CD28: Direct and Critical Receptor for Superantigen Toxins

Abstract
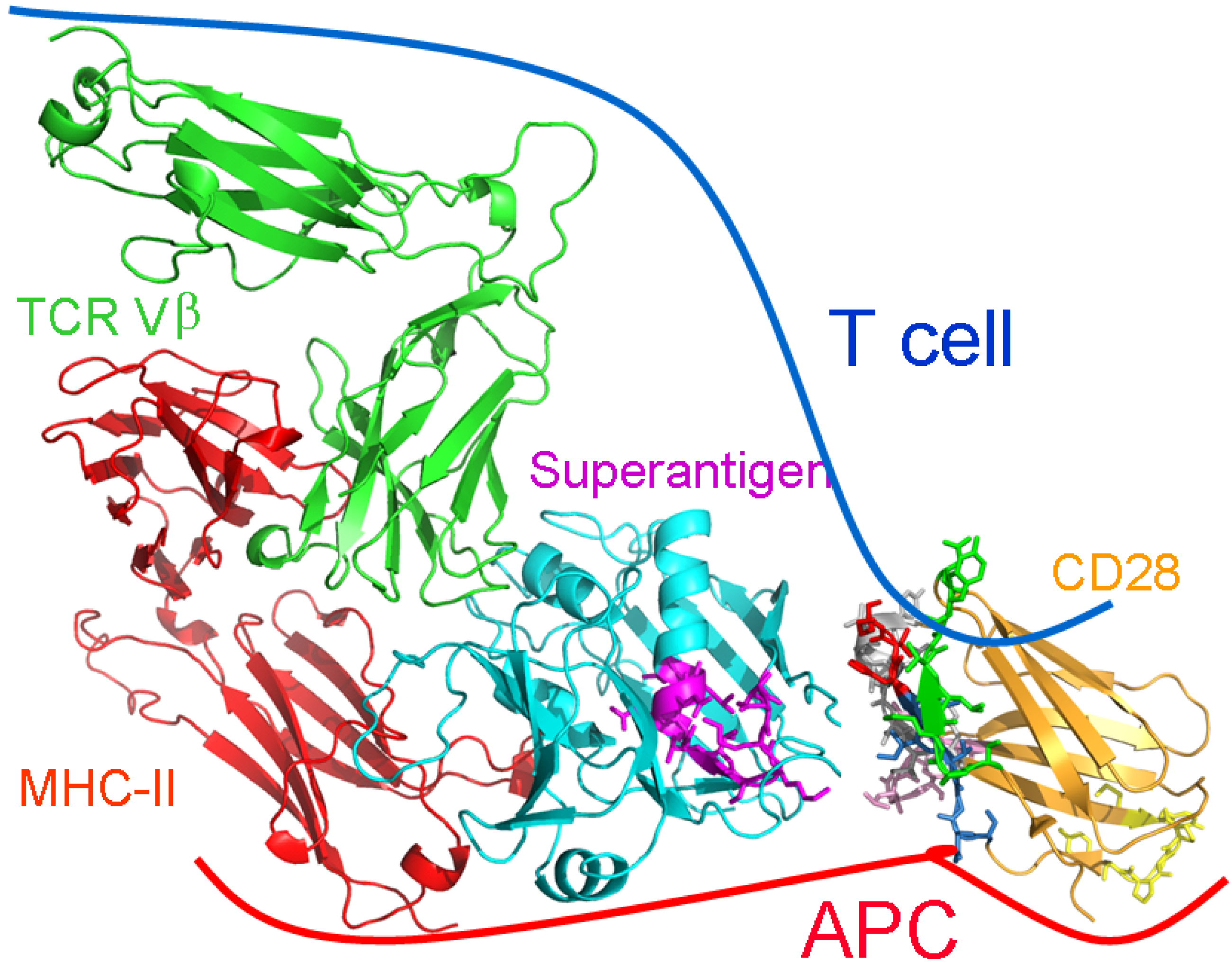
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
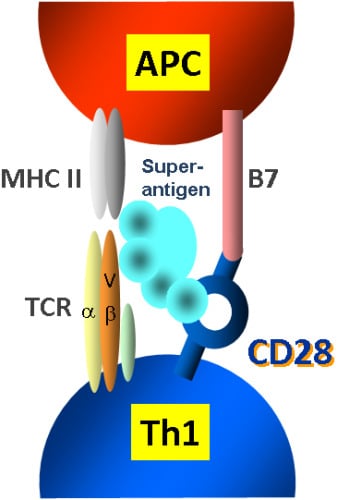
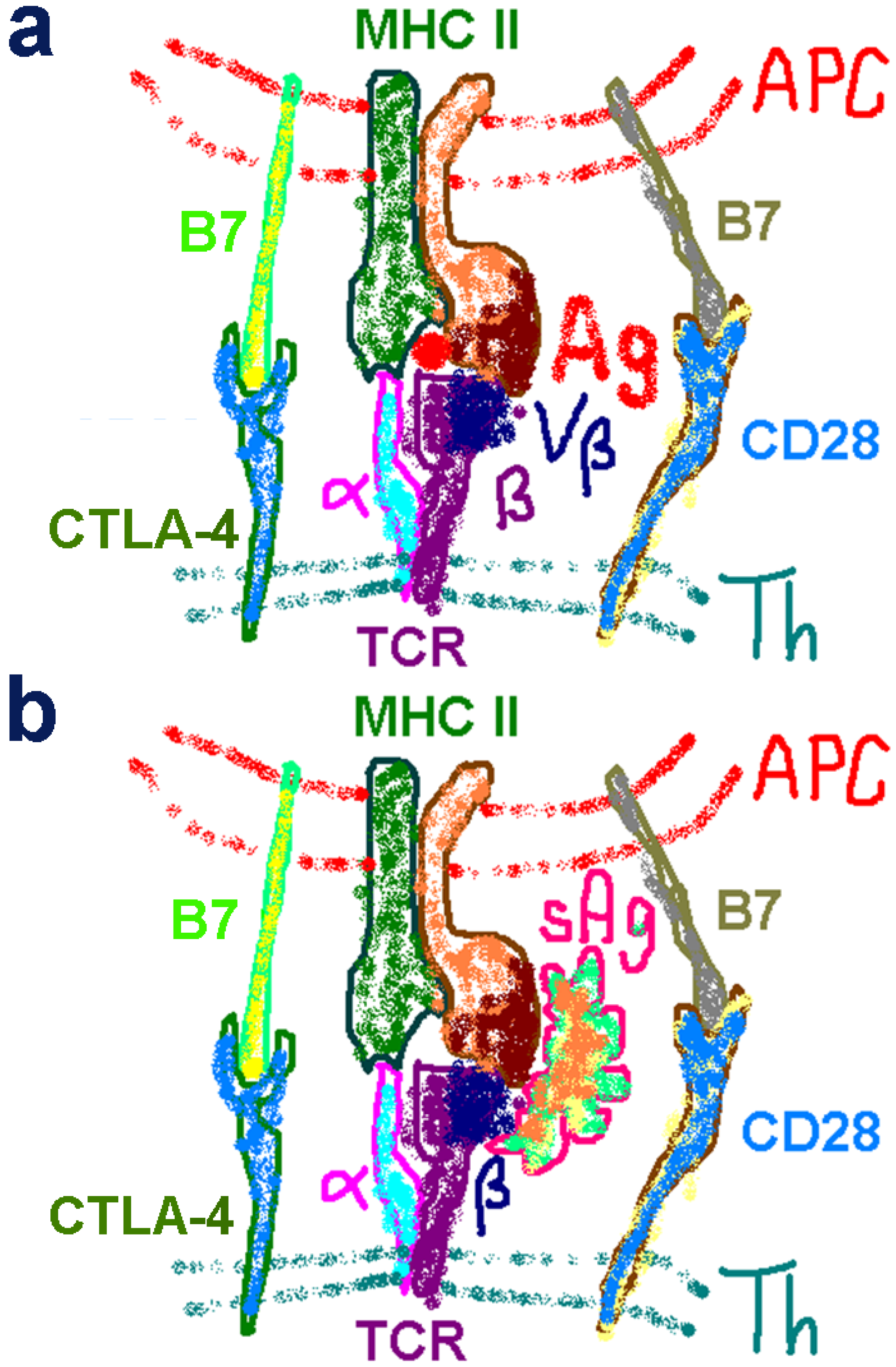
2.1. The Classical View of Superantigen Receptor Engagement
2.2. A Novel and Critical Domain in Superantigens of Unknown Function
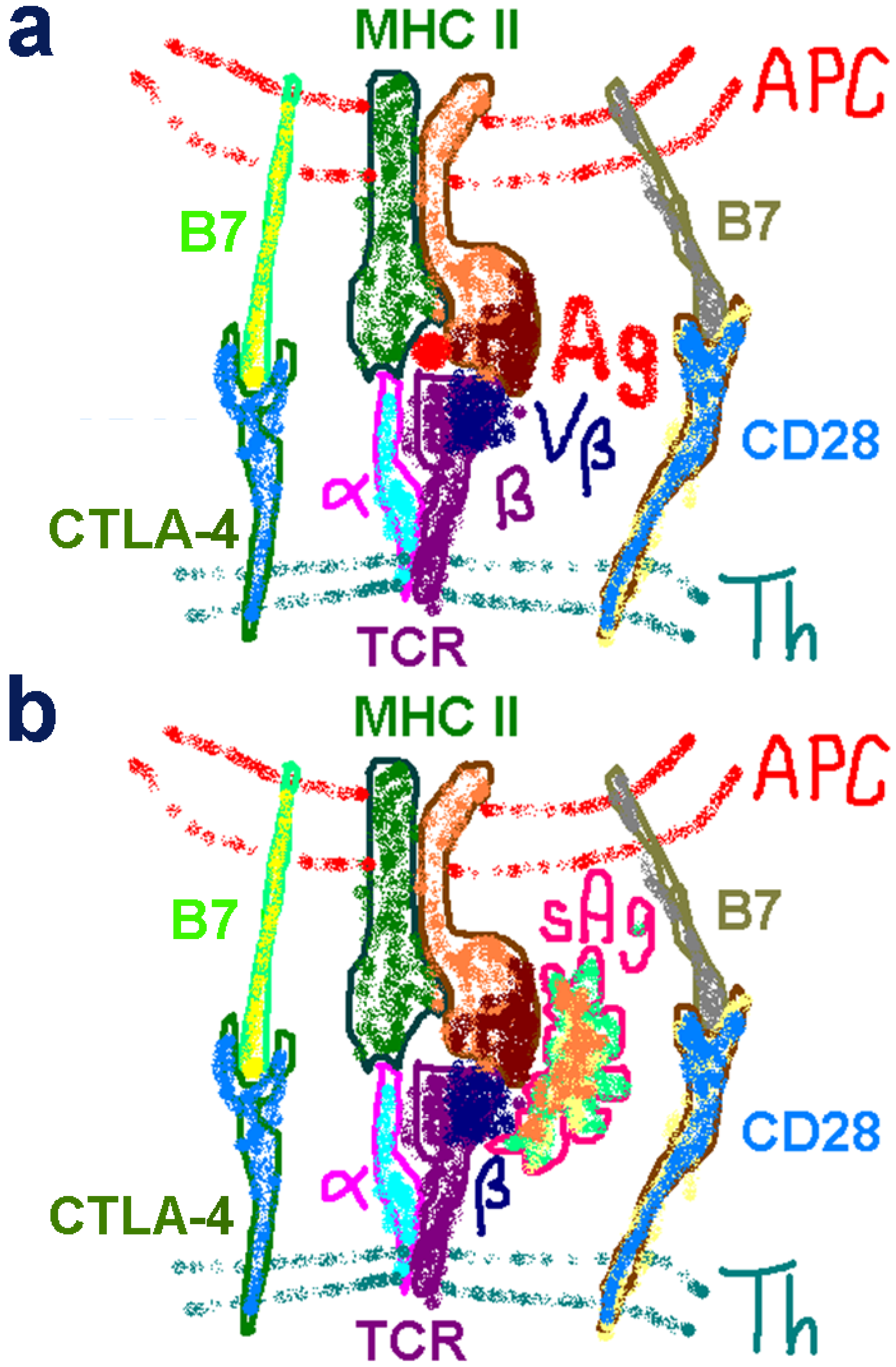

2.3. The Novel Superantigen Domain Engages CD28
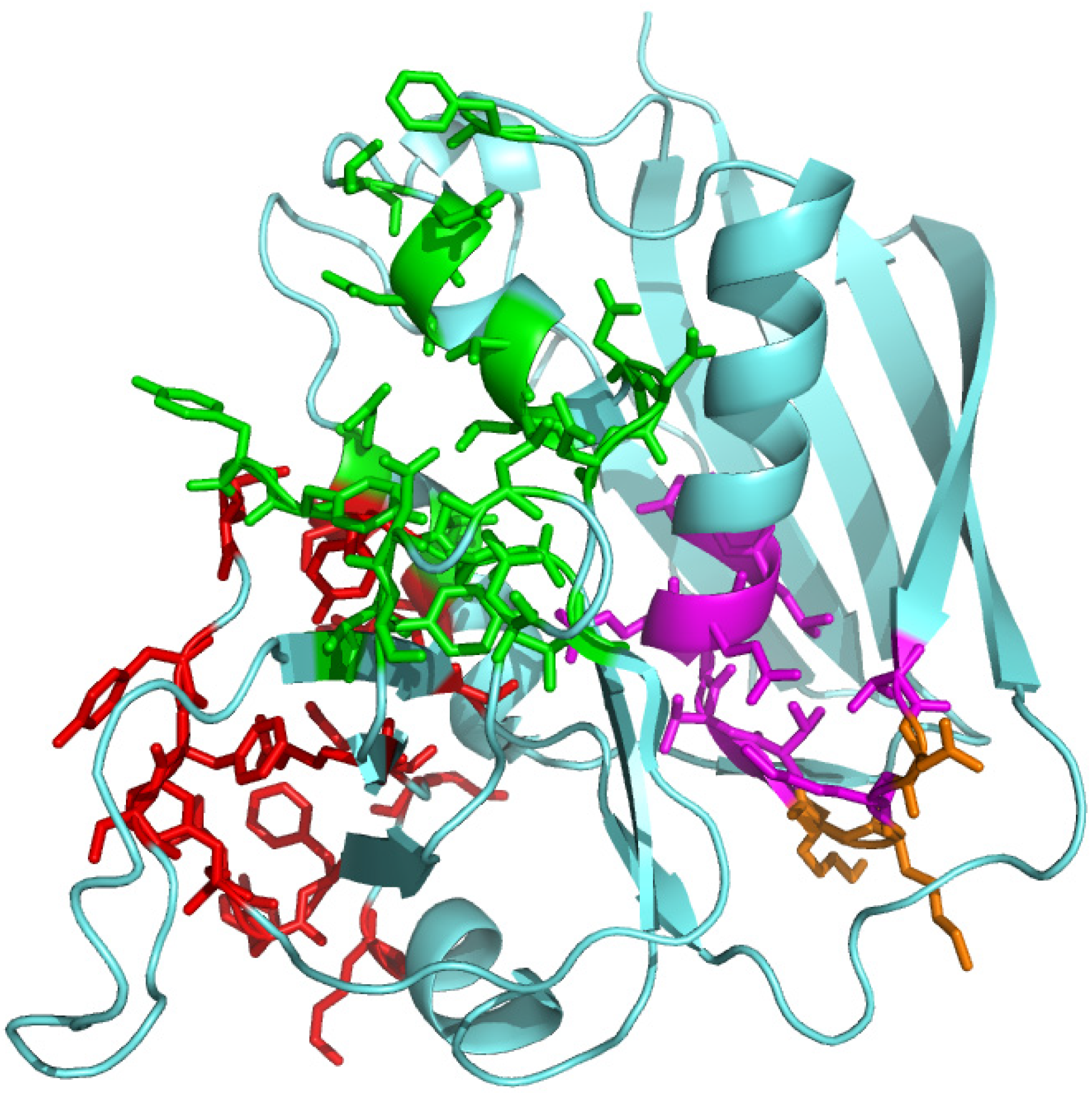
2.4. The Superantigen Binding Site in CD28 is the Homodimer Interface
2.5. The New Superantigen Synapse

2.6. The Importance of Moderate Binding Affinity
2.7. The Remarkable Ability of Short Peptides to Compete with Intact Proteins
2.8. Potential Clinical Utility of a Superantigen Antagonist Peptide
3. Experimental Section
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Arad, G.; Levy, R.; Hillman, D.; Kaempfer, R. Superantigen antagonist protects against lethal shock and defines a new domain for T-cell activation. Nat. Med. 2000, 6, 414–421. [Google Scholar] [CrossRef]
- Kaempfer, R.; Arad, G.; Levy, R.; Hillman, D. Defense against biologic warfare with superantigen toxins. Isr. Med. Assoc. J. 2002, 4, 520–523. [Google Scholar]
- Woods, J.B. (Ed.) Medical Management of Biological Casualties Handbook; United States Army Medical Research Institute of Infectious Diseases: Fort Detrick, MD, USA, 2005, 6th ed. Available online: http://www.dhhr.wv.gov/oeps/disease/Documents/USAMRIID_BlueBook.pdf (accessed on 2 August 2013).
- Schlievert, P.M.; Bohach, G.A. Staphylococcal and Streptococcal Superantigens: An Update. In Superantigens: Molecular Basis for Their Role in Human Diseases; Kotb, M.A., Fraser, J.D., Eds.; ASM Press: Washington, DC, USA, 2007; pp. 21–36. [Google Scholar]
- Marrack, P.; Blackman, M.; Kushnir, E.; Kappler, J. The toxicity of staphylococcal enterotoxin B in mice is mediated by T cells. J. Exp. Med. 1990, 171, 455–464. [Google Scholar] [CrossRef]
- Miethke, T.; Wahl, C.; Heeg, K.; Echtenacher, B.; Krammer, P.H.; Wagner, H. T cell-mediated lethal shock triggered in mice by the superantigen staphylococcal enterotoxin B: Critical role of tumor necrosis factor. J. Exp. Med. 1992, 175, 91–98. [Google Scholar] [CrossRef]
- Leder, L.; Llera, A.; Lavoie, P.M.; Lebedeva, M.I.; Li, H.; Sékaly, R.P.; Bohach, G.A.; Gahr, P.J.; Schlievert, P.M.; Karjalainen, K.; et al. A mutational analysis of the binding of staphylococcal enterotoxins B and C3 to the T cell receptor beta chain and major histocompatibility complex class II. J. Exp. Med. 1998, 187, 823–833. [Google Scholar] [CrossRef]
- Arad, G.; Levy, R.; Nasie, I.; Hillman, D.; Rotfogel, Z.; Barash, U.; Supper, E.; Shpilka, T.; Minis, A.; Kaempfer, R. Binding of superantigen toxins into CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. PLoS Biol. 2011, 9, e1001149. [Google Scholar] [CrossRef]
- Schwartz, J.C.; Zhang, X.; Fedorov, A.A.; Nathenson, S.G.; Almo, S.C. Structural basis for co-stimulation by the human CTLA-4/B7-2 complex. Nature 2001, 410, 604–608. [Google Scholar] [CrossRef]
- Sharpe, A.H.; Freeman, G.J. The B7-CD28 superfamily. Nat. Rev. Immunol. 2002, 2, 116–126. [Google Scholar] [CrossRef]
- Riley, J.L.; June, C.H. The CD28 family: A T-cell rheostat for therapeutic control of T-cell activation. Blood 2005, 105, 13–21. [Google Scholar] [CrossRef]
- Lindsten, T.; Lee, K.P.; Harris, E.S.; Petryniak, B.; Craighead, N.; Reynolds, P.J.; Lombard, D.B.; Freeman, G.J.; Nadler, L.M.; Gray, G.S.; et al. Characterization of CTLA-4 structure and expression on human T cells. J. Immunol. 1993, 151, 3489–3499. [Google Scholar]
- Collins, A.V.; Brodie, D.W.; Gilbert, R.J.; Iaboni, A.; Manso-Sancho, R.; Walse, B.; Stuart, D.I.; van der Merwe, P.A.; Davis, S.J. The interaction properties of costimulatory molecules revisited. Immunity 2002, 17, 201–210. [Google Scholar] [CrossRef]
- Lenschow, D.J.; Su, G.H.; Zuckerman, L.A.; Nabavi, N.; Jellis, C.L.; Gray, G.S.; Miller, J.; Bluestone, J.A. Expression and functional significance of an additional ligand for CTLA-4. Proc. Natl. Acad. Sci. USA 1993, 90, 11054–11058. [Google Scholar] [CrossRef]
- Greenwald, R.J.; Freeman, G.J.; Sharpe, A.H. The B7 family revisited. Annu. Rev. Immunol. 2005, 23, 515–548. [Google Scholar] [CrossRef]
- Bhatia, S.; Edidin, M.; Almo, S.C.; Nathenson, S.G. B7-1 and B7-2: Similar costimulatory ligands with different biochemical, oligomeric and signaling properties. Immunol. Lett. 2006, 104, 70–75. [Google Scholar] [CrossRef]
- Saha, B.; Harlan, D.M.; Lee, K.P.; June, C.H.; Abe, R. Protection against lethal toxic shock by targeted disruption of the CD28 gene. J. Exp. Med. 1996, 183, 2675–2680. [Google Scholar] [CrossRef]
- Mittrucker, H.W.; Shahinian, A.; Bouchard, D.; Kundig, T.M.; Mak, T.W. Induction of unresponsiveness and impaired T cell expansion by staphylococcal enterotoxin B in CD28-deficient mice. J. Exp. Med. 1996, 183, 2481–2488. [Google Scholar] [CrossRef]
- Muraille, E.; De Smedt, T.; Urbain, J.; Moser, M.; Leo, O. B7.2 provides co-stimulatory functions in vivo in response to staphylococcal enterotoxin B. Eur. J. Immunol. 1995, 25, 2111–2114. [Google Scholar] [CrossRef]
- Fraser, J.; Newton, M.; Weiss, A. CD28 and T-cell antigen receptor signal transduction coordinately regulates interleukin 2 gene expression in response to superantigen stimulation. J. Exp. Med. 1992, 175, 1131–1134. [Google Scholar] [CrossRef]
- Krakauer, T. Co-stimulatory receptors for the superantigen staphyloccoccal enterotoxin B on human vascular endothelial cells and T cells. J. Leukoc. Biol. 1994, 56, 458–463. [Google Scholar]
- Swaminathan, S.; Furey, W.; Pletcher, J.; Sax, M. Crystal structure of staphylococcal enterotoxin B, a superantigen. Nature 1992, 359, 801–806. [Google Scholar] [CrossRef]
- Jardetzky, T.S.; Brown, J.H.; Gorga, J.C.; Stern, L.J.; Urban, R.G.; Chi, Y.I.; Stauffacher, C.; Strominger, J.L.; Wiley, D.C. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature 1994, 368, 711–718. [Google Scholar] [CrossRef]
- Fields, B.A.; Malchiodi, E.L.; Li, H.; Ysern, X.; Stauffacher, C.V.; Schlievert, P.M.; Karjalainen, K.; Mariuzza, R.A. Crystal structure of a T-cell receptor beta-chain complexed with a superantigen. Nature 1996, 384, 188–192. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Tranter, H.S.; Acharya, K.R. Crystal structure of microbial superantigen staphylococcal enterotoxin B at 15 Å resolution: implications for superantigen recognition by MHC class II molecules and T-cell receptors. J. Mol. Biol. 1998, 277, 61–79. [Google Scholar] [CrossRef]
- Arad, G.; Hillman, D.; Levy, R.; Kaempfer, R. Superantigen antagonist blocks Th1 cytokine gene induction and lethal shock. J. Leukoc. Biol. 2001, 69, 921–927. [Google Scholar]
- Arad, G.; Hillman, D.; Levy, R.; Kaempfer, R. Broad-spectrum immunity against superantigens is elicited in mice protected from lethal shock by a superantigen antagonist peptide. Immunol. Lett. 2004, 91, 141–145. [Google Scholar] [CrossRef]
- Kaempfer, R. Peptide antagonists of superantigen toxins. Mol. Divers. 2004, 8, 113–120. [Google Scholar] [CrossRef]
- Evans, E.J.; Esnouf, R.M.; Manso-Sancho, R.; Gilbert, R.J.; James, J.R.; Yu, C.; Fennelly, J.A.; Vowles, C.; Hanke, T.; Walse, B.; et al. Crystal structure of a soluble CD28-Fab complex. Nat. Immunol. 2005, 6, 271–279. [Google Scholar]
- Ramachandran, G.; Tulapurkar, M.E.; Harris, K.M.; Arad, G.; Shirvan, A.; Shemesh, R.; Detolla, L.J.; Benazzi, C.; Opal, S.M.; Kaempfer, R.; et al. A peptide antagonist of CD28 signaling attenuates toxic shock and necrotizing soft-tissue infection induced by Streptococcus pyogenes. J. Infect. Dis. 2013, 207, 1869–1877. [Google Scholar] [CrossRef]
- Cunningham, B.C.; Ultsch, M.; De Vos, A.M.; Mulkerrin, M.G.; Clauser, K.R.; Wells, J.A. Dimerization of the extracellular domain of the human growth hormone receptor by a single hormone molecule. Science 1991, 254, 821–825. [Google Scholar]
- Livnah, O.; Stura, E.A.; Johnson, D.L.; Middleton, S.A.; Mulcahy, L.S.; Wrighton, N.C.; Dower, W.J.; Jolliffe, L.K.; Wilson, I.A. Functional mimicry of a protein hormone by a peptide agonist: the EPO receptor complex at 2.8 Å. Science 1996, 273, 464–471. [Google Scholar]
- Seth, A.; Stern, L.J.; Ottenhoff, T.H.; Engel, I.; Owen, M.J.; Lamb, J.R.; Klausner, R.D.; Wiley, D.C. Binary and ternary complexes between T-cell receptor, class II MHC and superantigen in vitro. Nature 1994, 369, 324–327. [Google Scholar]
- Redpath, S.; Alam, S.M.; Lin, C.M.; O’Rourke, A.M.; Gascoigne, N.R. Cutting edge: trimolecular interaction of TCR with MHC class II and bacterial superantigen shows a similar affinity to MHC:peptide ligands. J. Immunol. 1999, 163, 6–10. [Google Scholar]
- Andersen, P.S.; Geisler, C.; Buus, S.; Mariuzza, R.A.; Karjalainen, K. Role of the T cell receptor ligand affinity in T cell activation by bacterial superantigens. J. Biol. Chem. 2001, 276, 33452–33457. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kaempfer, R.; Arad, G.; Levy, R.; Hillman, D.; Nasie, I.; Rotfogel, Z. CD28: Direct and Critical Receptor for Superantigen Toxins. Toxins 2013, 5, 1531-1542. https://doi.org/10.3390/toxins5091531
Kaempfer R, Arad G, Levy R, Hillman D, Nasie I, Rotfogel Z. CD28: Direct and Critical Receptor for Superantigen Toxins. Toxins. 2013; 5(9):1531-1542. https://doi.org/10.3390/toxins5091531
Chicago/Turabian StyleKaempfer, Raymond, Gila Arad, Revital Levy, Dalia Hillman, Iris Nasie, and Ziv Rotfogel. 2013. "CD28: Direct and Critical Receptor for Superantigen Toxins" Toxins 5, no. 9: 1531-1542. https://doi.org/10.3390/toxins5091531