Effects of Clostridium difficile Toxin A and B on Human T Lymphocyte Migration


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
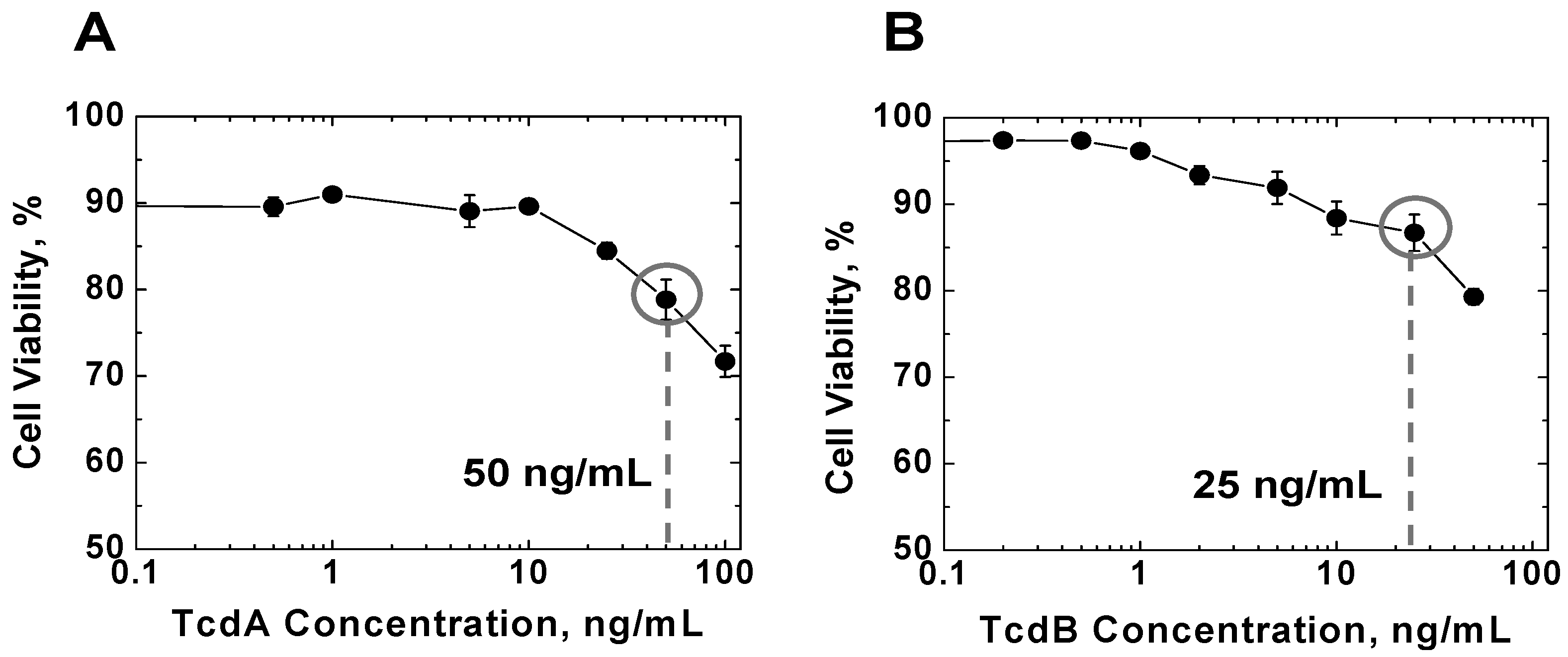
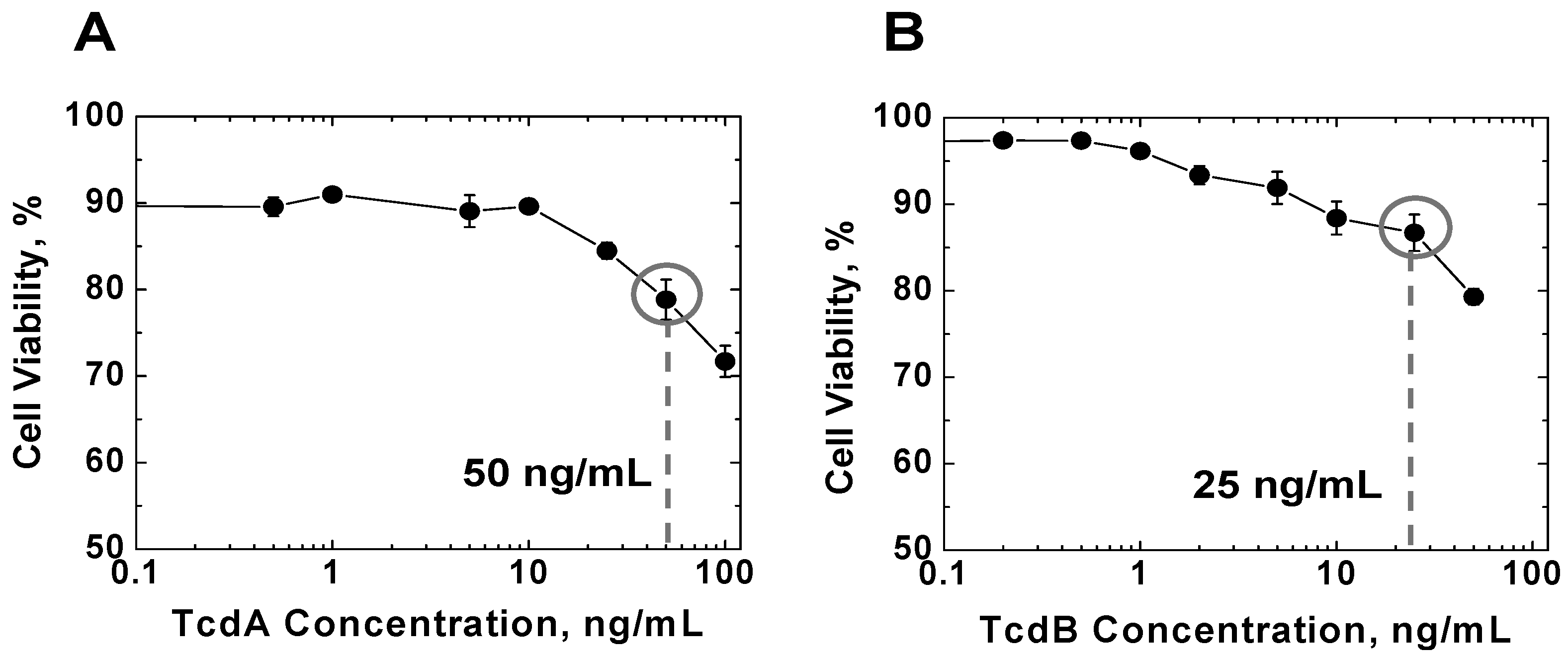
2.1. Effects of TcdA and TcdB on T Cell Viability
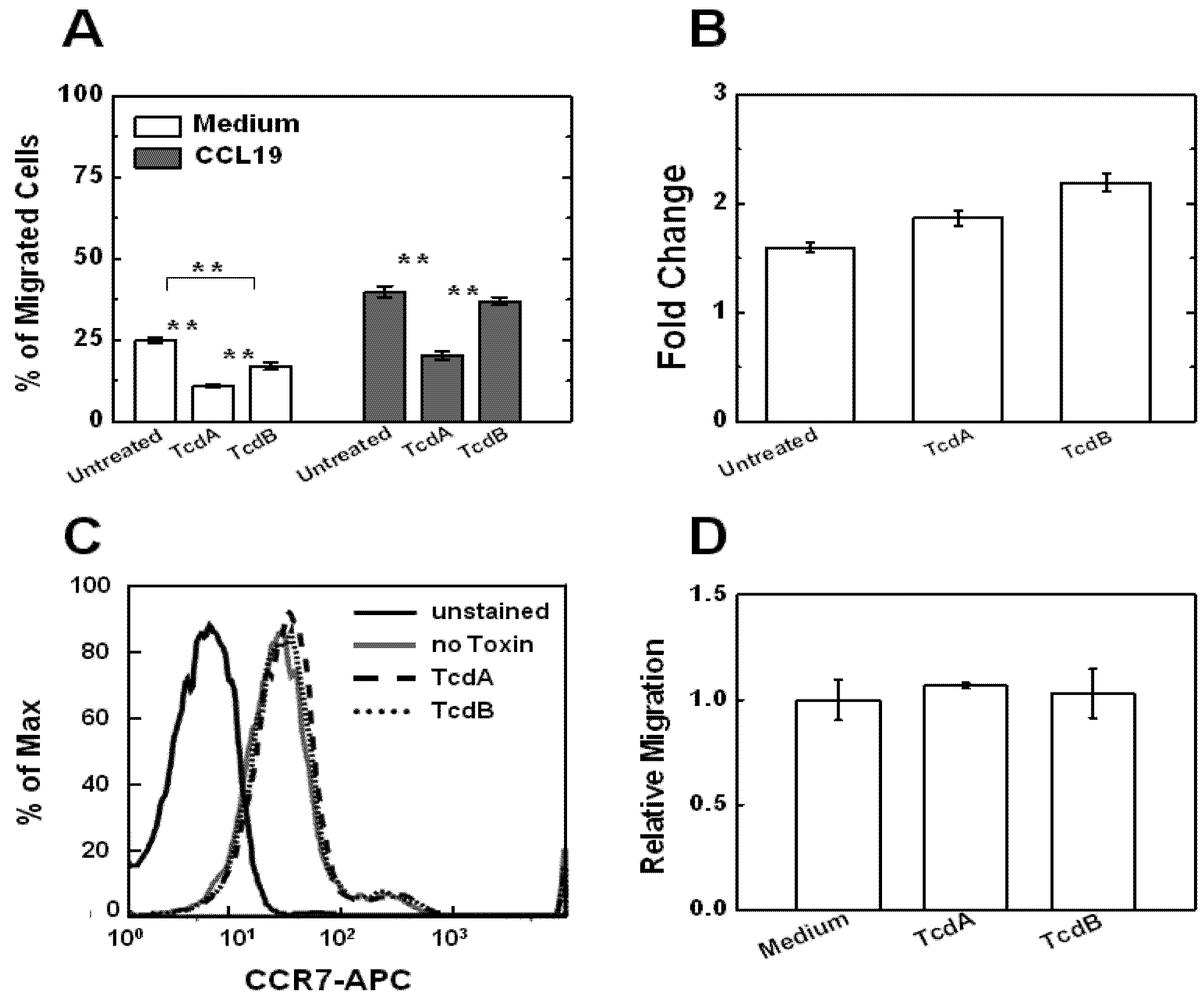
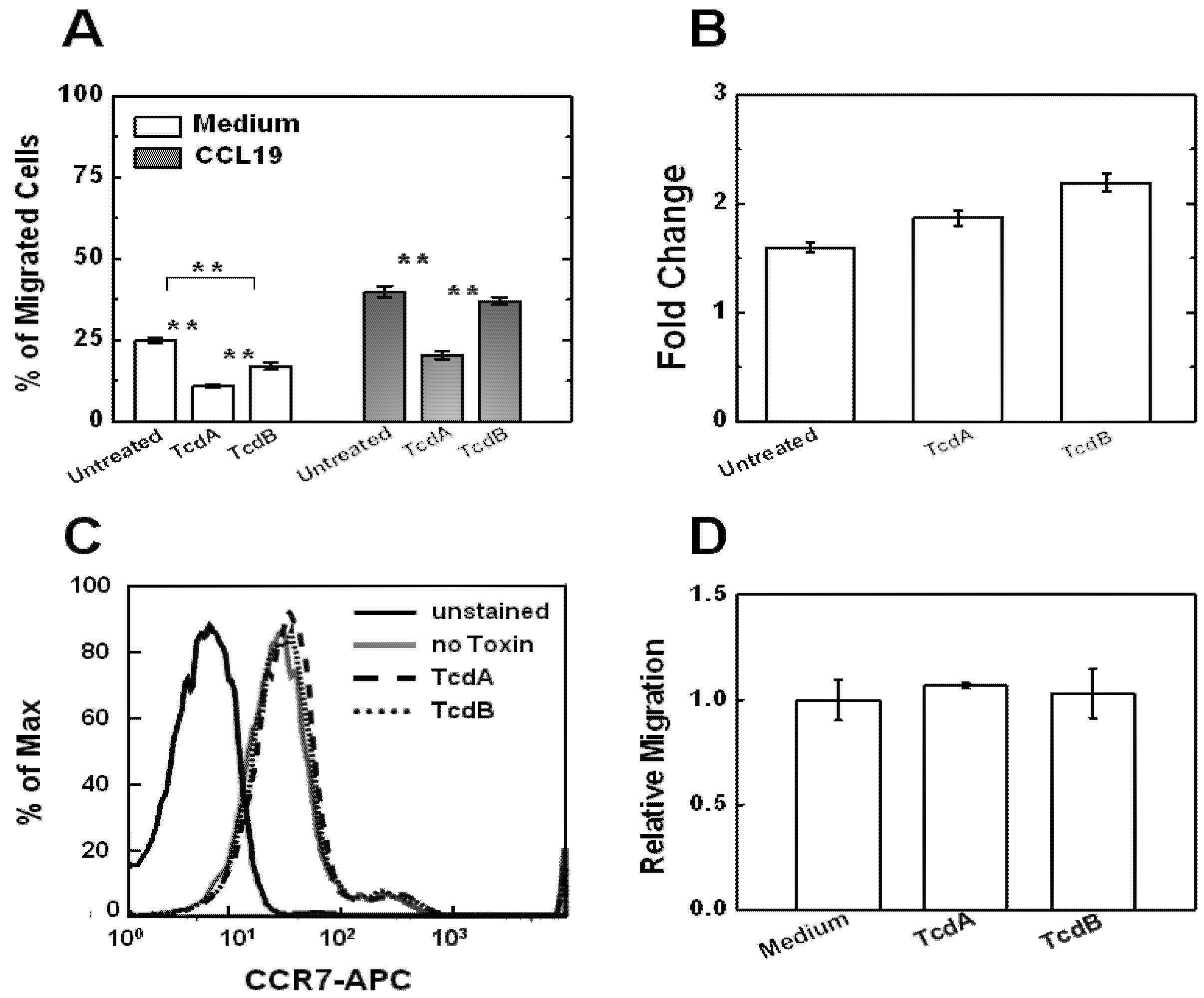
2.2. Effects of TcdA and TcdB on T Cell Motility Measured by Transwell Assays
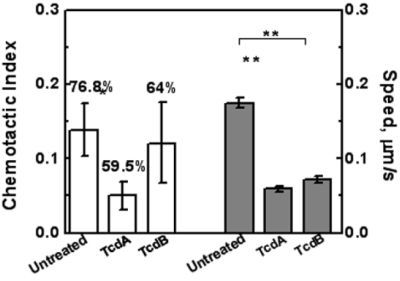
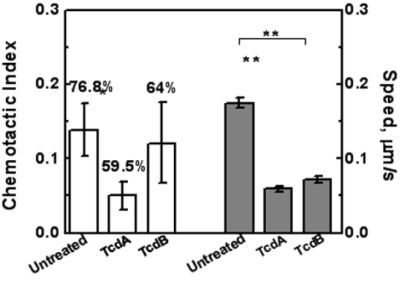
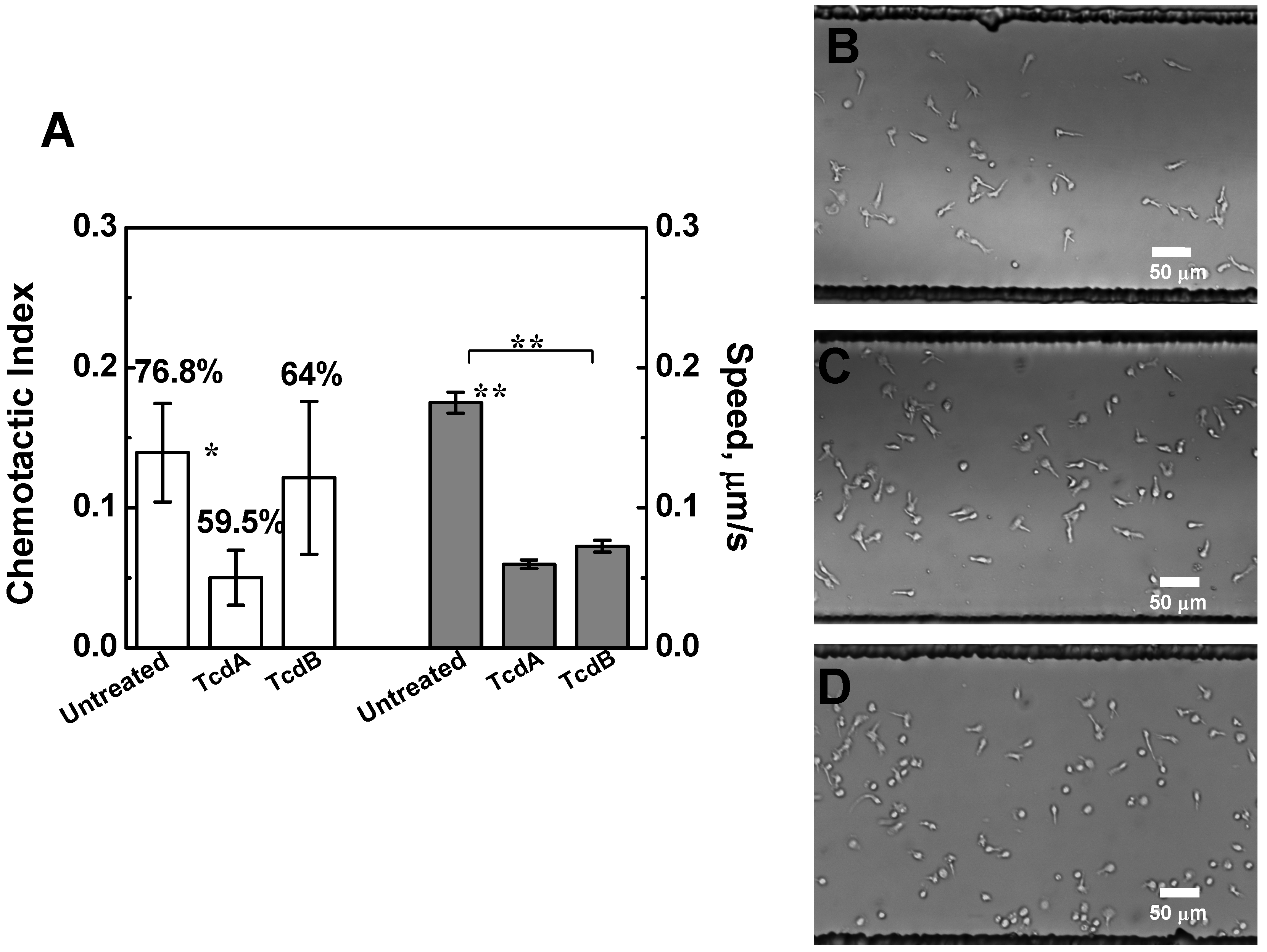
2.3. Effects of TcdA and TcdB on T Cell Chemotaxis
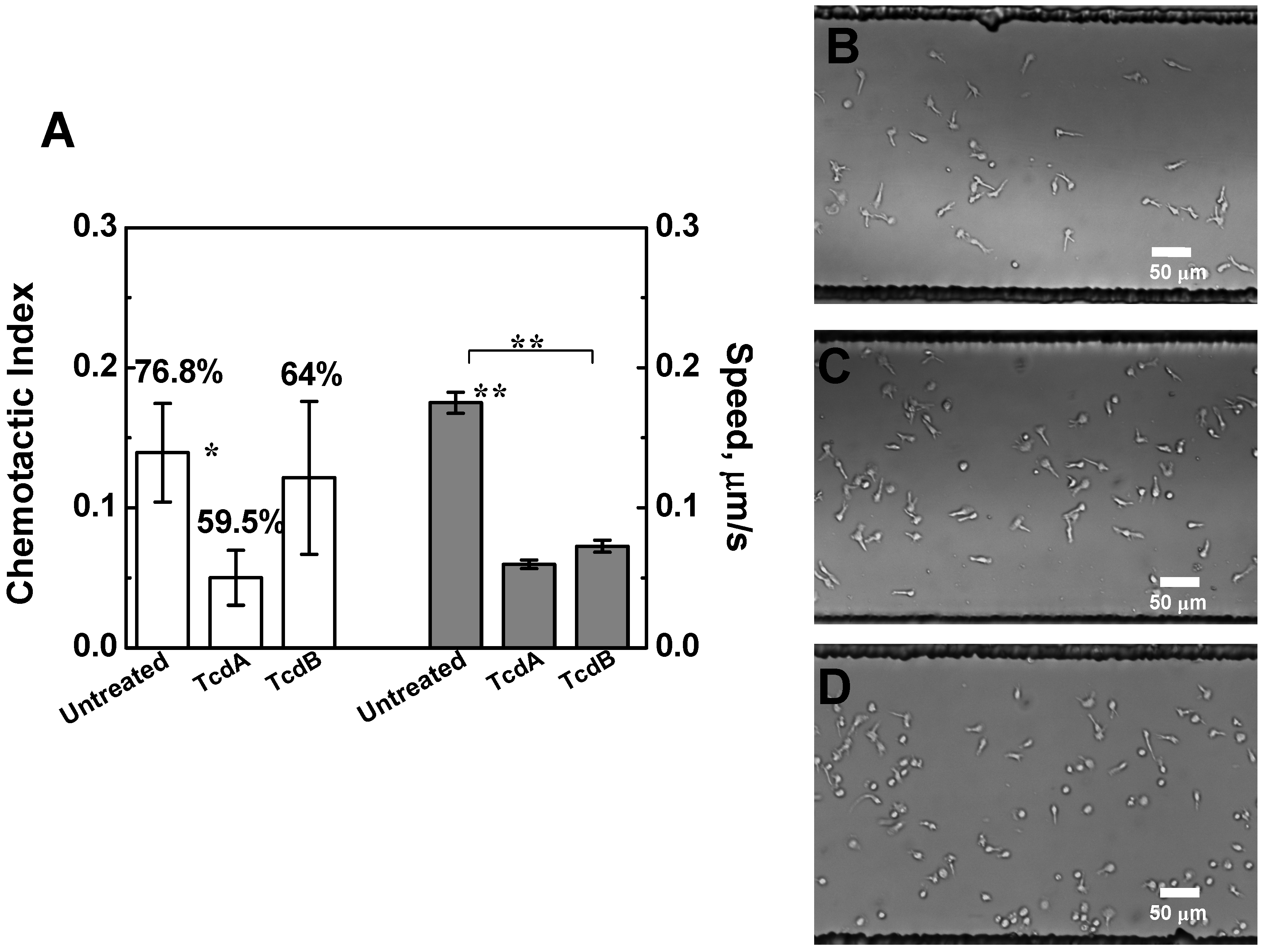
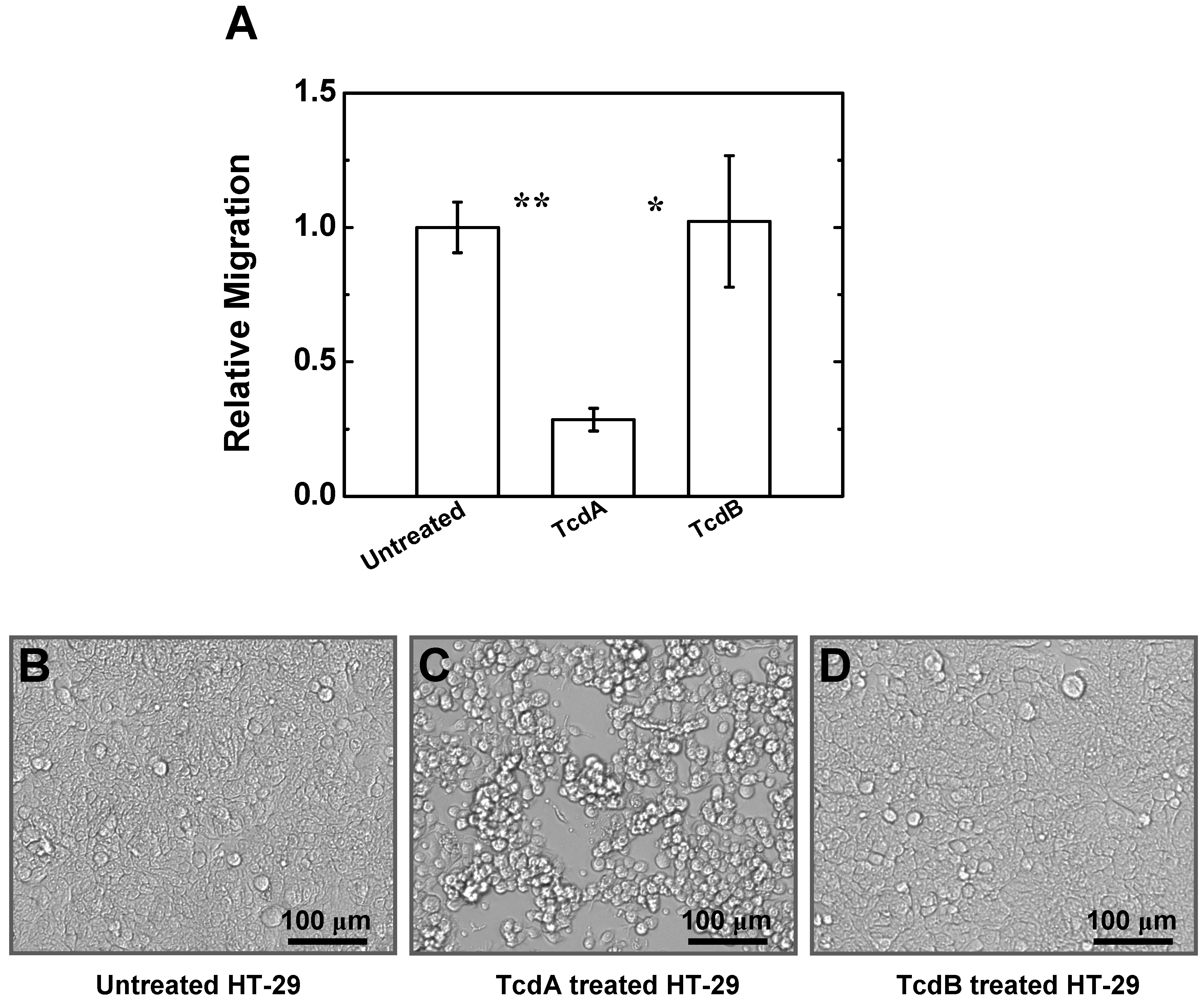
2.4. Effects of TcdA and TcdB on T Cell Migration to HT-29 Cell Culture
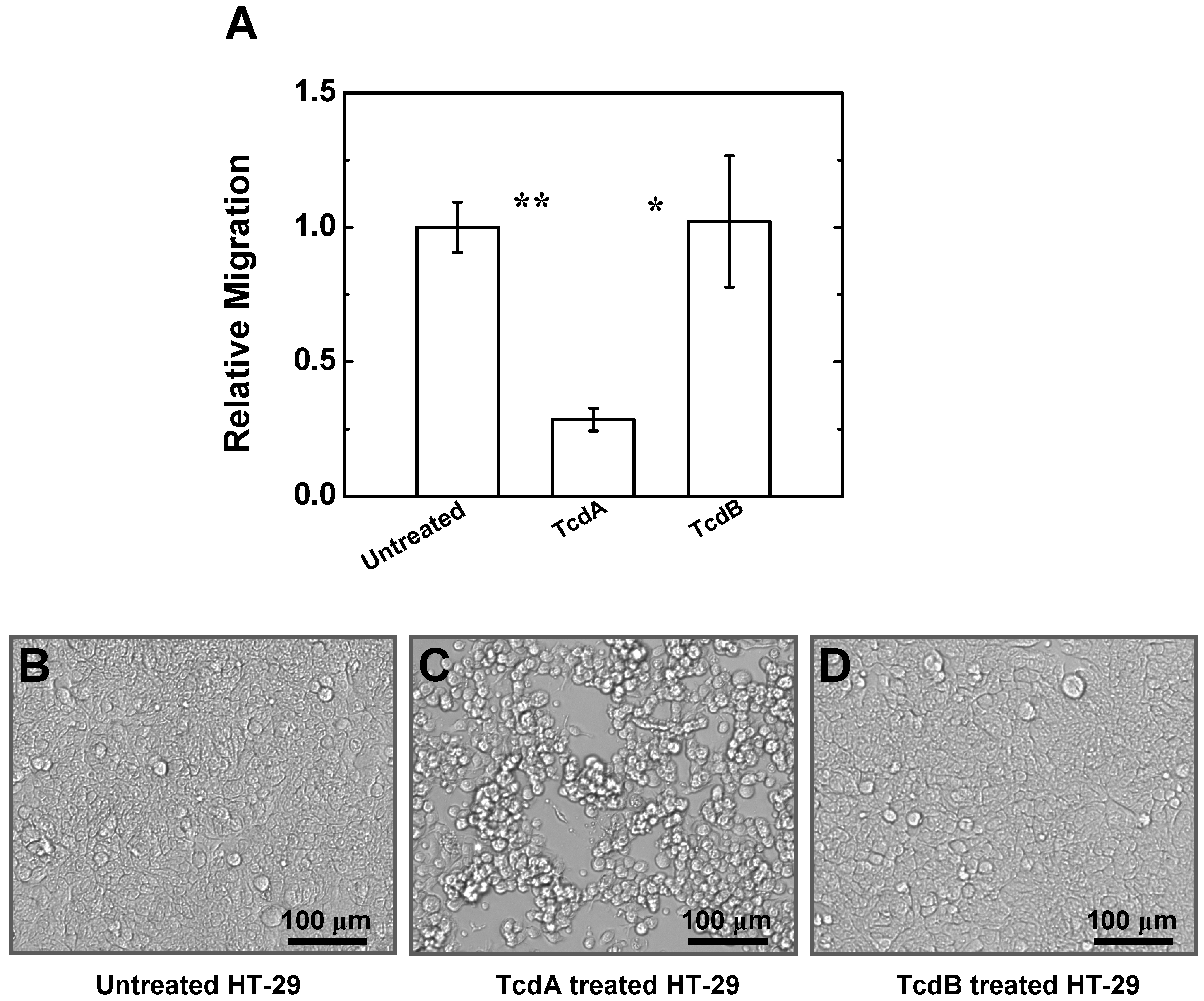
3. Experimental Section
3.1. Reagents
3.2. Cells
3.3. Flow Cytometric Analysis of Surface CCR7 Expression
3.4. Transwell Cell Migration Assays
3.5. Microfluidic Device and Gradient Generation
3.6. Cell Migration Experiments Using Microfluidic Device
3.7. Analysis of Microfluidic-Based Cell Migration Data
4. Discussion and Conclusions
Acknowledgments
Conflict of Interest
References
- Voth, D.E.; Ballard, J.D. Clostridium difficile toxins: Mechanism of action and role in disease. Clin. Microbiol. Rev. 2005, 18, 247–263. [Google Scholar] [CrossRef]
- Poutanen, S.M.; Simor, A.E. Clostridium difficile-associated diarrhea in adults. CMAJ 2004, 171, 51–58. [Google Scholar] [CrossRef]
- Elliott, B.; Chang, B.J.; Golledge, C.L.; Riley, T.V. Clostridium difficile-associated diarrhoea. Intern. Med. J. 2007, 37, 561–568. [Google Scholar] [CrossRef]
- Wilkins, T.D.; Lyerly, D.M. Clostridium difficile testing: After 20 years, still challenging. J. Clin. Microbiol. 2003, 41, 531–534. [Google Scholar] [CrossRef]
- Rupnik, M.; Dupuy, B.; Fairweather, N.F.; Gerding, D.N.; Johnson, S.; Just, I.; Lyerly, D.M.; Popoff, M.R.; Rood, J.I.; Sonenshein, A.L.; et al. Revised nomenclature of Clostridium difficile toxins and associated genes. J. Med. Microbiol. 2005, 54, 113–117. [Google Scholar] [CrossRef]
- Greco, A.; Ho, J.G.; Lin, S.J.; Palcic, M.M.; Rupnik, M.; Ng, K.K. Carbohydrate recognition by Clostridium difficile toxin A. Nat. Struct. Mol. Biol. 2006, 13, 460–461. [Google Scholar] [CrossRef]
- Lima, A.A.; Lyerly, D.M.; Wilkins, T.D.; Innes, D.J.; Guerrant, R.L. Effects of Clostridium difficile toxins A and B in rabbit small and large intestine in vivo and on cultured cells in vitro. Infect. Immun. 1988, 56, 582–588. [Google Scholar]
- Kuehne, S.A.; Cartman, S.T.; Heap, J.T.; Kelly, M.L.; Cockayne, A.; Minton, N.P. The role of toxin A and toxin B in Clostridium difficile infection. Nature 2010, 467, 711–713. [Google Scholar] [CrossRef]
- Just, I.; Gerhard, R. Large clostridial cytotoxins. Rev. Physiol. Biochem. Pharmacol. 2004, 152, 23–47. [Google Scholar]
- Brito, G.A.; Carneiro-Filho, B.; Oriá, R.B.; Destura, R.V.; Lima, A.A.; Guerrant, R.L. Clostridium difficile toxin A induces intestinal epithelial cell apoptosis and damage: Role of Gln and Ala-Gln in toxin A effects. Dig. Dis. Sci. 2005, 50, 1271–1278. [Google Scholar] [CrossRef]
- Brito, G.A.; Fujji, J.; Carneiro-Filho, B.A.; Lima, A.A.; Obrig, T.; Guerrant, R.L. Mechanism of Clostridium difficile toxin A-induced apoptosis in T84 cells. J. Infect. Dis. 2002, 186, 1438–1447. [Google Scholar] [CrossRef]
- Carneiro, B.A.; Fujii, J.; Brito, G.A.; Alcantara, C.; Oriá, R.B.; Lima, A.A.; Obrig, T.; Guerrant, R.L. Caspase and bid involvement in Clostridium difficile toxin A-induced apoptosis and modulation of toxin A effects by glutamine and alanyl-glutamine in vivo and in vitro. Infect. Immun. 2006, 74, 81–87. [Google Scholar] [CrossRef]
- Liu, T.S.; Musch, M.W.; Sugi, K.; Walsh-Reitz, M.M.; Ropeleski, M.J.; Hendrickson, B.A.; Pothoulakis, C.; Lamont, J.T.; Chang, E.B. Protective role of HSP72 against Clostridium difficile toxin A-induced intestinal epithelial cell dysfunction. Am. J. Physiol. Cell. Physiol. 2003, 284, C1073–C1082. [Google Scholar]
- Fiorentini, C.; Fabbri, A.; Falzano, L.; Fattorossi, A.; Matarrese, P.; Rivabene, R.; Donelli, G. Clostridium difficile toxin B induces apoptosis in intestinal cultured cells. Infect. Immun. 1998, 66, 2660–2665. [Google Scholar]
- Mahida, Y.R.; Galvin, A.; Makh, S.; Hyde, S.; Sanfilippo, L.; Borriello, S.P.; Sewell, H.F. Effect of Clostridium difficile toxin A on human colonic lamina propria cells: Early loss of macrophages followed by T-cell apoptosis. Infect. Immun. 1998, 66, 5462–5469. [Google Scholar]
- Hippenstiel, S.; Schmeck, B.; N’Guessan, P.D.; Seybold, J.; Krüll, M.; Preissner, K.; Eichel-Streiber, C.V.; Suttorp, N. Rho protein inactivation induced apoptosis of cultured human endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L830–L838. [Google Scholar]
- Warny, M.; Kelly, C.P. Monocytic cell necrosis is mediated by potassium depletion and caspase-like proteases. Am. J. Physiol. 1999, 276, C717–C724. [Google Scholar]
- Kim, J.M.; Kim, J.S.; Jun, H.C.; Oh, Y.K.; Song, I.S.; Kim, C.Y. Differential expression and polarized secretion of CXC and CC chemokines by human intestinal epithelial cancer cell lines in response to Clostridium difficile toxin A. Microbiol. Immunol. 2002, 46, 333–342. [Google Scholar]
- Kelly, C.P.; Becker, S.; Linevsky, J.K.; Joshi, M.A.; O’Keane, J.C.; Dickey, B.F.; LaMont, J.T.; Pothoulakis, C. Neutrophil recruitment in Clostridium difficile toxin A enteritis in the rabbit. J. Clin. Invest. 1994, 93, 1257–1265. [Google Scholar] [CrossRef]
- Lica, M.; Schulz, F.; Schelle, I.; May, M.; Just, I.; Genth, H. Difference in the biological effects of Clostridium difficile toxin B in proliferating and non-proliferating cells. Naunyn. Schmiedebergs. Arch. Pharmacol. 2011, 383, 275–283. [Google Scholar] [CrossRef]
- Tournier, J.N.; Rossi Paccani, S.; Quesnel-Hellmann, A.; Baldari, C.T. Anthrax toxins: A weapon to systematically dismantle the host immune defenses. Mol. Aspects Med. 2009, 30, 456–466. [Google Scholar] [CrossRef]
- Paccani, S.R.; Tonello, F.; Ghittoni, R.; Natale, M.; Muraro, L.; D’Elios, M.M.; Tang, W.J.; Montecucco, C.; Baldari, C.T. Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J. Exp. Med. 2005, 201, 325–331. [Google Scholar]
- Lin, F.; Butcher, E.C. T cell chemotaxis in a simple microfluidic device. Lab. Chip 2006, 6, 1462–1469. [Google Scholar] [CrossRef]
- Lin, F.; Baldessari, F.; Gyenge, C.C.; Sato, T.; Chambers, R.D.; Santiago, J.G.; Butcher, E.C. Lymphocyte electrotaxis in vitro and in vivo. J. Immunol. 2008, 181, 2465–2471. [Google Scholar]
- Nandagopal, S.; Wu, D.; Lin, F. Combinatorial guidance by CCR7 ligands for T lymphocytes migration in co-existing chemokine fields. PLoS One 2011, 6, e18183. [Google Scholar] [CrossRef]
- Viola, A.; Contento, R.L.; Molon, B. T cells and their partners: The chemokine dating agency. Trends Immunol. 2006, 27, 421–427. [Google Scholar] [CrossRef]
- Kunkel, E.J.; Butcher, E.C. Chemokines and the tissue-specific migration of lymphocytes. Immunity 2002, 16, 1–4. [Google Scholar] [CrossRef]
- Kunkel, E.J.; Butcher, E.C. Plasma-cell homing. Nat. Rev. Immunol. 2003, 3, 822–829. [Google Scholar] [CrossRef]
- Foxman, E.F.; Campbell, J.J.; Butcher, E.C. Multistep navigation and the combinatorial control of leukocyte chemotaxis. J. Cell. Biol. 1997, 139, 1349–1360. [Google Scholar] [CrossRef]
- Cyster, J.G. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu. Rev. Immunol. 2005, 23, 127–159. [Google Scholar] [CrossRef]
- Weninger, W.; von Andrian, U.H. Chemokine regulation of naïve T cell traffic in health and disease. Semin. Immunol. 2003, 15, 257–270. [Google Scholar] [CrossRef]
- Forster, R.; Davalos-Misslitz, A.C.; Rot, A. CCR7 and its ligands: Balancing immunity and tolerance. Nat. Rev. Immunol. 2008, 8, 362–371. [Google Scholar] [CrossRef]
- Otero, C.; Groettrup, M.; Legler, D.F. Opposite fate of endocytosed CCR7 and its ligands: Recycling versus degradation. J. Immunol. 2006, 177, 2314–2323. [Google Scholar]
- Schumann, K.; Lammermann, T.; Bruckner, M.; Legler, D.F.; Polleux, J.; Spatz, J.P.; Schuler, G.; Forster, R.; Lutz, M.B.; Sorokin, L.; et al. Immobilized chemokine fields and soluble chemokine gradients cooperatively shape migration patterns of dendritic cells. Immunity 2010, 32, 703–713. [Google Scholar] [CrossRef]
- Gerhard, R.; Nottrott, S.; Schoentaube, J.; Tatge, H.; Olling, A.; Just, I. Glucosylation of Rho GTPases by Clostridium difficile toxin A triggers apoptosis in intestinal epithelial cells. J. Med. Microbiol. 2008, 57, 765–770. [Google Scholar] [CrossRef]
- Kyne, L.; Warny, M.; Qamar, A.; Kelly, C.P. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N. Engl. J. Med. 2000, 342, 390–397. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wu, D.; Joyee, A.G.; Nandagopal, S.; Lopez, M.; Ma, X.; Berry, J.; Lin, F. Effects of Clostridium difficile Toxin A and B on Human T Lymphocyte Migration. Toxins 2013, 5, 926-938. https://doi.org/10.3390/toxins5050926
Wu D, Joyee AG, Nandagopal S, Lopez M, Ma X, Berry J, Lin F. Effects of Clostridium difficile Toxin A and B on Human T Lymphocyte Migration. Toxins. 2013; 5(5):926-938. https://doi.org/10.3390/toxins5050926
Chicago/Turabian StyleWu, Dan, Antony George Joyee, Saravanan Nandagopal, Marianela Lopez, Xiuli Ma, Jody Berry, and Francis Lin. 2013. "Effects of Clostridium difficile Toxin A and B on Human T Lymphocyte Migration" Toxins 5, no. 5: 926-938. https://doi.org/10.3390/toxins5050926