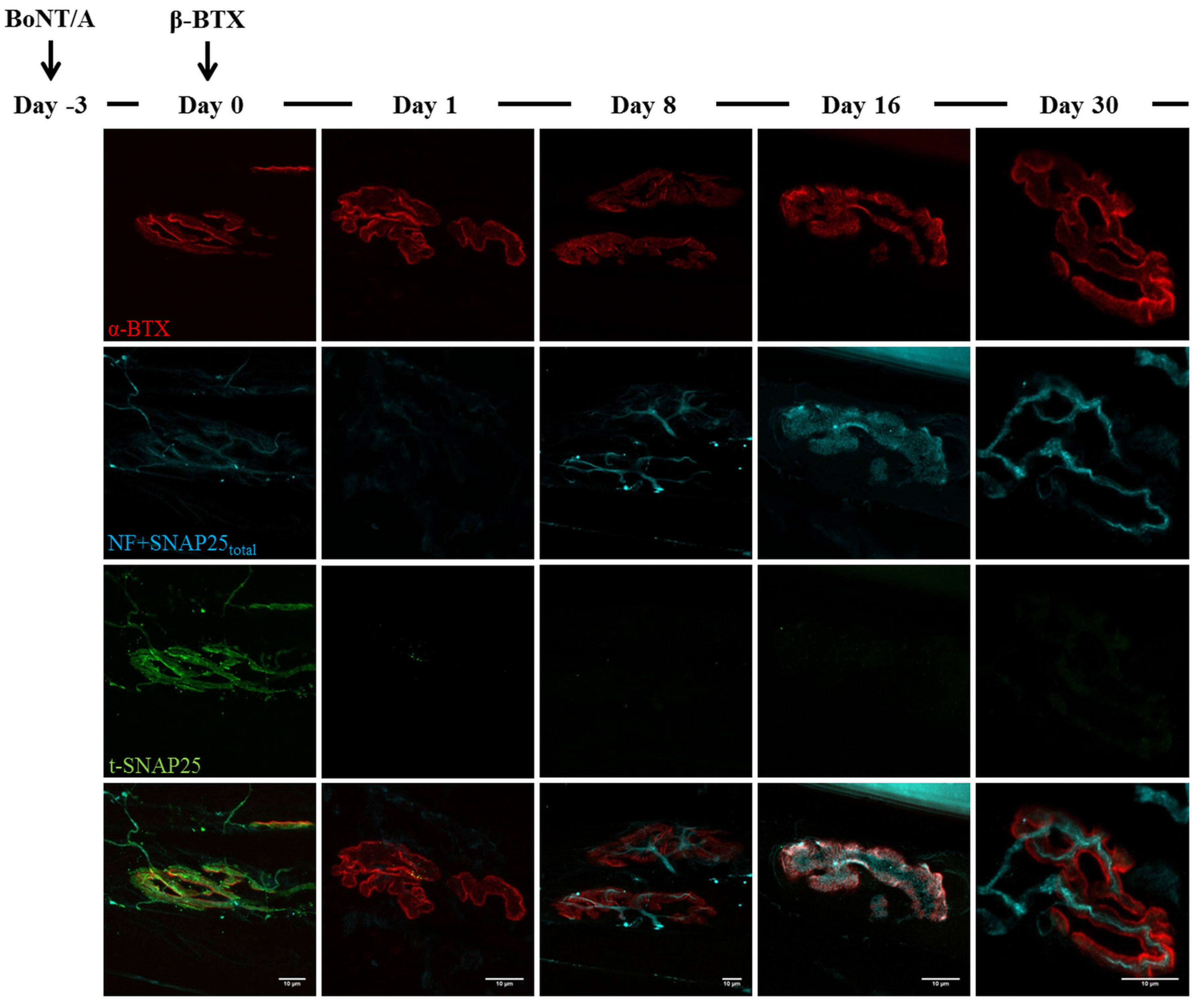

3. Discussion
4. Experimental Section
4.1. Animals and Toxins
4.2. Digit Abduction Score Assay (DAS)
4.3. Electrophysiological Recordings (ER)
4.4. NMJ Immunohistochemistry (IHC)
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Johnson, E.A.; Montecucco, C. Botulism. Handb. Clin. Neurol. 2008, 91, 333–368. [Google Scholar] [PubMed]
- Rossetto, O.; Pirazzini, M.; Montecucco, C. Botulinum neurotoxins: Genetic, structural and mechanistic insights. Nat. Rev. Microbiol. 2014, 12, 535–549. [Google Scholar] [CrossRef] [PubMed]
- Binz, T.; Sikorra, S.; Mahrhold, S. Clostridial neurotoxins: Mechanism of snare cleavage and outlook on potential substrate specificity reengineering. Toxins 2010, 2, 665–682. [Google Scholar] [CrossRef] [PubMed]
- Keller, J.E.; Neale, E.A.; Oyler, G.; Adler, M. Persistence of botulinum neurotoxin action in cultured spinal cord cells. FEBS Lett. 1999, 456, 137–142. [Google Scholar] [CrossRef]
- Pantano, S.; Montecucco, C. The blockade of the neurotransmitter release apparatus by botulinum neurotoxins. Cell Mol. Life Sci. 2014, 71, 793–811. [Google Scholar] [CrossRef] [PubMed]
- Eleopra, R.; Tugnoli, V.; Rossetto, O.; Montecucco, C.; de Grandis, D. Botulinum neurotoxin serotype C: A novel effective botulinum toxin therapy in human. Neurosci. Lett. 1997, 224, 91–94. [Google Scholar] [CrossRef]
- Eleopra, R.; Tugnoli, V.; Rossetto, O.; de Grandis, D.; Montecucco, C. Different time courses of recovery after poisoning with botulinum neurotoxin serotypes A and E in humans. Neurosci. Lett. 1998, 256, 135–138. [Google Scholar] [CrossRef]
- Eleopra, R.; Tugnoli, V.; Quatrale, R.; Rossetto, O.; Montecucco, C. Different types of botulinum toxin in humans. Mov. Disord. 2004, 19 (Suppl. 8), S53–S59. [Google Scholar] [CrossRef] [PubMed]
- Meunier, F.A.; Lisk, G.; Sesardic, D.; Dolly, J.O. Dynamics of motor nerve terminal remodeling unveiled using snare-cleaving botulinum toxins: The extent and duration are dictated by the sites of SNAP-25 truncation. Mol. Cell Neurosci. 2003, 22, 454–466. [Google Scholar] [CrossRef]
- Torii, Y.; Goto, Y.; Takahashi, M.; Ishida, S.; Harakawa, T.; Sakamoto, T.; Kaji, R.; Kozaki, S.; Ginnaga, A. Quantitative determination of biological activity of botulinum toxins utilizing compound muscle action potentials (cmap), and comparison of neuromuscular transmission blockage and muscle flaccidity among toxins. Toxicon 2010, 55, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Duregotti, E.; Negro, S.; Scorzeto, M.; Zornetta, I.; Dickinson, B.C.; Chang, C.J.; Montecucco, C.; Rigoni, M. Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic schwann cells. Proc. Natl. Acad. Sci. USA 2015, 112, E497–E505. [Google Scholar] [CrossRef] [PubMed]
- Ushkaryov, Y.A.; Rohou, A.; Sugita, S. Alpha-latrotoxin and its receptors. Handb. Exp. Pharmacol. 2008, 184, 171–206. [Google Scholar]
- Südhof, T.C. Alpha-latrotoxin and its receptors: Neurexins and cirl/latrophilins. Annu. Rev. Neurosci. 2001, 24, 933–962. [Google Scholar] [CrossRef] [PubMed]
- Dixon, R.W.; Harris, J.B. Nerve terminal damage by beta-bungarotoxin: Its clinical significance. Am. J. Pathol. 1999, 154, 447–455. [Google Scholar] [CrossRef]
- Rigoni, M.; Caccin, P.; Gschmeissner, S.; Koster, G.; Postle, A.D.; Rossetto, O.; Schiavo, G.; Montecucco, C. Equivalent effects of snake PLA2 neurotoxins and lysophospholipid-fatty acid mixtures. Science 2005, 310, 1678–1680. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Matteoli, M.; Montecucco, C. Neurotoxins affecting neuroexocytosis. Physiol. Rev. 2000, 80, 717–766. [Google Scholar] [PubMed]
- Tedesco, E.; Rigoni, M.; Caccin, P.; Grishin, E.; Rossetto, O.; Montecucco, C. Calcium overload in nerve terminals of cultured neurons intoxicated by alpha-latrotoxin and snake PLA2 neurotoxins. Toxicon 2009, 54, 138–144. [Google Scholar] [PubMed]
- Pungercar, J.; Krizaj, I. Understanding the molecular mechanism underlying the presynaptic toxicity of secreted phospholipases A2. Toxicon 2007, 50, 871–892. [Google Scholar] [CrossRef] [PubMed]
- Rigoni, M.; Pizzo, P.; Schiavo, G.; Weston, A.E.; Zatti, G.; Caccin, P.; Rossetto, O.; Pozzan, T.; Montecucco, C. Calcium influx and mitochondrial alterations at synapses exposed to snake neurotoxins or their phospholipid hydrolysis products. J. Biol. Chem. 2007, 282, 11238–11245. [Google Scholar] [CrossRef] [PubMed]
- Duregotti, E.; Tedesco, E.; Montecucco, C.; Rigoni, M. Calpains participate in nerve terminal degeneration induced by spider and snake presynaptic neurotoxins. Toxicon 2013, 64, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Kularatne, S.A.; Senanayake, N. Venomous snake bites, scorpions, and spiders. Handb. Clin. Neurol. 2014, 120, 987–1001. [Google Scholar] [PubMed]
- Duchen, L.W.; Gomez, S.; Queiroz, L.S. The neuromuscular junction of the mouse after black widow spider venom. J. Physiol. 1981, 316, 279–291. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.T.; Sanes, J.R.; Lichtman, J.W. Pre-existing pathways promote precise projection patterns. Nat. Neurosci. 2002, 5, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.J.; Trachtenberg, J.T.; Thompson, W.J. Schwann cells induce and guide sprouting and reinnervation of neuromuscular junctions. Trends Neurosci. 1996, 19, 280–285. [Google Scholar] [CrossRef]
- Pearn, J.H. Survival after snake-bite with prolonged neurotoxic envenomation. Med. J. Aust. 1971, 2, 259–261. [Google Scholar] [PubMed]
- Trevett, A.J.; Lalloo, D.G.; Nwokolo, N.C.; Naraqi, S.; Kevau, I.H.; Theakston, R.D.; Warrell, D.A. Electrophysiological findings in patients envenomed following the bite of a papuan taipan (oxyuranus scutellatus canni). Trans R. Soc. Trop. Med. Hyg. 1995, 89, 415–417. [Google Scholar] [CrossRef]
- Connolly, S.; Trevett, A.J.; Nwokolo, N.C.; Lalloo, D.G.; Naraqi, S.; Mantle, D.; Schofield, I.S.; Fawcett, P.R.; Harris, J.B.; Warrell, D.A. Neuromuscular effects of papuan taipan snake venom. Ann. Neurol. 1995, 38, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Kularatne, S.A. Common krait (bungarus caeruleus) bite in anuradhapura, sri lanka: A prospective clinical study, 1996–1998. Postgrad. Med. J. 2002, 78, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, C.; Molgó, J. Botulinal neurotoxins: Revival of an old killer. Curr. Opin. Pharmacol. 2005, 5, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, O.; Morbiato, L.; Caccin, P.; Rigoni, M.; Montecucco, C. Presynaptic enzymatic neurotoxins. J. Neurochem. 2006, 97, 1534–1545. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Rossi, C.; Gianfranceschi, L.; Rossetto, O.; Caleo, M. Long-distance retrograde effects of botulinum neurotoxin A. J. Neurosci. 2008, 28, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Roger Aoki, K. Botulinum neurotoxin serotypes A and B preparations have different safety margins in preclinical models of muscle weakening efficacy and systemic safety. Toxicon 2002, 40, 923–928. [Google Scholar] [CrossRef]
- Simpson, L. The life history of a botulinum toxin molecule. Toxicon 2013, 68, 40–59. [Google Scholar] [CrossRef] [PubMed]
- Fagan, R.P.; McLaughlin, J.B.; Middaugh, J.P. Persistence of botulinum toxin in patients’ serum: Alaska, 1959–2007. J. Infect. Dis. 2009, 199, 1029–1031. [Google Scholar] [CrossRef] [PubMed]
- Megighian, A.; Zordan, M.; Pantano, S.; Scorzeto, M.; Rigoni, M.; Zanini, D.; Rossetto, O.; Montecucco, C. Evidence for a radial snare super-complex mediating neurotransmitter release at the drosophila neuromuscular junction. J. Cell Sci. 2013, 126, 3134–3140. [Google Scholar] [CrossRef] [PubMed]
- Montecucco, C.; Schiavo, G.; Pantano, S. Snare complexes and neuroexocytosis: How many, how close? Trends Biochem. Sci. 2005, 30, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, O.; Gorza, L.; Schiavo, G.; Schiavo, N.; Scheller, R.H.; Montecucco, C. Vamp/synaptobrevin isoforms 1 and 2 are widely and differentially expressed in nonneuronal tissues. J. Cell Biol. 1996, 132, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Edelmann, L.; Jahn, R.; Dahlström, A. Axonal transport and distribution of synaptobrevin I and II in the rat peripheral nervous system. J. Neurosci. 1996, 16, 137–147. [Google Scholar] [PubMed]
- de Paiva, A.; Meunier, F.A.; Molgó, J.; Aoki, K.R.; Dolly, J.O. Functional repair of motor endplates after botulinum neurotoxin type A poisoning: Biphasic switch of synaptic activity between nerve sprouts and their parent terminals. Proc. Natl. Acad. Sci. USA 1999, 96, 3200–3205. [Google Scholar] [CrossRef] [PubMed]
- Vinsant, S.; Mansfield, C.; Jimenez-Moreno, R.; del Gaizo Moore, V.; Yoshikawa, M.; Hampton, T.G.; Prevette, D.; Caress, J.; Oppenheim, R.W.; Milligan, C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of als: Part II, results and discussion. Brain Behav. 2013, 3, 431–457. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.B.; de Winter, F.; Verhaagen, J. Als as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Front. Neurosci. 2014, 8, 252. [Google Scholar] [CrossRef] [PubMed]
- Plomp, J.J.; Willison, H.J. Pathophysiological actions of neuropathy-related anti-ganglioside antibodies at the neuromuscular junction. J. Physiol. 2009, 587, 3979–3999. [Google Scholar] [CrossRef] [PubMed]
- Kaida, K.; Kusunoki, S. Antibodies to gangliosides and ganglioside complexes in guillain-barré syndrome and fisher syndrome: Mini-review. J. Neuroimmunol. 2010, 223, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Brashear, A.; Lew, M.F.; Dykstra, D.D.; Comella, C.L.; Factor, S.A.; Rodnitzky, R.L.; Trosch, R.; Singer, C.; Brin, M.F.; Murray, J.J.; et al. Safety and efficacy of neurobloc (botulinum toxin type B) in type A-responsive cervical dystonia. Neurology 1999, 53, 1439–1446. [Google Scholar] [CrossRef] [PubMed]
- Abrams, S.B.; Hallett, M. Clinical utility of different botulinum neurotoxin preparations. Toxicon 2013, 67, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Dressler, D. Clinical applications of botulinum toxin. Curr. Opin. Microbiol. 2012, 15, 325–336. [Google Scholar] [CrossRef] [PubMed]
- Sutton, R.B.; Fasshauer, D.; Jahn, R.; Brunger, A.T. Crystal structure of a snare complex involved in synaptic exocytosis at 2.4 Å resolution. Nature 1998, 395, 347–353. [Google Scholar] [PubMed]
- Jahn, R.; Fasshauer, D. Molecular machines governing exocytosis of synaptic vesicles. Nature 2012, 490, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez, J.M.; Lomonte, B. Phospholipases A2: Unveiling the secrets of a functionally versatile group of snake venom toxins. Toxicon 2013, 62, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Stern, R.; Valjevac, K.; Dursum, K.; Ducic, V. Increased survival time in botulinum toxin poisoning by treatment with a venom gland extract from the black widow spider. Toxicon 1975, 13, 197–198. [Google Scholar] [CrossRef]
- Gomez, S.; Queiroz, L.S. The effects of black widow spider venom on the innervation of muscles paralysed by botulinum toxin. Q. J. Exp. Physiol. 1982, 67, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Thesleff, S.; Zelena, J.; Hofmann, W.W. Restoration of function in botulinum paralysis by experimental nerve regeneration. Proc. Soc. Exp. Biol. Med. 1964, 116, 19–20. [Google Scholar] [CrossRef] [PubMed]
- Duchen, L.W. The effects in the mouse of nerve crush and regneration on the innervation of skeletal muscles paralysed by clostridium botulinum toxin. J. Pathol. 1970, 102, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Mesngon, M.; McNutt, P. Alpha-latrotoxin rescues SNAP-25 from BoNT/A-mediated proteolysis in embryonic stem cell-derived neurons. Toxins 2011, 3, 489–503. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.; Montecucco, C. Tetanus and botulism neurotoxins: Isolation and assay. Methods Enzymol. 1995, 248, 643–652. [Google Scholar] [PubMed]
- Shone, C.C.; Tranter, H.S. Growth of clostridia and preparation of their neurotoxins. Curr. Top. Microbiol. Immunol. 1995, 195, 143–160. [Google Scholar] [PubMed]
- Rummel, A.; Mahrhold, S.; Bigalke, H.; Binz, T. The hcc-domain of botulinum neurotoxins A and B exhibits a singular ganglioside binding site displaying serotype specific carbohydrate interaction. Mol. Microbiol. 2004, 51, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.R. A comparison of the safety margins of botulinum neurotoxin serotypes A, B, and F in mice. Toxicon 2001, 39, 1815–1820. [Google Scholar] [CrossRef]
- Broide, R.S.; Rubino, J.; Nicholson, G.S.; Ardila, M.C.; Brown, M.S.; Aoki, K.R.; Francis, J. The rat digit abduction score (DAS) assay: A physiological model for assessing botulinum neurotoxin-induced skeletal muscle paralysis. Toxicon 2013, 71, 18–24. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duregotti, E.; Zanetti, G.; Scorzeto, M.; Megighian, A.; Montecucco, C.; Pirazzini, M.; Rigoni, M. Snake and Spider Toxins Induce a Rapid Recovery of Function of Botulinum Neurotoxin Paralysed Neuromuscular Junction. Toxins 2015, 7, 5322-5336. https://doi.org/10.3390/toxins7124887
Duregotti E, Zanetti G, Scorzeto M, Megighian A, Montecucco C, Pirazzini M, Rigoni M. Snake and Spider Toxins Induce a Rapid Recovery of Function of Botulinum Neurotoxin Paralysed Neuromuscular Junction. Toxins. 2015; 7(12):5322-5336. https://doi.org/10.3390/toxins7124887
Chicago/Turabian StyleDuregotti, Elisa, Giulia Zanetti, Michele Scorzeto, Aram Megighian, Cesare Montecucco, Marco Pirazzini, and Michela Rigoni. 2015. "Snake and Spider Toxins Induce a Rapid Recovery of Function of Botulinum Neurotoxin Paralysed Neuromuscular Junction" Toxins 7, no. 12: 5322-5336. https://doi.org/10.3390/toxins7124887