Mutagenicity of Ochratoxin A and Its Hydroquinone Metabolite in the SupF Gene of the Mutation Reporter Plasmid Ps189

Abstract
:1. Introduction
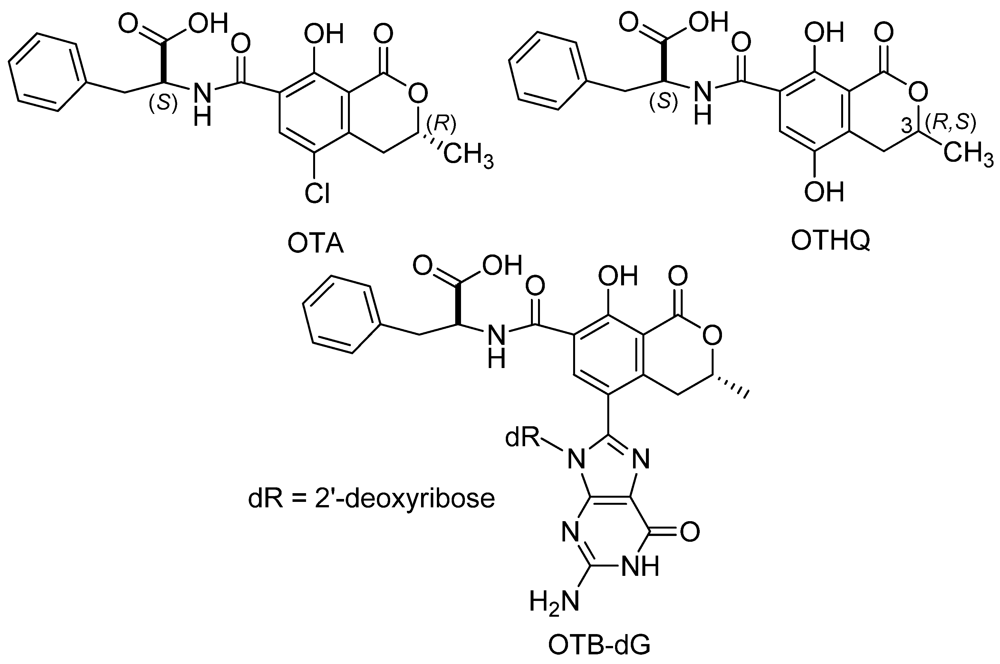
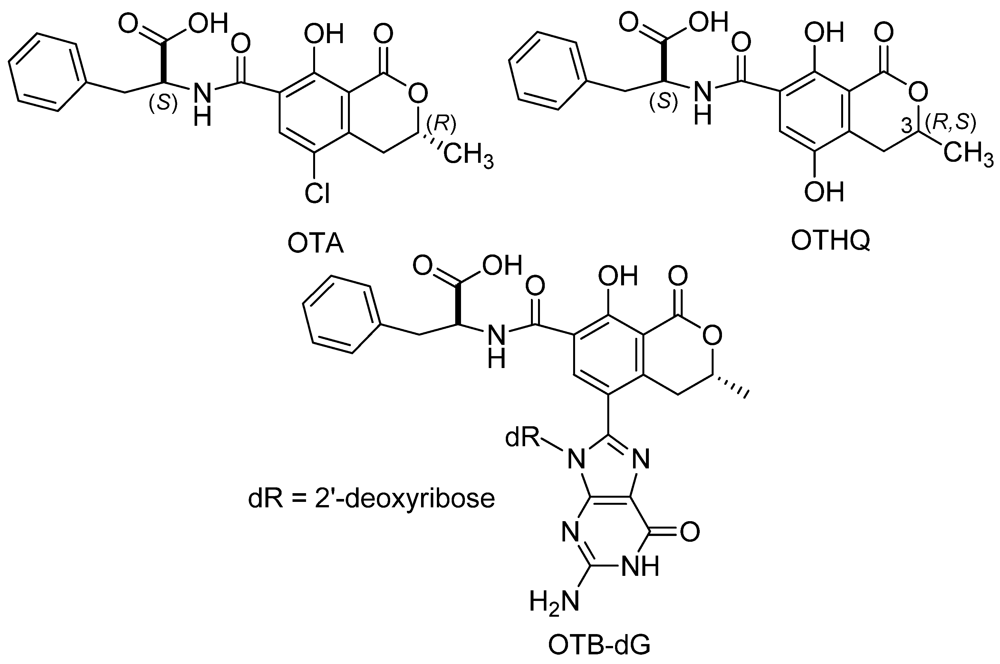
2. Experimental Section
2.1. Reagents
2.2. Treatment of Plasmid PSP189 with OTA
2.3. Transfection of Target Cells
2.4. Mutation Analysis
2.5. Statistical Analysis
3. Results
3.1. Activation with Rat Liver Microsomes

{kind=link}
{kind=link}
| Expt. | Treatment | Colonies | M Colonies a | MF (×104) b | Mean ± SE |
|---|---|---|---|---|---|
| 1 | Plasmid alone | 211267 | 2 | 0.09 | 0.035 ± 0.025 |
| 2 | 81533 | 0 | 0.00 | ||
| 3 | 274100 | 0 | 0.00 | ||
| 1 | 1 mM OTA | 145467 | 1 | 0.07 | 0.061 ± 0.035 |
| 2 | 48367 | 1 | 0.21 | ||
| 3 | 295233 | 1 | 0.04 | ||
| 1 | RLM + 0.5 mM OTA | 165233 | 3 | 0.18 | 0.31 ± 0.087 |
| 2 | 81767 | 2 | 0.24 | ||
| 3 | 169300 | 8 | 0.47 | ||
| 1 | RLM + 1 mM OTA | 104167 | 2 | 0.19 | 0.23 ± 0.072 |
| 2 | 57767 | 2 | 0.35 | ||
| 3 | 275867 | 6 | 0.21 | ||
| 1 | RLM + 5 mM OTA | 88900 | 2 | 0.22 | 0.35 ± 0.093 |
| 2 | 31333 | 2 | 0.64 | ||
| 3 | 281700 | 10 | 0.36 | ||
| 1 | Boiled RLM + 1 mM OTA | 56067 | 0 | 0.00 | 0.032 ± 0.032 |
| 2 | 51567 | 1 | 0.19 | ||
| 3 | 204900 | 0 | 0.00 |
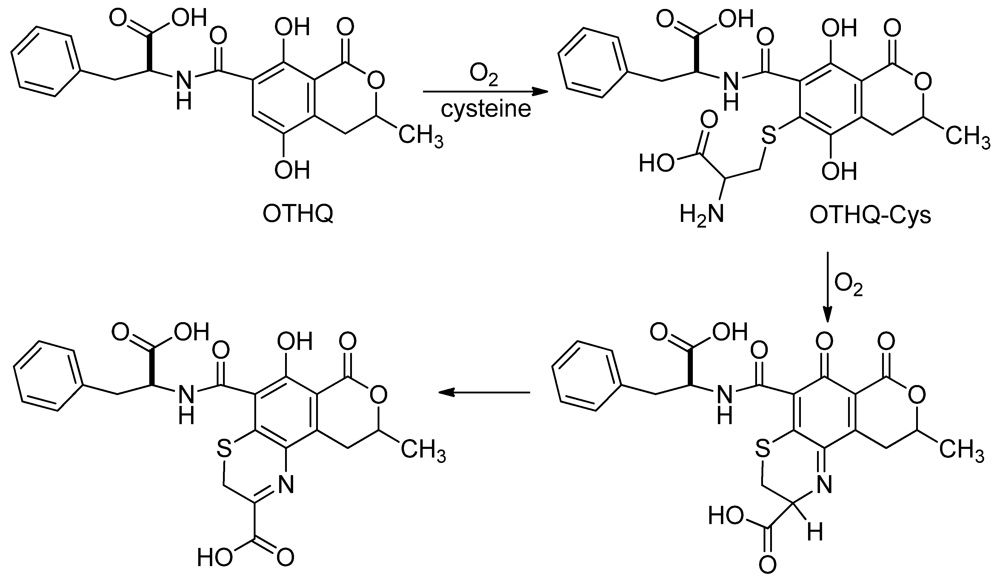
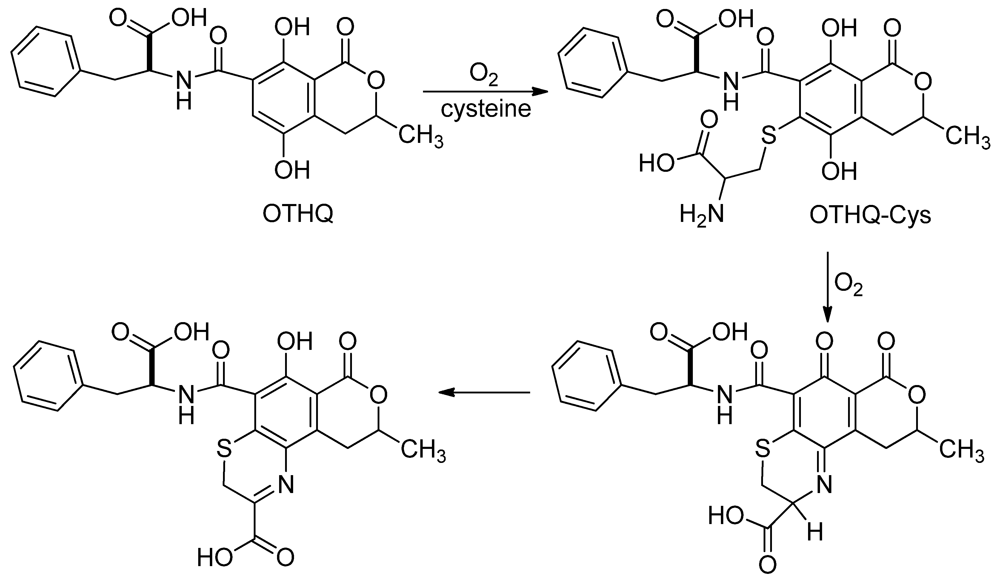
3.2. Mutagenicity of OTHQ
| Expt. | Treatment | Colonies | M Colonies | MF (× 104) | Mean ± SE |
|---|---|---|---|---|---|
| 1 | Plasmid alone | 40150 | 2 | 0.50 | 0.39 ± 0.22 |
| 2 | 18050 | 1 | 0.55 | ||
| 3 | 19600 | 0 | 0.00 | ||
| 1 | 1 mM cysteine | 30500 | 3 | 0.98 | 0.73 ± 0.28 |
| 2 | 32700 | 3 | 0.92 | ||
| 3 | 32700 | 1 | 0.32 | ||
| 1 | 1 mM OTHQ | 12400 | 4 | 3.2 | 1.41 ± 0.45 |
| 2 | 29200 | 3 | 1.0 | ||
| 3 | 29350 | 3 | 1.0 | ||
| 1 | 1 mM OTHQ + 1 mM cysteine | 11200 | 6 | 5.4 | 2.45 ± 0.60 |
| 2 | 29050 | 7 | 2.4 | ||
| 3 | 29050 | 4 | 1.4 |
3.3. Mutagenicity of OTA-Transition Metal Ion Complexes
| Expt. | Treatment | Colonies | M Colonies | MF (× 104) | Mean ± SE |
|---|---|---|---|---|---|
| 1 | Plasmid alone | 12100 | 0 | 0.00 | 0.080 ± 0.080 |
| 2 | 37200 | 0 | 0.00 | ||
| 3 | 76050 | 1 | 0.13 | ||
| 1 | 1 mM Cu(II)/2 mM OTA | 10067 | 1 | 0.99 | 0.71 ± 0.27 |
| 2 | 50700 | 3 | 0.59 | ||
| 3 | 38350 | 3 | 0.78 | ||
| 1 | 1 mM Fe(III)/2 mM OTA | 24800 | 12 | 4.8 | 2.52 ± 0.52 |
| 2 | 35600 | 4 | 1.1 | ||
| 3 | 34650 | 8 | 2.3 |
4. Discussion
Acknowledgments
References
- van der Merwe, K.J.; Steyn, P.S.; Fourie, L.; Scott, D.B.; Theron, J.J. Ochratoxin A, a toxic metabolite produced by Aspergillus ochraceus Wilh. Nature 1965, 205, 1112–1113. [Google Scholar] [CrossRef]
- Pohland, A.E.; Nesheim, S.; Friedman, L. Ochratoxin A, a review. Pure Appl. Chem. 1992, 64, 1029–1046. [Google Scholar]
- Jørgensen, K. Survey of pork, poultry, coffee, beer and pulses for ochratoxin A. Food Addit. Contam. 1998, 15, 550–554. [Google Scholar] [CrossRef]
- Trucksess, M.; Giler, J.; Young, K.; White, K.D.; Page, S.W. Determination and survey of ochratoxin A in wheat, barley, and coffee—1997. J. AOAC Int. 1999, 82, 85–89. [Google Scholar]
- Pfohl-Leszkowicz, A.; Manderville, R.A. Ochratoxin A: An overview on toxicity and carcinogenicity in animals and humans. Mol. Nutr. Food Res. 2007, 51, 61–99. [Google Scholar]
- Stoev, S.D. The role of ochratoxin A as a possible cause of Balkan endemic nephropathy and its risk evaluation. Vet. Human Toxicol. 1998, 40, 352–360. [Google Scholar]
- Pfohl-Leszkowicz, A.; Petkova-Bocharova, T.; Chernozemsky, I.N.; Castegnaro, M. Balkan endemic nephropathy and the associated urinary tract tumours: Review on etiological causes, potential role of mycotoxins. Food Add. Contam. 2002, 19, 282–302. [Google Scholar]
- Grosso, F.; Saïd, S.; Mabrouk, I.; Fremy, J.M.; Castegnaro, M.; Jemmali, M.; Dragacci, S. New data on the occurrence of ochratoxin A in human sera from patients affected or not by renal diseases in Tunisia. Food Chem. Toxicol. 2003, 41, 1133–1140. [Google Scholar]
- IARC, Monographs on the evaluation of carcinogenic risks to humans. In Some Naturally Occurring Substances: Food Items and Constituents, Heterocyclic Aromatic Amines and Mycotoxins, No. 56, Ochratoxin A; International Agency for Research on Cancer: Lyon, France, 1993; pp. 489–521.
- Boorman, G.A. Toxicology and Carcinogenesis Studies of Ochratoxin A (CAS No 303-47-9)in F344/N Rats; Technical Report for National Toxicology Program, NIH Publication No. 89-2813; US Department of Health and Human Services, National Institutes of Health: Research Triangle Park, NC, USA, 1989. [Google Scholar]
- Stoev, S.D. Studies on carcinogenic and toxic effects of ochratoxin A in chicks. Toxins 2010, 2, 649–664. [Google Scholar]
- Schwartz, G.G. Hypothesis: Does ochratoxin A cause testicular cancer? Cancer Causes Control 2002, 13, 91–100. [Google Scholar] [CrossRef]
- Jennings-Gee, J.E.; Tozlovanu, M.; Manderville, R.; Miller, M.S.; Pfohl-Leszkowicz, A.; Schwartz, G.G. Ochratoxin A: In utero exposure in mice induces adducts in testicular DNA. Toxins 2010, 2, 1428–1444. [Google Scholar]
- Gillman, I.G.; Yezek, J.M.; Manderville, R.A. Ochratoxin A acts as a photoactivatable DNA cleaving agent. Chem.Commun. 1998, 647–648. [Google Scholar]
- Il’ichev, Y.V.; Perry, J.L.; Manderville, R.A.; Chignell, C.F.; Simon, J.D. The pH-dependent primary photoreactions of ochratoxin A. J. Phys. Chem. B 2001, 105, 11369–11376. [Google Scholar]
- Brow, M.E.; Dai, J.; Park, G.; Wright, M.W.; Gillman, I.G.; Manderville, R.A. Photochemically catalyzed reaction of ochratoxin A with D- and L-cysteine. Photochem. Photobiol. 2002, 76, 649–656. [Google Scholar] [CrossRef]
- Calcutt, M.W.; Gillman, I.G.; Noftle, R.E.; Manderville, R.A. Electrochemical oxidation of ochratoxin A: Correlation with 4-chlorophenol. Chem.Res.Toxicol. 2001, 14, 1266–1272. [Google Scholar]
- Ardus, J.A.; Gillman, I.G.; Manderville, R.A. On the role of copper and iron in DNA cleavage by ochratoxin A. Structure-activity relationships in metal binding and copper-mediated DNA cleavage. Can J. Chem. 1998, 76, 907–918. [Google Scholar] [CrossRef]
- Manderville, R.A.; Calcutt, M.W.; Dai, J.; Park, G.; Gillman, I.G.; Noftle, R.E.; Mohammed, A.K.; Dizdaroglu, M.; Rodriguez, H.; Akman, S.A. Stoichiometric preference in copper-promoted oxidative DNA damage by ochratoxin A. J.Inorg.Biochem. 2003, 95, 87–96. [Google Scholar]
- Stormer, F.C.; Storen, O.; Hansen, C.E.; Pedersen, J.I.; Aasen, A.J. Formation of (4R)- and (4S)-4-hydroxyochratoxin A and 10-hydroxyochratoxin A from ochratoxin A by rabbit liver microsomes. Appl. Environ. Microbiol. 1983, 45, 1183–1191. [Google Scholar]
- Omar, R.F.; Hasinoff, B.B.; Mejilla, F.; Rahimtula, A.D. Effect of cytochrome P450 induction on the metabolism and toxicity of ochratoxin A. Biochem. Pharmac. 1996, 40, 1183–1191. [Google Scholar]
- Gillman, I.G.; Clark, T.N.; Manderville, R.A. Oxidation of ochratoxin A by an Fe-porphyrin system: Model for enzymatic activation and DNA cleavage. Chem.Res.Toxicol. 1999, 12, 1066–1076. [Google Scholar]
- Pfohl-Leszkowicz, A.; Grosse, Y.; Kane, A.; Creppy, E.E.; Dirheimer, G. Differential DNA adduct formation and disappearance in three mouse tissues after treatment with the mycotoxin ochratoxin A. Mutat. Res. 1993, 289, 265–273. [Google Scholar]
- Pinelli, E.; Adlouni, C.E.; Pipy, B.; Quartulli, F.; Pfohl-Leszkowicz, A. Roles of cyclooxygenase and lipoxygenases in ochratoxin A genotoxicity in human epithelial lung cells. Environ.Toxicol. Pharmacol. 1999, 7, 95–107. [Google Scholar] [CrossRef]
- Kamp, H.G.; Eisenbrand, G.; Schlatter, J.; Würth, K.; Janzowski, C. Ochratoxin A: Induction of (oxidative) DNA damage, cytotoxicity and apoptosis in mammalian cell lines and primary cells. Toxicology 2005, 206, 413–425. [Google Scholar]
- Faucet-Marquis, V.; Pont, F.; Størmer, F.; Rizk, T.; Castegnaro, M.; Pfohl-Leszkowicz, A. Evidence of a new dechlorinated OTA derivative formed in opossum kidney cell cultures after pre-treatment by modulators of glutathione pathways. Correlation with DNA adducts formation. Mol. Nutr. Food Res. 2006, 50, 531–542. [Google Scholar]
- Arbillaga, L.; Azqueta, A.; Ezpeleta, O.; Lopez de Cerain, A. Oxidative DNA damage induced by ochratoxin A in the HK-2 human kidney cell line: evidence of the relationship with cytotoxicity. Mutagenesis 2007, 22, 35–42. [Google Scholar]
- Ali, R.; Mittelstaedt, R.A.; Shaddock, J.G.; Ding, W.; Bhalli, J.A.; Khan, Q.M.; Heflich, R.H. Comparative analysis of micronuclei and DNA damage induced by ochratoxin A in two mammalian cell lines. Mutat.Res. 2011, 723, 58–64. [Google Scholar]
- Baudrimont, I.; Betbeder, A.M.; Gharbi, A.; Pfohl-Leszkowicz, A.; Dirheimer, G.; Creppy, E.E. Effect of superoxide dismutase and catalase on the nephrotoxicity of subchronical administration of ochratoxin A in rats. Toxicology 1994, 89, 101–111. [Google Scholar]
- Gautier, J.C.; Holzhaeuser, D.; Markovic, J.; Gremaud, E.; Schilter, B.; Turesky, R.J. Oxidative damage and stress response from ochratoxin A exposure in rats. Free Rad. Biol. Med. 2001, 30, 1089–1098. [Google Scholar] [CrossRef]
- Kamp, H.G.; Eisenbrand, G.; Janzowski, C.; Kiossev, J.; Latendresse, J.R.; Schlatter, J.; Turesky, R.J. Ochratoxin A induces oxidative DNA damage in liver and kidney after oral dosing to rats. Mol. Nutr. Food Res. 2005, 49, 1160–1167. [Google Scholar] [CrossRef]
- Wurgler, F.E.; Friederich, U.; Schlatter, J. Lack of mutagenicity of ochratoxin A and B, citrinin, patulin and cnestine in Salmonella typhimurium TA102. Mutat. Res. 1991, 261, 209–216. [Google Scholar] [CrossRef]
- De Groene, E.M.; Hassing, I.G.A.M.; Blom, M.J.; Seinen, W.; Fink-Gremmels, J.; Horbach, G.J. Development of human cytochrome P450-expressing cell lines: Application in mutagenicity testing of ochratoxin A. Cancer Res. 1996, 56, 299–304. [Google Scholar]
- De Groene, E.M.; Jahn, A.; Horbach, G.J.; Fink-Gremmels, J. Mutagenicity and genotoxicity of the mycotoxin ochratoxin A. Environ. Toxicol. Pharmacol. 1996, 1, 21–26. [Google Scholar] [CrossRef]
- Obrecht-Plumio, S.; Chassat, T.; Dirheimer, G.; Marzin, D. Genotoxicity of ochratoxin A by Salmonella mutagenicity test after bioactivation by mouse kidney microsomes. Mutat. Res. 1999, 446, 95–102. [Google Scholar]
- Zepnik, H.; Pähler, A.; Schauer, U.; Dekant, W. Ochratoxin A-induced tumor formation: Is there a role of reactive ochratoxin A metabolites? Toxicol. Sci. 2001, 59, 59–67. [Google Scholar] [CrossRef]
- Palma, N.; Cinelli, S.; Sapora, O.; Wilson, S.H.; Dogliotti, E. Ochratoxin A-induced mutagenesis in mammalian cells is consistent with the production of oxidative stress. Chem. Res.Toxicol. 2007, 20, 1031–1037. [Google Scholar] [CrossRef]
- Gross-Steinmayer, K.; Weymann, J.; Hege, H.C.; Metzler, M. Metabolism and lack of DNA reactivity of the mycotoxin ochratoxin A in cultured rat and human primary hepatocytes. J. Agric. Food Chem. 2002, 50, 938–945. [Google Scholar] [CrossRef]
- Mally, A.; Zepnik, H.; Wanek, P.; Eder, E.; Dingley, K.; Ihmels, H.; Völkel, W.; Dekant, W. Ochratoxin A: Lack of formation of covalent DNA adducts. Chem. Res. Toxicol. 2004, 17, 234–242. [Google Scholar] [CrossRef]
- Mally, A.; Pepe, G.; Ravoori, S.; Fiore, M.; Gupta, R.C.; Dekant, W.; Mosesso, P. Ochratoxin A causes DNA damage and cytogenetic effects but no DNA adducts in rats. Chem. Res.Toxicol. 2005, 18, 1253–1261. [Google Scholar] [CrossRef] [Green Version]
- Marin-Kuan, M.; Ehrlich, V.; Delatour, T.; Cavin, C.; Schilter, B. Evidence for a role of oxidative stress in the carcinogenicity of ochratoxin A. J.Toxicol. 2011, 1–15. [Google Scholar]
- Faucet, V.; Pfohl-Leszkowicz, A.; Dai, J.; Castegnaro, M.; Manderville, R.A. Evidence for covalent DNA adduction by ochratoxin A following chronic exposure to rat and subacute exposure to pig. Chem.Res. Toxicol. 2004, 17, 1289–1296. [Google Scholar]
- Tozlovanu, M.; Faucet-Marquis, V.; Pfohl-Leszkowicz, A.; Manderville, R.A. Genotoxicity of the hydroquinone metabolite of ochratoxin A: Structure-activity relationships for covalent DNA adduction. Chem. Res.Toxicol. 2006, 19, 1241–1247. [Google Scholar] [CrossRef]
- Mantle, P.G.; Faucet-Marquis, V.; Manderville, R.A.; Squillaci, B.; Pfohl-Leszkowicz, A. Structures of covalent adducts between DNA and ochratoxin A: A new factor in debate about genotoxicity and human risk assessment. Chem. Res. Toxicol. 2010, 23, 89–98. [Google Scholar] [CrossRef]
- Hibi, D.; Suzuki, Y.; Ishii, Y.; Jin, M.; Watanabe, M.; Sugita-Konishi, Y.; Yanai, T.; Nohmi, T.; Nishikawa, A.; Umemura, T. Site-specific in vivo mutagenicity in the kidney of gpt delta rats given a carcinogenic dose of ochratoxin A. Toxicol. Sci. 2011, 122, 406–414. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Manderville, R.A. An update on direct genotoxicity as a molecular mechanism of ochratoxin A carcinogenicity. Chem. Res. Toxicol. 2012, 25, 252–262. [Google Scholar]
- Seidman, M.M.; Dixon, K.; Razzaque, A.; Zagursky, R.J.; Berman, M.L. A shuttle vector plasmid for studying carcinogen-induced point mutations in mammalian cells. Gene 1985, 38, 233–237. [Google Scholar]
- Parris, C.N.; Seidman, M.M. A signature element distinguishes sibling and independent mutations in a shuttle vector plasmid. Gene 1992, 117, 1–5. [Google Scholar]
- Gillman, I.G.; Day, C.S.; Manderville, R.A. Stepwise formation of a nonsymmetric dinuclear copper complex of ochratoxin A. Inorg. Chem. 1998, 37, 6385–6388. [Google Scholar]
- McCuthen, J.A.; Pagano, J.S. Enhancement of the infectivity of SV40 deoxyribonucleic acid with diethylaminoethyl dextran. J. Natl. Cancer Inst. 1968, 41, 351–357. [Google Scholar]
- Hirt, B. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 1967, 20, 365–369. [Google Scholar] [CrossRef]
- Waters, S.B.; Akman, S.A. A new assay to quantify in vivo repair of G:T mispairs by base excision repair. Mutat. Res. 2001, 487, 109–119. [Google Scholar]
- Dai, J.; Wright, M.W.; Manderville, R.A. Ochratoxin A forms a carbon-bonded C8-deoxyguanosine nucleoside adduct: Implication for C8-reactivity by a phenolic radical. J. Am. Chem. Soc. 2003, 125, 3716–3717. [Google Scholar] [CrossRef]
- Omar, R.F.; Hasinoff, B.B.; Mejilla, F.; Rahimtula, A.D. Mechanism of ochratoxin A stimulated lipid peroxidation. Biochem. Pharmacol. 1990, 40, 1183–1191. [Google Scholar]
- Akman, S.A.; Forrest, G.P.; Doroshow, J.H.; Dizdaroglu, M. Mutation of potassium permanganate-and hydrogen peroxide-treated plasmid pZ189 replicating in CV-1 monkey kidney cells. Mutat. Res. 1991, 261, 123–130. [Google Scholar]
- Akman, S.A.; Doroshow, J.H.; Kensler, T.W. Copper-dependent site-specific mutagenesis by benzoyl peroxide in the supF gene of the mutation reporter plasmid pS189. Carcinogenesis 1992, 13, 1783–1787. [Google Scholar]
- Manderville, R.A. A case for the genotoxicity of ochratoxin A by bioactivation and covalent DNA adduction. Chem. Res. Toxicol. 2005, 18, 1091–1097. [Google Scholar] [CrossRef]
- Turesky, R.J. Perspective: Ochratoxin A is not a genotoxic carcinogen. Chem Res.Toxicol. 2005, 18, 1082–1090. [Google Scholar]
- Dai, J.; Park, G.; Wright, M.W.; Adams, M.; Akman, S.A.; Manderville, R.A. Detection and characterization of a glutathione conjugate of ochratoxin A. Chem. Res.Toxicol. 2002, 15, 1581–1588. [Google Scholar] [CrossRef]
- Pfohl-Leszkowicz, A.; Grosse, Y.; Kane, A.; Gharbi, A.; Baudrimont, I.; Obrecht, S.; Creppy, E.E.; Dirheimer, G. Is the oxidative pathway implicated in the genotoxicity of ochratoxin A? In Human Ochratoxicosis and Its Pathologies; Creppy, E.E., Castegnaro, M., Dirheimer, G., Eds.; John Libbey Euro-Text: Montrouge, France, 1993; Volume 231, pp. 177–187. [Google Scholar]
- Hadjeba-Medjdoub, K.; Tozlovanu, M.; Pfohl-Leszkowicz, A.; Frenette, C.; Paugh, R.J.; Manderville, R.A. Structure-activity relationships imply different mechanisms of action for ochratoxin A-mediated cytotoxicity and genotoxicity. Chem. Res.Toxicol. 2012, 25, 181–190. [Google Scholar] [CrossRef]
- Monks, T.J.; Lau, S.S. Biological reactivity of polyphenolic-glutathione conjugates. Chem. Res. Toxicol. 1997, 10, 1296–1313. [Google Scholar] [CrossRef]
- Rahman, K.M.; James, C.H.; Thurston, D.E. Observation of the reversibility of a covalent pyrrolobenzodiazepine (PDB) DNA adduct by HPLC/MS and CD spectroscopy. Org. Biomol. Chem. 2011, 9, 1632–1641. [Google Scholar]
- Obrecht-Pflumio, S.; Dirheimer, G. In vitro DNA and dGMP adducts formation caused by ochratoxin A. Chem. Biol. Interact. 2000, 127, 29–44. [Google Scholar] [CrossRef]
- Obrecht-Pflumio, S.; Dirheimer, G. Horseradish peroxidase mediates DNA and deoxyguanosine 3’-monophosphate adduct formation in the presence of ochratoxin A. Arch. Toxicol. 2001, 75, 583–590. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Akman, S.A.; Adams, M.; Case, D.; Park, G.; Manderville, R.A. Mutagenicity of Ochratoxin A and Its Hydroquinone Metabolite in the SupF Gene of the Mutation Reporter Plasmid Ps189. Toxins 2012, 4, 267-280. https://doi.org/10.3390/toxins4040267
Akman SA, Adams M, Case D, Park G, Manderville RA. Mutagenicity of Ochratoxin A and Its Hydroquinone Metabolite in the SupF Gene of the Mutation Reporter Plasmid Ps189. Toxins. 2012; 4(4):267-280. https://doi.org/10.3390/toxins4040267
Chicago/Turabian StyleAkman, Steven A., Marissa Adams, Doug Case, Gyungse Park, and Richard A. Manderville. 2012. "Mutagenicity of Ochratoxin A and Its Hydroquinone Metabolite in the SupF Gene of the Mutation Reporter Plasmid Ps189" Toxins 4, no. 4: 267-280. https://doi.org/10.3390/toxins4040267