Signaling Cascades of Pasteurella multocida Toxin in Immune Evasion


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
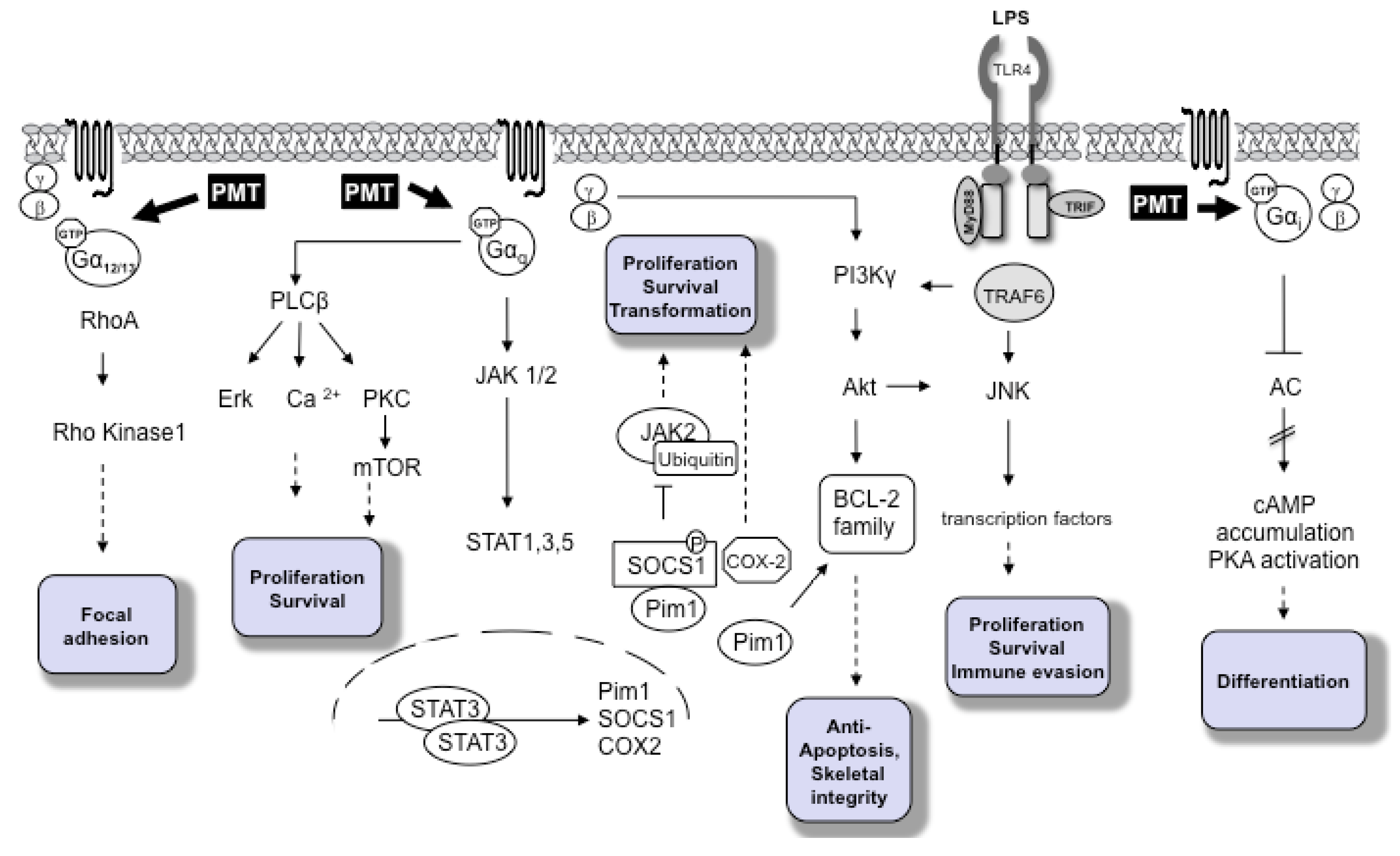
2. Molecular Pathways
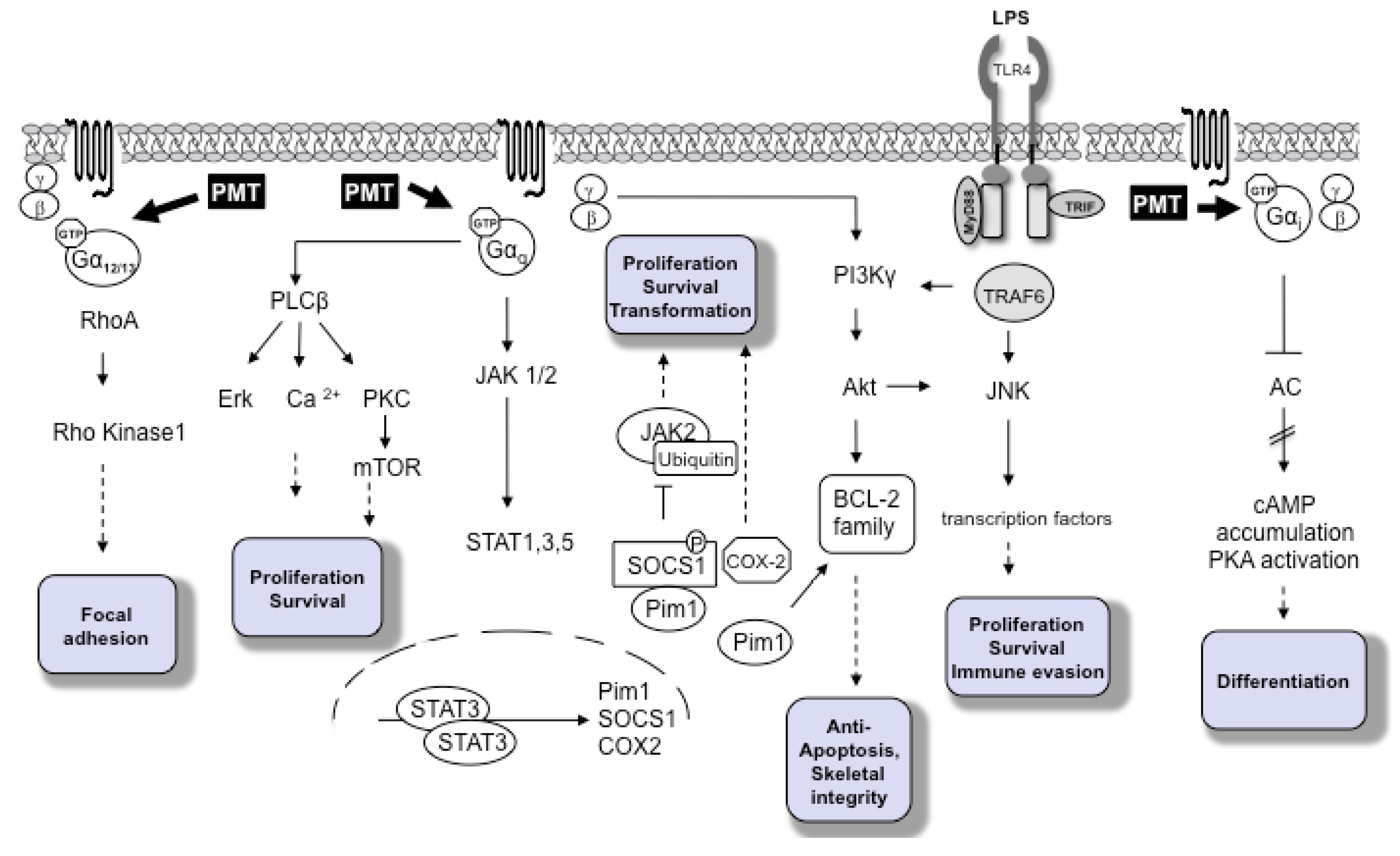
3. PMT as A Potential Carcinogen
3.1. Inflammation and Cancer
3.2. PMT and Cancer
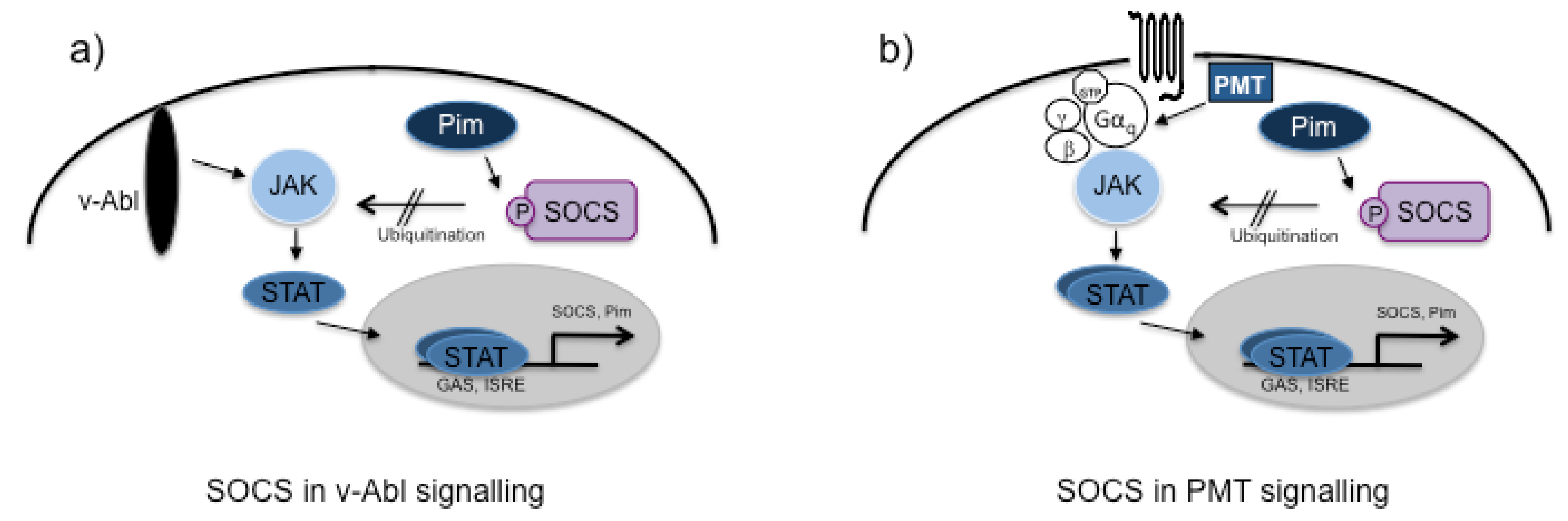
4. PMT-Mediated Pathways in Immune Cells
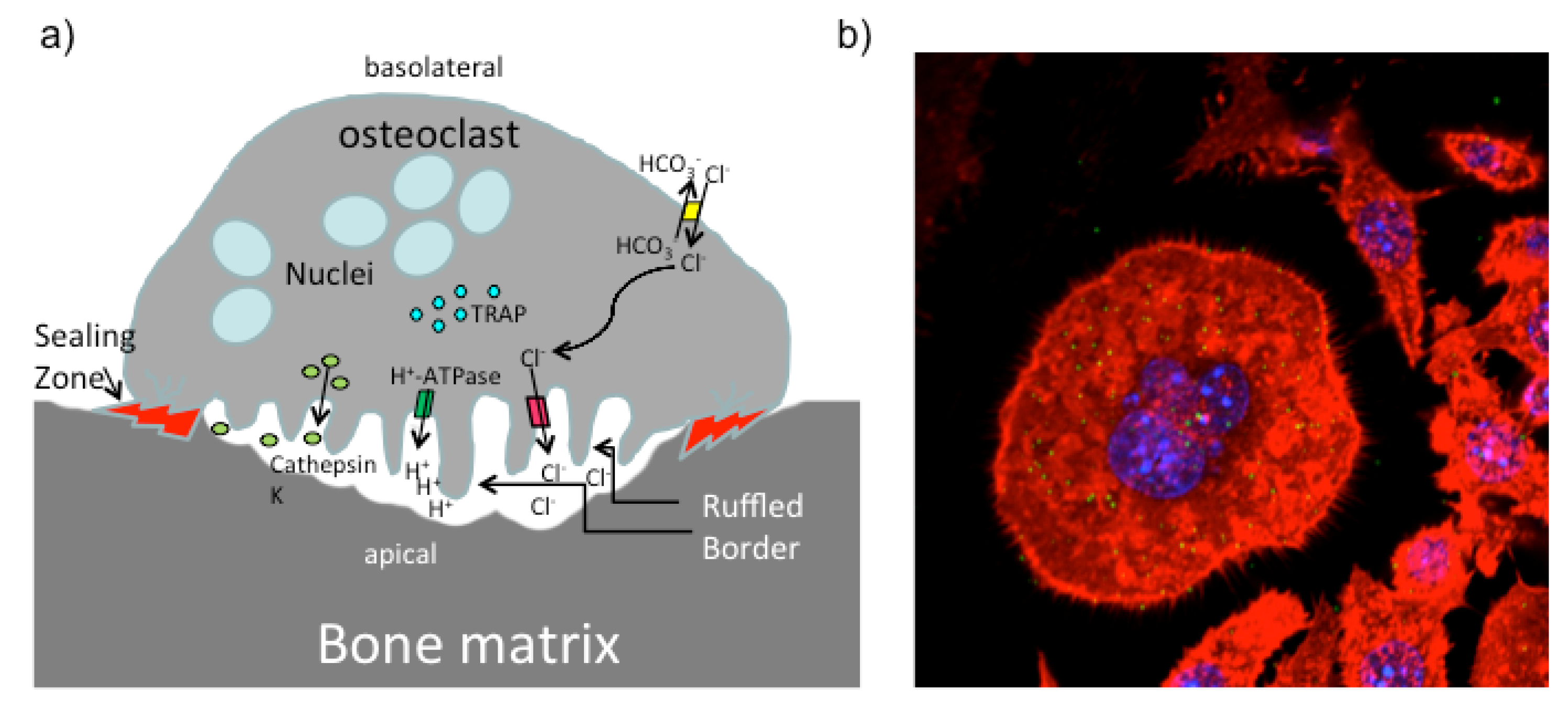
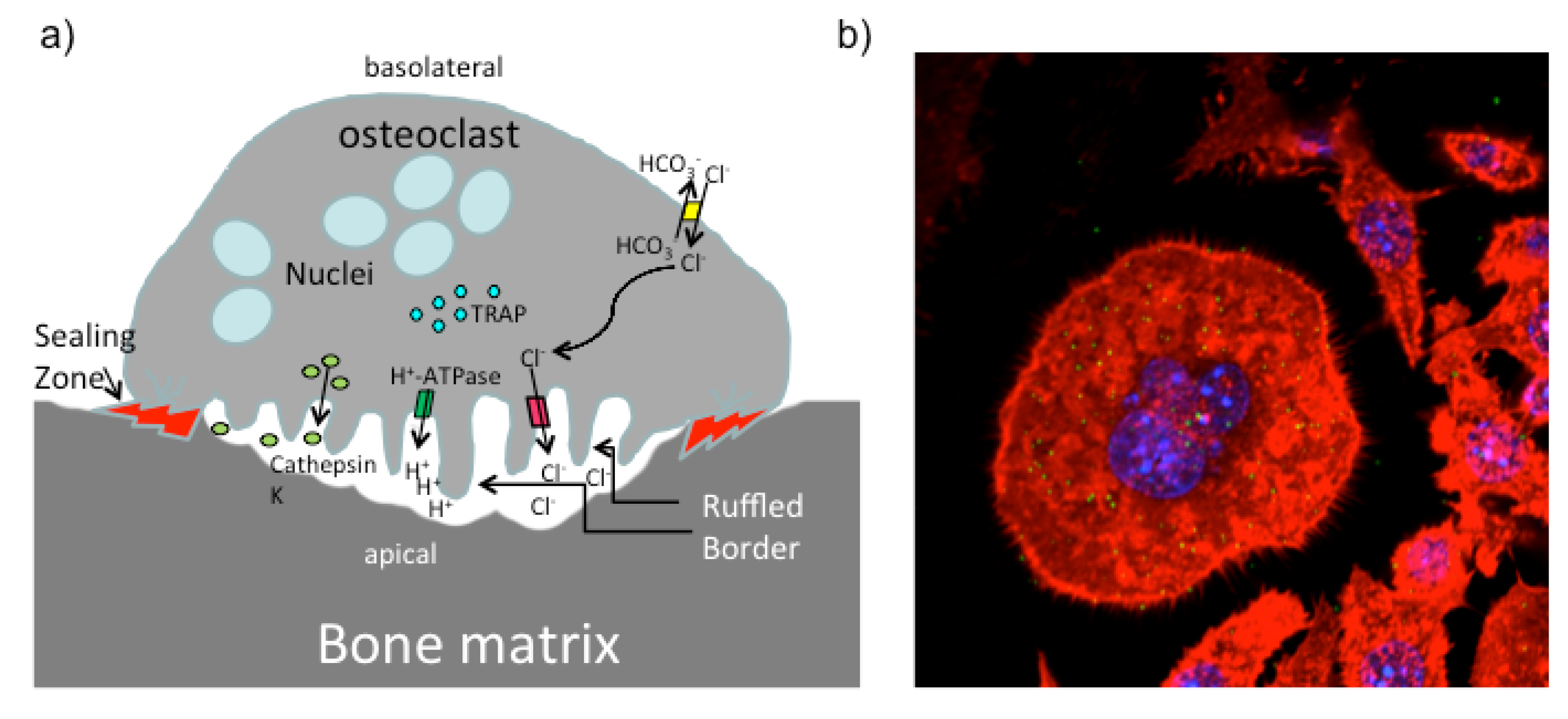
5. PMT and the Skeletal System
5.1. PMT and Osteoclasts
5.2. PMT and Osteoblasts
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Wilkie, I.W.; Harper, M.; Boyce, J.D.; Adler, B. Pasteurella multocida: Diseases and pathogenesis. Curr. Top. Microbiol. Immunol. 2012, 361, 1–22. [Google Scholar] [CrossRef]
- Amigot, J.A.; Torremorell, M.; Pijoan, C. Evaluation of techniques for the detection of toxigenic Pasteurella multocida strains from pigs. J. Vet. Diagn. Investig. 1998, 10, 169–173. [Google Scholar] [CrossRef]
- Martineau-Doize, B.; Frantz, J.C.; Martineau, G.P. Effects of purified Pasteurella multocida dermonecrotoxin on cartilage and bone of the nasal ventral conchae of the piglet. Anat. Rec. 1990, 228, 237–246. [Google Scholar] [CrossRef]
- Horiguchi, Y. Swine atrophic rhinitis caused by Pasteurella multocida toxin and bordetella dermonecrotic toxin. Curr. Top. Microbiol. Immunol. 2012, 361, 113–129. [Google Scholar]
- De Jong, M.F.; Nielsen, J.P. Definition of progressive atrophic rhinitis. Vet. Rec. 1990, 126, 93. [Google Scholar]
- Nakai, T.; Sawata, A.; Tsuji, M.; Kume, K. Characterization of dermonecrotic toxin produced by serotype D strains of Pasteurella multocida. Am. J. Vet. Res. 1984, 45, 2410–2413. [Google Scholar]
- Kubatzky, K.F. Pasteurella multocida and immune cells. Curr. Top. Microbiol. Immunol. 2012, 361, 53–72. [Google Scholar] [CrossRef]
- Sansonetti, P. Host-pathogen interactions: The seduction of molecular cross talk. Gut 2002. [Google Scholar] [CrossRef]
- Barth, K.; Remick, D.G.; Genco, C.A. Disruption of immune regulation by microbial pathogens and resulting chronic inflammation. J. Cell. Physiol. 2013, 228, 1413–1422. [Google Scholar] [CrossRef]
- Coombes, B.K.; Valdez, Y.; Finlay, B.B. Evasive maneuvers by secreted bacterial proteins to avoid innate immune responses. Curr. Biol. 2004, 14, R856–R867. [Google Scholar] [CrossRef]
- Finlay, B.B.; McFadden, G. Anti-immunology: Evasion of the host immune system by bacterial and viral pathogens. Cell 2006, 124, 767–782. [Google Scholar] [CrossRef]
- Repella, T.L.; Ho, M.; Chong, T.P.; Bannai, Y.; Wilson, B.A. Arf6-dependent intracellular trafficking of Pasteurella multocida toxin and pH-dependent translocation from late endosomes. Toxins 2011, 3, 218–241. [Google Scholar] [CrossRef]
- Orth, J.H.; Preuss, I.; Fester, I.; Schlosser, A.; Wilson, B.A.; Aktories, K. Pasteurella multocida toxin activation of heterotrimeric G proteins by deamidation. Proc. Natl. Acad. Sci. USA 2009, 106, 7179–7184. [Google Scholar]
- Orth, J.H.; Fester, I.; Siegert, P.; Weise, M.; Lanner, U.; Kamitani, S.; Tachibana, T.; Wilson, B.A.; Schlosser, A.; Horiguchi, Y.; et al. Substrate specificity of Pasteurella multocida toxin for alpha subunits of heterotrimeric G proteins. FASEB J. 2013, 27, 832–842. [Google Scholar] [CrossRef]
- Babb, R.C.; Homer, K.A.; Robbins, J.; Lax, A.J. Modification of heterotrimeric G-proteins in Swiss 3T3 cells stimulated with Pasteurella multocida toxin. PLoS One 2012, 7, e47188. [Google Scholar]
- Orth, J.H.; Fester, I.; Preuss, I.; Agnoletto, L.; Wilson, B.A.; Aktories, K. Activation of Galpha (i) and subsequent uncoupling of receptor-Galpha(i) signaling by Pasteurella multocida toxin. J. Biol. Chem. 2008, 283, 23288–23294. [Google Scholar] [CrossRef]
- Orth, J.H.C.; Aktories, K. Pasteurella multocida toxin activates various heterotrimeric G proteins by deamidation. Toxins 2010, 2, 205–214. [Google Scholar] [CrossRef]
- Orth, J.H.; Lang, S.; Aktories, K. Action of Pasteurella multocida toxin depends on the helical domain of Galphaq. J. Biol. Chem. 2004, 279, 34150–34155. [Google Scholar] [CrossRef]
- Preuss, I.; Kurig, B.; Nurnberg, B.; Orth, J.H.; Aktories, K. Pasteurella multocida toxin activates Gbetagamma dimers of heterotrimeric G proteins. Cell. Signal. 2009, 21, 551–558. [Google Scholar] [CrossRef]
- Wilson, B.A.; Ho, M. Cellular and molecular action of the mitogenic protein-deamidating toxin from Pasteurella multocida. FEBS J. 2011, 278, 4616–4632. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Shishodia, S.; Sandur, S.K.; Pandey, M.K.; Sethi, G. Inflammation and cancer: How hot is the link? Biochem. Pharmacol. 2006, 72, 1605–1621. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Dalton-Griffin, L.; Kellam, P. Infectious causes of cancer and their detection. J. Biol. 2009, 8, 67. [Google Scholar] [CrossRef]
- Kipanyula, M.J.; Seke Etet, P.F.; Vecchio, L.; Farahna, M.; Nukenine, E.N.; Nwabo Kamdje, A.H. Signaling pathways bridging microbial-triggered inflammation and cancer. Cell. Signal. 2013, 25, 403–416. [Google Scholar] [CrossRef]
- Lax, A.J.; Thomas, W. How bacteria could cause cancer: One step at a time. Trends Microbiol. 2002, 10, 293–299. [Google Scholar] [CrossRef]
- Grote, V.A.; Kaaks, R.; Nieters, A.; Tjonneland, A.; Halkjaer, J.; Overvad, K.; Skjelbo Nielsen, M.R.; Boutron-Ruault, M.C.; Clavel-Chapelon, F.; Racine, A.; et al. Inflammation marker and risk of pancreatic cancer: A nested case-control study within the EPIC cohort. Br. J. Cancer 2012, 106, 1866–1874. [Google Scholar] [CrossRef] [Green Version]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Compare, D.; Nardone, G. Contribution of gut microbiota to colonic and extracolonic cancer development. Dig. Dis. 2011, 29, 554–561. [Google Scholar] [CrossRef]
- Oswald, E.; Nougayrede, J.P.; Taieb, F.; Sugai, M. Bacterial toxins that modulate host cell-cycle progression. Curr. Opin. Microbiol. 2005, 8, 83–91. [Google Scholar]
- Lax, A.J. Opinion: Bacterial toxins and cancer—A case to answer? Nat. Rev. Microbiol. 2005, 3, 343–349. [Google Scholar] [CrossRef]
- Lax, A. The Pasteurella multocida toxin: A new paradigm for the link between bacterial infection and cancer. Curr. Top. Microbiol. Immunol. 2012, 361, 131–144. [Google Scholar] [CrossRef]
- Hoskins, I.C.; Thomas, L.H.; Lax, A.J. Nasal infection with Pasteurella multocida causes proliferation of bladder epithelium in gnotobiotic pigs. Vet. Rec. 1997, 140, 22. [Google Scholar] [CrossRef]
- Rozengurt, E.; Higgins, T.; Chanter, N.; Lax, A.J.; Staddon, J.M. Pasteurella multocida toxin: Potent mitogen for cultured fibroblasts. Proc. Natl. Acad. Sci. USA 1990, 87, 123–127. [Google Scholar] [CrossRef]
- Martineau-Doize, B.; Caya, I.; Gagne, S.; Jutras, I.; Dumas, G. Effects of Pasteurella multocida toxin on the osteoclast population of the rat. J. Comp. Pathol. 1993, 108, 81–91. [Google Scholar] [CrossRef]
- Nougayrede, J.P.; Taieb, F.; de Rycke, J.; Oswald, E. Cyclomodulins: Bacterial effectors that modulate the eukaryotic cell cycle. Trends Microbiol. 2005, 13, 103–110. [Google Scholar] [CrossRef]
- Orth, J.H.; Aktories, K.; Kubatzky, K.F. Modulation of host cell gene expression through activation of STAT transcription factors by Pasteurella multocida toxin. J. Biol. Chem. 2007, 282, 3050–3057. [Google Scholar]
- Seo, B.; Choy, E.W.; Maudsley, S.; Miller, W.E.; Wilson, B.A.; Luttrell, L.M. Pasteurella multocida toxin stimulates mitogen-activated protein kinase via G(q/11)-dependent transactivation of the epidermal growth factor receptor. J. Biol. Chem. 2000, 275, 2239–2245. [Google Scholar]
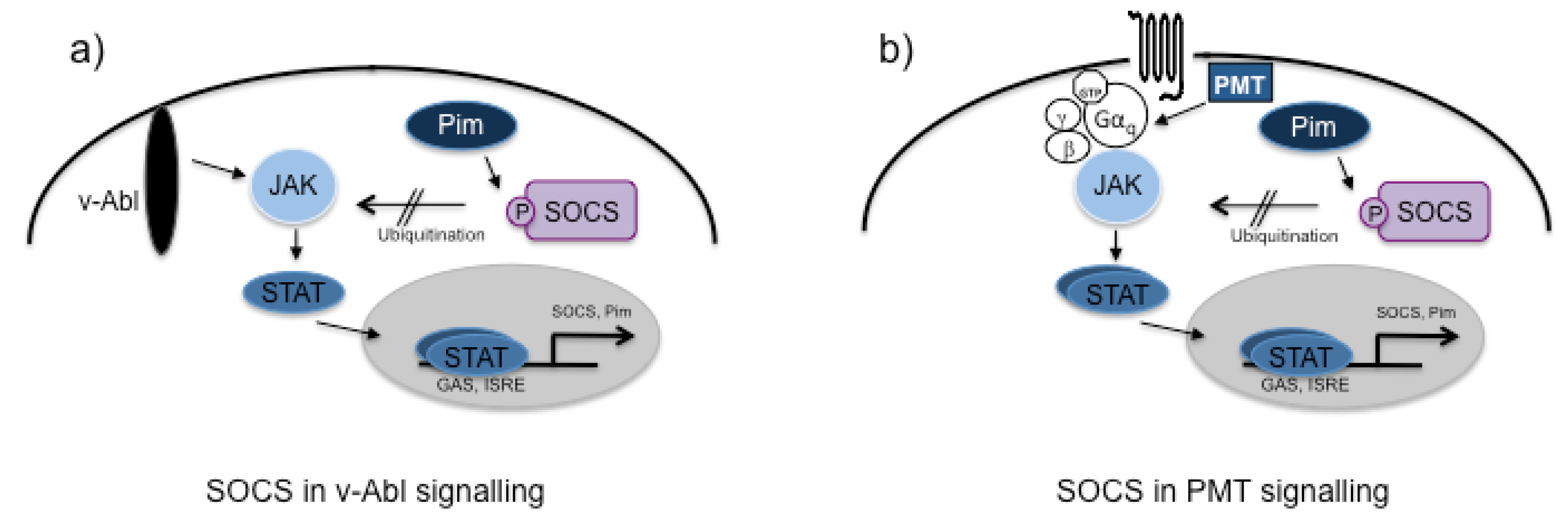
- Yoshimura, A.; Suzuki, M.; Sakaguchi, R.; Hanada, T.; Yasukawa, H. SOCS, inflammation, and autoimmunity. Front. Immunol. 2012, 3, 20. [Google Scholar]
- Hildebrand, D.; Walker, P.; Dalpke, A.; Heeg, K.; Kubatzky, K.F. Pasteurella multocida Toxin-induced Pim-1 expression disrupts suppressor of cytokine signaling (SOCS)-1 activity. Cell. Microbiol. 2010, 12, 1732–1745. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef]
- Chen, J.L.; Limnander, A.; Rothman, P.B. Pim-1 and Pim-2 kinases are required for efficient pre-B-cell transformation by v-Abl oncogene. Blood 2008, 111, 1677–1685. [Google Scholar]
- Limnander, A.; Danial, N.N.; Rothman, P.B. v-Abl signaling disrupts SOCS-1 function in transformed pre-B cells. Mol. Cell. 2004, 15, 329–341. [Google Scholar] [CrossRef]
- Al-Haddawi, M.H.; Jasni, S.; Zamri-Saad, M.; Mutalib, A.R.; Son, R.; Sheikh-Omar, A.R. Ultrastructural observation of nasal and pulmonary intracellular Pasteurella multocida A:3 in rabbits. Vet. Res. Commun. 2000, 24, 153–167. [Google Scholar] [CrossRef]
- Al-haj Ali, H.; Sawada, T.; Hatakeyama, H.; Katayama, Y.; Ohtsuki, N.; Itoh, O. Invasion of chicken embryo fibroblast cells by avian Pasteurella multocida. Vet. Microbiol. 2004, 104, 55–62. [Google Scholar] [CrossRef]
- Galdiero, M.; de Martino, L.; Pagnini, U.; Pisciotta, M.G.; Galdiero, E. Interactions between bovine endothelial cells and Pasteurella multocida: Association and invasion. Res. Microbiol. 2001, 152, 57–65. [Google Scholar] [CrossRef]
- Hundt, M.J.; Ruffolo, C.G. Interaction of Pasteurella multocida with free-living amoebae. Appl. Environ. Microbiol. 2005, 71, 5458–5464. [Google Scholar] [CrossRef]
- Othman, S.; Parton, R.; Coote, J. Interaction between mammalian cells and Pasteurella multocida B:2. Adherence, invasion and intracellular survival. Microbial. Pathog. 2012, 52, 353–358. [Google Scholar] [CrossRef]
- Rabier, M.J.; Tyler, N.K.; Walker, N.J.; Hansen, L.M.; Hirsh, D.C.; Tablin, F. Pasteurella multocida enters polarized epithelial cells by interacting with host F-actin. Vet. Microbiol. 1997, 54, 343–355. [Google Scholar] [CrossRef]
- Lee, M.D.; Wooley, R.E.; Glisson, J.R. Invasion of epithelial cell monolayers by turkey strains of Pasteurella multocida. Avian Dis. 1994, 38, 72–77. [Google Scholar] [CrossRef]
- Nawijn, M.C.; Alendar, A.; Berns, A. For better or for worse: The role of Pim oncogenes in tumorigenesis. Nat. Rev. Cancer 2011, 11, 23–34. [Google Scholar] [CrossRef]
- Preuss, I.; Hildebrand, D.; Orth, J.H.; Aktories, K.; Kubatzky, K.F. Pasteurella multocida toxin is a potent activator of anti-apoptotic signaling pathways. Cell. Microbiol. 2010, 12, 1174–1185. [Google Scholar] [CrossRef]
- Nagase, Y.; Iwasawa, M.; Akiyama, T.; Kadono, Y.; Nakamura, M.; Oshima, Y.; Yasui, T.; Matsumoto, T.; Hirose, J.; Nakamura, H.; et al. Anti-apoptotic molecule Bcl-2 regulates the differentiation, activation, and survival of both osteoblasts and osteoclasts. J. Biol. Chem. 2009, 284, 36659–36669. [Google Scholar] [CrossRef]
- Yamashita, J.; Datta, N.S.; Chun, Y.H.; Yang, D.Y.; Carey, A.A.; Kreider, J.M.; Goldstein, S.A.; McCauley, L.K. Role of Bcl2 in osteoclastogenesis and PTH anabolic actions in bone. J. Bone Miner. Res. 2008, 23, 621–632. [Google Scholar]
- Faustin, B.; Chen, Y.; Zhai, D.; le Negrate, G.; Lartigue, L.; Satterthwait, A.; Reed, J.C. Mechanism of Bcl-2 and Bcl-X(L) inhibition of NLRP1 inflammasome: Loop domain-dependent suppression of ATP binding and oligomerization. Proc. Natl. Acad. Sci. USA 2009, 106, 3935–3940. [Google Scholar]
- Bruey, J.M.; Bruey-Sedano, N.; Luciano, F.; Zhai, D.; Balpai, R.; Xu, C.; Kress, C.L.; Bailly-Maitre, B.; Li, X.; Osterman, A.; et al. Bcl-2 and Bcl-XL regulate proinflammatory caspase-1 activation by interaction with NALP1. Cell 2007, 129, 45–56. [Google Scholar] [CrossRef]
- Oubrahim, H.; Wong, A.; Wilson, B.A.; Chock, P.B. Mammalian target of rapamycin complex 1 (mTORC1) plays a role in Pasteurella multocida toxin (PMT)-induced protein synthesis and proliferation in Swiss 3T3 cells. J. Biol. Chem. 2013, 288, 2805–2815. [Google Scholar] [CrossRef]
- Alayev, A.; Holz, M.K. mTOR signaling for biological control and cancer. J. Cell. Physiol. 2013, 228, 1658–1664. [Google Scholar] [CrossRef]
- Thiem, S.; Pierce, T.P.; Palmieri, M.; Putoczki, T.L.; Buchert, M.; Preaudet, A.; Farid, R.O.; Love, C.; Catimel, B.; Lei, Z.; et al. mTORC1 inhibition restricts inflammation-associated gastrointestinal tumorigenesis in mice. J. Clin. Invest. 2013, 123, 767–781. [Google Scholar]
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell. Biol. 2011, 12, 21–35. [Google Scholar] [CrossRef] [Green Version]
- Oubrahim, H.; Wong, A.; Wilson, B.A.; Chock, P.B. Pasteurella multocida toxin (PMT) upregulates CTGF which leads to mTORC1 activation in Swiss 3T3 cells. Cell. Signal. 2013, 25, 1136–1148. [Google Scholar] [CrossRef]
- Kular, L.; Pakradouni, J.; Kitabgi, P.; Laurent, M.; Martinerie, C. The CCN family: A new class of inflammation modulators? Biochimie 2011, 93, 377–388. [Google Scholar] [CrossRef]
- Cheville, N.F.; Rimler, R.B. A protein toxin from Pasteurella multocida type D causes acute and chronic hepatic toxicity in rats. Vet. Pathol. 1989, 26, 148–157. [Google Scholar] [CrossRef]
- Al-Haddawi, M.H.; Jasni, S.; Israf, D.A.; Zamri-Saad, M.; Mutalib, A.R.; Sheikh-Omar, A.R. Ultrastructural pathology of nasal and tracheal mucosa of rabbits experimentally infected with Pasteurella multocida serotype D:1. Res. Vet. Sci. 2001, 70, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Hildebrand, D.; Heeg, K.; Kubatzky, K.F. Pasteurella multocida toxin-stimulated osteoclast differentiation is B cell dependent. Infect. Immun. 2011, 79, 220–228. [Google Scholar] [CrossRef]
- Hildebrand, D.; Sahr, A.; Wolfle, S.J.; Heeg, K.; Kubatzky, K.F. Regulation of Toll-like receptor 4-mediated immune responses through Pasteurella multocida toxin-induced G protein signaling. Cell. Commun. Signal. 2012, 10, 22. [Google Scholar] [CrossRef]
- Wolfle, S.J.; Strebovsky, J.; Bartz, H.; Sahr, A.; Arnold, C.; Kaiser, C.; Dalpke, A.H.; Heeg, K. PD-L1 expression on tolerogenic APCs is controlled by STAT-3. Eur. J. Immunol. 2011, 41, 413–424. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef]
- Guha, M.; Mackman, N. LPS induction of gene expression in human monocytes. Cell. Signal. 2001, 13, 85–94. [Google Scholar] [CrossRef]
- Schmittel, A.; Scheibenbogen, C.; Keilholz, U. Lipopolysaccharide effectively up-regulates B7-1 (CD80) expression and costimulatory function of human monocytes. Scand. J. Immunol. 1995, 42, 701–704. [Google Scholar] [CrossRef]
- Bagley, K.C.; Abdelwahab, S.F.; Tuskan, R.G.; Fouts, T.R.; Lewis, G.K. Cholera toxin and heat-labile enterotoxin activate human monocyte-derived dendritic cells and dominantly inhibit cytokine production through a cyclic AMP-dependent pathway. Infect. Immun. 2002, 70, 5533–5539. [Google Scholar] [CrossRef]
- Braun, M.C.; Kelsall, B.L. Regulation of interleukin-12 production by G-protein-coupled receptors. Microbes Infect. 2001, 3, 99–107. [Google Scholar] [CrossRef]
- Bagley, K.C.; Abdelwahab, S.F.; Tuskan, R.G.; Lewis, G.K. Pasteurella multocida toxin activates human monocyte-derived and murine bone marrow-derived dendritic cells in vitro but suppresses antibody production in vivo. Infect. Immun. 2005, 73, 413–421. [Google Scholar] [CrossRef]
- Trinchieri, G. Interleukin-12: A cytokine at the interface of inflammation and immunity. Adv. Immunol. 1998, 70, 83–243. [Google Scholar] [CrossRef]
- Kubin, M.; Kamoun, M.; Trinchieri, G. Interleukin 12 synergizes with B7/CD28 interaction in inducing efficient proliferation and cytokine production of human T cells. J. Exp. Med. 1994, 180, 211–222. [Google Scholar] [CrossRef]
- Wolf, S.F.; Temple, P.A.; Kobayashi, M.; Young, D.; Dicig, M.; Lowe, L.; Dzialo, R.; Fitz, L.; Ferenz, C.; Hewick, R.M.; et al. Cloning of cDNA for natural killer cell stimulatory factor, a heterodimeric cytokine with multiple biologic effects on T and natural killer cells. J. Immunol. 1991, 146, 3074–3081. [Google Scholar]
- Blocker, D.; Berod, L.; Fluhr, J.W.; Orth, J.; Idzko, M.; Aktories, K.; Norgauer, J. Pasteurella multocida toxin (PMT) activates RhoGTPases, induces actin polymerization and inhibits migration of human dendritic cells, but does not influence macropinocytosis. Int. Immunol. 2006, 18, 459–464. [Google Scholar] [CrossRef]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef]
- Aktories, K.; Schwan, C.; Papatheodorou, P.; Lang, A.E. Bidirectional attack on the actin cytoskeleton. Bacterial protein toxins causing polymerization or depolymerization of actin. Toxicon 2012, 60, 572–581. [Google Scholar] [CrossRef]
- Barbieri, J.T.; Riese, M.J.; Aktories, K. Bacterial toxins that modify the actin cytoskeleton. Annu. Rev. Cell Dev. Biol. 2002, 18, 315–344. [Google Scholar] [CrossRef]
- Van Diemen, P.M.; de Vries Reilingh, G.; Parmentier, H.K. Immune responses of piglets to Pasteurella multocida toxin and toxoid. Vet. Immunol. Immunopathol. 1994, 41, 307–321. [Google Scholar] [CrossRef]
- Bording, A.; Foged, N.T. Characterization of the immunogenicity of formaldehyde detoxified Pasteurella multocida toxin. Vet. Microbiol. 1991, 29, 267–280. [Google Scholar] [CrossRef]
- Suckow, M.A.; Bowersock, T.L.; Nielsen, K.; Chrisp, C.E.; Frandsen, P.L.; Janovitz, E.B. Protective immunity to Pasteurella multocida heat-labile toxin by intranasal immunization in rabbits. Lab. Anim. Sci. 1995, 45, 526–532. [Google Scholar]
- To, H.; Someno, S.; Nagai, S. Development of a genetically modified nontoxigenic Pasteurella multocida toxin as a candidate for use in vaccines against progressive atrophic rhinitis in pigs. Am. J. Vet. Res. 2005, 66, 113–118. [Google Scholar] [CrossRef]
- Arron, J.R.; Choi, Y. Bone versus immune system. Nature 2000, 408, 535–536. [Google Scholar] [CrossRef]
- Takayanagi, H. New developments in osteoimmunology. Nat. Rev. Rheumatol. 2012, 8, 684–689. [Google Scholar] [CrossRef]
- Kamp, E.M.; Kimman, T.G. Induction of nasal turbinate atrophy in germ-free pigs, using Pasteurella multocida as well as bacterium-free crude and purified dermonecrotic toxin of P. multocida. Am. J. Vet. Res. 1988, 49, 1844–1849. [Google Scholar]
- Jutras, I.; Martineau-Doize, B. Stimulation of osteoclast-like cell formation by Pasteurella multocida toxin from hemopoietic progenitor cells in mouse bone marrow cultures. Can. J. Vet. Res. 1996, 60, 34–39. [Google Scholar]
- Gwaltney, S.M.; Galvin, R.J.; Register, K.B.; Rimler, R.B.; Ackermann, M.R. Effects of Pasteurella multocida toxin on porcine bone marrow cell differentiation into osteoclasts and osteoblasts. Vet. Pathol. 1997, 34, 421–430. [Google Scholar] [CrossRef]
- Mullan, P.B.; Lax, A.J. Pasteurella multocida toxin stimulates bone resorption by osteoclasts via interaction with osteoblasts. Calcif. Tissue Int. 1998, 63, 340–345. [Google Scholar] [CrossRef]
- Sterner-Kock, A.; Lanske, B.; Uberschar, S.; Atkinson, M.J. Effects of the Pasteurella multocida toxin on osteoblastic cells in vitro. Vet. Pathol. 1995, 32, 274–279. [Google Scholar] [CrossRef]
- Mullan, P.B.; Lax, A.J. Pasteurella multocida toxin is a mitogen for bone cells in primary culture. Infect. Immun. 1996, 64, 959–965. [Google Scholar]
- Siegert, P.; Schmidt, G.; Papatheodorou, P.; Wieland, T.; Aktories, K.; Orth, J.H. Pasteurella multocida toxin prevents osteoblast differentiation by transactivation of the MAP-kinase cascade via the Galpha(q/11)-p63RhoGEF-RhoA axis. PLoS Pathog. 2013, 9, e1003385. [Google Scholar] [CrossRef]
- Suganami, T.; Tanaka, M.; Ogawa, Y. Adipose tissue inflammation and ectopic lipid accumulation. Endocr. J. 2012, 59, 849–857. [Google Scholar] [CrossRef]
- Fresno, M.; Alvarez, R.; Cuesta, N. Toll-like receptors, inflammation, metabolism and obesity. Arch. Physiol. Biochem. 2011, 117, 151–164. [Google Scholar] [CrossRef]
- Marie, P.J. Transcription factors controlling osteoblastogenesis. Arch. Biochem. Biophys. 2008, 473, 98–105. [Google Scholar] [CrossRef]
- Orth, J.H.; Lang, S.; Taniguchi, M.; Aktories, K. Pasteurella multocida toxin-induced activation of RhoA is mediated via two families of G{alpha} proteins, G{alpha}q and G{alpha}12/13. J. Biol. Chem. 2005, 280, 36701–36707. [Google Scholar] [CrossRef]
- Harmey, D.; Stenbeck, G.; Nobes, C.D.; Lax, A.J.; Grigoriadis, A.E. Regulation of osteoblast differentiation by Pasteurella multocida toxin (PMT): A role for Rho GTPase in bone formation. J. Bone Miner. Res. 2004, 19, 661–670. [Google Scholar] [CrossRef]
- Haniffa, M.A.; Collin, M.P.; Buckley, C.D.; Dazzi, F. Mesenchymal stem cells: The fibroblasts’ new clothes? Haematologica 2009, 94, 258–263. [Google Scholar] [CrossRef]
- Ren, G.; Zhang, L.; Zhao, X.; Xu, G.; Zhang, Y.; Roberts, A.I.; Zhao, R.C.; Shi, Y. Mesenchymal stem cell-mediated immunosuppression occurs via concerted action of chemokines and nitric oxide. Cell. Stem Cell 2008, 2, 141–150. [Google Scholar] [CrossRef]
- Flavell, S.J.; Hou, T.Z.; Lax, S.; Filer, A.D.; Salmon, M.; Buckley, C.D. Fibroblasts as novel therapeutic targets in chronic inflammation. Br. J. Pharmacol. 2008, 153, S241–S246. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kubatzky, K.F.; Kloos, B.; Hildebrand, D. Signaling Cascades of Pasteurella multocida Toxin in Immune Evasion. Toxins 2013, 5, 1664-1681. https://doi.org/10.3390/toxins5091664
Kubatzky KF, Kloos B, Hildebrand D. Signaling Cascades of Pasteurella multocida Toxin in Immune Evasion. Toxins. 2013; 5(9):1664-1681. https://doi.org/10.3390/toxins5091664
Chicago/Turabian StyleKubatzky, Katharina F., Bianca Kloos, and Dagmar Hildebrand. 2013. "Signaling Cascades of Pasteurella multocida Toxin in Immune Evasion" Toxins 5, no. 9: 1664-1681. https://doi.org/10.3390/toxins5091664