Binding Modes of Two Scorpion Toxins to the Voltage-Gated Potassium Channel Kv1.3 Revealed from Molecular Dynamics

Abstract
:1. Introduction
2. Results and Discussion
2.1. Binding of MgTx
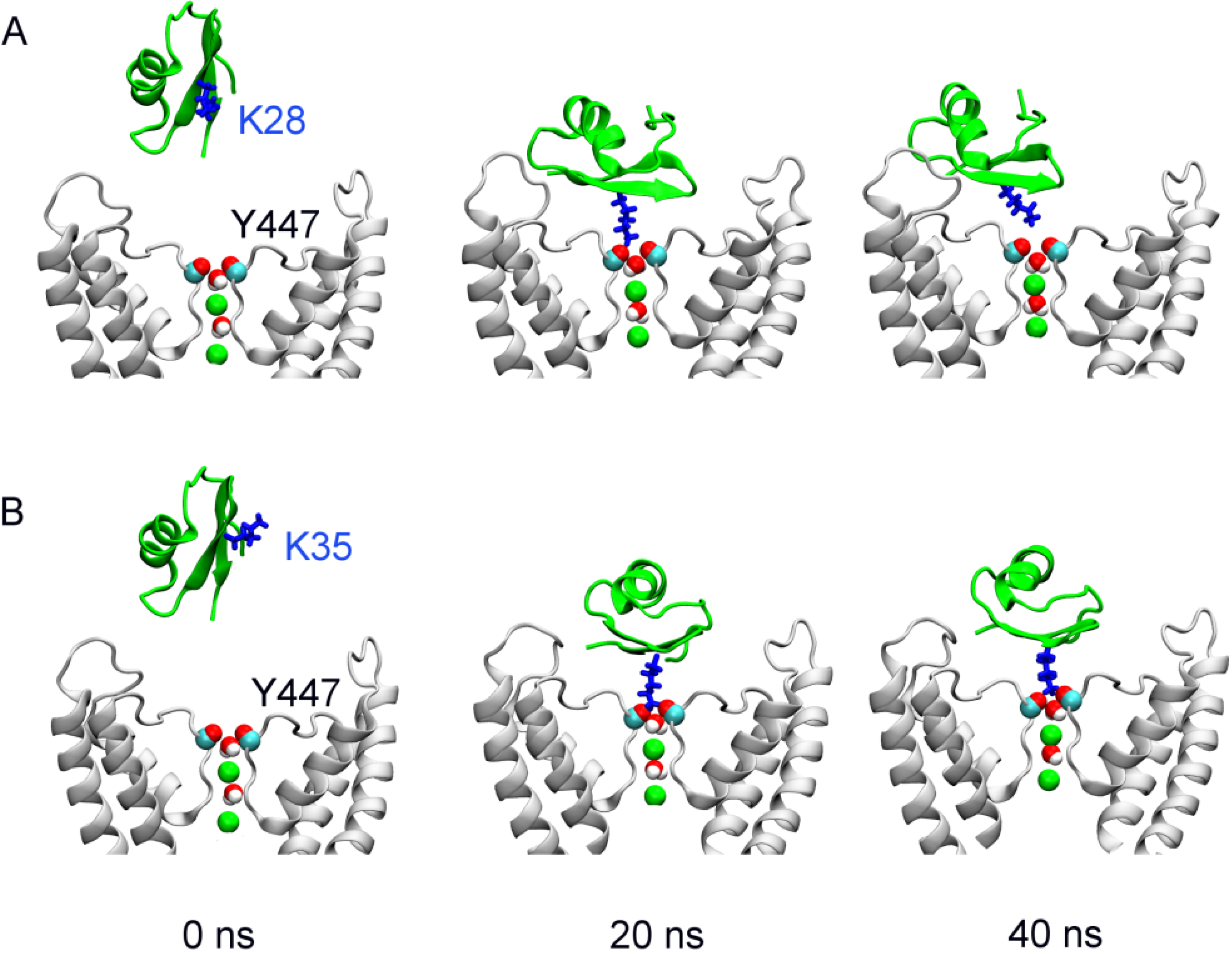

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MgTx-Kv1.3 | Average distance | HgTx-Kv1.3 (R24-D433) | Average distance | HgTx-Kv1.3 (R24-E420) | Average distance |
|---|---|---|---|---|---|
| I2-H451 | 2.3 ± 0.3 | I23-T425 | 2.6 ± 0.3 | P10-H451 | 2.7 ± 0.6 |
| K28-D449 | 1.7 ± 0.1 | R24-D433 | 1.7 ± 0.1 | R24-E420 | 1.8 ± 0.3 |
| M30-M450 | 2.3 ± 0.2 | K28-Y447 | 1.8 ± 0.2 | K28-Y447 | 1.8 ± 0.2 |
| K33-D449 | 2.0 ± 0.4 | M30-H451 | 2.5 ± 0.3 | M30-H451 | 2.4 ± 0.3 |
| K35-Y447 | 1.8 ± 0.1 | K35-D449 | 1.8 ± 0.3 | K35-D449 | 1.8 ± 0.4 |
| Y37-F428 | 2.8 ± 0.4 | Y37-H451 | 2.5 ± 0.3 | Y37-H451 | 2.4 ± 0.3 |
| P38-V453 | 3.3 ± 0.7 | H39-V453 | 2.4 ± 0.3 | H39-M450 | 2.5 ± 0.3 |

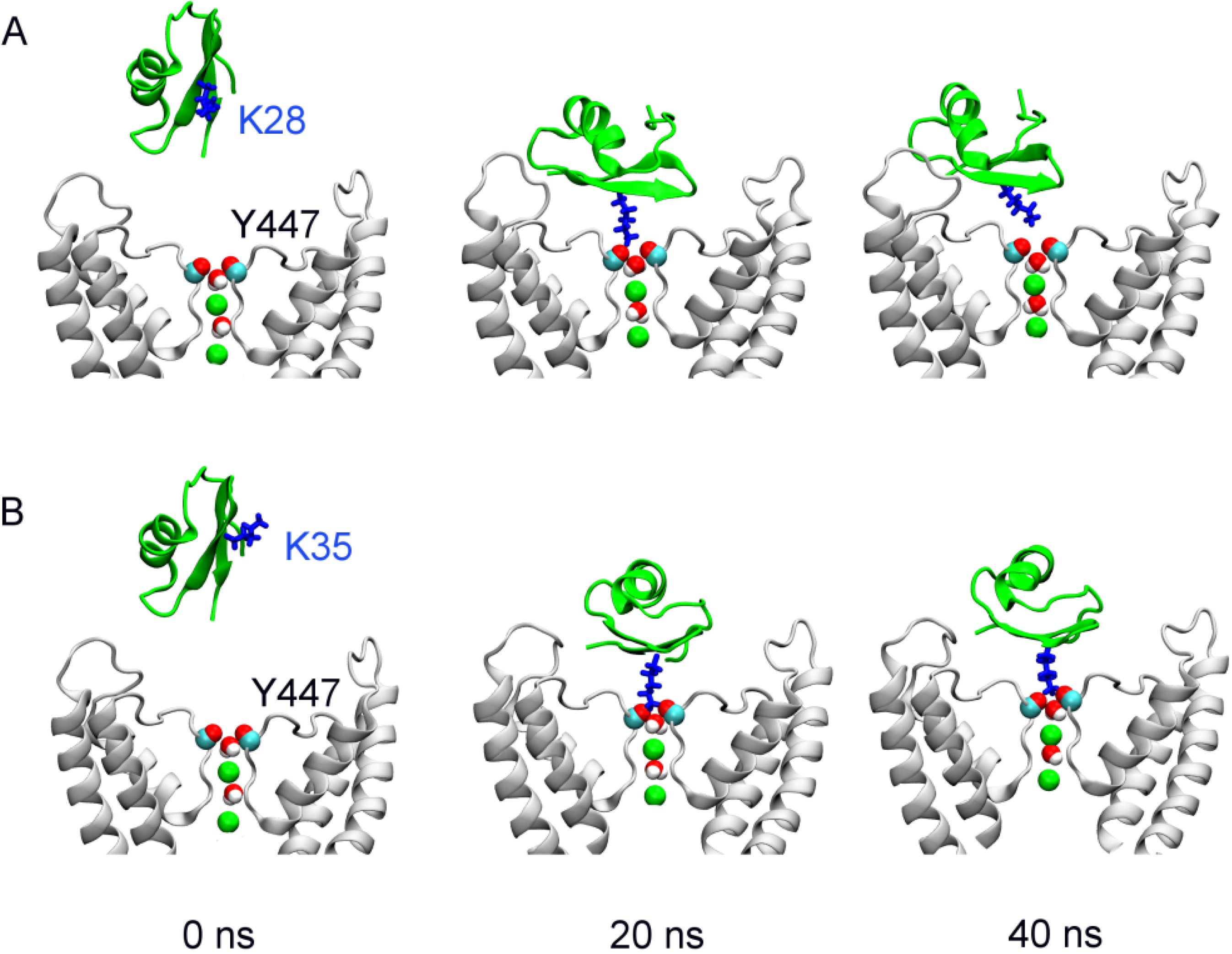
2.2. Binding of HgTx
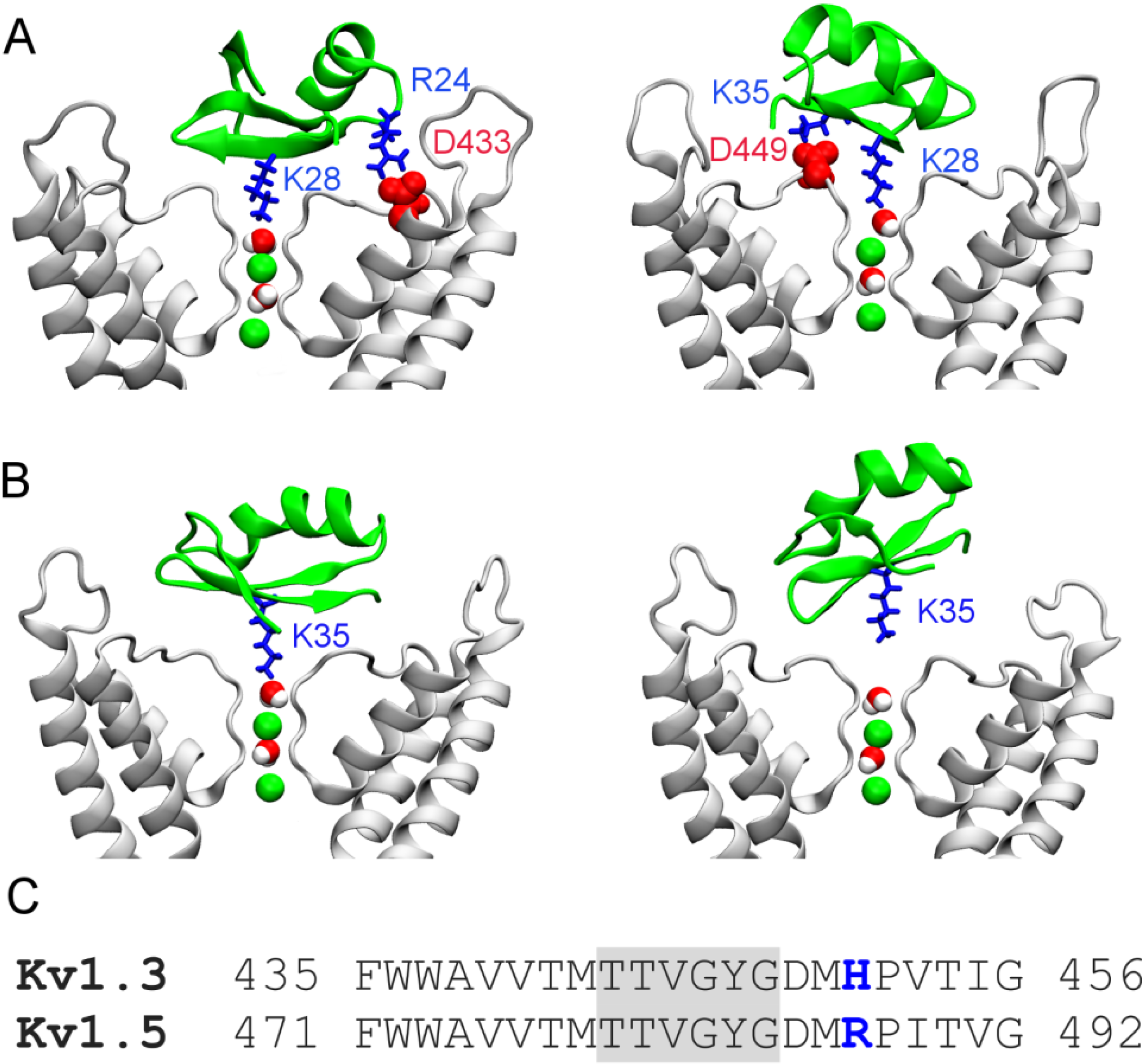
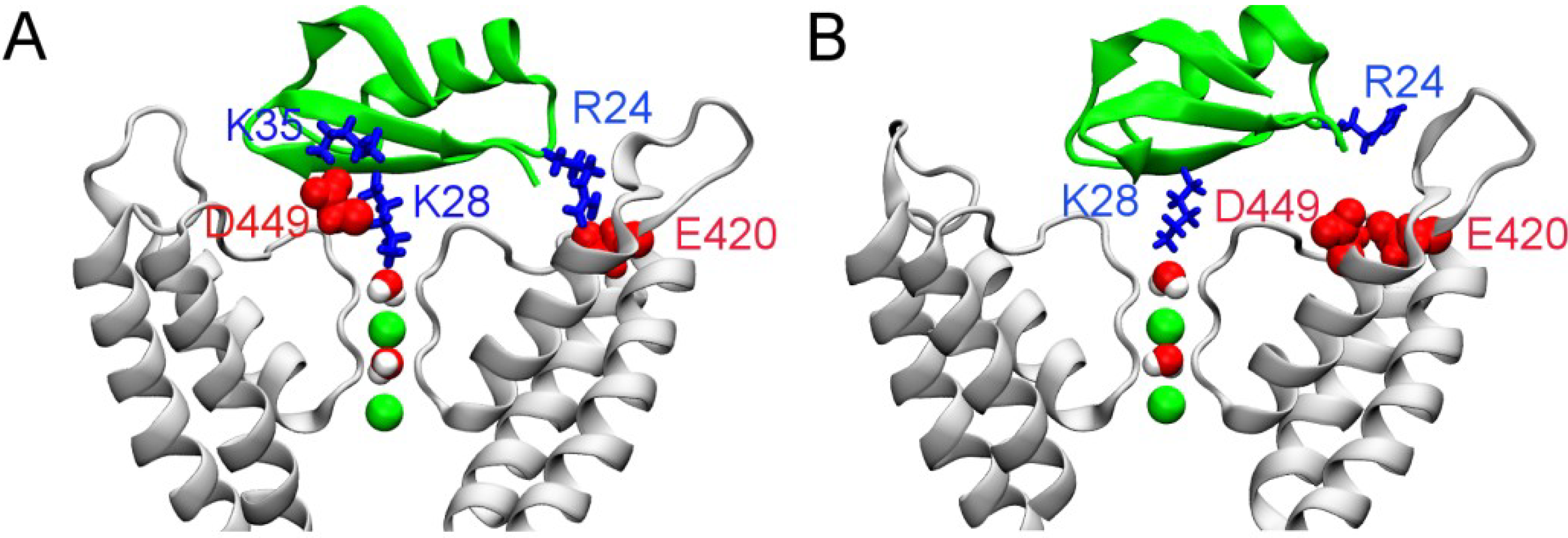
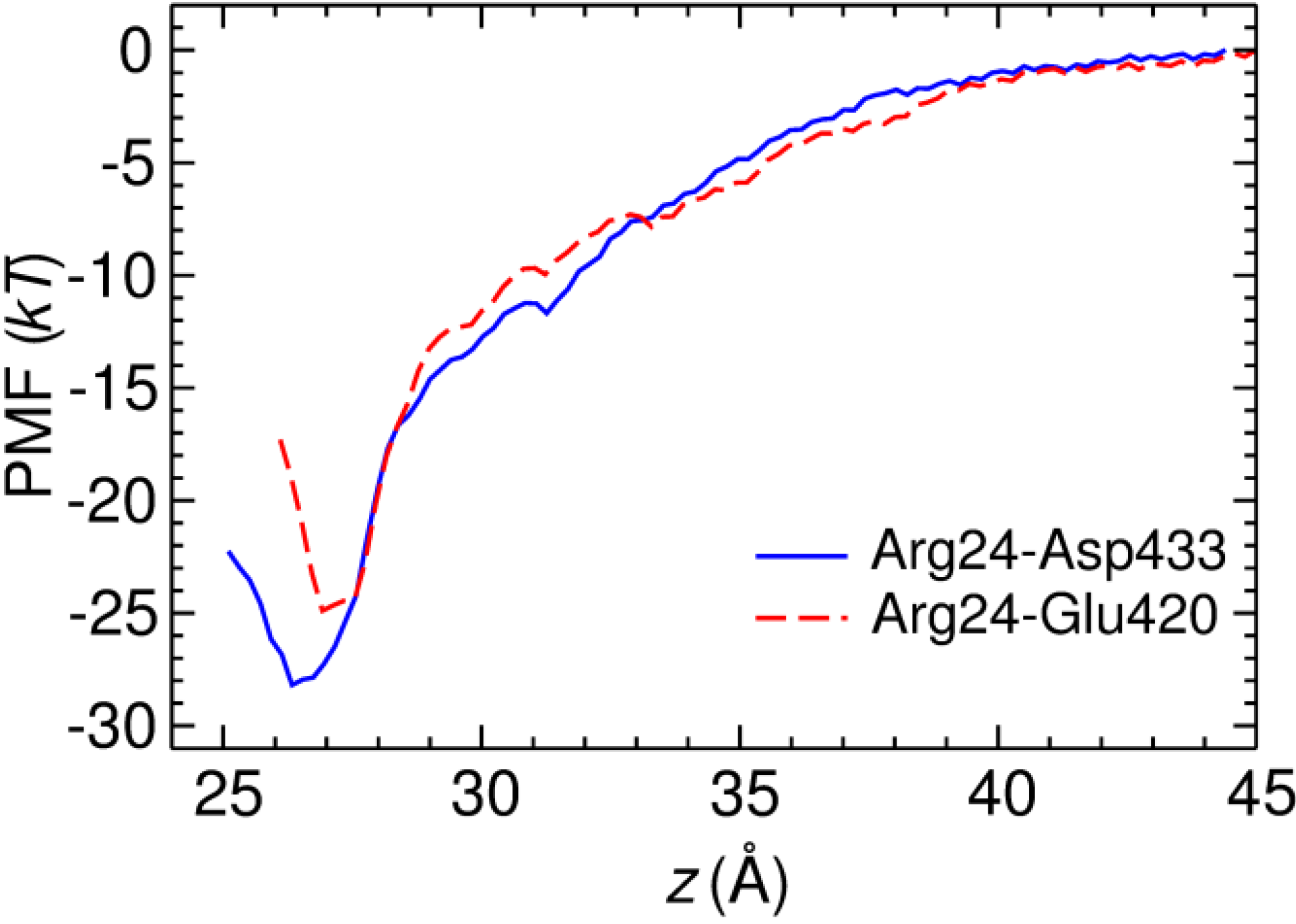
2.3. Two Binding Modes of HgTx-Kv1.3
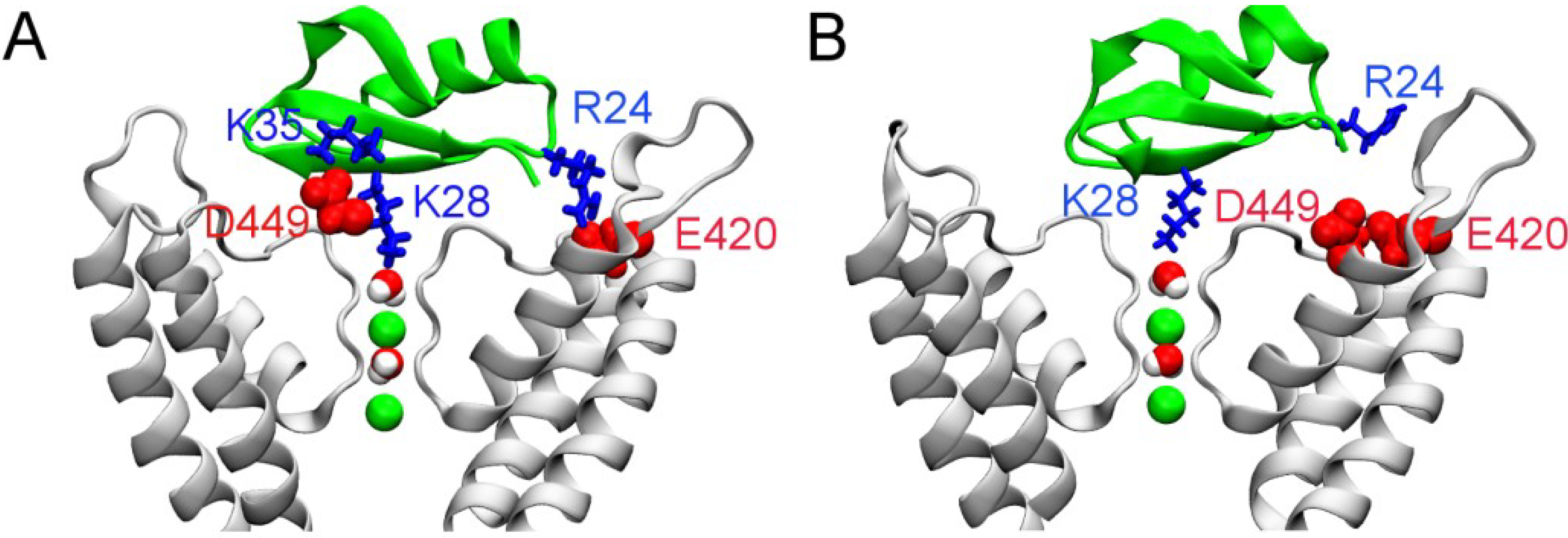
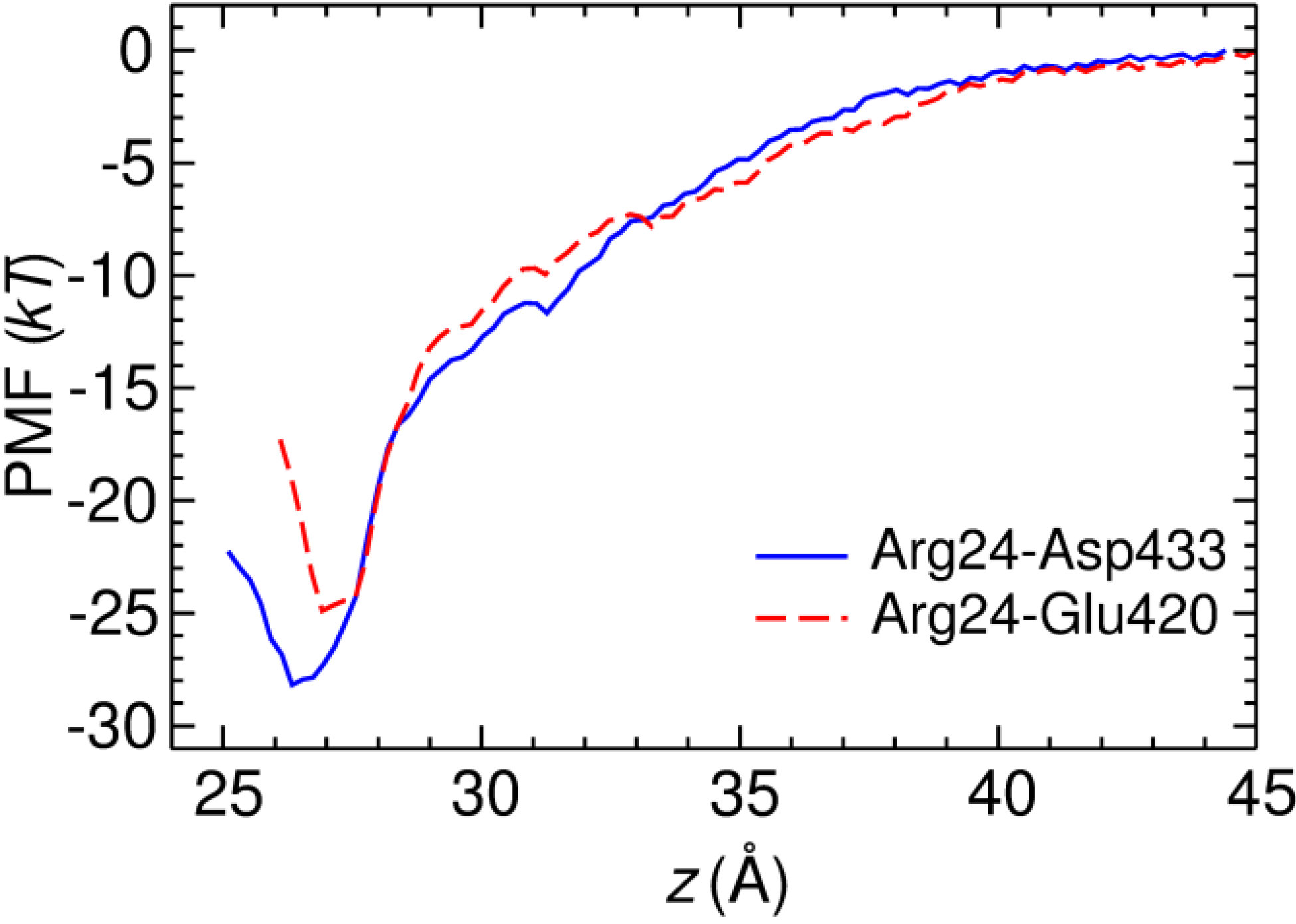
3. Experimental Section
3.1. Molecular Dynamics Simulations
3.2. Potential of Mean Force Calculations
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rangaraju, S.; Chi, V.; Pennington, M.W.; Chandy, K.G. Kv1.3 potassium channels as a therapeutic target in multiple sclerosis. Expert Opin. Ther. Tar. 2009, 13, 909–924. [Google Scholar] [CrossRef]
- Wulff, H.; Pennington, M. Targeting effector memory T-cells with Kv1.3 blockers. Curr. Opin. Drug Discov. Devel. 2007, 10, 438–445. [Google Scholar]
- Wulff, H.; Calabresi, P.A.; Allie, R.; Yun, S.; Pennington, M.; Beeton, C.; Chandy, K.G. The voltage-gated Kv1.3 K+ channel in effector memory T cells as new target for MS. J. Clin. Invest. 2003, 111, 1703–1713. [Google Scholar] [CrossRef]
- Beeton, C.; Wulff, H.; Barbaria, J.; Clot-Faybesse, O.; Pennington, M.; Bernard, D.; Cahalan, M.D.; Chandy, K.G.; Béraud, E. Selective blockade of T lymphocyte K+ channels ameliorates experimental autoimmune encephalomyelitis, a model for multiple sclerosis. Proc. Natl. Acad. Sci. USA. 2001, 98, 13942–13947. [Google Scholar] [CrossRef]
- Chandy, K.G.; Wulff, H.; Beeton, C.; Pennington, M.; Gutman, G.A.; Cahalan, M. K+ channels as targets for specific immunomodulation. Trends Pharmacol. Sci. 2004, 25, 280–289. [Google Scholar] [CrossRef]
- Norton, R.S.; Pallaghy, P.K. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon 1998, 36, 1573–1583. [Google Scholar] [CrossRef]
- Gordon, D.; Chen, R.; Chung, S.H. Computational methods of studying the binding of toxins from venomous animals to biological ion channels: theory and applications. Physiol. Rev. 2013, 93, 767–802. [Google Scholar] [CrossRef]
- Koschak, A.; Bugianesi, R.M.; Mitterdorfer, J.; Kaczorowski, G.J.; Garcia, M.L.; Knaus, H.G. Subunit composition of brain voltage-gated potassium channels determined by hongotoxin-1, a novel peptide derived from Centruroides limbatus venom. J. Biol. Chem. 1998, 273, 2639–2644. [Google Scholar]
- Garciacalvo, M.; Leonard, R.J.; Novick, J.; Stevens, S.P.; Schmalhofer, W.; Kaczorowski, G.J.; Garcia, M.L. Purification, characterization, and biosynthesis of margatoxin, a component of Centruroides Margaritatus venom that selectively inhibits voltage-dependent potassium channels. J. Biol. Chem. 1993, 268, 18866–18874. [Google Scholar]
- Anangi, R.; Koshy, S.; Huq, R.; Beeton, C.; Chuang, W.J.; King, G.F. Recombinant expression of margatoxin and agitoxin-2 in Pichia pastoris: An efficient method for production of Kv1.3 channel blockers. PLoS One 2012, 7, e52965. [Google Scholar]
- Yi, H.; Cao, Z.J.; Yin, S.J.; Dai, C.; Wu, Y.L.; Li, W.X. Interaction simulation of hERG K+ channel with its specific BeKm-1 peptide: Insights into the selectivity of molecular recognition. J. Proteome Res. 2007, 6, 611–620. [Google Scholar] [CrossRef]
- Han, S.; Yin, S.J.; Yi, H.; Mouhat, S.; Qiu, S.; Cao, Z.J.A.; Sabatier, J.M.; Wu, Y.L.; Li, W.X. Protein-protein recognition control by modulating electrostatic interactions. J. Proteome Res. 2010, 9, 3118–3125. [Google Scholar]
- Yi, H.; Qiu, S.; Cao, Z.; Wu, Y.; Li, W. Molecular basis of inhibitory peptide maurotoxin recognizing Kv1.2 channel explored by ZDOCK and molecular dynamic simulations. Proteins 2008, 70, 844–854. [Google Scholar]
- Gan, G.; Yi, H.; Chen, M.; Sun, L.; Li, W.; Wu, Y.; Ding, J. Structural basis for toxin resistance of β4-associated calcium-activated potassium (BK) channels. J. Biol. Chem. 2008, 283, 24177–24184. [Google Scholar]
- Han, S.; Yi, H.; Yin, S.J.; Chen, Z.Y.; Liu, H.; Cao, Z.J.; Wu, Y.L.; Li, W.X. Structural basis of a potent peptide inhibitor designed for Kv1.3 channel, a therapeutic target of autoimmune disease. J. Biol. Chem. 2008, 283, 19058–19065. [Google Scholar]
- Yin, S.J.; Jiang, L.; Yi, H.; Han, S.; Yang, D.W.; Liu, M.L.; Liu, H.; Cao, Z.J.; Wu, Y.L.; Li, W.X. Different residues in channel turret determining the selectivity of ADWX-1 inhibitor peptide between Kv1.1 and Kv1.3 channels. J. Proteome Res. 2008, 7, 4890–4897. [Google Scholar] [CrossRef]
- Rashid, M.H.; Kuyucak, S. Affinity and selectivity of ShK toxin for the Kv1 potassium channels from free energy simulations. J. Phys. Chem. B 2012, 116, 4812–4822. [Google Scholar] [CrossRef]
- Mahdavi, S.; Kuyucak, S. Why the Drosophila Shaker K+ channel is not a good model for ligand binding to voltage-gated Kv1 channels. Biochemistry 2013, 59, 1631–1640. [Google Scholar] [CrossRef]
- Rashid, M.H.; Heinzelmann, G.; Huq, R.; Tajhya, R.B.; Chang, S.C.; Chhabra, S.; Pennington, M.W.; Beeton, C.; Norton, R.S.; Kuyucak, S. A potent and selective peptide blocker of the Kv1.3 channel: prediction from free-energy simulations and experimental confirmation. PLoS One 2013, 8, e78712. [Google Scholar]
- Khabiri, M.; Nikouee, A.; Cwiklik, L.; Grissmer, S.; Ettrich, R. Charybdotoxin unbinding from the mKv1.3 potassium channel: A combined computational and experimental study. J. Phys. Chem. B 2011, 115, 11490–11500. [Google Scholar] [CrossRef]
- Eriksson, M.A.; Roux, B. Modeling the structure of agitoxin in complex with the Shaker K+ channel: A computational approach based on experimental distance restraints extracted from thermodynamic mutant cycles. Biophys. J. 2002, 83, 2595–2609. [Google Scholar] [CrossRef]
- Pragl, B.; Koschak, A.; Trieb, M.; Obermair, G.; Kaufmann, W.A.; Gerster, U.; Blanc, E.; Hahn, C.; Prinz, H.; Schutz, G.; et al. Synthesis, characterization, and application of cy-dye- and alexa-dye-labeled hongotoxin1 analogues. The first high affinity fluorescence probes for voltage-gated K+ channels. Bioconjugate Chem. 2002, 13, 416–425. [Google Scholar] [CrossRef]
- Rashid, M.H.; Mahdavi, S.; Kuyucak, S. Computational studies of marine toxins targeting ion channels. Mar. Drugs 2013, 11, 848–869. [Google Scholar] [CrossRef]
- Banerjee, A.; Lee, A.; Campbell, E.; Mackinnon, R. Structure of a pore-blocking toxin in complex with a eukaryotic voltage-dependent K+ channel. Elife 2013, 2, e00594. [Google Scholar]
- Yu, L.; Sun, C.; Song, D.; Shen, J.; Xu, N.; Gunasekera, A.; Hajduk, P.J.; Olejniczak, E.T. Nuclear magnetic resonance structural studies of a potassium channel-charybdotoxin complex. Biochemistry 2005, 44, 15834–15841. [Google Scholar] [CrossRef]
- Lange, A.; Giller, K.; Hornig, S.; Martin-Eauclaire, M.F.; Pongs, O.; Becker, S.; Baldus, M. Toxin-induced conformational changes in a potassium channel revealed by solid-state NMR. Nature 2006, 440, 959–962. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Hu, Y.T.; Yang, W.S.; He, Y.W.; Feng, J.; Wang, B.; Zhao, R.M.; Ding, J.P.; Cao, Z.J.; Li, W.X.; et al. Hg1, novel peptide inhibitor specific for Kv1.3 channels from first scorpion Kunitz-type potassium channel toxin family. J. Biol. Chem. 2012, 287, 13813–13821. [Google Scholar] [CrossRef]
- Chen, Z.Y.; Zeng, D.Y.; Hu, Y.T.; He, Y.W.; Pan, N.; Ding, J.P.; Cao, Z.J.; Liu, M.L.; Li, W.X.; Yi, H.; et al. Structural and functional diversity of acidic scorpion potassium channel toxins. PLoS One 2012, 7, e35154. [Google Scholar]
- Feng, J.; Hu, Y.; Yi, H.; Yin, S.; Han, S.; Hu, J.; Chen, Z.; Yang, W.; Cao, Z.; de Waard, M.; et al. Two conserved arginine residues from the SK3 potassium channel outer vestibule control selectivity of recognition by scorpion toxins. J. Biol. Chem. 2013, 288, 12544–12553. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, Y.; Hu, J.; Yang, W.; Sabatier, J.M.; De Waard, M.; Cao, Z.; Li, W.; Han, S.; Wu, Y. Unusual binding mode of scorpion toxin BmKTX onto potassium channels relies on its distribution of acidic residues. Biochem. Biophys. Res. Commun. 2014, 447, 70–76. [Google Scholar]
- Chen, R.; Robinson, A.; Gordon, D.; Chung, S.H. Modeling the binding of three toxins to the voltage-gated potassium channel (Kv1.3). Biophys. J. 2011, 101, 2652–2660. [Google Scholar] [CrossRef]
- Johnson, B.A.; Stevens, S.P.; Williamson, J.M. Determination of the three-dimensional structure of margatoxin by 1H, 13C, 15N triple-resonance nuclear magnetic resonance spectroscopy. Biochemistry 1994, 33, 15061–15070. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Structural basis of the selective block of Kv1.2 by maurotoxin from computer simulations. PLoS One 2012, 7, e47253. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef]
- Shoup, D.; Szabo, A. Role of diffusion in ligand binding to macromolecules and cell-bound receptors. Biophys. J. 1982, 40, 33–39. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Molecular dynamics simulations of scorpion toxin recognition by the Ca2+-activated potassium channel KCa3.1. Biophys. J. 2013, 105, 1829–1837. [Google Scholar] [CrossRef]
- Dawson, R.J.; Benz, J.; Stohler, P.; Tetaz, T.; Joseph, C.; Huber, S.; Schmid, G.; Hugin, D.; Pflimlin, P.; Trube, G.; et al. Structure of the Acid-sensing ion channel 1 in complex with the gating modifier Psalmotoxin 1. Nat. Commun. 2012, 3, 936. [Google Scholar] [CrossRef]
- Baconguis, I.; Bohlen, C.J.; Goehring, A.; Julius, D.; Gouaux, E. X-ray structure of acid-sensing ion channel 1-snake toxin complex reveals open state of a Na+-selective channel. Cell 2014, 156, 717–729. [Google Scholar] [CrossRef]
- Lew, M.J.; Flinn, J.P.; Pallaghy, P.K.; Murphy, R.; Whorlow, S.L.; Wright, C.E.; Norton, R.S.; Angus, J.A. Structure-function relationships of ω-conotoxin GVIA. Synthesis, structure, calcium channel binding, and functional assay of alanine-substituted analogues. J. Biol. Chem. 1997, 272, 12014–12023. [Google Scholar] [CrossRef]
- Karbat, I.; Frolow, F.; Froy, O.; Gilles, N.; Cohen, L.; Turkov, M.; Gordon, D.; Gurevitz, M. Molecular basis of the high insecticidal potency of scorpion α-toxins. J. Biol. Chem. 2004, 279, 31679–31686. [Google Scholar]
- Nielsen, K.J.; Schroeder, T.; Lewis, R. Structure-activity relationships of ω-conotoxins at N-type voltage-sensitive calcium channels. J. Mol. Recognit. 2000, 13, 55–70. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Binding modes of μ-conotoxin to the bacterial sodium channel (NaVAb). Biophys. J. 2012, 102, 483–488. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Complex structures between the N-type calcium channel (CaV2.2) and ω-conotoxin GVIA predicted via molecular dynamics. Biochemistry 2013, 52, 3765–3772. [Google Scholar] [CrossRef]
- Chen, R.; Robinson, A.; Chung, S.H. Mechanism of µ-conotoxin PIIIA binding to the voltage-gated Na+ channel NaV1.4. PLoS One 2014, 9, e93267. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, R.; Chung, S.-H. Binding Modes of Two Scorpion Toxins to the Voltage-Gated Potassium Channel Kv1.3 Revealed from Molecular Dynamics. Toxins 2014, 6, 2149-2161. https://doi.org/10.3390/toxins6072149
Chen R, Chung S-H. Binding Modes of Two Scorpion Toxins to the Voltage-Gated Potassium Channel Kv1.3 Revealed from Molecular Dynamics. Toxins. 2014; 6(7):2149-2161. https://doi.org/10.3390/toxins6072149
Chicago/Turabian StyleChen, Rong, and Shin-Ho Chung. 2014. "Binding Modes of Two Scorpion Toxins to the Voltage-Gated Potassium Channel Kv1.3 Revealed from Molecular Dynamics" Toxins 6, no. 7: 2149-2161. https://doi.org/10.3390/toxins6072149