
Computational Studies of Venom Peptides Targeting Potassium Channels

Research School of Biology, Australian National University, Acton ACT 2601, Australia
*
Author to whom correspondence should be addressed.
Toxins 2015, 7(12), 5194-5211; https://doi.org/10.3390/toxins7124877
Submission received: 15 October 2015
/
Revised: 13 November 2015
/
Accepted: 20 November 2015
/
Published: 1 December 2015
(This article belongs to the Special Issue Animal Toxins and Biological Ion Channels)
Abstract
:Small peptides isolated from the venom of animals are potential scaffolds for ion channel drug discovery. This review article mainly focuses on the computational studies that have advanced our understanding of how various toxins interfere with the function of K+ channels. We introduce the computational tools available for the study of toxin-channel interactions. We then discuss how these computational tools have been fruitfully applied to elucidate the mechanisms of action of a wide range of venom peptides from scorpions, spiders, and sea anemone.

1. Overview of Ion Channels and Venom Peptides
Ion channels allow certain ions to move across cell membranes. They are involved in a range of physiological processes such as electrical signaling in muscles and neurons, and their malfunction is implicated in various human diseases. As such, ion channels are one of the most important drug targets [1,2,3]. For example, two isoforms of K+ channels, Kv1.3 and KCa3.1, are promising targets for the treatment of autoimmune diseases [4], whereas local anesthetics primarily target Na+ channels involved in pain pathways [5].
Animal venoms provide a rich resource from which modulators of a wide array of ion channels can be isolated. Many short peptides from scorpion venoms are potent and specific inhibitors of certain K+ channels. For example, charybdotoxin (ChTx) from the deathstalker scorpion is a potent blocker of two isoforms of K+ channels, Kv1.3 and KCa3.1, which are potential targets for immunosuppression [6]. The toxin ADWX-1 (Autoimmune Drug from Wen-Xin Group), developed by Li and coworkers [7], has picomolar affinities for Kv1.3 and yet it is more than 300-fold selective for this channel over two highly related channels, Kv1.1 and Kv1.2. Similarly, a family of small peptides isolated from the venom of cone snails, referred to as µ-conotoxins, are potent inhibitors of Na+ channels, which are targets for the treatment of pain [8,9]. These venom peptides are promising leads for drug development [10,11,12].
The structure-activity relationship of various toxins, especially the ones that are specific for K+ and Na+ channels, has been an area of intensive study. Several research teams have focused on the isolation and characterization of toxins from venomous animals, focusing on marine invertebrates [13], arachnids [14,15,16,17,18,19,20], and reptiles [21,22]. Li and coworkers have made many seminal contributions to our understanding of the mechanisms of action of scorpion toxins on voltage-gated K+ (Kv) channels [7,23,24,25,26,27,28,29,30,31]. They have isolated and characterized a large number of scorpion toxins and showed how one such toxin could be modified to enhance its potency and selectivity to Kv1.3 [7].
The direct interactions between a toxin and a channel are difficult to probe using available experimental methods but can be examined in atomic detail using computational methods. Therefore, computational tools have been used extensively in recent years to aid the interpretation of experimental data and guide the design of new experiments. Most of the computational studies reported have focused on K+ channels whose structures are better understood than other cation channels [32,33,34].
This review article primarily focuses on the computational approaches used to aid our understanding of how various toxins interfere with the function of Kv channels. The paper is organized as follows. We first describe briefly the general architecture of K+ channels and introduce computational tools commonly used to study toxin-channel interactions. We then show how these computational tools have been applied to elucidate the mechanisms of action of venom peptides on Kv channels. Finally, we discuss how the principles of toxin-channel interactions learned from those studies may be used for the development of toxin analogs as novel drug scaffolds.
2. Structure of K+ Channels
In the past two decades, the molecular structures of several isoforms of K+ channels, such as the bacterial K+ channel KcsA [35], the calcium-gated K+ channel MthK [36], the voltage-gated K+ channels KvAP [37] and Kv1.2 [38], and the inward-rectifying channel Kir2.2 [39], have been determined from crystallographic analyses. These structures allow the homology modeling of a broad range of K+ channels and enable detailed analysis of toxin-channel interactions using computational methods.
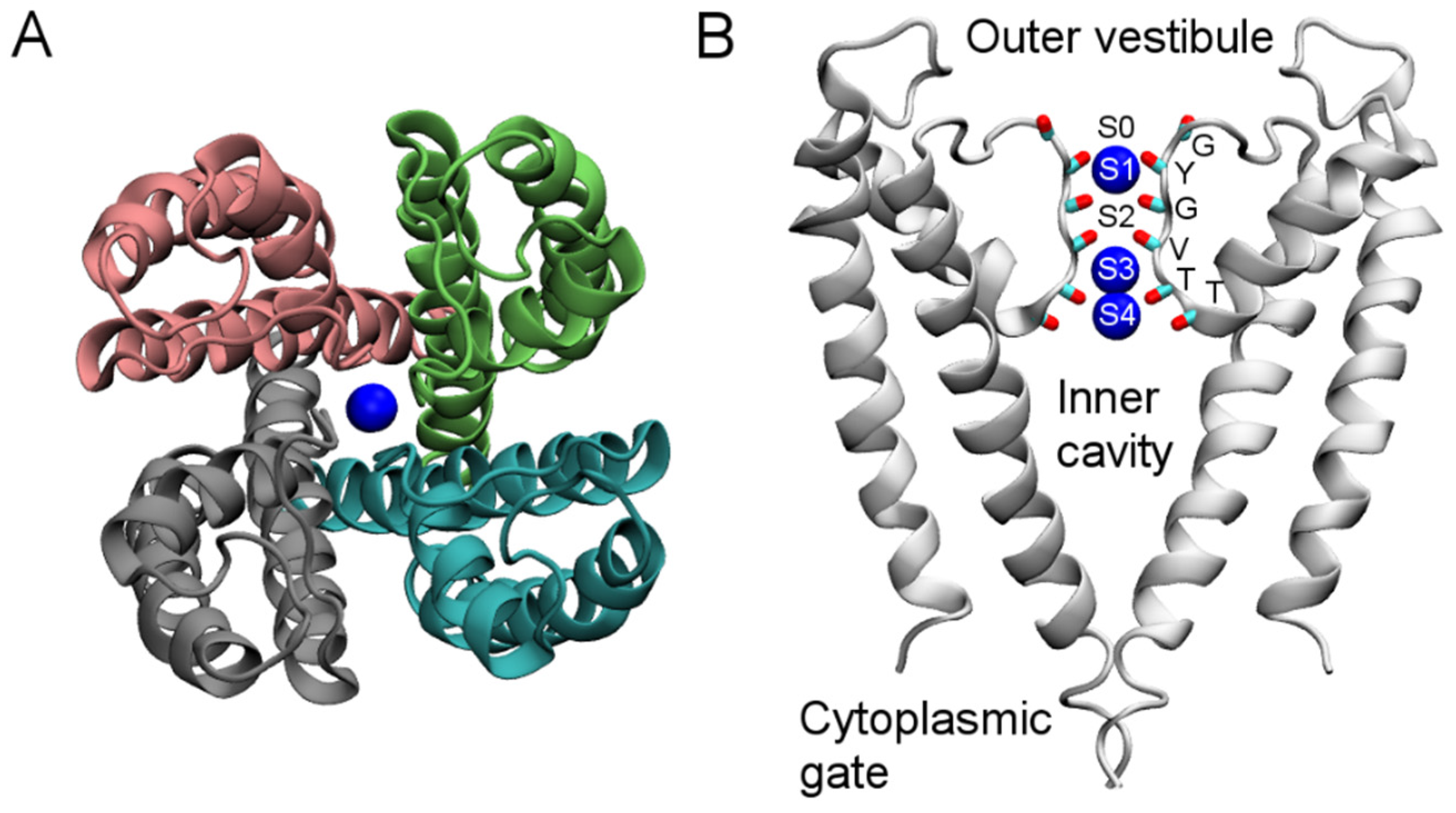
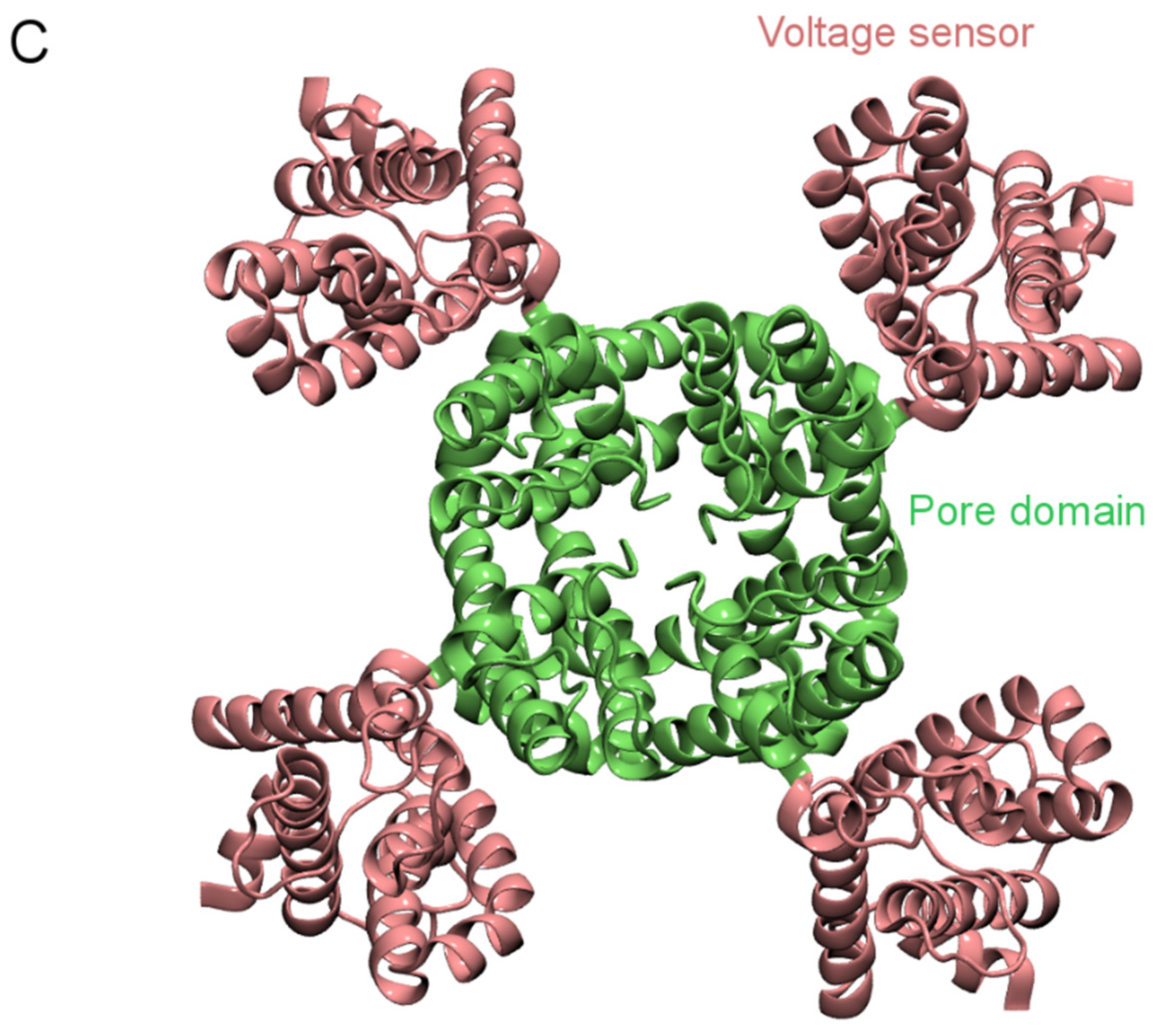
KcsA only has a pore domain consisting of four identical subunits (Figure 1A). The channel is composed of three segments. In the middle of the pore domain is the selectivity filter, approximately 12 Å in length and 2.8 Å in diameter. The filter is lined with the backbone carbonyl groups of six residues, TTVGYG, which is a fingerprint sequence and highly conserved across K+ channels (Figure 1B). The space between the planes of the six residues forms five K+ binding sites, designated as S0–S4. In the crystal structure, the S1, S3, and S4 sites are occupied by K+ ions (Figure 1B). Just below the selectivity filter is a water-filled inner cavity of ~5 Å in radius that extends toward the intracellular space (Figure 1B). The pore domain of Kv1.2 is similar to that of KcsA, but each subunit of Kv1.2 also carries a voltage sensor attached to the pore domain (Figure 1C). The voltage sensor moves within the membrane in response to changes in membrane potential, resulting in channel closing or opening. Venom peptides thus can interact with K+ channels in two different ways. They either bind to the wall of the external vestibule and the selectivity filter, thereby physically occluding the ionic pathway, or target the voltage sensor, thus interfering with the gating behavior of the channels. Recently, the first crystal structure of a pore-blocking scorpion toxin bound to a Kv channel was solved [40], providing excellent reference for both computational and further experimental studies.
Figure 1.
Structure of KcsA (PDB ID 1BL8 [35]) viewed parallel to the channel axis from the periplasmic side (A) and perpendicular to the channel axis (B). Blue spheres represent K+ ions. In (B), only two channel subunits are shown for clarity. In (C), the crystal structure of the Kv1.2-Kv2.1 paddle chimera channel (PDB ID 2R9R [41]) viewed from the periplasmic side along the channel axis is shown.
Figure 1.
Structure of KcsA (PDB ID 1BL8 [35]) viewed parallel to the channel axis from the periplasmic side (A) and perpendicular to the channel axis (B). Blue spheres represent K+ ions. In (B), only two channel subunits are shown for clarity. In (C), the crystal structure of the Kv1.2-Kv2.1 paddle chimera channel (PDB ID 2R9R [41]) viewed from the periplasmic side along the channel axis is shown.
3. Computational Methods
Parallel to the advances in understanding channel structures in atomic detail, there have also been important advances in computational biophysics. As new analytical methods have been developed and the available computational power increased, it became possible to unravel the direct interactions between toxins and ion channels using the state-of-the-art computational tools. Interacting residue pairs of a specific toxin and an ion channel can be determined, and their binding affinity can be calculated accurately.
The study of toxin-channel interactions typically consists of two steps. First, the most favorable binding modes between a toxin and a channel are predicted using different methods such as molecular docking [30], Brownian dynamics (BD) [42] and molecular dynamics (MD) [43], often with the help of available experimental data. Then, the dissociation constant (Kd) is calculated as a means to validate the binding modes predicted. In electrophysiological experiments, the half-maximal inhibitory concentration (IC50) or the half-maximal effective concentration (EC50), instead of Kd, is typically determined. IC50 and EC50 measure how strongly a toxin modulates the gating of a channel, whereas Kd reflects how strongly a toxin binds to a channel. Thus, IC50 (or EC50) and Kd are different quantities by definition. However, the binding of a toxin to a channel usually implies modulation to the channel activity. Therefore, IC50 (or EC50) and Kd are expected to be in the same order in these systems, and have been used interchangeably in literature.
3.1. Prediction of Binding Modes
The three most widely used computational methods for the prediction of toxin-channel complex structures, namely, molecular docking, Brownian dynamics (BD), and molecular dynamics (MD), are compared in Table 1. Molecular docking is a computational algorithm that aims to predict complexes between the ligand (toxin) and the receptor (ion channel). Compared with MD, docking methods utilize various ligand sampling techniques such as shape matching, systematic search, and stochastic sampling to enable the rapid prediction of a large number of likely toxin-channel structures with low computational cost. The flexibility of channel and toxin is ignored in rigid-body docking for computational efficiency, although some docking methods allow limited flexibility [44]. The output of docking calculations is a list of docked complexes or poses of the toxin with the channel protein, and the poses are ranked according to an empirical function. For a detailed review of molecular docking algorithms, the reader is referred to Huang et al. [45].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Comparison of three computational methods: docking, Brownian dynamics (BD) and molecular dynamics (MD).
| Items | Docking | BD | MD |
|---|---|---|---|
| Water | Implicit | Implicit | Explicit |
| Ions | Ignored | Explicit | Explicit |
| Membrane | Ignored | Implicit | Explicit |
| Flexibility | Ignored/Limited | Ignored | Allowed |
| Output trajectory | No | Yes | Yes |
| Time scale | - | Microseconds | Nanoseconds |
In BD, two simplifying assumptions are made. First, the water is treated as a continuum. Instead of simulating water molecules explicitly, as in MD, the effects of incessant collisions between ions and water molecules are lumped together, and are represented as frictional and random forces. Second, the internal degrees of freedom in the ion channel and the toxin are ignored. The channel is placed at a fixed position, while the tumbling and translocation but not conformational changes of the toxin are allowed. Therefore, similar to docking methods, the predictions of BD are influenced by the structures of the channel and the toxin used.
In classic MD, the trajectories of N particles interacting via a many-body potential are traced using Newton’s equation of motion. All the molecules in the simulation assembly, including water molecules and ions, are represented explicitly. The interactions between all atoms or particles in the system are described by an empirical potential energy function. This potential function contains harmonic terms for bonds and angles, a cosine expansion for torsion angles, and Lennard-Jones and Coulomb interactions for non-bonded interactions. The potential energy function, together with a set of parameters such as equilibrium bond lengths and angles required for describing interatomic interactions, are referred to as a force field. The accuracy of the force field is essential for a realistic simulation of a biomolecular system. The primary result of MD simulations is a trajectory of all particles in time, from which specific details of the assembly can be analyzed. Thus, the motion of each particle can be followed as it moves in real time on a timescale up to a microsecond, although longer simulations have also been reported.
Once the toxin-channel bound complex is determined, using any one of the three procedures described above, the system is further equilibrated with MD to allow the complex to evolve to a stable state. Atoms can be followed as they move in real time and thus it is possible to identify amino acid side chains on the toxin forming hydrogen bonds with those on the channel, as well as the residue pairs making hydrophobic interactions. This information can be used to validate the binding mode predicted computationally against available experimental data and guide the design of further mutagenesis experiments. Furthermore, the binding affinity (Kd) can be calculated and compared to the IC50 or EC50 values determined experimentally to further verify the predictions.
Although MD allows the interactions and dynamics in a molecular system to be examined in atomic detail, it has several important limitations. First, it requires sufficient experimental data to be available for constructing the molecular system and validating the computational predictions. However, our knowledge on the structure of ion channels is still limited. For example, no crystal structure for a mammalian Na+ and Ca2+ channel has been reported. Second, the accuracy of its predictions is limited by the quality of the force field used to describe the interatomic forces in the system. Although the force fields for biomolecular simulations have evolved over the past decade, certain aspects such as the treatment of atomic polarizability are yet to be implemented in any of the most widely used biomolecular force fields such as CHARMM and GROMOS. Third, the time scale of the MD simulation that can be achieved is limited by the computational resources available. For a comprehensive discussion on the limitations of MD, see van Gunsteren et al. [46].
3.2. Calculation of Kd
One of the major aims of computational studies is to calculate Kd accurately, because it is not only a central quantity for describing toxin block, but also the only quantity that is directly accessible to available experimental techniques. The potential of mean force (PMF) profile of toxin binding to a channel, which provides the work required to move a toxin along the channel axis, can be used to derive the Kd value of toxin binding. Typically, the well in the PMF profile points to the binding site in the channel and the depth of the profile largely determine the Kd. The formal link between the PMF profile and Kd has been derived using different statistical mechanical methods [32,47]. The Kd can be calculated from the PMF, W(z), using the following equation:
where πR2 is the cross-sectional area sampled by the center of mass (COM) of the toxin, NA is Avogadro’s number, zmin and zmax are the z positions of the toxin COM when the toxin is fully bound to the channel and in the bulk, respectively.
The PMF can be calculated most simply from the definition of work as a line integral of force. In the simulation the COM of the toxin is constrained with a spring constant to a reference point that moves slowly with a constant velocity. The work performed by the spring as a function of the toxin position is obtained. The simulation is then repeated many times and the ensemble average of the work is used to construct the PMF profile according to Jarzynski’s equality [48]. The reference point must move slow enough such that the work performed is reversible and can be used to estimate the PMF accurately. If the reference point moves too fast, the work would be too large and not measure the PMF correctly [49].
In order to construct the PMF profile efficiently, the umbrella sampling technique is widely used. From the bound position to the channel, the toxin is slowly pulled out along the channel axis. A series of structures with the COM of the toxin spaced at small intervals (typically 0.5 Å) is generated. Each of these structures is viewed as an umbrella window and simulated independently. In each umbrella window the COM of the toxin is harmonically restrained to the starting position, so that the sampling of toxin COM is focused on a narrow region. The COM of the toxin is also restrained to a small cylinder (typically 8 Å in radius) centered on the channel axis using a flat-bottom harmonic potential. The cylinder must be wide enough such that the restraining potential is always zero when the toxin is bound to the channel and does not interfere with sampling. The distribution of toxin COM can be derived from each simulation and then unbiased to obtain the correct PMF profile [50].
At the fully bound position, the degrees of freedom of the toxin are restricted by the toxin-channel interactions. Therefore, the PMF of the toxin would largely be determined by the interactions in the complex predicted. If the complex predicted is the most favorable one, the Kd value predicted from the PMF profile should be in close proximity to the IC50 value determined experimentally. On the other hand, if the complex is not favorable, the experimental IC50 value would not be reproduced. Thus, calculations of IC50 can help verify the binding mode predicted.
4. Venom Peptides
4.1. Scorpion Toxins
Many small peptides of 30–70 residues from scorpion venoms have been isolated and characterized. These peptides can be broadly classified into two classes: pore blockers of K+ channels and gating modifiers of Na+ channels. The pore blocking peptides are typically 30–40 residues in length. They bind to the outer vestibule and physically occlude the selectivity filter of the channel, thereby inhibiting ion permeation. On the other hand, scorpion α- and β-toxins, which are well characterized gating modifiers of Na+ channels, typically comprise of 60–70 residues. Scorpion α- and β-toxins bind to the voltage sensor of the channel and modulate the movement of the voltage sensor in response to changes in membrane potential.
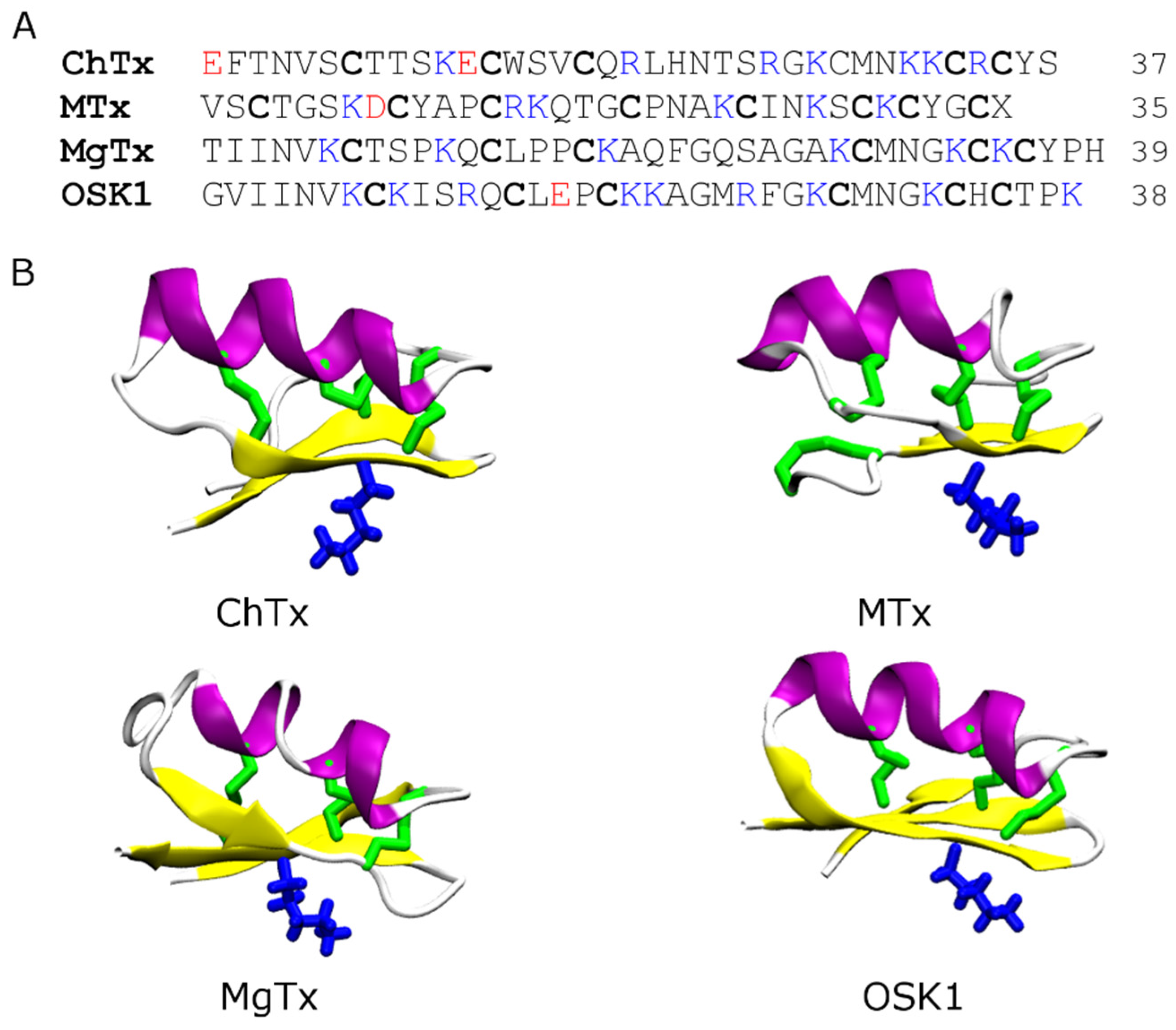
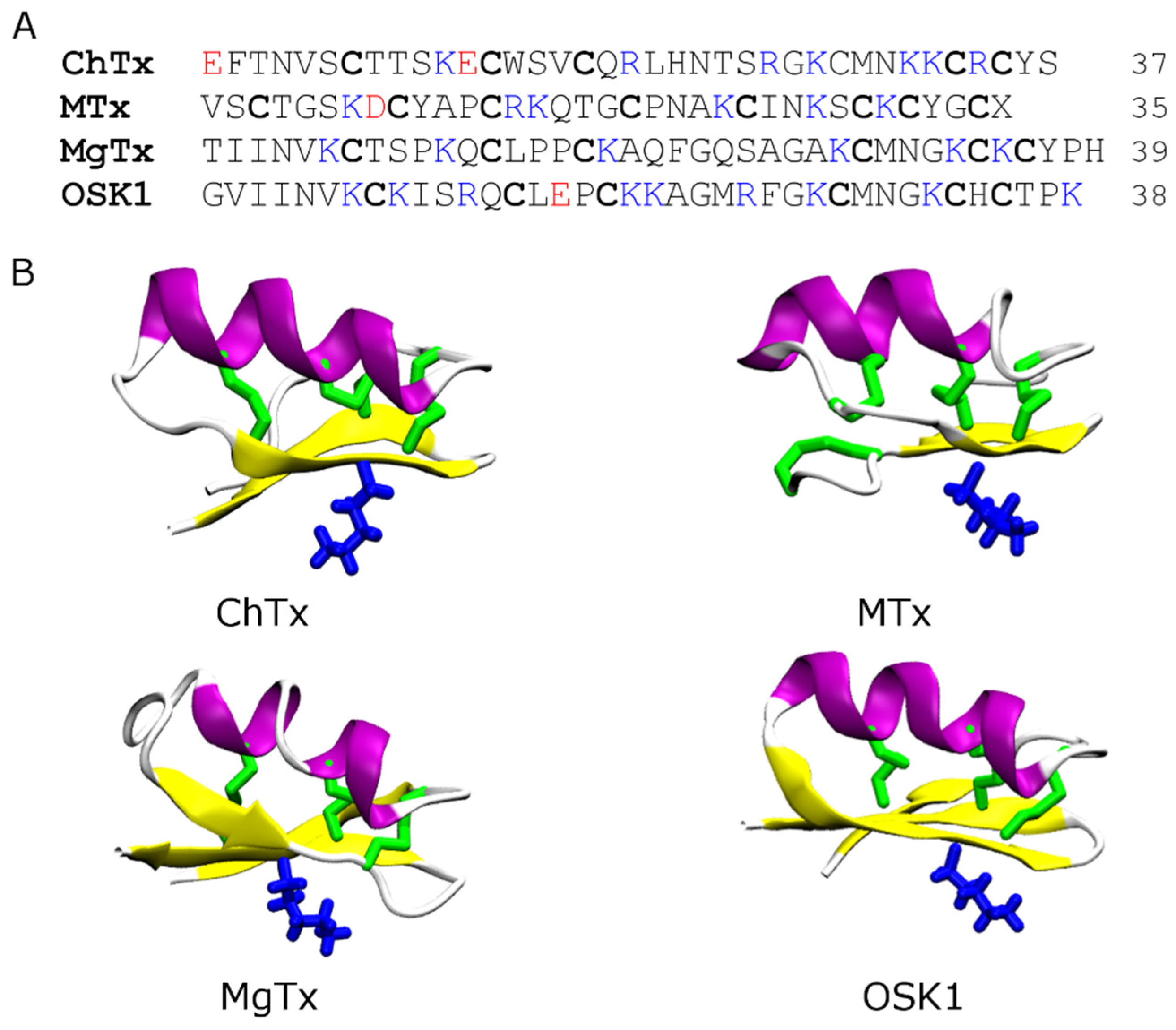
The majority of the scorpion toxins that have been tested on ion channels belong to the pore blockers category. Among the most well studied are charybdotoxin (ChTx) [51], maurotoxin (MTx) [52], margatoxin (MgTx) [53], OSK1 [54], and ADWX-1 [7]. All these toxins are potent blockers of Kv1.3, a target for autoimmune diseases [6]. They share a common structural scaffold consisting of an α-helix and two or three anti-parallel β-strands (Figure 2). They all carry six or eight cysteine residues forming three or four disulfide bridges, and multiple lysine and arginine residues that are positively charged at neutral pH. However, several scorpion toxins of different structural folds have also been shown to have inhibitory effects on K+ channels [25,55]. For example, Li and coworkers discovered that the scorpion toxin Hg1 [25], carrying two anti-parallel β-strands and one α-helix consistent with the Kunitz type fold, is a potent inhibitor of Kv1.3 (Kd = 6 nM).
Figure 2.
Primary (A) and secondary (B) structures of four scorpion toxins: charybdotoxin (ChTx, PDB ID 2CRD [56]), maurotoxin (MTx, PDB ID 1TXM [57]), margatoxin (MgTx, PDB ID 1MTX [58]) and OSK1 (PDB ID 1SCO [59]). The disulfide bridges are shown in green and the key pore-blocking lysine residue in blue. The α-helix is shown in purple and β-strand in yellow.
Figure 2.
Primary (A) and secondary (B) structures of four scorpion toxins: charybdotoxin (ChTx, PDB ID 2CRD [56]), maurotoxin (MTx, PDB ID 1TXM [57]), margatoxin (MgTx, PDB ID 1MTX [58]) and OSK1 (PDB ID 1SCO [59]). The disulfide bridges are shown in green and the key pore-blocking lysine residue in blue. The α-helix is shown in purple and β-strand in yellow.
The pore-blocking mechanisms of scorpion toxins for K+ channels have been examined extensively using computational methods [29,30,42,43,60,61,62,63,64,65,66,67,68,69,70]. In these studies different methods such as molecular docking [30,66,69], Brownian dynamics [42,60], and MD simulation with distance restraints [43,67,68,70] were used to predict the binding modes between the toxin and the channel. Although the methods are of a different level of complexity and accuracy, the general principles for toxin-channel association predicted from these methods were consistent. In general, the key lysine residue from the middle of the β-strand of the toxin, which is preceded by the α-helix (Figure 2), protrudes into the selectivity filter of the channel, thereby physically occluding the permeation pathway. Additional hydrophobic and electrostatic contacts are also formed to stabilize the toxin-channel complex (Figure 3).
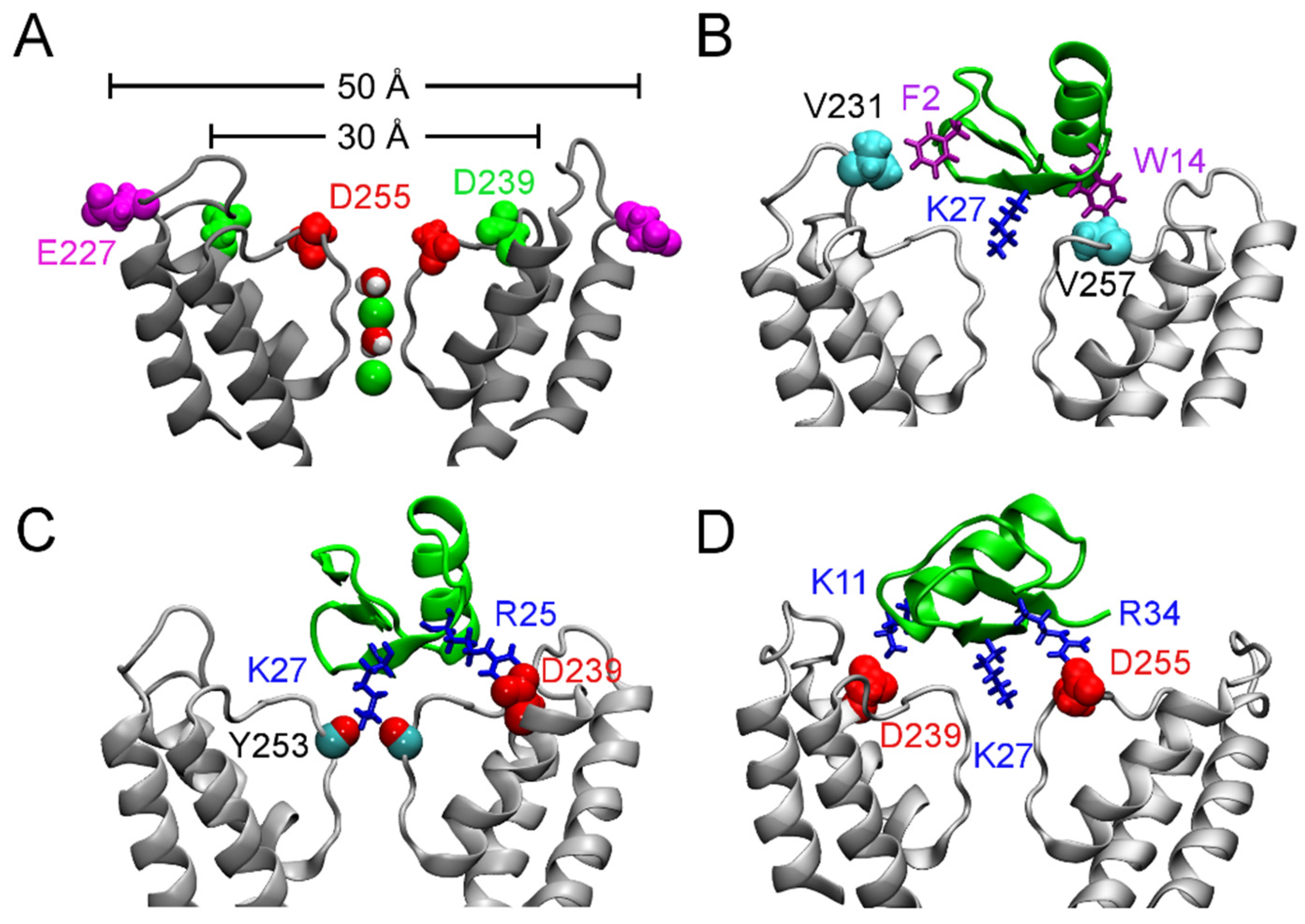
Figure 3.
Binding of ChTx to the Ca2+-activated K+ channel of intermediate conductance, KCa3.1. (A) Structure of KCa3.1 modeled on Kv1.2 viewed perpendicular to the channel axis. Only two of the four subunits are shown for clarity. The position of three acidic residues relevant for scorpion toxin binding are highlighted. (B–D) ChTx (green) bound to KCa3.1, with the most important contacts highlighted. Adapted from Chen et al. [70], Copyright 2013, Elsevier.
Figure 3.
Binding of ChTx to the Ca2+-activated K+ channel of intermediate conductance, KCa3.1. (A) Structure of KCa3.1 modeled on Kv1.2 viewed perpendicular to the channel axis. Only two of the four subunits are shown for clarity. The position of three acidic residues relevant for scorpion toxin binding are highlighted. (B–D) ChTx (green) bound to KCa3.1, with the most important contacts highlighted. Adapted from Chen et al. [70], Copyright 2013, Elsevier.
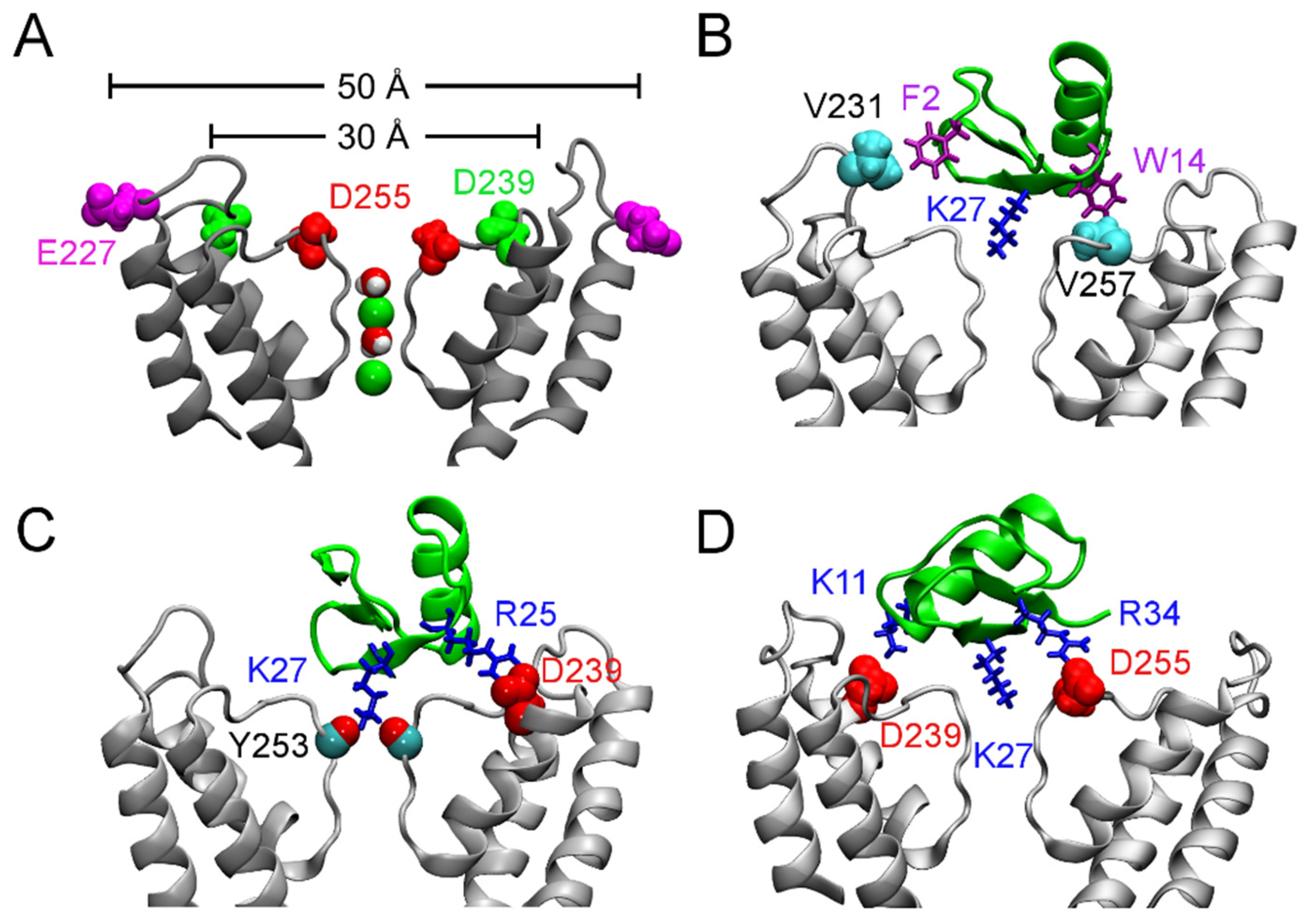
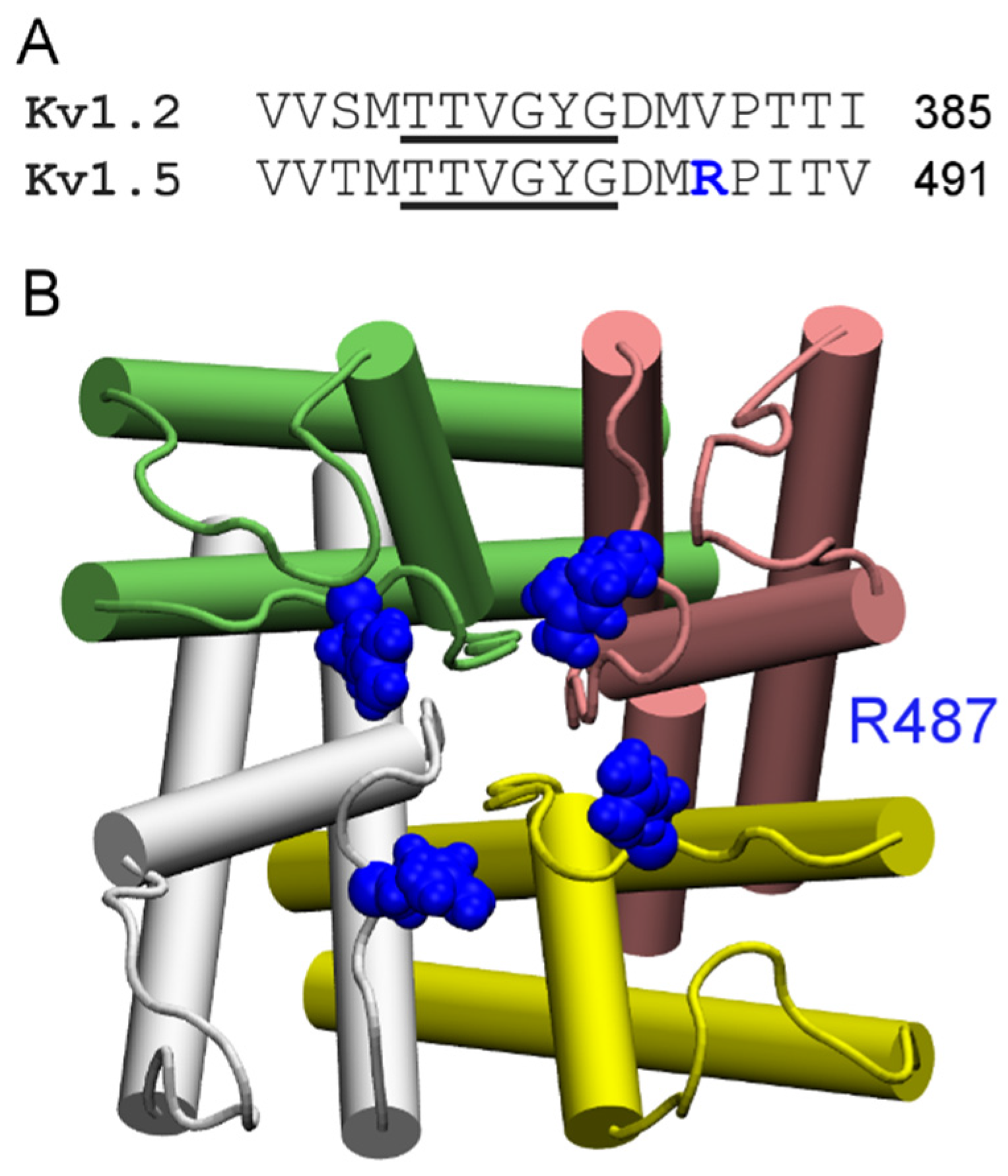
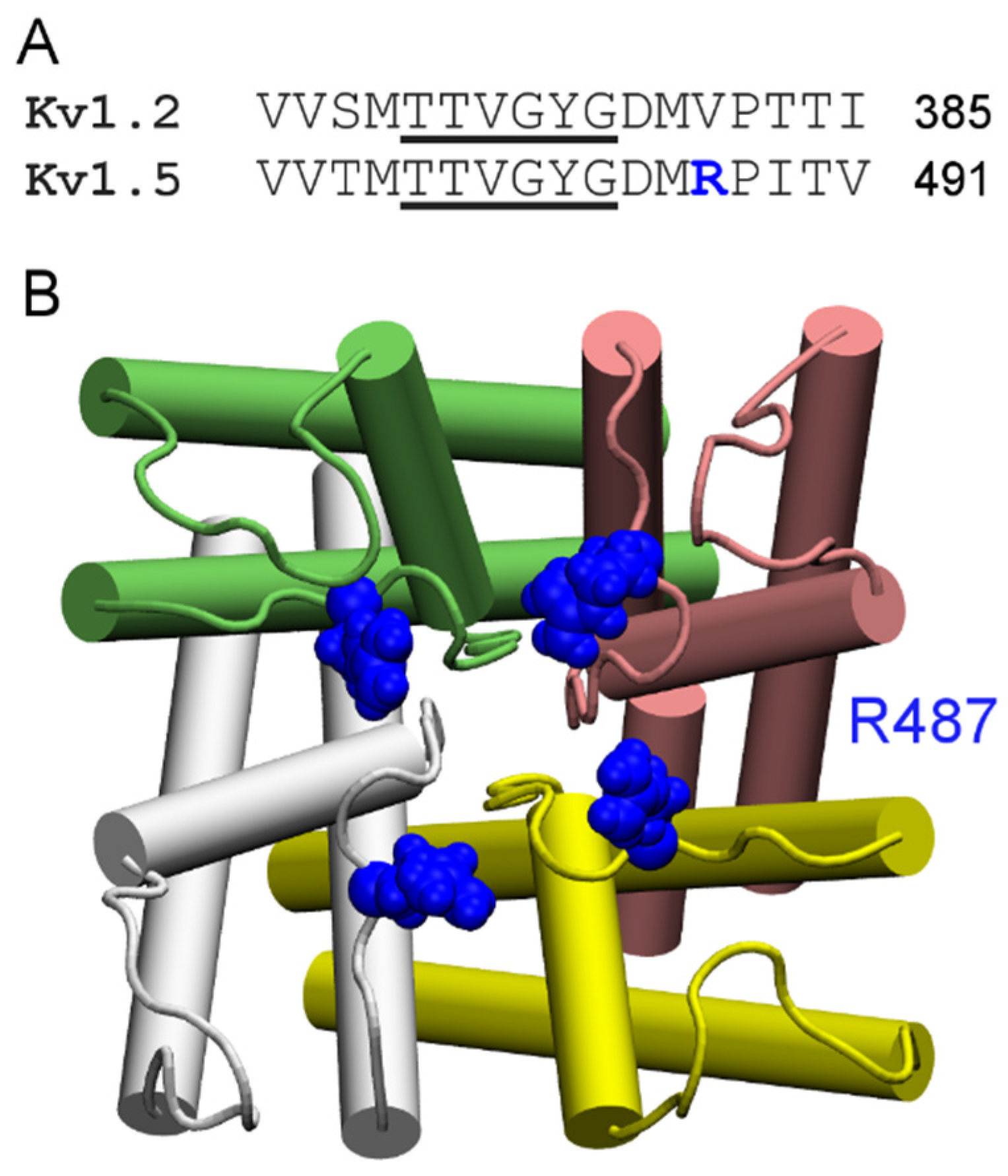
According to their sensitivity to the classic scorpion toxin ChTx, K+ channels can be classified into two groups. The first group comprises of K+ channels such as Kv1.1, Kv1.2, Kv1.3, and KCa3.1 that are extremely sensitive to ChTx, with IC50 values in the nanomolar range. In contrast, KCNQ1, Kv1.5, and Kv4.3, which are channels of the second group, are insensitive to ChTx. Channels of the first group generally carry several rings of acidic residues in the outer vestibule which interact favorably with the highly basic toxins. For example, the outer wall of KCa3.1 carries three rings of acidic residues (Figure 3A). Once ChTx is bound, in addition to hydrophobic interactions (Figure 3B), several hydrogen bonds are formed between the acidic residue of the channel and basic residues of the toxin (Figure 3C,D). These hydrogen bonds play crucial role in stabilizing the complex. However, Kv1.5 carries a ring of arginine residues just outside the filter, at the position corresponding to V381 of Kv1.2 (Figure 4). The positive charges carried by the arginines could render the channel insensitive to ChTx, which carries a net charge of +5 e. Thus, toxins active on channels of the second group must carry acidic residues. Consistent with this prediction, several toxins with micromolar affinities for KCNQ1 discovered by Li et al. [26] all carry multiple acidic residues.
Figure 4.
Sequence and structure of the ChTx-insensitive channel Kv1.5. (A) Sequence alignment of human Kv1.2 and human Kv1.5 in the filter (underscored) region; (B) Structure of Kv1.5 modeled on the crystal structure of Kv1.2 (PDB ID 3LUT [71]) viewed from the periplasmic side along the channel axis. The Arg487 residue just outside the filter is highlighted. The four channel subunits are shown in the Cartoon representation and different colors.
Figure 4.
Sequence and structure of the ChTx-insensitive channel Kv1.5. (A) Sequence alignment of human Kv1.2 and human Kv1.5 in the filter (underscored) region; (B) Structure of Kv1.5 modeled on the crystal structure of Kv1.2 (PDB ID 3LUT [71]) viewed from the periplasmic side along the channel axis. The Arg487 residue just outside the filter is highlighted. The four channel subunits are shown in the Cartoon representation and different colors.
4.2. Conotoxins
In over 50 million years of evolution, marine cone snails have developed a vast array of toxins for prey capture and defense. Only a small fraction of conotoxins has been characterized so far. There are over 500 species of cone snails, each species carrying over 100 different conotoxins [72]. A major component of the cone snail venom is peptide toxins. Based on primary structure, these toxins have been classified into seven superfamilies, each of which comprise of several subfamilies acting on different types of ion channels [72]. For example, µ-conotoxins belonging to the M superfamily are potent blockers of Na+ channels [73], whereas ω-conotoxins of the O superfamily are Ca2+ channel blockers [74]. Although the majority of conotoxins that have been characterized are inhibitors of Na+ channels, Ca2+ channels and nicotinic acetylcholine receptors, several conotoxins, such as ViTx [75] and PVIIA [76], have been shown to block Kv channel currents. The mechanism of action by ViTx is unclear, but PVIIA likely acts on the channel pore directly [77].
The primary structure of PVIIA is CRIPN-QKCFQ-HLDDC-CSRKC-NRFNK-CV and the C-terminus of the toxin is amidated. The toxin carries six lysine and arginine residues, and thus is positively charged at neutral pH. The tertiary structure of PVIIA is given in Figure 5. It is evident that PVIIA does not carry the α-helix in the N-terminus typical to ChTx-like scorpion toxins. Despite differences in structural folds, PVIIA and scorpion toxins are believed to block K+ channels in a similar manner. The binding modes of PVIIIA to the Shaker K+ channel have been examined in detail using docking and MD by Mahdavi and Kuyucak [78]. The lysine residue at position 7 of PVIIA was found to protrude into the channel filter and physically occlude the permeation pathway, in a way similar to that of scorpion toxins [78,79].
Figure 5.
Solution structure of PVIIA (PDB ID 1KCP [77]). The three disulfide bonds are shown in green and β-strands in yellow.
Figure 5.
Solution structure of PVIIA (PDB ID 1KCP [77]). The three disulfide bonds are shown in green and β-strands in yellow.
4.3. Spider Toxins
Spider venoms are also a rich resource for the isolation of small peptides as potential drug scaffolds [10]. It has been estimated that spider venoms as a whole can provide more than 16 million unique peptides, with each venom of approximately 80,000 species containing 200 unique peptides [80]. However, only a small number of those peptides have been characterized.
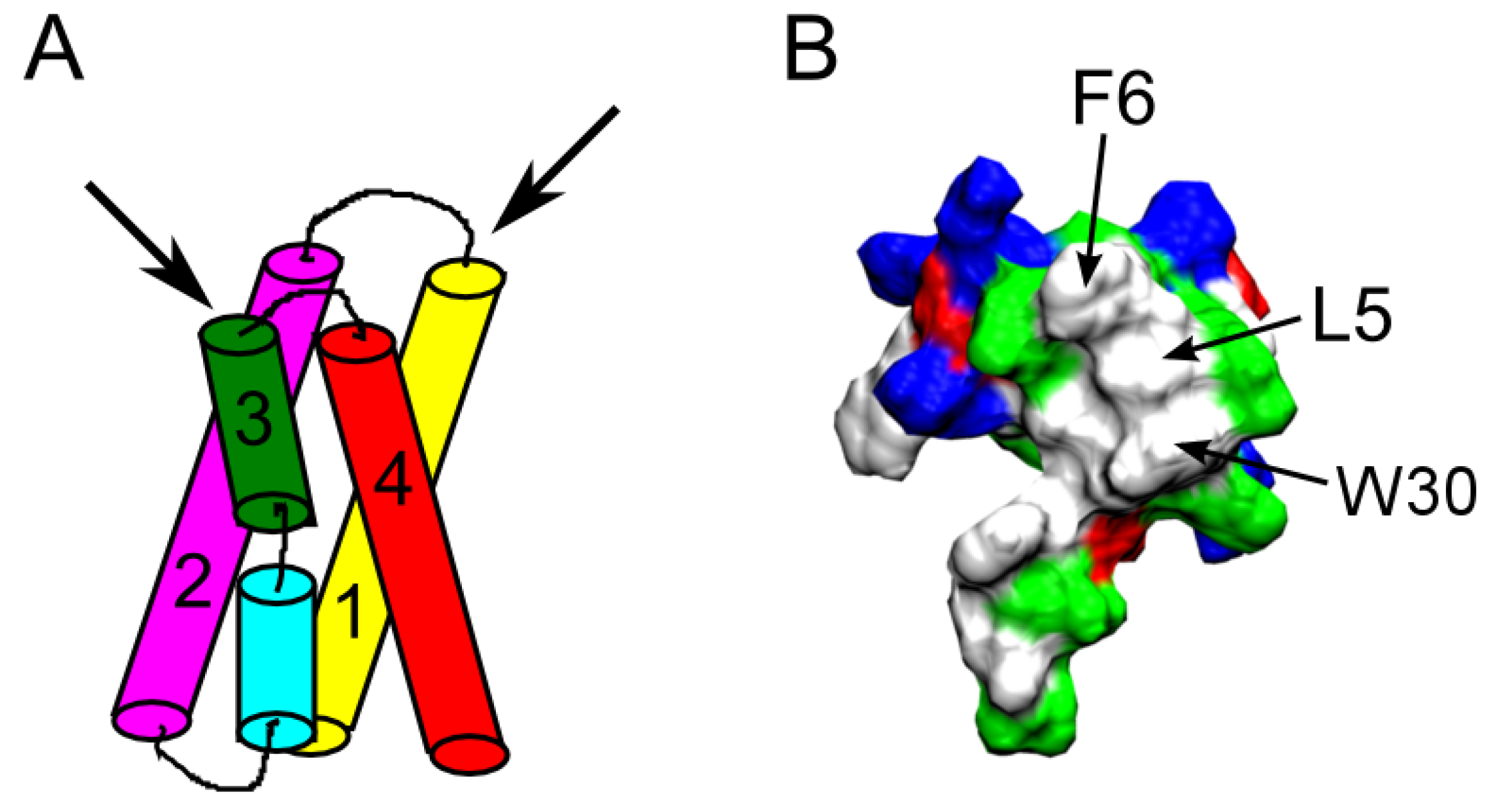
Some of the most well characterized spider toxins are Hanatoxin (HaTx) [81], Scodra griseipes toxin (SGTx1) [82], heteropodatoxin (HpTx) [83], phrixotoxin (PaTx) [84], and the voltage sensor toxin (VSTx) [85]. All these toxins target the voltage sensor domain, formed by four helices S1–S4 (Figure 6A), within the membrane [86]. This is different from ChTx-like toxins that bind to the pore domain. As such, these toxins are referred to as gating-modifiers. The backbones of these toxins contain three disulfide bonds forming the inhibitor cysteine knot motif, which is also common to ChTx and other pore-blockers [87]. A hydrophobic face primarily formed by phenylalanine, leucine, methionine, and tryptophan residues is conserved across these gating-modifiers (Figure 6B) [88,89,90,91]. This hydrophobic face is believed to be critical for toxin function [86,92,93].
The detailed process by which one of the gating-modifier toxins penetrates into the membrane has been examined in detail using MD simulations [94,95,96,97]. These simulations suggested that the toxins bind preferentially to head group region of the bilayer, consistent with recent experiments [98]. Once bound, the hydrophobic face of the toxin would interact favorably with the hydrophobic core of the bilayer, and the polar region of the toxin with lipid head groups and water molecules. However, the interaction between the toxin and the voltage sensor domain within the membrane is less well studied. A toxin may bind to the extracellular end of the voltage sensor from the side of S2 and S3 helices, or S1 and S4 helices (Figure 6A). MD simulations suggested that HaTx1 may bind to the voltage sensor of Kv2.1 from the S2 and S3 helices [99]. This proposal is in accord with the recent study by Gupta et al. [91]. Using the ROSETTA program [100], they constructed the complex structure of the spider toxin Guangxitoxin and the Kv1.2/2.1 paddle chimera channel, and found that this toxin also binds primarily to the S2 and S3 helices [91].
A globular polypeptide toxin, known as GsMTx4, extracted from the tarantula Grammostola spatulata, influences the gating kinetics of gramicidin [101] and mechanosensitive channels [102,103]. The mechanism by which this toxin interferes with the gating behavior of the two cationic channels is different from that of other spider toxins we discussed above. Instead of binding to the channel protein, GsMTx4 partitions into the membrane and interfere with the gating mechanisms indirectly through changes in membrane properties. MD simulations suggest that GsMTx4 binds to the interfacial region of the membrane and induces membrane thinning [104]. Notably, experimental evidence indicates that VSTx1 also causes membrane thinning when bound to the headgroup region of the lipid membrane [98].
Figure 6.
(A) Schematic of the voltage sensor domain of voltage-gated K+ channels. The four helices forming the voltage sensor, S1-S4, are indicated. The two black arrows denote the two possible directions a gating-modifier toxin may bind to the crevice between the S1-S2 and S3-S4 linking loops at the extracellular end of the voltage sensor. (B) Molecular surface of HaTx1 (PDB ID 1D1H [88]) showing three key hydrophobic residues.
Figure 6.
(A) Schematic of the voltage sensor domain of voltage-gated K+ channels. The four helices forming the voltage sensor, S1-S4, are indicated. The two black arrows denote the two possible directions a gating-modifier toxin may bind to the crevice between the S1-S2 and S3-S4 linking loops at the extracellular end of the voltage sensor. (B) Molecular surface of HaTx1 (PDB ID 1D1H [88]) showing three key hydrophobic residues.
4.4. Other Animal Toxins
Several sea anemone toxins such as BgK [105,106] and ShK [107] have been shown to be potent inhibitors of K+ channels. BgK blocks Kv1.1, Kv1.2, and Kv1.3 effectively at nanomolar concentrations [108]. ShK is an extremely potent blocker of Kv1.3, with an IC50 value in the picomolar range [107]. A domain of the matrix metalloprotease 23 protein from mammals, MMP23TxD, shows high sequence similarity with BgK and ShK. However, MMP23TxD from rat is only a weak inhibitor of Kv1.3 (IC50 = 2.8 µM) [109].
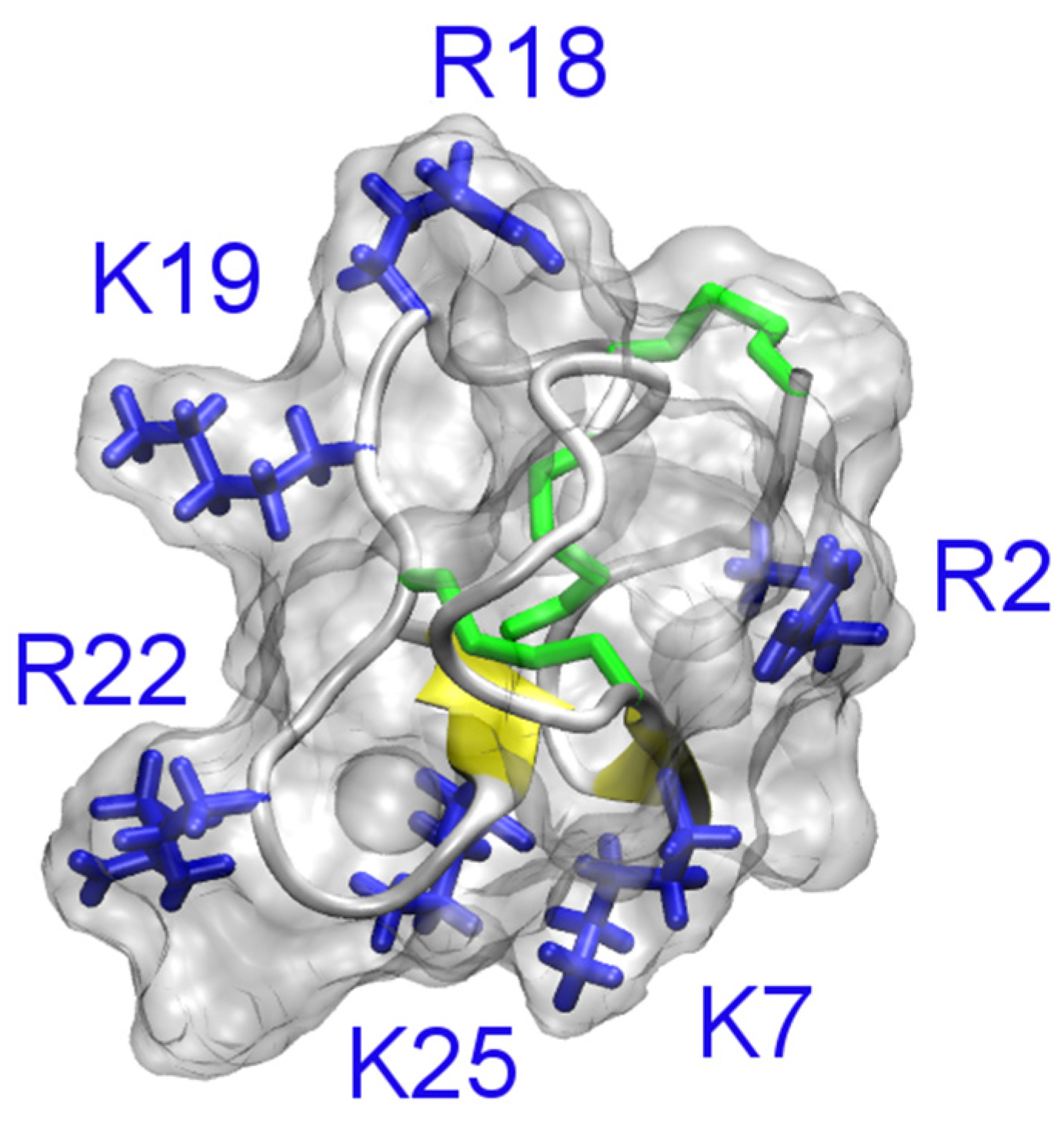
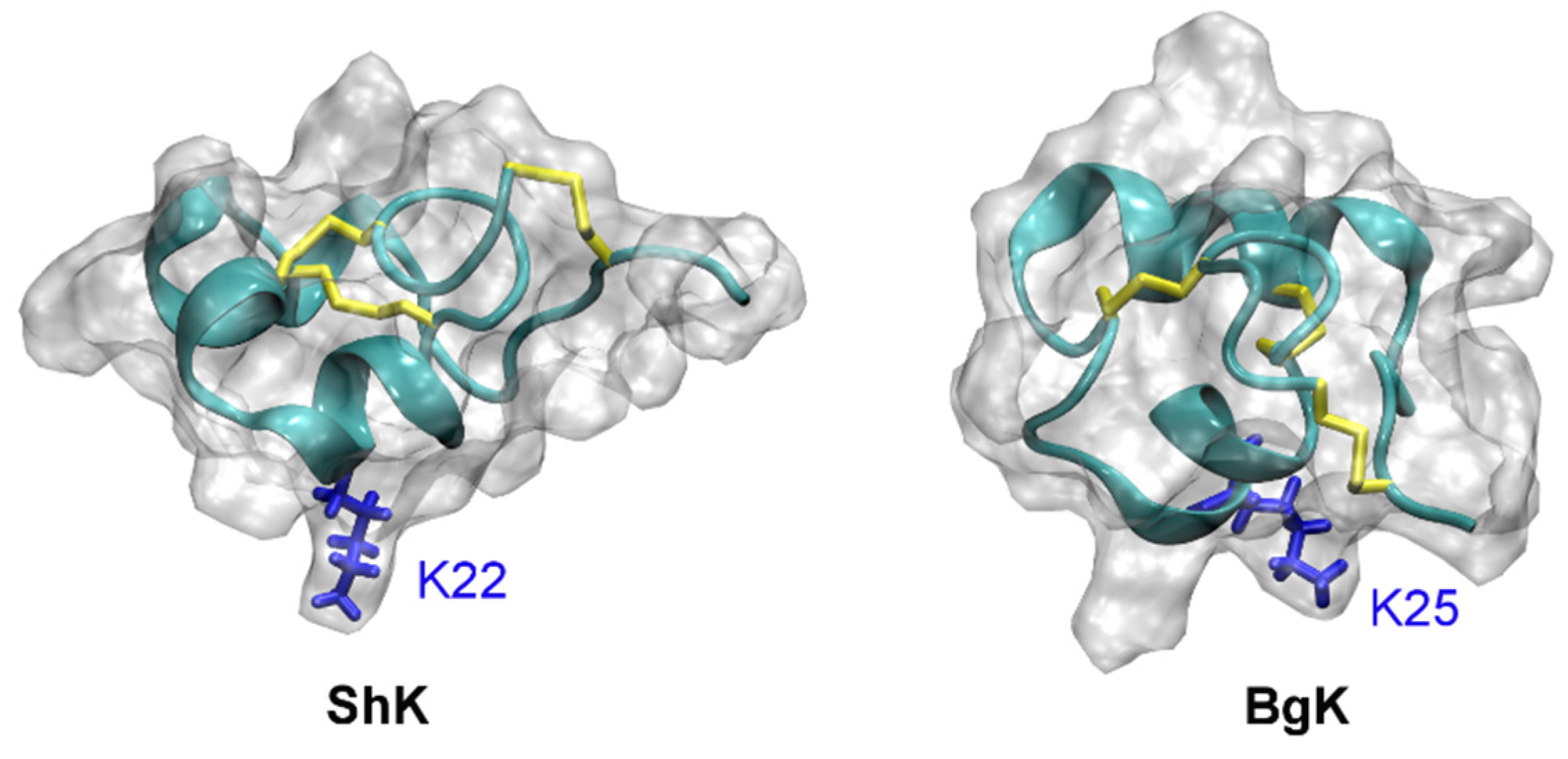
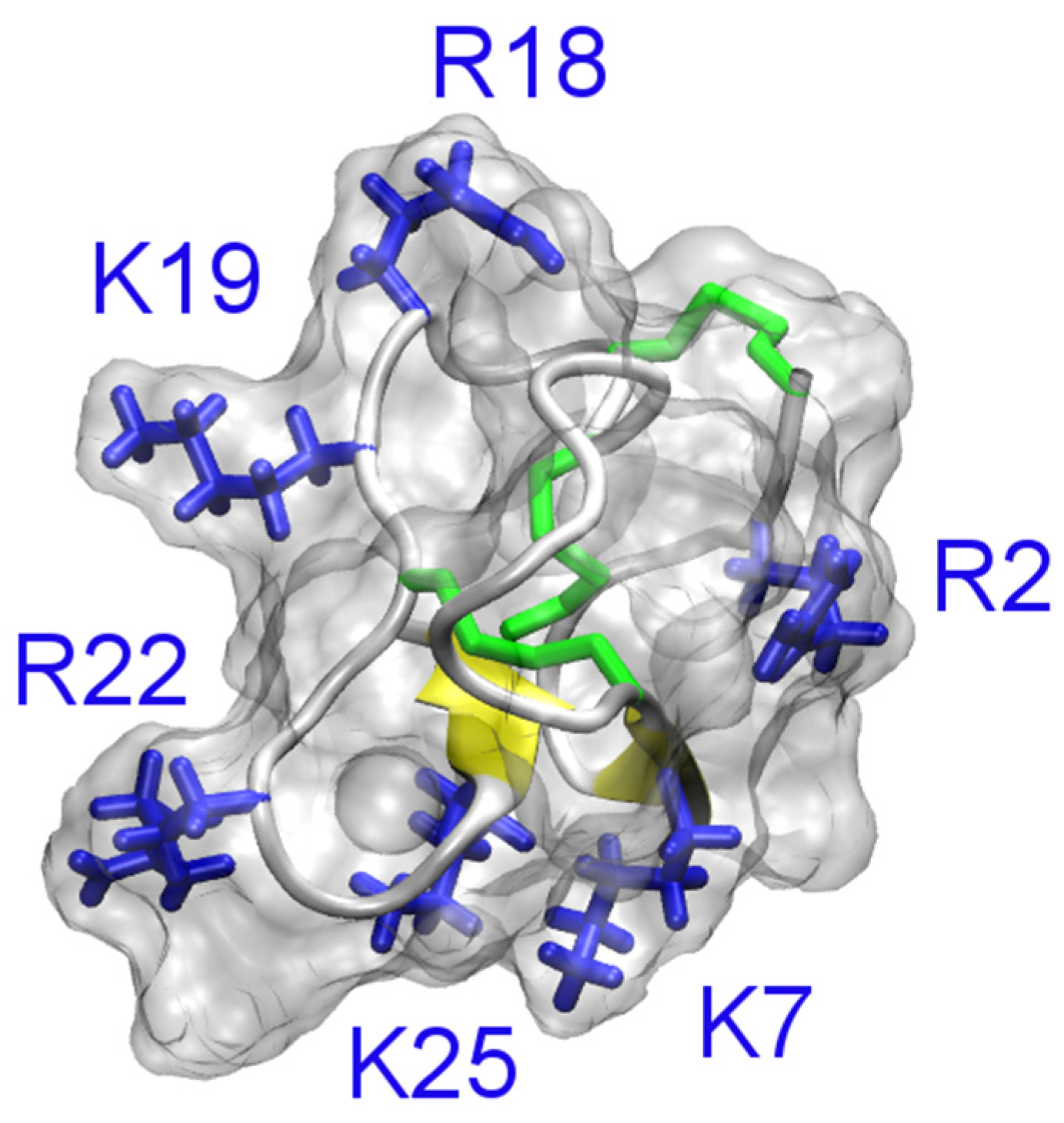
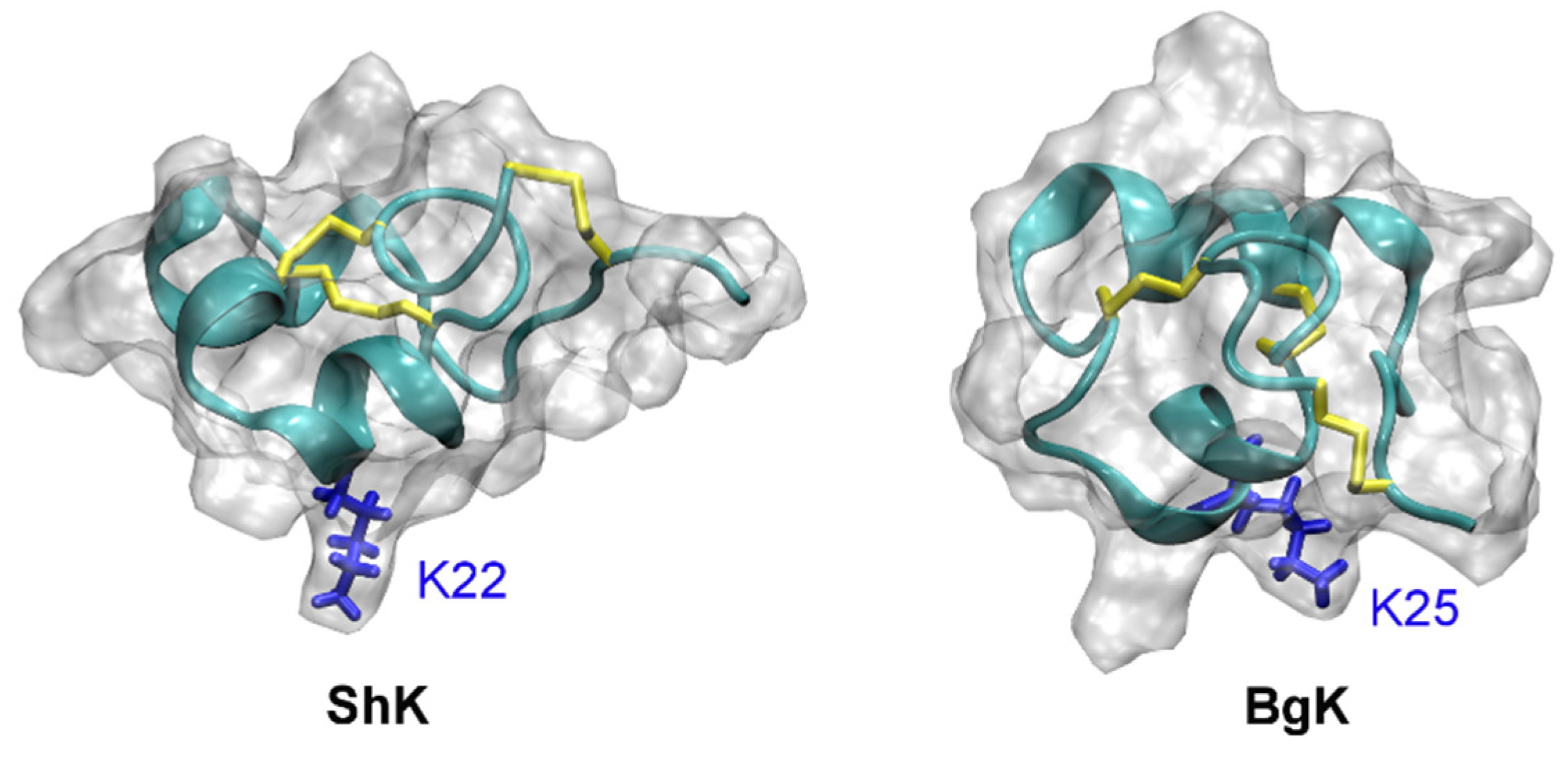
The solution structures of ShK (PDB ID 1ROO [110]) and BgK (PDB ID 1BGK [106]) are shown in Figure 7. The position of the pore-blocking lysine residue for K+ channels (Lys22 for ShK and Lys25 for BgK), as inferred from experimental data [111,112], is also indicated in the figure. It is evident that ShK and BgK share a common structural fold, which is significantly different from that of the scorpion toxin ChTx. In these two toxins, two α-helices are connected with three disulfide bonds but no β-strand structure is evident.
Figure 7.
Solution structures of ShK (PDB ID 1ROO [110]) and BgK (PDB ID 1BGK [113]). The three disulfide bonds in each toxin are shown in yellow. The side chain of the key lysine residue is highlighted.
ShK has been studied extensively in an attempt to develop novel drugs for the treatment of autoimmune diseases, because it blocks Kv1.3 extremely potently [114,115,116,117]. Several derivatives of ShK, such as ShK-Dap22, ShK-186 and ShK192, highly specific for Kv1.3 over other Kv channel isoforms have been reported [114]. For example, ShK-192 is about 1,000-fold selective for Kv1.3 over Kv1.1, while the wild type toxin only has three-fold selectivity.
Several computational studies are consistent with the Lys22 residue of ShK being the pore-blocking residue for Kv1.3 [64,69]. However, whether Arg11 of the toxin interacts with the aspartate just outside the filter (Asp449) or on the outer vestibule about 10 Å from the filter (Asp433) is unclear [64,69]. Since mutagenesis experiments do not probe the interacting residue pairs in the toxin-channel complex directly, it is difficult to conclude which one of the models is more correct. It should be noted that binding of biomolecules is a dynamic process and different microstates likely co-exist at equilibrium. It has been shown that different binding modes are possible for hongotoxin and Kv1.3 when the same pore-blocking lysine (Lys28) occludes the entrance of the filter [68]. Therefore, it remains to be determined whether multiple binding modes would be required to describe the binding of ShK to Kv1.3.
Recently, several computational studies have focused on tertiapin (TPN), which is isolated from honey bee venoms, and inward-rectifying K+ channels [118,119,120,121]. In all these studies, a rigid-body docking method was used for the prediction of toxin-channel complex structures. However, results from rigid-body docking methods are sensitive to the initial structures used. Therefore, inconsistent models of toxin-channel complexes have been proposed in these studies. For example, the 12th conformer of the 21 solution structures of TPN was proposed to dock rKir1.1 most favorably [121]. The Lys17 residue of TPN was predicted to occlude the filter of Kir3.2 [119], whereas different residues (His12 and Lys21) were proposed to be the pore-blocking residue for Kir1.1 [118,120,121]. Whether TPN can block Kir3.2 or Kir1.1 by inserting alternative residues into the channel pore remains to be elucidated. Such a multiple-binding-mode mechanism has been demonstrated for µ-conotoxins and Na+ channels [122], but not yet for K+ channels.
5. Toxin Analogues
Wild type toxins usually lack specificity and act on a range of targets, as specificity is not required for venomous animals to paralyze prey. On the other hand, high specificity is desired for the toxins to be used as drug candidates. Therefore, toxin analogs selectively acting on the drug target must be designed. At least two approaches for developing more specific toxin analogs are possible. Point mutations can be introduced to the toxins that are potent but nonspecific, or molecules that broadly mimick the action of venom peptides on ion channels can be designed de novo.
Several mutant toxins with high specificity for Kv1.3 have been successfully developed. For example, OSK1, a peptide of 38 residues isolated from the venom of Orthochirun scrobiculosus, is one of the most potent toxins for Kv1.3 (IC50 = 14 pM), but it inhibits Kv1.1 equally well (IC50 = 14 pM) [54]. The K16D20 double mutant of OSK1 is more than 100-fold selective for Kv1.3 (IC50 = 3 pM) over Kv1.1 (IC50 = 400 pM) and Kv1.2 (IC50 = 3 nM) [54]. A synthetic analog of ShK, ShK(L5), is also more than 100-fold selective for Kv1.3 (IC50 = 0.07 nM) over other Kv1.x channels [123]. ShK-Dap22, in which the Lys22 residue of ShK is replaced with a diaminopropionic acid, is about 80-fold selective for Kv1.3 (IC50 = 23 pM) over Kv1.1 (IC50 = 1.8 nM) [116]. A triple-point mutation of a kaliotoxin enhances the toxin’s specificity for Kv1.3 over Kv1.1 to over 300-fold [31]. Rashid et al. computationally designed a mutant HsTx1, HsTx1-[R14A], which was shown experimentally to be more than 2000-fold selective for Kv1.3 over Kv1.1 [124]. Thus it is possible to design highly selective toxin derivatives. The selectivity of a toxin may also be tuned by repositioning its acidic residues, according to the recent results by Wu and coworkers [125].
6. Concluding Remarks
The mechanisms of action of a variety of venom peptides on ion channels have been elucidated with the help of experimental and computational methods. Using the available computational tools, it is possible to predict the modes of binding of a toxin to a channel, and compute the binding affinity (Kd) accurately. For example, the Kd value for the binding of µ-conotoxin PIIIA to the bacterial sodium channel NaVAb was computed to be 20–30 pM [122], within six-fold to the value of 5 pM determined in subsequent experiments [126]. With the advance in the understanding of toxin action on a molecular level, rational design of highly specific toxin analogs will become possible. It is expected that computational methods will continue to play an important role in the development of ion channel drugs from venom peptides.
Acknowledgments
Supported by the National Health and Medical research Council of Australia.
Author Contributions
R.C and S.-H.C. wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bagal, S.; Brown, A.D.; Cox, P.J.; Omoto, K.; Owen, R.M.; Pryde, D.C.; Sidders, B.; Skerratt, S.E.; Stevens, E.B.; Storer, R.I.; et al. Ion channels as therapeutic targets: A drug discovery perspective. J. Med. Chem. 2013, 56, 593–624. [Google Scholar] [CrossRef] [PubMed]
- Clare, J.J. Targeting ion channels for drug discovery. Discov. Med. 2010, 9, 253–260. [Google Scholar] [PubMed]
- Xie, M.; Holmqvist, M.H.; Hsia, A.Y. Ion channel drug discovery expands into new disease areas. Curr. Drug Discov. 2004, 31, 31–33. [Google Scholar]
- Wulff, H.; Castle, N.A.; Pardo, L.A. Voltage-gated potassium channels as therapeutic targets. Nat. Rev. Drug Discov. 2009, 8, 982–1001. [Google Scholar] [CrossRef] [PubMed]
- Ragsdale, D.S.; McPhee, J.C.; Scheuer, T.; Catterall, W.A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994, 265, 1724–1728. [Google Scholar] [CrossRef] [PubMed]
- Chandy, K.G.; Wulff, H.; Beeton, C.; Pennington, M.; Gutman, G.A.; Cahalan, M. K+ channels as targets for specific immunomodulation. Trends Pharmacol. Sci. 2004, 25, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Yi, H.; Yin, S.J.; Chen, Z.Y.; Liu, H.; Cao, Z.J.; Wu, Y.L.; Li, W.X. Structural basis of a potent peptide inhibitor designed for Kv1.3 channel, a therapeutic target of autoimmune disease. J. Biol. Chem. 2008, 283, 19058–19065. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S. µ-Conotoxins as leads in the development of new analgesics. Molecules 2010, 15, 2825–2844. [Google Scholar] [CrossRef] [PubMed]
- Knapp, O.; McArthur, J.R.; Adams, D.J. Conotoxins targeting neuronal voltage-gated sodium channel subtypes: Potential analgesics? Toxins 2012, 4, 1236–1260. [Google Scholar] [CrossRef] [PubMed]
- Estrada, G.; Villegas, E.; Corzo, G. Spider venoms: A rich source of acylpolyamines and peptides as new leads for CNS drugs. Nat. Prod. Rep. 2007, 24, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M. Conus venom peptides, receptor and ion channel targets, and drug design: 50 million years of neuropharmacology. Mol. Biol. Cell 1997, 8, 2101–2109. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, E.; Gurrola, G.B.; Schwartz, E.F.; Possani, L.D. Scorpion venom components as potential candidates for drug development. Toxicon 2015, 93, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Lewis, R.J.; Nielsen, K.J.; Craik, D.J.; Loughnan, M.L.; Adams, D.A.; Sharpe, I.A.; Luchian, T.; Adams, D.J.; Bond, T.; Thomas, L.; et al. Novel ω-conotoxins from Conus catus discriminate among neuronal calcium channel subtypes. J. Biol. Chem. 2000, 275, 35335–35344. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Smith, J.J.; Vetter, I.; Rupasinghe, D.B.; Er, S.Y.; Senff, S.; Herzig, V.; Mobli, M.; Lewis, R.J.; Bosmans, F.; et al. Seven novel modulators of the analgesic target NaV1.7 uncovered using a high-throughput venom-based discovery approach. Br. J. Pharmacol. 2015, 172, 2445–2458. [Google Scholar] [PubMed]
- Gur, M.; Kahn, R.; Karbat, I.; Regev, N.; Wang, J.T.; Catterall, W.A.; Gordon, D.; Gurevitz, M. Elucidation of the molecular basis of selective recognition uncovers the interaction site for the core domain of scorpion α-toxins on sodium channels. J. Biol. Chem. 2011, 286, 35209–35217. [Google Scholar] [CrossRef] [PubMed]
- Gurevitz, M. Mapping of scorpion toxin receptor sites at voltage-gated sodium channels. Toxicon 2012, 60, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Z.; Yarov-Yarovoy, V.; Scheuer, T.; Karbat, I.; Cohen, L.; Gordon, D.; Gurevitz, M.; Catterall, W.A. Mapping the interaction site for a β-scorpion toxin in the pore module of domain III of voltage-gated Na+ channels. J. Biol. Chem. 2012, 287, 30719–30728. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.T.; Yarov-Yarovoy, V.; Kahn, R.; Gordon, D.; Gurevitz, M.; Scheuer, T.; Catterall, W.A. Mapping the receptor site for α-scorpion toxins on a Na+ channel voltage sensor. Proc. Natl. Acad. Sci. USA 2011, 108, 15426–15431. [Google Scholar] [CrossRef] [PubMed]
- Possani, L.D.; Becerril, B.; Delepierre, M.; Tytgat, J. Scorpion toxins specific for Na+-channels. Eur. J. Biochem. 1999, 264, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Bosmans, F.; Tytgat, J. Voltage-gated sodium channel modulation by scorpion α-toxins. Toxicon 2007, 49, 142–158. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Robertson, B. Dendrotoxins: Structure-activity relationships and effects on potassium ion channels. Curr. Med. Chem. 2004, 11, 3065–3072. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kini, R.M. From snake venom toxins to therapeutics—Cardiovascular examples. Toxicon 2012, 59, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Cao, Z.J.; Li, W.X.; Wu, Y.L. Cloning and characterization of a novel Kunitz-type inhibitor from scorpion with unique cysteine framework. Toxicon 2013, 72, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Hu, Y.T.; Han, S.; Yin, S.J.; He, Y.W.; Wu, Y.L.; Cao, Z.J.; Li, W.X. ImKTx1, a new Kv1.3 channel blocker with a unique primary structure. J. Biochem. Mol. Toxicol. 2011, 25, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.Y.; Hu, Y.T.; Yang, W.S.; He, Y.W.; Feng, J.; Wang, B.; Zhao, R.M.; Ding, J.P.; Cao, Z.J.; Li, W.X.; et al. Hg1, novel peptide inhibitor specific for Kv1.3 channels from first scorpion Kunitz-type potassium channel toxin family. J. Biol. Chem. 2012, 287, 13813–13821. [Google Scholar] [PubMed]
- Chen, Z.Y.; Zeng, D.Y.; Hu, Y.T.; He, Y.W.; Pan, N.; Ding, J.P.; Cao, Z.J.; Liu, M.L.; Li, W.X.; Yi, H.; et al. Structural and functional diversity of acidic scorpion potassium channel toxins. PLoS ONE 2012, 7, e35154. [Google Scholar] [PubMed]
- Han, S.; Yin, S.J.; Yi, H.; Mouhat, S.; Qiu, S.; Cao, Z.J.A.; Sabatier, J.M.; Wu, Y.L.; Li, W.X. Protein-protein recognition control by modulating electrostatic interactions. J. Proteome Res. 2010, 9, 3118–3125. [Google Scholar] [PubMed]
- Wu, Y.L.; Cao, Z.J.; Yi, H.; Jiang, D.H.; Mao, X.; Liu, H.; Li, W.X. Simulation of the interaction between ScyTx and small conductance calcium-activated potassium channel by docking and MM-PBSA. Biophys. J. 2004, 87, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Cao, Z.J.; Yin, S.J.; Dai, C.; Wu, Y.L.; Li, W.X. Interaction simulation of hERG K+ channel with its specific BeKm-1 peptide: Insights into the selectivity of molecular recognition. J. Proteome Res. 2007, 6, 611–620. [Google Scholar] [PubMed]
- Yi, H.; Qiu, S.; Wu, Y.; Li, W.X.; Wang, B. Differential molecular information of maurotoxin peptide recognizing IKCa and Kv1.2 channels explored by computational simulation. BMC Struct. Biol. 2011, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.J.; Jiang, L.; Yi, H.; Han, S.; Yang, D.W.; Liu, M.L.; Liu, H.; Cao, Z.J.; Wu, Y.L.; Li, W.X. Different residues in channel turret determining the selectivity of ADWX-1 inhibitor peptide between Kv1.1 and Kv1.3 channels. J. Proteome Res. 2008, 7, 4890–4897. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Chen, R.; Chung, S.H. Computational methods of studying the binding of toxins from venomous animals to biological ion channels: Theory and applications. Physiol. Rev. 2013, 93, 767–802. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Mahdavi, S.; Kuyucak, S. Computational studies of marine toxins targeting ion channels. Mar. Drugs 2013, 11, 848–869. [Google Scholar] [CrossRef] [PubMed]
- Kuyucak, S.; Norton, R.S. Computational approaches for designing potent and selective analogs of peptide toxins as novel therapeutics. Future Med. Chem. 2014, 6, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lee, A.; Chen, J.; Cadene, M.; Chait, B.T.; MacKinnon, R. Crystal structure and mechanism of a calcium-gated potassium channel. Nature 2002, 417, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Lee, A.; Chen, J.; Ruta, V.; Cadene, M.; Chait, B.T.; MacKinnon, R. X-ray structure of a voltage-dependent K+ channel. Nature 2003, 423, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Long, S.B.; Campbell, E.B.; Mackinnon, R. Voltage sensor of Kv1.2: Structural basis of electromechanical coupling. Science 2005, 309, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Avalos, J.L.; Chen, J.Y.; MacKinnon, R. Crystal structure of the eukaryotic strong inward-rectifier K+ channel Kir2.2 at 3.1 Å resolution. Science 2009, 326, 1668–1674. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Lee, A.; Campbell, E.; Mackinnon, R. Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K+ channel. eLife 2013, 2, e00594. [Google Scholar] [CrossRef] [PubMed]
- Long, S.B.; Tao, X.; Campbell, E.B.; MacKinnon, R. Atomic structure of a voltage-dependent K+ channel in a lipid membrane-like environment. Nature 2007, 450, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Shen, J.H.; Briggs, J.M.; Luo, X.M.; Tan, X.J.; Jiang, H.L.; Chen, K.X.; Ji, R.Y. Brownian dynamics simulations of interaction between scorpion toxin Lq2 and potassium ion channel. Biophys. J. 2001, 80, 1659–1669. [Google Scholar] [CrossRef]
- Eriksson, M.A.; Roux, B. Modeling the structure of agitoxin in complex with the Shaker K+ channel: A computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys. J. 2002, 83, 2595–2609. [Google Scholar] [CrossRef]
- Wang, C.; Bradley, P.; Baker, D. Protein-protein docking with backbone flexibility. J. Mol. Biol. 2007, 373, 503–519. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Advances and challenges in protein-ligand docking. Int. J. Mol. Sci. 2010, 11, 3016–3034. [Google Scholar] [CrossRef] [PubMed]
- Van Gunsteren, W.F.; Bakowies, D.; Baron, R.; Chandrasekhar, I.; Christen, M.; Daura, X.; Gee, P.; Geerke, D.P.; Glattli, A.; Hunenberger, P.H.; et al. Biomolecular modeling: Goals, problems, perspectives. Angew. Chem. Int. Ed. 2006, 45, 4064–4092. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.W.; Andersen, O.S.; Roux, B. Energetics of ion conduction through the gramicidin channel. Proc. Natl. Acad. Sci. USA 2004, 101, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Jarzynski, C. Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 1997, 78, 2690–2693. [Google Scholar] [CrossRef]
- Baştuğ, T.; Chen, P.C.; Patra, S.M.; Kuyucak, S. Potential of mean force calculations of ligand binding to ion channels from Jarzynski‘s equality and umbrella sampling. J. Chem. Phys. 2008, 128, 155104. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenberg, J.M. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
- Anderson, C.S.; MacKinnon, R.; Smith, C.; Miller, C. Charybdotoxin block of single Ca2+-activated K+ channels. Effects of channel gating, voltage, and ionic strength. J. Gen. Physiol. 1988, 91, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Kharrat, R.; Mabrouk, K.; Crest, M.; Darbon, H.; Oughideni, R.; MartinEauclaire, M.F.; Jacquet, G.; ElAyeb, M.; VanRietschoten, J.; Rochat, H.; et al. Chemical synthesis and characterization of maurotoxin, a short scorpion toxin with four disulfide bridges that acts on K+ channels. Eur. J. Biochem. 1996, 242, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Garciacalvo, M.; Leonard, R.J.; Novick, J.; Stevens, S.P.; Schmalhofer, W.; Kaczorowski, G.J.; Garcia, M.L. Purification, characterization, and biosynthesis of margatoxin, a component of Centruroides Margaritatus venom that selectively inhibits voltage-dependent potassium channels. J. Biol. Chem. 1993, 268, 18866–18874. [Google Scholar]
- Mouhat, S.; Visan, V.; Ananthakrishnan, S.; Wulff, H.; Andreotti, N.; Grissmer, S.; Darbon, H.; de Waard, M.; Sabatier, J.-M. K+ channel types targeted by synthetic OSK1, a toxin from Orthochirus scrobiculosus scorpion venom. Biochem. J. 2005, 385, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Nirthanan, S.; Pil, J.; Abdel-Mottaleb, Y.; Sugahara, Y.; Gopalakrishnakone, P.; Joseph, J.S.; Sato, K.; Tytgat, J. Assignment of voltage-gated potassium channel blocking activity to κ-KTx1.3, a non-toxic homologue of κ-hefutoxin-1, from Heterometrus spinifer venom. Biochem. Pharmacol. 2005, 69, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Bontems, F.; Roumestand, C.; Gilquin, B.; Menez, A.; Toma, F. Refined structure of charybdotoxin: Common motifs in scorpion toxins and insect defensins. Science 1991, 254, 1521–1523. [Google Scholar] [CrossRef] [PubMed]
- Blanc, E.; Sabatier, J.M.; Kharrat, R.; Meunier, S.; el Ayeb, M.; Van Rietschoten, J.; Darbon, H. Solution structure of maurotoxin, a scorpion toxin from Scorpio maurus, with high affinity for voltage-gated potassium channels. Proteins 1997, 29, 321–333. [Google Scholar] [CrossRef]
- Johnson, B.A.; Stevens, S.P.; Williamson, J.M. Determination of the three-dimensional structure of margatoxin by 1H, 13C, 15N triple-resonance nuclear magnetic resonance spectroscopy. Biochemistry 1994, 33, 15061–15070. [Google Scholar] [CrossRef] [PubMed]
- Jaravine, V.A.; Nolde, D.E.; Reibarkh, M.J.; Korolkova, Y.V.; Kozlov, S.A.; Pluzhnikov, K.A.; Grishin, E.V.; Arseniev, A.S. Three-dimensional structure of toxin OSK1 from Orthochirus scrobiculosus scorpion venom. Biochemistry 1997, 36, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Cui, M.; Briggs, J.M.; Huang, X.; Xiong, B.; Zhang, Y.; Luo, X.; Shen, J.; Ji, R.; Jiang, H.; et al. Brownian dynamics simulations of the recognition of the scorpion toxin maurotoxin with the voltage-gated potassium ion channels. Biophys. J. 2002, 83, 2370–2385. [Google Scholar] [CrossRef]
- Jin, L.; Wu, Y. Molecular mechanism of the sea anemone toxin ShK recognizing the Kv1.3 channel explored by docking and molecular dynamic simulations. J. Chem. Inf. Model. 2007, 47, 1967–1972. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Wu, Y. Molecular mechanism of δ-dendrotoxin-potassium channel recognition explored by docking and molecular dynamic simulations. J. Mol. Recognit. 2011, 24, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Heinzelmann, G.; Huq, R.; Tajhya, R.B.; Chang, S.C.; Chhabra, S.; Pennington, M.W.; Beeton, C.; Norton, R.S.; Kuyucak, S. A potent and selective peptide blocker of the Kv1.3 channel: Prediction from free-energy simulations and experimental confirmation. PLoS ONE 2013, 8, e78712. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Kuyucak, S. Affinity and selectivity of ShK toxin for the Kv1 potassium channels from free energy simulations. J. Phys. Chem. B 2012, 116, 4812–4822. [Google Scholar] [CrossRef] [PubMed]
- Khabiri, M.; Nikouee, A.; Cwiklik, L.; Grissmer, S.; Ettrich, R. Charybdotoxin unbinding from the mKv1.3 potassium channel: A combined computational and experimental study. J. Phys. Chem. B 2011, 115, 11490–11500. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Qiu, S.; Cao, Z.; Wu, Y.; Li, W. Molecular basis of inhibitory peptide maurotoxin recognizing Kv1.2 channel explored by ZDOCK and molecular dynamic simulations. Proteins 2008, 70, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.H. Structural basis of the selective block of Kv1.2 by maurotoxin from computer simulations. PLoS ONE 2012, 7, e47253. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.H. Binding modes of two scorpion toxins to the voltage-gated potassium channel Kv1.3 revealed from molecular dynamics. Toxins 2014, 6, 2149–2161. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Robinson, A.; Gordon, D.; Chung, S.H. Modeling the binding of three toxins to the voltage-gated potassium channel (Kv1.3). Biophys. J. 2011, 101, 2652–2660. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.H. Molecular dynamics simulations of scorpion toxin recognition by the Ca2+-activated potassium channel KCa3.1. Biophys. J. 2013, 105, 1829–1837. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Q.; Ni, F.; Ma, J. Structure of the full-length Shaker potassium channel Kv1.2 by normal-mode-based X-ray crystallographic refinement. Proc. Natl. Acad. Sci. USA 2010, 107, 11352–11357. [Google Scholar] [CrossRef] [PubMed]
- Terlau, H.; Olivera, B.M. Conus venoms: A rich source of novel ion channel-targeted peptides. Physiol. Rev. 2004, 84, 41–68. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Ishida, Y.; Wakamatsu, K.; Kato, R.; Honda, H.; Ohizumi, Y.; Nakamura, H.; Ohya, M.; Lancelin, J.-M.; Kohda, D.; et al. Active site of μ-conotoxin GIIIA, a peptide blocker of muscle sodium channels. J. Biol. Chem. 1991, 266, 16989–16991. [Google Scholar] [PubMed]
- Reynolds, I.J.; Wagner, J.A.; Snyder, S.H.; Thayer, S.A.; Olivera, B.M.; Miller, R.J. Brain voltage-sensitive calcium channel subtypes differentiated by ω-conotoxin fraction GVIA. Proc. Natl. Acad. Sci. USA 1986, 83, 8804–8807. [Google Scholar] [CrossRef] [PubMed]
- Kauferstein, S.; Huys, I.; Lamthanh, H.; Stocklin, R.; Sotto, F.; Menez, A.; Tytgat, J.; Mebs, D. A novel conotoxin inhibiting vertebrate voltage-sensitive potassium channels. Toxicon 2003, 42, 43–52. [Google Scholar] [CrossRef]
- Shon, K.J.; Stocker, M.; Terlau, H.; Stühmer, W.; Jacobsen, R.; Walker, C.; Grilley, M.; Watkins, M.; Hillyard, D.R.; Gray, W.R.; et al. κ-Conotoxin PVIIA is a peptide inhibiting the Shaker K+ channel. J. Biol. Chem. 1998, 273, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Savarin, P.; Guenneugues, M.; Gilquin, B.; Lamthanh, H.; Gasparini, S.; Zinn-Justin, S.; Menez, A. Three-dimensional structure of κ-conotoxin PVIIA, a novel potassium channel-blocking toxin from cone snails. Biochemistry 1998, 37, 5407–5416. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, S.; Kuyucak, S. Why the Drosophila Shaker K+ channel is not a good model for ligand binding to voltage-gated Kv1 channels. Biochemistry 2013, 59, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Dong, F.; Zhou, H.X. Electrostatic recognition and induced fit in the κ-PVIIA toxin binding to Shaker potassium channel. J. Am. Chem. Soc. 2005, 127, 6836–6849. [Google Scholar] [CrossRef] [PubMed]
- Escoubas, P.; Sollod, B.; King, G.F. Venom landscapes: Mining the complexity of spider venoms via a combined cDNA and mass spectrometric approach. Toxicon 2006, 47, 650–663. [Google Scholar] [CrossRef] [PubMed]
- Swartz, K.J.; MacKinnon, R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron 1995, 15, 941–949. [Google Scholar] [CrossRef]
- Marvin, L.; De, E.; Cosette, P.; Gagnon, J.; Molle, G.; Lange, C. Isolation, amino acid sequence and functional assays of SGTx1. The first toxin purified from the venom of the spider scodra griseipes. Eur. J. Biochem. 1999, 265, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.C.; Johnson, J.H.; Hammerland, L.G.; Kelbaugh, P.R.; Volkmann, R.A.; Saccomano, N.A.; Mueller, A.L. Heteropodatoxins: Peptides isolated from spider venom that block Kv4.2 potassium channels. Mol. Pharmacol. 1997, 51, 491–498. [Google Scholar] [PubMed]
- Diochot, S.; Drici, M.D.; Moinier, D.; Fink, M.; Lazdunski, M. Effects of phrixotoxins on the Kv4 family of potassium channels and implications for the role of Ito1 in cardiac electrogenesis. Br. J. Pharmacol. 1999, 126, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Ruta, V.; Jiang, Y.; Lee, A.; Chen, J.; MacKinnon, R. Functional analysis of an archaebacterial voltage-dependent K+ channel. Nature 2003, 422, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Swartz, K.J. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon 2007, 49, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S.; Pallaghy, P.K. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 1998, 36, 1573–1583. [Google Scholar] [CrossRef]
- Takahashi, H.; Kim, J.I.; Min, H.J.; Sato, K.; Swartz, K.J.; Shimada, I. Solution structure of hanatoxin1, a gating modifier of voltage-dependent K+ channels: Common surface features of gating modifier toxins. J. Mol. Biol. 2000, 297, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.W.; Kim, S.; Roh, S.H.; Endoh, H.; Kodera, Y.; Maeda, T.; Kohno, T.; Wang, J.M.; Swartz, K.J.; Kim, J.I. Solution structure and functional characterization of SGTx1, a modifier of Kv2.1 channel gating. Biochemistry 2004, 43, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.J.; Lee, J.Y.; Kim, S.H.; Eu, Y.J.; Shin, S.Y.; Milescu, M.; Swartz, K.J.; Kim, J.I. Solution structure and lipid membrane partitioning of VSTx1, an inhibitor of the KvAP potassium channel. Biochemistry 2005, 44, 6015–6023. [Google Scholar] [CrossRef] [PubMed]
- Gupta, K.; Zamanian, M.; Bae, C.; Milescu, M.; Krepkiy, D.; Tilley, D.C.; Sack, J.T.; Yarov-Yarovoy, V.; Kim, J.I.; Swartz, K.J. Tarantula toxins use common surfaces for interacting with Kv and ASIC ion channels. eLife 2015, 4, e06774. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.M.; Roh, S.H.; Kim, S.; Lee, C.W.; Kim, J.I.; Swartz, K.J. Molecular surface of tarantula toxins interacting with voltage sensors in KV channels. J. Gen. Physiol. 2004, 123, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Milescu, M.; Vobecky, J.; Roh, S.H.; Kim, S.H.; Jung, H.J.; Kim, J.I.; Swartz, K.J. Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 2007, 130, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Nishizawa, M.; Nishizawa, K. Interaction between K+ channel gate modifier hanatoxin and lipid bilayer membranes analyzed by molecular dynamics simulation. Eur. Biophys. J. 2006, 35, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Wee, C.L.; Bemporad, D.; Sands, Z.A.; Gavaghan, D.; Sansom, M.S.P. SGTx1, a Kv channel gating-modifier toxin, binds to the interfacial region of lipid bilayers. Biophys. J. 2007, 92, L07–L09. [Google Scholar] [CrossRef] [PubMed]
- Bemporad, D.; Sands, Z.A.; Wee, C.L.; Grottesi, A.; Sansom, M.S.P. Vstx1, a modifier of Kv channel gating, localizes to the interfacial region of lipid bilayers. Biochemistry 2006, 45, 11844–11855. [Google Scholar] [CrossRef] [PubMed]
- Wee, C.L.; Gavaghan, D.; Sansom, M.S.P. Interactions between a voltage sensor and a toxin via multiscale simulations. Biophys. J. 2010, 98, 1558–1565. [Google Scholar] [CrossRef] [PubMed]
- Mihailescu, M.; Krepkiy, D.; Milescu, M.; Gawrisch, K.; Swartz, K.J.; White, S. Structural interactions of a voltage sensor toxin with lipid membranes. Proc. Natl. Acad. Sci. USA 2014, 111, E5463–E5470. [Google Scholar] [CrossRef]
- Chen, R.; Robinson, A.; Chung, S.H. Binding of hanatoxin to the voltage sensor of Kv2.1. Toxins 2012, 4, 1552–1564. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.; Vernon, R.; Thompson, J.; Tyka, M.; Sadreyev, R.; Pei, J.M.; Kim, D.; Kellogg, E.; DiMaio, F.; Lange, O.; et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins 2009, 77, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Suchyna, T.M.; Tape, S.E.; Koeppe, R.E.; Andersen, O.S.; Sachs, F.; Gottlieb, P.A. Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature 2004, 430, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Hurst, A.C.; Gottlieb, P.A.; Martinac, B. Concentration dependent effect of GsMTx4 on mechanosensitive channels of small conductance in E. coli spheroplasts. Eur. Biophys. J. 2009, 38, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Kamaraju, K.; Gottlieb, P.A.; Sachs, F.; Sukharev, S. Effects of GsMTx4 on bacterial mechanosensitive channels in inside-out patches from giant spheroplasts. Biophys. J. 2010, 99, 2870–2878. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Chung, S.H. Effect of gating modifier toxins on membrane thickness: Implications for toxin effect on gramicidin and mechanosensitive channels. Toxins 2013, 5, 456–471. [Google Scholar] [CrossRef] [PubMed]
- Aneiros, A.; Garcia, I.; Martinez, J.R.; Harvey, A.L.; Anderson, A.J.; Marshall, D.L.; Engstrom, A.; Hellman, U.; Karlsson, E. A potassium channel toxin from the secretion of the sea anemone Bunodosoma granulifera. Isolation, amino acid sequence and biological activity. Biochim. Biophys. Acta 1993, 1157, 86–92. [Google Scholar] [CrossRef]
- Dauplais, M.; Lecoq, A.; Song, J.; Cotton, J.; Jamin, N.; Gilquin, B.; Roumestand, C.; Vita, C.; de Medeiros, C.L.; Rowan, E.G.; et al. On the convergent evolution of animal toxins. Conservation of a diad of functional residues in potassium channel-blocking toxins with unrelated structures. J. Biol. Chem. 1997, 272, 4302–4309. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.W.; Byrnes, M.E.; Zaydenberg, I.; Khaytin, I.; de Chastonay, J.; Krafte, D.S.; Hill, R.; Mahnir, V.M.; Volberg, W.A.; Gorczyca, W.; et al. Chemical synthesis and characterization of ShK toxin: A potent potassium channel inhibitor from a sea anemone. Int. J. Peptide Protein Res. 1995, 46, 354–358. [Google Scholar] [CrossRef]
- Alessandri-Haber, N.; Lecoq, A.; Gasparini, S.; Grangier-Macmath, G.; Jacquet, G.; Harvey, A.L.; de Medeiros, C.; Rowan, E.G.; Gola, M.; Menez, A.; et al. Mapping the functional anatomy of BgK on Kv1.1, Kv1.2, and Kv1.3. Clues to design analogs with enhanced selectivity. J. Biol. Chem. 1999, 274, 35653–35661. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, S.; Khoo, K.K.; Feng, Z.P.; Crossley, G.; Nugent, D.; Khaytin, I.; Chi, V.; Pham, C.; Calabresi, P.; Pennington, M.W.; et al. Potassium channel modulation by a toxin domain in matrix metalloprotease 23. J. Biol. Chem. 2010, 285, 9124–9136. [Google Scholar] [CrossRef] [PubMed]
- Tudor, J.E.; Pallaghy, P.K.; Pennington, M.W.; Norton, R.S. Solution structure of ShK toxin, a novel potassium channel inhibitor from a sea anemone. Nat. Struct. Biol. 1996, 3, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Gilquin, B.; Racape, J.; Wrisch, A.; Visan, V.; Lecoq, A.; Grissmer, S.; Menez, A.; Gasparini, S. Structure of the BgK-Kv1.1 complex based on distance restraints identified by double mutant cycles. Molecular basis for convergent evolution of Kv1 channel blockers. J. Biol. Chem. 2002, 277, 37406–37413. [Google Scholar] [CrossRef] [PubMed]
- Pennington, M.W.; Mahnir, V.M.; Khaytin, I.; Zaydenberg, I.; Byrnes, M.E.; Kem, W.R. An essential binding surface for ShK toxin interaction with rat brain potassium channels. Biochemistry 1996, 35, 16407–16411. [Google Scholar] [CrossRef] [PubMed]
- Dauplais, M.; Gilquin, B.; Possani, L.D.; Gurrola-Briones, G.; Roumestand, C.; Menez, A. Determination of the three-dimensional solution structure of noxiustoxin: Analysis of structural differences with related short-chain scorpion toxins. Biochemistry 1995, 34, 16563–16573. [Google Scholar] [CrossRef] [PubMed]
- Chi, V.; Pennington, M.W.; Norton, R.S.; Tarcha, E.J.; Londono, L.M.; Sims-Fahey, B.; Upadhyay, S.K.; Lakey, J.T.; Iadonato, S.; Wulff, H.; et al. Development of a sea anemone toxin as an immunomodulator for therapy of autoimmune diseases. Toxicon 2012, 59, 529–546. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Smith, B.J.; Sabo, J.K.; Crossley, G.; Nugent, D.; Khaytin, I.; Chi, V.; Chandy, K.G.; Pennington, M.W.; Norton, R.S. The D-diastereomer of ShK toxin selectively blocks voltage-gated K+ channels and inhibits T lymphocyte proliferation. J. Biol. Chem. 2008, 283, 988–997. [Google Scholar] [CrossRef] [PubMed]
- Kalman, K.; Pennington, M.W.; Lanigan, M.D.; Nguyen, A.; Rauer, H.; Mahnir, V.; Paschetto, K.; Kem, W.R.; Grissmer, S.; Gutman, G.A.; et al. ShK-Dap22, a potent Kv1.3-specific immunosuppressive polypeptide. J. Biol. Chem. 1998, 273, 32697–32707. [Google Scholar] [CrossRef] [PubMed]
- Lanigan, M.D.; Kalman, K.; Lefievre, Y.; Pennington, M.W.; Chandy, K.G.; Norton, R.S. Mutating a critical lysine in ShK toxin alters its binding configuration in the pore-vestibule region of the voltage-gated potassium channel, Kv1.3. Biochemistry 2002, 41, 11963–11971. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Qiu, S.; Yang, F.; Cao, Z.J.; Li, W.X.; Wu, Y.L. Unique mechanism of the interaction between honey bee toxin TPNQ and rKir1.1 potassium channel explored by computational simulations: Insights into the relative insensitivity of channel towards animal toxins. PLoS ONE 2013, 8, e67213. [Google Scholar] [CrossRef] [PubMed]
- Hilder, T.A.; Chung, S.H. Conductance properties of the inwardly rectifying channel, Kir3.2: Molecular and Brownian dynamics study. Biochim. Biophys. Acta 2013, 1828, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Hilder, T.A.; Chung, S.H. Conduction and block of inward rectifier K+ channels: Predicted structure of a potent blocker of Kir2.1. Biochemistry 2013, 52, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Doupnik, C.A.; Parra, K.C.; Guida, W.C. A computational design approach for virtual screening of peptide interactions across K+ channel families. Comput. Struct. Biotechnol. J. 2015, 13, 85–94. [Google Scholar] [PubMed]
- Chen, R.; Chung, S.H. Binding modes of μ-conotoxin to the bacterial sodium channel (NaVAb). Biophys. J. 2012, 102, 483–488. [Google Scholar] [CrossRef] [PubMed]
- Beeton, C.; Pennington, M.W.; Wulff, H.; Singh, S.; Nugent, D.; Crossley, G.; Khaytin, I.; Calabresi, P.A.; Chen, C.Y.; Gutman, G.A.; et al. Targeting effector memory T cells with a selective peptide inhibitor of Kv1.3 channels for therapy of autoimmune diseases. Mol. Pharmacol. 2005, 67, 1369–1381. [Google Scholar] [CrossRef] [PubMed]
- Rashid, M.H.; Huq, R.; Tanner, M.R.; Chhabra, S.; Khoo, K.K.; Estrada, R.; Dhawan, V.; Chauhan, S.; Pennington, M.W.; Beeton, C.; et al. A potent and Kv1.3-selective analogue of the scorpion toxin HsTX1 as a potential therapeutic for autoimmune diseases. Sci. Rep. 2014, 4, 4509. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, Y.; Hong, J.; Hu, J.; Yang, W.; Xiang, F.; Yang, F.; Xie, Z.; Cao, Z.; Li, W.; et al. Toxin acidic residue evolutionary function-guided design of de novo peptide drugs for the immunotherapeutic target, the Kv1.3 channel. Sci. Rep. 2015, 5, 9881. [Google Scholar] [CrossRef] [PubMed]
- Finol-Urdaneta, R.K.; Glavica, R.; McArthur, J.R.; French, R.J. Polymodal, high affinity actions of μ-conotoxins on a bacterial voltage-gated sodium channel. Biophys. J. 2013, 104, 136a–137a. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chen, R.; Chung, S.-H. Computational Studies of Venom Peptides Targeting Potassium Channels. Toxins 2015, 7, 5194-5211. https://doi.org/10.3390/toxins7124877
AMA Style
Chen R, Chung S-H. Computational Studies of Venom Peptides Targeting Potassium Channels. Toxins. 2015; 7(12):5194-5211. https://doi.org/10.3390/toxins7124877
Chicago/Turabian StyleChen, Rong, and Shin-Ho Chung. 2015. "Computational Studies of Venom Peptides Targeting Potassium Channels" Toxins 7, no. 12: 5194-5211. https://doi.org/10.3390/toxins7124877