Effect of Gating Modifier Toxins on Membrane Thickness: Implications for Toxin Effect on Gramicidin and Mechanosensitive Channels

Abstract
:
1. Introduction
2. Results and Discussion
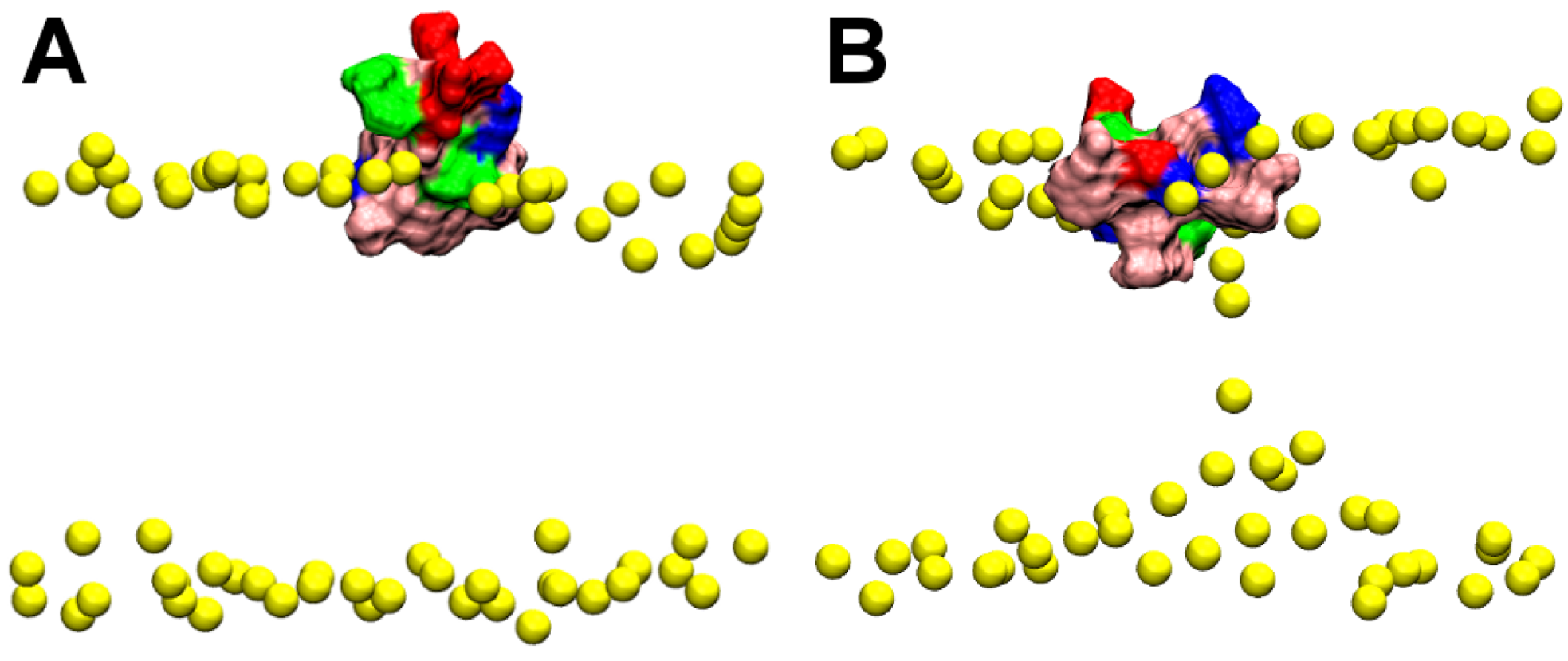
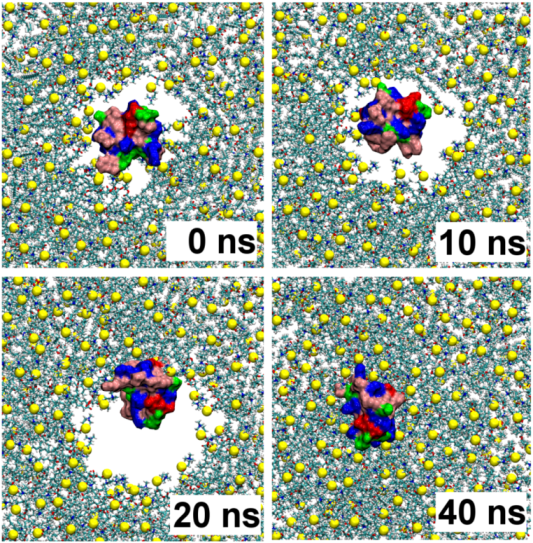
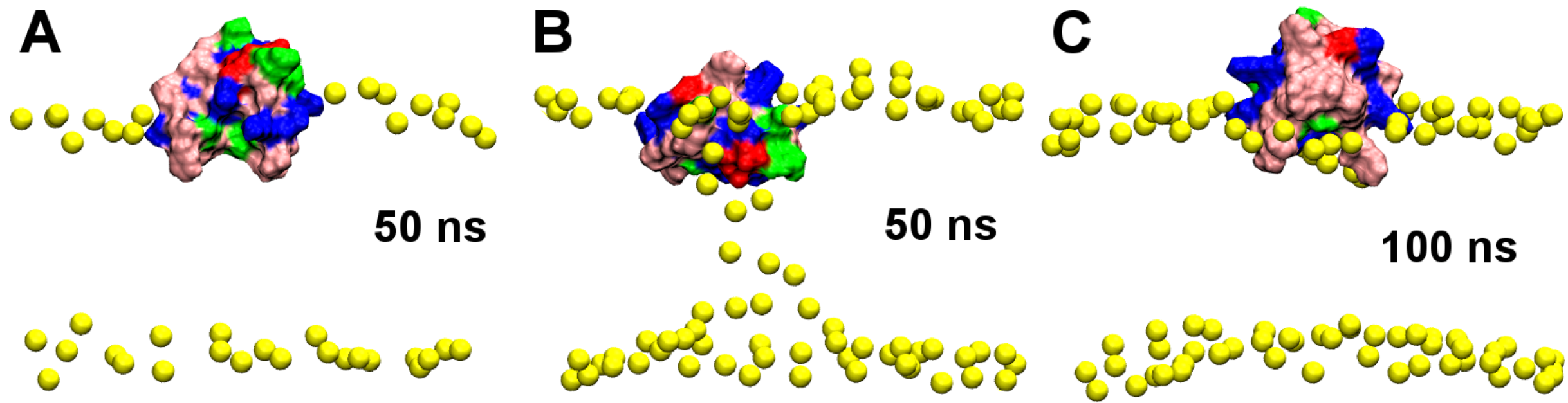
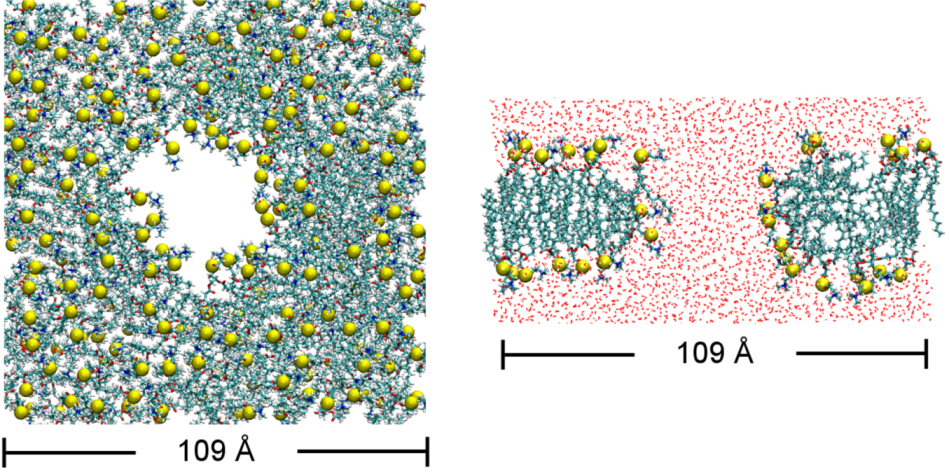
2.1. Binding of GsMTx4
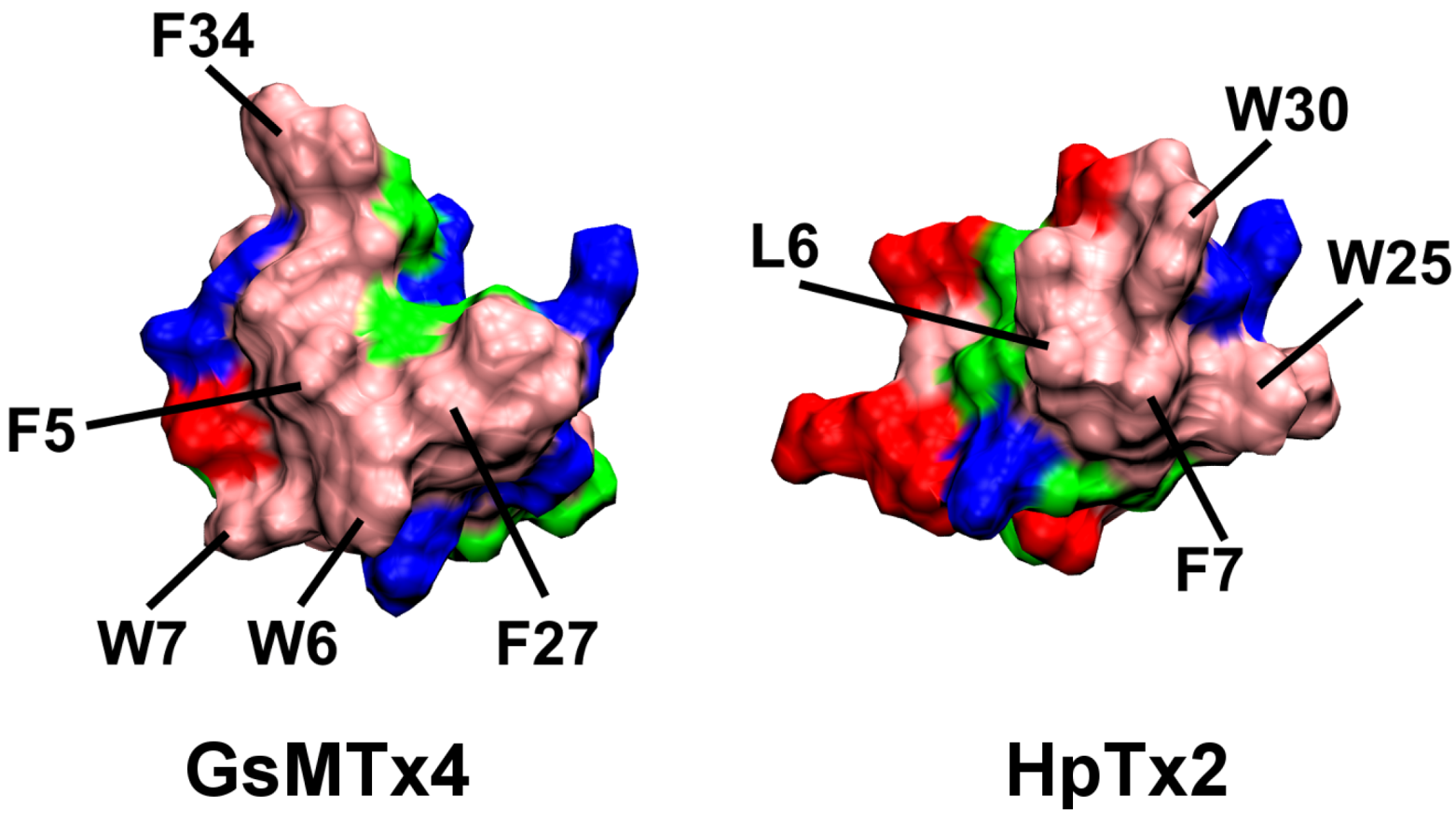
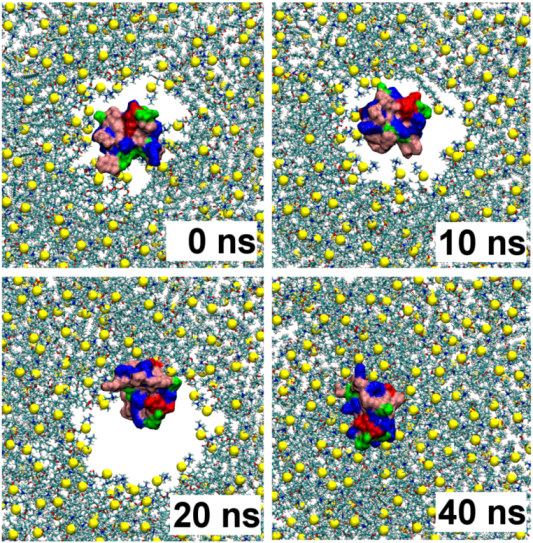

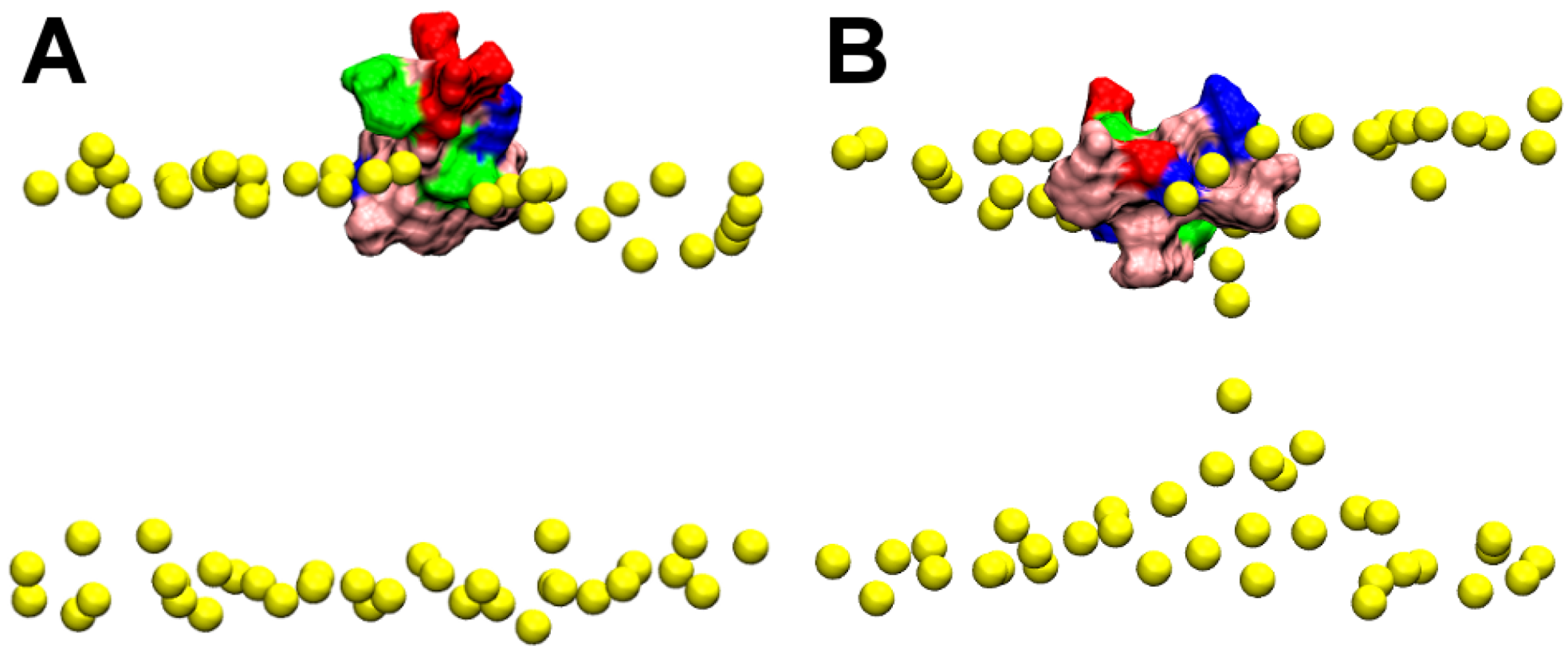
2.2. Binding of HpTx2
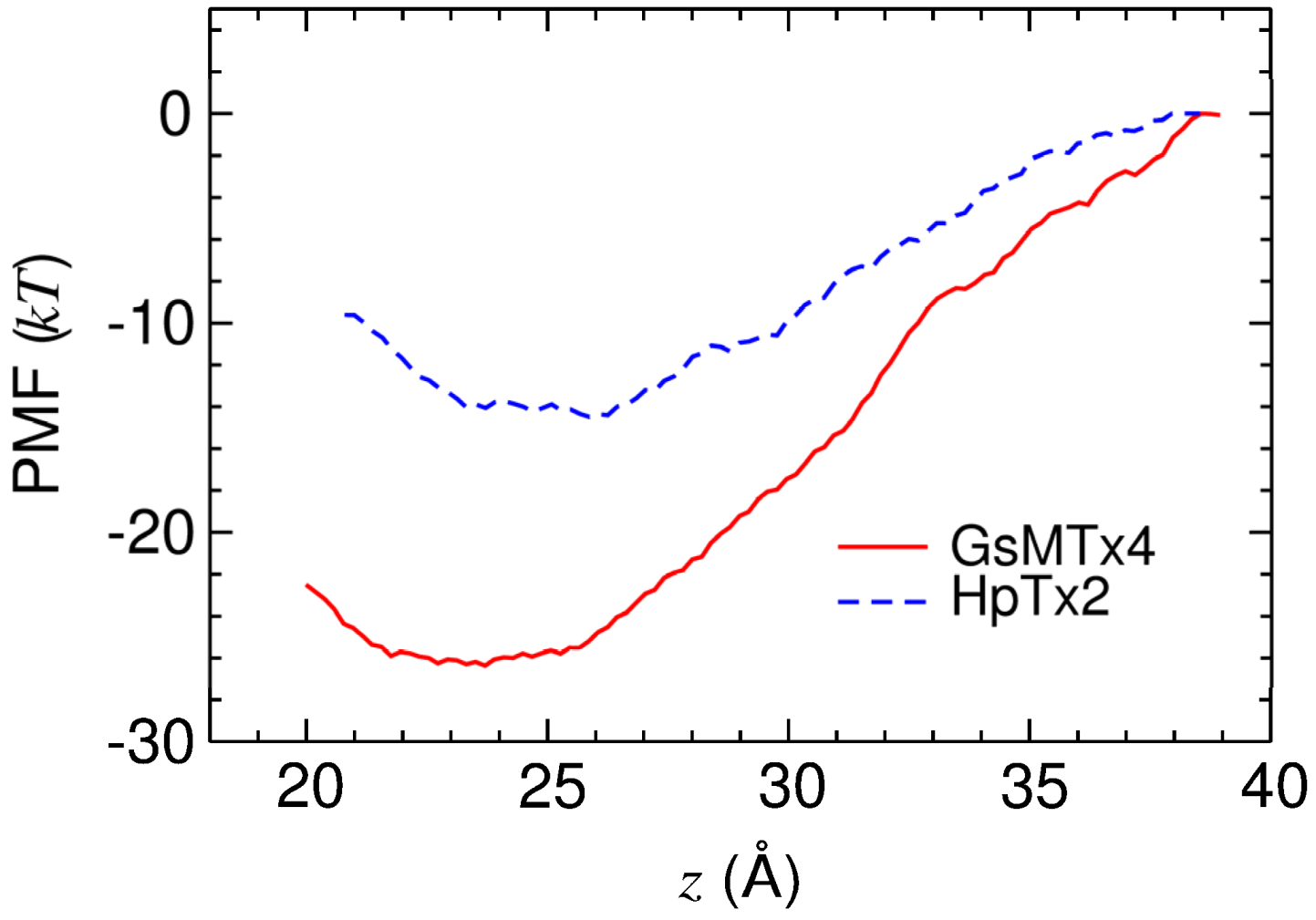
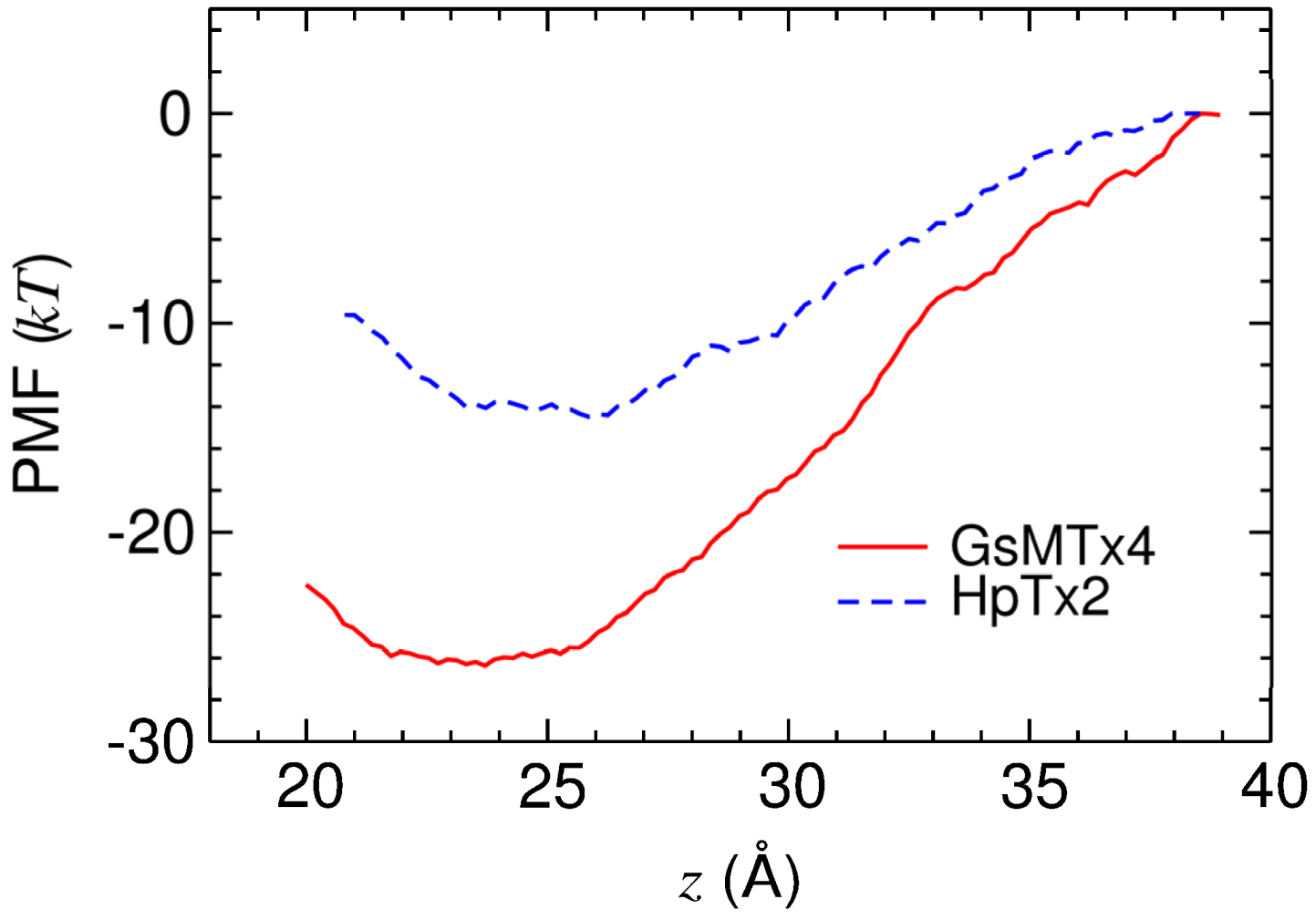
2.3. Energetics of Toxin Binding
2.4. Bilayer Thickness

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance from toxin center (Å) | No. of lipids in one leaflet | dH (Å) |
|---|---|---|
| <15 | 8.3 ± 2.6 | 27.9 ± 1.6 |
| 15–25 | 16.1 ± 3.7 | 27.2 ± 1.0 |
| 25–35 | 29.4 ± 4.9 | 26.0 ± 0.5 |
| >35 | 71.2 ± 4.1 | 25.8 ± 0.5 |
| Total | 125 | 26.3 ± 0.2 |
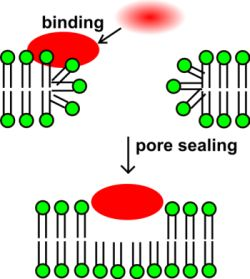
2.5. Implications for Toxin Effect on Ion Channels
3. Methods
3.1. Simulation Protocol
| Label | Toxin | Start | Time scale (ns) | Lipid distribution | dH (Å) § |
|---|---|---|---|---|---|
| PC-sim1 * | - | - | 20 | 128/128 | - |
| PC-sim2 | - | 40 | 128/128 | 28.3 ± 0.3 | |
| PC-sim3 | GT-sim3 | 40 | 126/130 | 27.3 ± 0.2 | |
| GT-sim1 † | GsMTx4 | PC-sim1 | 80 | 125/131 | 26.3 ± 0.2 |
| GT-sim2 † | PC-sim1 | 100 | 128/128 | 27.2 ± 0.3 | |
| GT-sim3 † | PC-sim1 | 50 | 126/130 | 26.0 ± 0.4 | |
| GT-sim4 ‡ | GT-sim1 | 30 | 116/131 | 25.5 ± 0.2 | |
| HT-sim1 † | HpTx2 | PC-sim1 | 80 | 128/128 | 27.6 ± 0.4 |
| HT-sim2 † | PC-sim1 | 50 | - | - | |
| HT-sim3 | HT-sim1 | 40 | 124/128 | 26.8 ± 0.3 |
3.2. Molecular Dynamics Simulations
3.3. Umbrella Sampling
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Cestèle, S.; Qu, Y.; Rogers, J.C.; Rochat, H.; Scheuer, T.; Catterall, W.A. Voltage sensor-trapping: Enhanced activation of sodium channels by β-scorpion toxin bound to the S3–S4 loop in domain II. Neuron 1998, 21, 919–931. [Google Scholar] [CrossRef]
- Catterall, W.A.; Cestele, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [Green Version]
- Bosmans, F.; Swartz, K.J. Targeting voltage sensors in sodium channels with spider toxins. Trends Pharmacol. Sci. 2010, 31, 175–182. [Google Scholar] [CrossRef]
- Windley, M.J.; Herzig, V.; Dziemborowicz, S.A.; Hardy, M.C.; King, G.F.; Nicholson, G.M. Spider-venom peptides as bioinsecticides. Toxins 2012, 4, 191–227. [Google Scholar] [CrossRef]
- Swartz, K.J. Tarantula toxins interacting with voltage sensors in potassium channels. Toxicon 2007, 49, 213–230. [Google Scholar] [CrossRef]
- Lee, C.W.; Bae, C.; Lee, J.; Ryu, J.H.; Kim, H.H.; Kohno, T.; Swartz, K.J.; Kim, J.I. Solution structure of Kurtoxin: A gating modifier selective for Cav3 voltage-gated Ca2+ channels. Biochemistry 2012, 51, 1862–1873. [Google Scholar] [CrossRef]
- Koh, C.Y.; Kini, R.M. From snake venom toxins to therapeutics—Cardiovascular examples. Toxicon 2012, 59, 497–506. [Google Scholar] [CrossRef]
- Bosmans, F.; Tytgat, J. Sea anemone venom as a source of insecticidal peptides acting on voltage-gated Na+ channels. Toxicon 2007, 49, 550–560. [Google Scholar] [CrossRef]
- Moran, Y.; Gordon, D.; Gurevitz, M. Sea anemone toxins affecting voltage-gated sodium channels—Molecular and evolutionary features. Toxicon 2009, 54, 1089–1101. [Google Scholar] [CrossRef]
- Leipold, E.; Hansel, A.; Olivera, B.M.; Terlau, H.; Heinemann, S.H. Molecular interaction of δ-conotoxins with voltage-gated sodium channels. FEBS Lett. 2005, 579, 3881–3884. [Google Scholar] [CrossRef]
- West, P.J.; Bulaj, G.; Yoshikami, D. Effects of δ-conotoxins PVIA and SVIE on sodium channels in the amphibian sympathetic nervous system. J. Neurophysiol. 2005, 94, 3916–3924. [Google Scholar] [CrossRef]
- Pennington, M.W.; Byrnes, M.E.; Zaydenberg, I.; Khaytin, I.; de Chastonay, J.; Krafte, D.S.; Hill, R.; Mahnir, V.M.; Volberg, W.A.; Gorczyca, W.; Kem, W.R. Chemical synthesis and characterization of ShK toxin: A potent potassium channel inhibitor from a sea anemone. Int. J. Peptide Protein Res. 1995, 46, 354–358. [Google Scholar]
- Hillyard, D.R.; Olivera, B.M.; Woodward, S.; Corpuz, G.P.; Gray, W.R.; Ramilo, C.A.; Cruz, L.J. A molluscivorous Conus toxin: Conserved frameworks in conotoxins. Biochemistry 1989, 28, 358–361. [Google Scholar]
- Kharrat, R.; Mabrouk, K.; Crest, M.; Darbon, H.; Oughideni, R.; MartinEauclaire, M.F.; Jacquet, G.; ElAyeb, M.; VanRietschoten, J.; Rochat, H.; Sabatier, J.M. Chemical synthesis and characterization of maurotoxin, a short scorpion toxin with four disulfide bridges that acts on K+ channels. Eur. J. Biochem. 1996, 242, 491–498. [Google Scholar]
- Jaravine, V.A.; Nolde, D.E.; Reibarkh, M.J.; Korolkova, Y.V.; Kozlov, S.A.; Pluzhnikov, K.A.; Grishin, E.V.; Arseniev, A.S. Three-dimensional structure of toxin OSK1 from Orthochirus scrobiculosus scorpion venom. Biochemistry 1997, 36, 1223–1232. [Google Scholar]
- Bosmans, F.; Tytgat, J. Voltage-gated sodium channel modulation by scorpion α-toxins. Toxicon 2007, 49, 142–158. [Google Scholar] [CrossRef]
- Gurevitz, M.; Karbat, I.; Cohen, L.; Ilan, N.; Kahn, R.; Turkov, M.; Stankiewicz, M.; Stühmer, W.; Dong, K.; Gordon, D. The insecticidal potential of scorpion β-toxins. Toxicon 2007, 49, 473–489. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Conserved functional surface of anti-mammalian scorpion β-toxins. J. Phys. Chem. B 2012, 116, 4796–4800. [Google Scholar] [CrossRef]
- Chen, R.; Chung, S.H. Binding modes and functional surface of anti-mammalian scorpion α-toxins to sodium channels. Biochemistry 2012, 51, 7775–7782. [Google Scholar] [CrossRef]
- Marvin, L.; De, E.; Cosette, P.; Gagnon, J.; Molle, G.; Lange, C. Isolation, amino acid sequence and functional assays of SGTx1. The first toxin purified from the venom of the spider scodra griseipes. Eur. J. Biochem. 1999, 265, 572–579. [Google Scholar]
- Swartz, K.J.; MacKinnon, R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron 1995, 15, 941–949. [Google Scholar] [CrossRef]
- Lee, S.Y.; MacKinnon, R. A membrane-access mechanism of ion channel inhibition by voltage sensor toxins from spider venom. Nature 2004, 430, 232–235. [Google Scholar]
- Sanguinetti, M.C.; Johnson, J.H.; Hammerland, L.G.; Kelbaugh, P.R.; Volkmann, R.A.; Saccomano, N.A.; Mueller, A.L. Heteropodatoxins: Peptides isolated from spider venom that block Kv4.2 potassium channels. Mol. Pharmacol. 1997, 51, 491–498. [Google Scholar]
- Lee, C.W.; Kim, S.; Roh, S.H.; Endoh, H.; Kodera, Y.; Maeda, T.; Kohno, T.; Wang, J.M.; Swartz, K.J.; Kim, J.I. Solution structure and functional characterization of SGTx1, a modifier of Kv2.1 channel gating. Biochemistry 2004, 43, 890–897. [Google Scholar]
- Zarayskiy, V.V.; Balasubramanian, G.; Bondarenko, V.E.; Morales, M.J. Heteropoda toxin 2 is a gating modifier toxin specific for voltage-gated K+ channels of the Kv4 family. Toxicon 2005, 45, 431–442. [Google Scholar] [CrossRef]
- Alabi, A.A.; Bahamonde, M.I.; Jung, H.J.; Kim, J.I.; Swartz, K.J. Portability of paddle motif function and pharmacology in voltage sensors. Nature 2007, 450, 370–376. [Google Scholar] [CrossRef]
- Milescu, M.; Vobecky, J.; Roh, S.H.; Kim, S.H.; Jung, H.J.; Kim, J.I.; Swartz, K.J. Tarantula toxins interact with voltage sensors within lipid membranes. J. Gen. Physiol. 2007, 130, 497–511. [Google Scholar]
- Posokhov, Y.O.; Gottlieb, P.A.; Morales, M.J.; Sachs, F.; Ladokhin, A.S. Is lipid bilayer binding a common property of inhibitor cysteine knot ion-channel blockers? Biophys. J. 2007, 93, L20–L22. [Google Scholar] [CrossRef]
- Phillips, L.R.; Milescu, M.; Li-Smerin, Y.Y.; Mindell, J.A.; Kim, J.I.; Swartz, K.J. Voltage-sensor activation with a tarantula toxin as cargo. Nature 2005, 436, 857–860. [Google Scholar] [CrossRef]
- Suchyna, T.M.; Johnson, J.H.; Hamer, K.; Leykam, J.F.; Gage, D.A.; Clemo, H.F.; Baumgarten, C.M.; Sachs, F. Identification of a peptide toxin from Grammostola spatulata spider venom that blocks cation-selective stretch-activated channels. J. Gen. Physiol. 2000, 115, 583–598. [Google Scholar] [CrossRef]
- Kamaraju, K.; Gottlieb, P.A.; Sachs, F.; Sukharev, S. Effects of GsMTx4 on bacterial mechanosensitive channels in inside-out patches from giant spheroplasts. Biophys. J. 2010, 99, 2870–2878. [Google Scholar] [CrossRef]
- Suchyna, T.M.; Tape, S.E.; Koeppe, R.E.; Andersen, O.S.; Sachs, F.; Gottlieb, P.A. Bilayer-dependent inhibition of mechanosensitive channels by neuroactive peptide enantiomers. Nature 2004, 430, 235–240. [Google Scholar]
- Bemporad, D.; Sands, Z.A.; Wee, C.L.; Grottesi, A.; Sansom, M.S.P. Vstx1, a modifier of Kv channel gating, localizes to the interfacial region of lipid bilayers. Biochemistry 2006, 45, 11844–11855. [Google Scholar]
- Wee, C.L.; Bemporad, D.; Sands, Z.A.; Gavaghan, D.; Sansom, M.S.P. SGTx1, a Kv channel gating-modifier toxin, binds to the interfacial region of lipid bilayers. Biophys. J. 2007, 92, L07–L09. [Google Scholar] [CrossRef]
- Wee, C.L.; Gavaghan, D.; Sansom, M.S.P. Lipid bilayer deformation and the free energy of interaction of a Kv channel gating-modifier toxin. Biophys. J. 2008, 95, 3816–3826. [Google Scholar] [CrossRef]
- Wee, C.L.; Ulmschneider, M.B.; Sansom, M.S.P. Membrane/toxin interaction energetics via serial multiscale molecular dynamics simulations. J. Chem. Theory Comput. 2010, 6, 966–976. [Google Scholar] [CrossRef]
- Nishizawa, M.; Nishizawa, K. Interaction between K+ channel gate modifier hanatoxin and lipid bilayer membranes analyzed by molecular dynamics simulation. Eur. Biophys. J. 2006, 35, 373–381. [Google Scholar] [CrossRef]
- Nishizawa, M.; Nishizawa, K. Molecular dynamics simulations of a stretch-activated channel inhibitor GsMTx4 with lipid membranes: Two binding modes and effects of lipid structure. Biophys. J. 2007, 92, 4233–4243. [Google Scholar] [CrossRef]
- Chen, R. Effect of External Conditions on Membrane-Protein Interactions. PhD Thesis, The University of Queensland, Brisbane, Australia, 2011. [Google Scholar]
- Wee, C.L.; Gavaghan, D.; Sansom, M.S.P. Interactions between a voltage sensor and a toxin via multiscale simulations. Biophys. J. 2010, 98, 1558–1565. [Google Scholar]
- Wee, C.L.; Sansom, M.S.; Reich, S.; Akhmatskaya, E. Improved sampling for simulations of interfacial membrane proteins: Application of generalized shadow hybrid Monte Carlo to a peptide toxin/bilayer system. J. Phys. Chem. B 2008, 112, 5710–5717. [Google Scholar] [CrossRef]
- Chen, R.; Mark, A.E. The effect of membrane curvature on the conformation of antimicrobial peptides: Implications for binding and the mechanism of action. Eur. Biophys. J. 2011, 40, 545–553. [Google Scholar] [CrossRef]
- Neale, C.; Bennett, W.F.D.; Tieleman, D.P.; Pomès, R. Statistical convergence of equilibrium properties in simulations of molecular solutes embedded in lipid bilayers. J. Chem. Theory Comput. 2011, 7, 4175–4188. [Google Scholar] [CrossRef]
- Lewis, B.A.; Engelman, D.M. Lipid bilayer thickness varies linearly with acyl chain length in fluid phosphatidylcholine vesicles. J. Mol. Biol. 1983, 166, 211–217. [Google Scholar] [CrossRef]
- Krepkiy, D.; Mihailescu, M.; Freites, J.A.; Schow, E.V.; Worcester, D.L.; Gawrisch, K.; Tobias, D.J.; White, S.H.; Swartz, K.J. Structure and hydration of membranes embedded with voltage-sensing domains. Nature 2009, 462, 473–479. [Google Scholar]
- Yoo, J.; Cui, Q. Membrane-mediated protein-protein interactions and connection to elastic models: A coarse-grained simulation analysis of gramicidin A association. Biophys. J. 2013, 104, 128–138. [Google Scholar]
- Poger, D.; van Gunsteren, W.F.; Mark, A.E. A new force field for simulating phosphatidylcholine bilayers. J. Comput. Chem. 2010, 31, 1117–1125. [Google Scholar] [CrossRef]
- Hurst, A.C.; Gottlieb, P.A.; Martinac, B. Concentration dependent effect of GsMTx4 on mechanosensitive channels of small conductance in E. coli spheroplasts. Eur. Biophys. J. 2009, 38, 415–425. [Google Scholar] [CrossRef]
- Elliott, J.R.; Needham, D.; Dilger, J.P.; Haydon, D.A. The effects of bilayer thickness and tension on gramicidin single-channel lifetime. Biochim. Biophys. Acta 1983, 735, 95–103. [Google Scholar] [CrossRef]
- Perozo, E.; Cortes, D.M.; Sompornpisut, P.; Kloda, A.; Martinac, B. Open channel structure of MscL and the gating mechanism of mechanosensitive channels. Nature 2002, 418, 942–948. [Google Scholar] [CrossRef]
- Bowman, C.L.; Gottlieb, P.A.; Suchyna, T.M.; Murphy, Y.K.; Sachs, F. Mechanosensitive ion channels and the peptide inhibitor GsMTx-4: History, properties, mechanisms and pharmacology. Toxicon 2007, 49, 249–270. [Google Scholar] [CrossRef]
- Gurtovenko, A.A.; Anwar, J.; Vattulainen, I. Defect-mediated trafficking across cell membranes: Insights from in silico modeling. Chem. Rev. 2010, 110, 6077–6103. [Google Scholar] [CrossRef]
- Heimburg, T. Lipid ion channels. Biophys. Chem. 2010, 150, 2–22. [Google Scholar] [CrossRef]
- Oswald, R.E.; Suchyna, T.M.; McFeeters, R.; Gottlieb, P.; Sachs, F. Solution structure of peptide toxins that block mechanosensitive ion channels. J. Biol. Chem. 2002, 277, 34443–34450. [Google Scholar]
- Bernard, C.; Legros, C.; Ferrat, G.; Bischoff, U.; Marquardt, A.; Pongs, O.; Darbon, H. Solution structure of HpTX2, a toxin from Heteropoda venatoria spider that blocks Kv4.2 potassium channel. Protein Sci. 2000, 9, 2059–2067. [Google Scholar]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar]
- Klauda, J.B.; Venable, R.M.; Freites, J.A.; O’Connor, J.W.; Tobias, D.J.; Mondragon-Ramirez, C.; Vorobyov, I.; MacKerell, A.D., Jr.; Pastor, R.W. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J. Phys. Chem. B 2010, 114, 7830–7843. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1982, 79, 926–935. [Google Scholar]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar]
- Miyamoto, S.; Kollman, P.A. SETTLE: An analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992, 13, 952–962. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Kumar, S.; Bouzida, D.; Swendsen, R.H.; Kollman, P.A.; Rosenberg, J.M. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 1992, 13, 1011–1021. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chen, R.; Chung, S.-H. Effect of Gating Modifier Toxins on Membrane Thickness: Implications for Toxin Effect on Gramicidin and Mechanosensitive Channels. Toxins 2013, 5, 456-471. https://doi.org/10.3390/toxins5020456
Chen R, Chung S-H. Effect of Gating Modifier Toxins on Membrane Thickness: Implications for Toxin Effect on Gramicidin and Mechanosensitive Channels. Toxins. 2013; 5(2):456-471. https://doi.org/10.3390/toxins5020456
Chicago/Turabian StyleChen, Rong, and Shin-Ho Chung. 2013. "Effect of Gating Modifier Toxins on Membrane Thickness: Implications for Toxin Effect on Gramicidin and Mechanosensitive Channels" Toxins 5, no. 2: 456-471. https://doi.org/10.3390/toxins5020456