PI3K/Akt/mTOR, a Pathway Less Recognized for Staphylococcal Superantigen-Induced Toxicity

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Staphylococcal Superantigen Structure and Binding

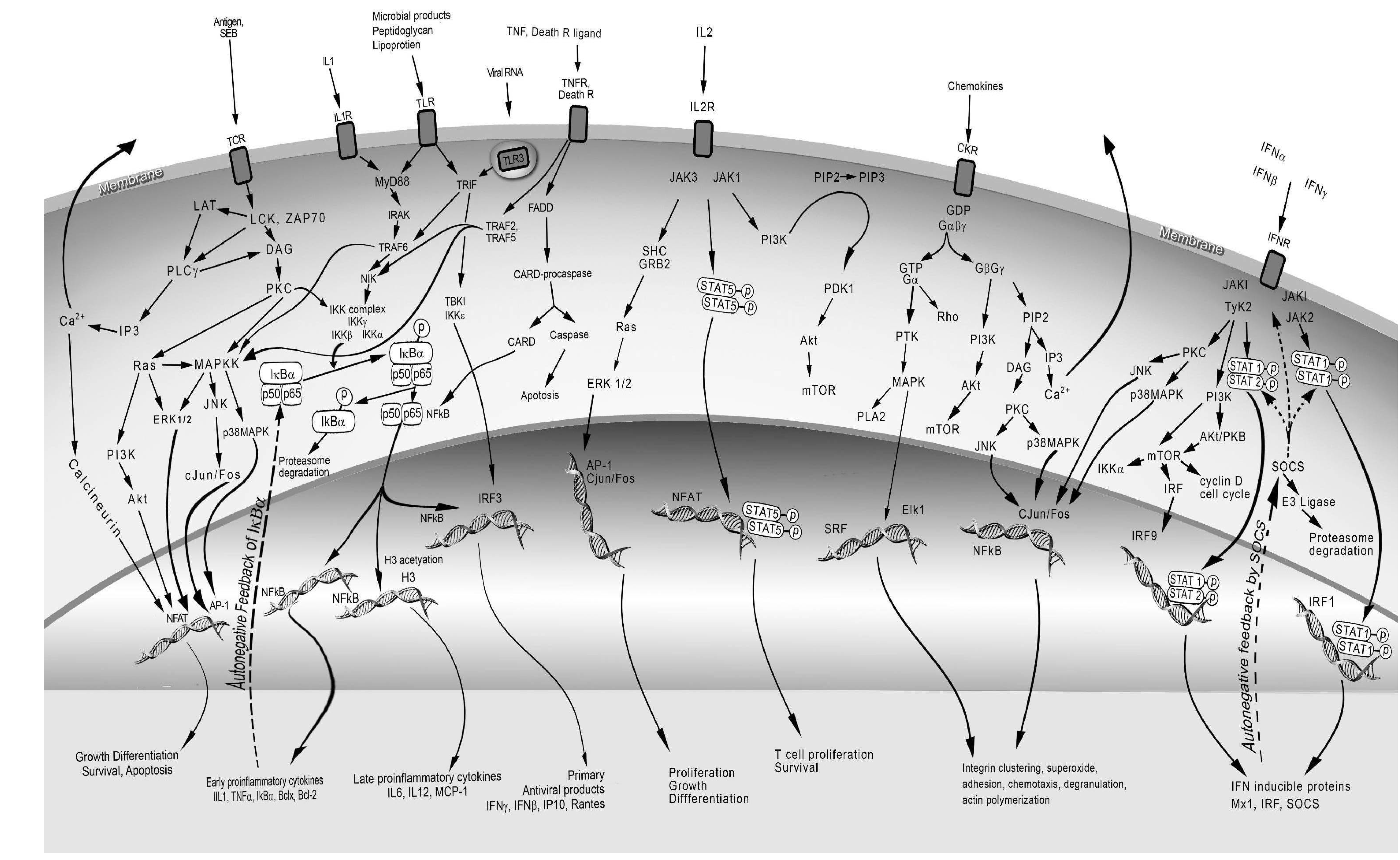
3. The Three Signals for T Cell Activation
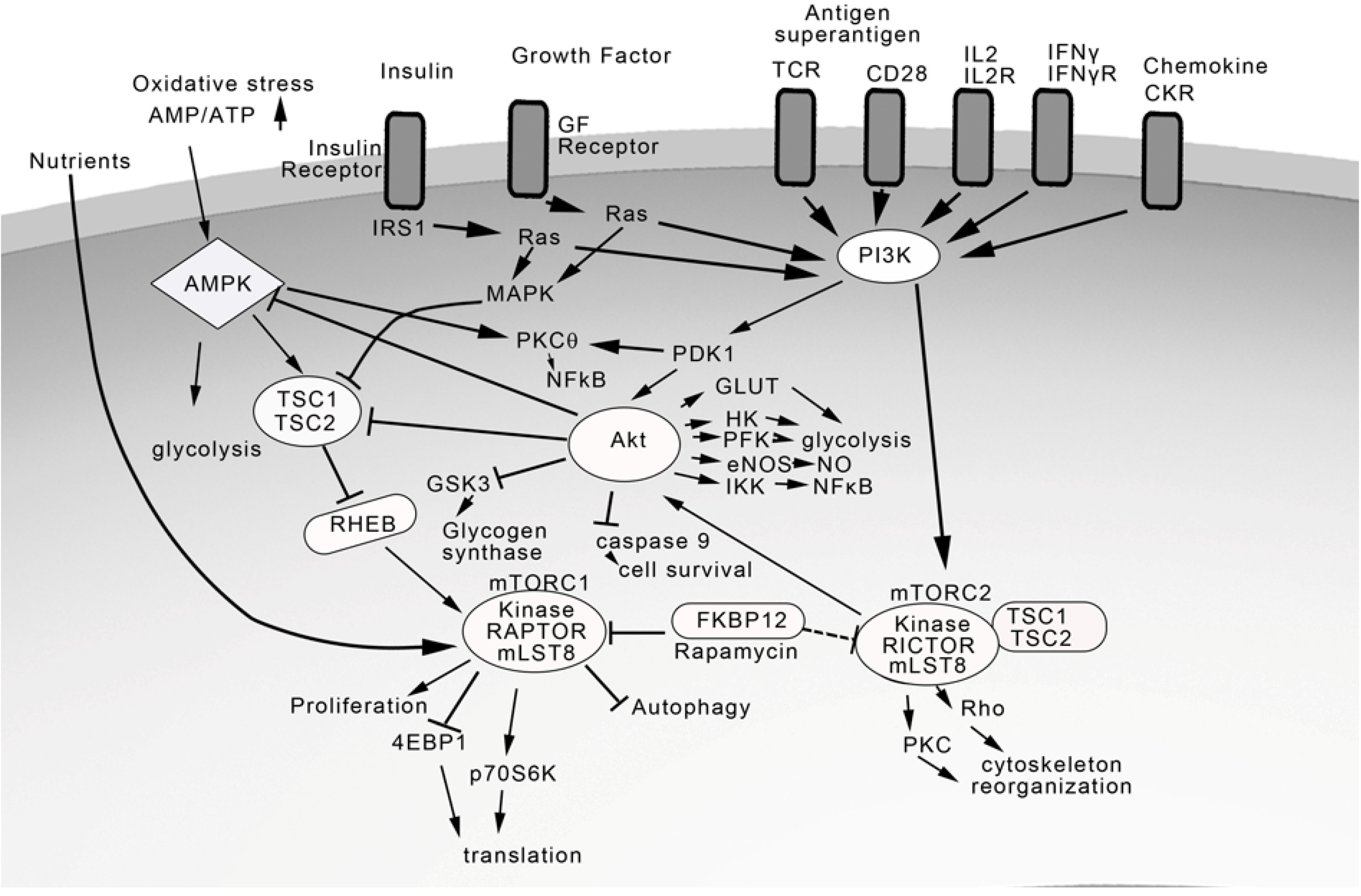
4. TCR and Costimulatory Receptors Activate the Phosphatidylinositol Pathway
5. Regulation of Akt and Mammalian Target of Rapamycin (mTOR)

6. Proinflammatory Mediators Signal via NFκB Activation
7. T Cell Cytokines and Chemokines Activate the PI3K/Akt/mTOR Pathway
8. Therapeutic Antibodies against SEB
9. Inhibitors of Cell Receptor-Toxin Interaction
10. Inhibitors of Signal Transduction
11. Inhibitors of Cytokines
12. Summary
Acknowledgments
Conflict of Interest
Disclaimer
References
- Chesney, P.J.; Davis, P.J.; Purdy, W.K.; Wand, P.J.; Chesney, R.W. Clinical manifestations of toxic shock syndrome. JAMA 1981, 246, 741–748. [Google Scholar]
- Marrack, P.; Kappler, J. The staphylococcal enterotoxins and their relatives. Science 1990, 248, 705–709. [Google Scholar]
- Kotzin, B.L.; Leung, D.Y.M.; Kappler, J.; Marrack, P. Superantigens and their potential role in human disease. Adv. Immunol. 1993, 54, 99–166. [Google Scholar] [CrossRef]
- Kotb, M. Bacterial pyrogenic exotoxins as superantigens. Clin. Microbiol. Rev. 1995, 8, 411–426. [Google Scholar]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Ann. Rev. Microbiol. 2001, 55, 77–104. [Google Scholar] [CrossRef]
- Proft, T.; Fraser, J.D. Bacterial superantigens. Clin. Exp. Immunol. 2003, 133, 299–306. [Google Scholar] [CrossRef]
- Fraser, J.D.; Proft, T. The bacterial superantigen and superantigen-like proteins. Immunol. Rev. 2008, 225, 226–243. [Google Scholar] [CrossRef]
- Choi, Y.; Kotzin, B.; Hernon, L.; Callahan, J.; Marrack, P.; Kappler, J. Interaction of Staphylococcus aureus toxin “superantigens” with human T cells. Proc. Natl. Acad. Sci. USA 1989, 86, 8941–8945. [Google Scholar] [CrossRef]
- Jupin, C.; Anderson, S.; Damais, C.; Alouf, J.E.; Parant, M. Toxic shock syndrome toxin 1 as an inducer of human tumor necrosis factors and gamma interferon. J. Exp. Med. 1988, 167, 752–761. [Google Scholar] [CrossRef]
- Trede, N.S.; Geha, R.S.; Chatila, T. Transcriptional activation of IL-1 beta and tumor necrosis factor-alpha genes by MHC class II ligands. J. Immunol. 1991, 146, 2310–2315. [Google Scholar]
- See, R.H.; Kum, W.W.; Chang, A.H.; Goh, S.H.; Chow, A.W. Induction of tumor necrosis factor and interleukin-1 by purified staphylococcal toxic shock syndrome toxin 1 requires the presence of both monocytes and T lymphocytes. Infect. Immun. 1992, 60, 2612–2618. [Google Scholar]
- Miethke, T.; Wahl, C.; Heeg, K.; Echtenacher, B.; Krammer, P.H.; Wagner, H. Superantigen mediated shock: A cytokine release syndrome. Immunobiology 1993, 189, 270–284. [Google Scholar] [CrossRef]
- Tessier, P.A.; Naccache, P.H.; Diener, K.R.; Gladue, R.P.; Neotem, K.S.; Clark-Lewis, I.; McColl, S.R. Induction of acute inflammation in vivo by staphylococcal superantigens. II. Critical role for chemokines, ICAM-1, and TNF-alpha. J. Immunol. 1998, 161, 1204–1211. [Google Scholar]
- Krakauer, T. The induction of CC chemokines in human peripheral blood mononuclear cells by staphylococcal exotoxins and its prevention by pentoxifylline. J. Leukco. Biol. 1999, 66, 158–164. [Google Scholar]
- Faulkner, L.; Cooper, A.; Fantino, C.; Altmann, D.M.; Sriskandan, S. The mechanism of superantigen-mediated toxic shock: Not a simple Th1 cytokine storm. J. Immunol. 2005, 175, 6870–6877. [Google Scholar]
- Krakauer, T.; Buckley, M.; Fisher, D. Proinflammatory mediators of toxic shock and their correlation to lethality. Mediators Inflamm. 2010. [Google Scholar] [CrossRef]
- Mattsson, E.; Herwald, H.; Egsten, A. Superantigen from Staphylococcus aureus induce procoagulant activity and monocyte tissue factor expression in whole blood and mononuclear cells via IL-1β. J. Thromb. Haemost. 2003, 1, 2569–2575. [Google Scholar] [CrossRef]
- Neumann, B.; Engelhardt, B.; Wagner, H.; Holzmann, B. Induction of acute inflammatory lung injury by staphylococcal enterotoxin B. J. Immunol. 1997, 158, 1862–1871. [Google Scholar]
- Miethke, T.; Wahl, C.; Heeg, K.; Echtenacher, B.; Krammer, P.H.; Wagner, H. T cell-mediated lethal shock triggered in mice by the superantigen staphylococcal enterotoxin B: Critical role of tumor necrosis factor. J. Exp. Med. 1992, 175, 91–98. [Google Scholar] [CrossRef]
- Stiles, B.G.; Bavari, S.; Krakauer, T.; Ulrich, R.G. Toxicity of staphylococcal enterotoxins potentiated by lipopolysaccharide: Major histocompatibility complex class II molecule dependency and cytokine release. Infect. Immun. 1993, 61, 5333–5338. [Google Scholar]
- Krakauer, T.; Vilcek, J.; Oppenheim, J.J. Proinflammatory Cytokines: TNF and IL-1 Families, Chemokines, TGFβ and Others. In Fundamental Immunology, 4th; Paul, W., Ed.; Lippincott-Raven: Philadelphia, PA, USA, 1998; pp. 775–811. [Google Scholar]
- Vial, T.; Descotes, J. Immune-mediated side-effects of cytokines in human. Toxicology 1995, 105, 31–57. [Google Scholar] [CrossRef]
- Holmberg, S.D.; Blake, P.A. Staphylococcal food poisoning in the United States. New facts and old misconceptions. JAMA 1984, 251, 487–489. [Google Scholar] [CrossRef]
- Schwab, J.H.; Brown, R.R.; Anderle, S.K.; Schlievert, P.M. Superantigen can reactivate bacterial cell wall-induced arthritis. J. Immunol. 1993, 150, 4151–4159. [Google Scholar]
- Brocke, S.; Hausmann, S.; Steinmam, L.; Wucherpfennig, K.W. Microbial peptides and superantigens in the pathogenesis of autoimmune diseases of the central nervous system. Semin. Immunol. 1998, 10, 57–67. [Google Scholar] [CrossRef]
- Yarwood, J.M.; Leung, D.Y.; Schlievert, P.M. Evidence for the involvement of bacterial superantigens in psoriasis, atopic dermatitis, and Kawasaki syndrome. FEMS Microbiol. Lett. 2000, 192, 1–7. [Google Scholar] [CrossRef]
- McKay, D.M. Bacterial superantigens: Provocateurs of gut dysfunction and inflammation? Trends Immunol. 2001, 22, 497–501. [Google Scholar] [CrossRef]
- Sugiyama, H.; McKissic, E.M.; Bergdoll, M.S.; Heller, B. Enhancement of bacterial endotoxin lethality by staphylococcal enterotoxin. J. Infect. Dis. 1964, 4, 111–118. [Google Scholar]
- Sarawar, S.R.; Blackman, B.A.; Doherty, P.C. Superantigen shock in mice with an inapparent viral infection. J. Infect. Dis. 1994, 170, 1189–1194. [Google Scholar] [CrossRef]
- Zhang, W.J.; Sarawar, S.; Nguyen, P.; Daly, K.; Rehig, J.E.; Doherty, P.C.; Woodland, D.L.; Blackman, M.A. Lethal synergism between influenza infection and staphylococcal enterotoxin B in mice. J. Immunol. 1996, 157, 5049–5060. [Google Scholar]
- Blank, C.; Luz, A.; Bendigs, S.; Erdmann, A.; Wagner, H.; Heeg, K. Superantigen and endotoxin synergize in the induction of lethal shock. Eur. J. Immunol. 1997, 27, 825–833. [Google Scholar] [CrossRef]
- Hopkins, P.A.; Fraser, J.D.; Pridmore, A.C.; Russell, H.H.; Read, R.C.; Sriskandan, S. Superantigen recognition by HLA class II on monocytes up-regulates toll-like receptor 4 and enhances proinflammatory responses to endotoxin. Blood 2005, 105, 3655–3662. [Google Scholar] [CrossRef]
- Hopkins, P.A.; Pridmore, A.C.; Ellmerich, S.; Fraser, J.D.; Russell, H.H.; Read, R.C.; Sriskandan, S. Increased surface toll-like receptor 2 expression in superantigen shock. Crit. Care Med. 2008, 36, 1267–1276. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Acharya, K.R. Microbial superantigens: From structure to function. Trends Microbiol. 2000, 8, 369–375. [Google Scholar] [CrossRef]
- Monday, S.R.; Bohach, G.A. Properties of Staphylococcus aureus Enterotoxins and Toxic Shock Syndrome Toxin-1. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J.E., Freer, J.H., Eds.; Academic: London, UK, 1999; pp. 589–610. [Google Scholar]
- Wang, X.; Xu, M.; Cai, Y.; Yang, H.; Zhang, H.; Zhang, C. Functional analysis of the disulphide loop mutant of staphylococcal enterotoxin C2. Appl. Microbiol. Biotechnol. 2009, 82, 861–871. [Google Scholar] [CrossRef]
- Bavari, S.; Ulrich, R.G.; LeClaire, R.D. Cross-reactive antibodies prevent the lethal effects of Staphylococcus aureus superantigens. J. Infect. Dis. 1999, 180, 1365–1369. [Google Scholar] [CrossRef]
- Kappler, J.W.; Herman, A.; Clements, J.; Marrack, P. Mutations defining functional regions of the superantigen staphylococcal enterotoxin B. J. Exp. Med. 1992, 175, 387–396. [Google Scholar] [CrossRef]
- Li, H.; Llera, A.; Tsuchiya, D.; Leder, L.; Ysern, X.; Schlievert, P.M.; Karjalainen, K.; Mariuzza, R.A. Three-dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B. Immunity 1998, 9, 807–816. [Google Scholar] [CrossRef]
- Mollick, J.A.; Chintagumpala, M.; Cook, R.G.; Rich, R.R. Staphylococcal exotoxin activation of T cells. Role of exotoxin-MHC class II binding affinity and class II isotype. J. Immunol. 1991, 146, 463–468. [Google Scholar]
- Chintagumpala, M.M.; Mollick, J.A.; Rich, R.R. Staphylococcal toxins bind to different sites on HLA-DR. J. Immunol. 1991, 147, 3876–3882. [Google Scholar]
- Ulrich, R.G.; Bavari, B.; Olson, M.A. Staphylococcal enterotoxins A and B share a common structural motif for binding class II major histocompatibility complex molecules. Nat. Struct. Biol. 1995, 2, 554–560. [Google Scholar] [CrossRef]
- Hudson, K.R.; Tiedemann, R.E.; Urban, R.G.; Lowe, S.C.; Strominger, J.L.; Fraser, J.D. Staphylococcal enterotoxin A has two cooperative binding sites on major histocompatibility complex class II. J. Exp. Med. 1995, 182, 711–720. [Google Scholar] [CrossRef]
- Tiedemann, R.E.; Urban, R.J.; Strominger, J.L.; Fraser, J.D. Isolation of HLA-DR1. (staphylococcal enterotoxins A) 2 trimers in solution. Proc. Natl. Acad. Sci. USA 1995, 92, 12156–12159. [Google Scholar] [CrossRef]
- Thibodeau, J.; Cloutier, I.; Lavoie, P.M.; Labrecque, N.; Mourad, W.; Jardetzky, T.; Sekaly, R.P. Subsets of HLA-DR1 molecules defined by SEB and TSST-1 binding. Science 1994, 266, 1874–1878. [Google Scholar]
- Herrmann, T.; Acolla, R.S.; MacDonald, H.R. Different staphylococcal enterotoxins bind preferentially to distinct MHC class II isotypes. Eur. J. Immunol. 1989, 19, 2171–2174. [Google Scholar] [CrossRef]
- Herman, A.; Croteau, G.; Sekaly, R.P.; Kappler, J.; Marrack, P. HLA-DR alleles differ in their ability to present staphylococcal enterotoxins to T cells. J. Exp. Med. 1990, 172, 709–712. [Google Scholar] [CrossRef]
- Scholl, P.; Sekaly, R.; Diez, A.; Glimcher, L.; Geha, R. Binding of toxic shock syndrome toxin-1 to murine major histocompatibility complex class II molecules. Eur. J. Immunol. 1990, 20, 1911–1916. [Google Scholar] [CrossRef]
- Seth, A.; Stern, L.J.; Ottenhoff, T.H.; Engel, I.; Owen, M.J.; Lamb, J.R.; Klausner, R.D.; Wiley, D.C. Binary and ternary complexes between T-cell receptor, class II MHC and superantigen in vitro. Nature 1994, 369, 324–27. [Google Scholar]
- Moza, B.; Varma, A.K.; Buonpane, R.A.; Zhu, P.; Herfst, C.A.; Nicholson, M.J.; Wilbuer, A.K.; Seth, N.P.; Wucherpfennig, K.W.; McCormick, J.K.; Kranz, D.M.; Sundberg, E.J. Structural basis of T-cell specificity and activation by the bacterial superantigen TSST-1. EMBO J. 2007, 26, 1187–1197. [Google Scholar]
- Ferry, T.; Thomas, D.; Perpoint, T.; Lina, G.; Monneret, G.; Mohammedi, I.; Chidiac, C.; Peyramond, D.; Vandenesch, F.; Etienne, J. Analysis of superantigenic toxin Vbeta T-cell signatures produced during cases of staphylococcal toxic shock syndrome and septic shock. Clin. Microbiol. Infect. 2008, 14, 546–554. [Google Scholar] [CrossRef]
- Arad, G.; Levy, R.; Nasie, I.; Hillman, D.; Rotfogel, Z.; Barash, U.; Supper, E.; Shpilka, T.; Minis, A.; Kaempfer, R. Binding of superantigen toxins into CD28 homodimer interface is essential for induction of cytokine genes that mediate lethal shock. Plos Biol. 2012, 9, e1001149. [Google Scholar]
- Germain, R.N. T-cell signaling: The importance of receptor clustering. Curr. Biol. 1997, 7, R640–R644. [Google Scholar] [CrossRef]
- Chatila, T.; Geha, R.S. Signal transduction by microbial superantigens via MHC class II molecules. Immunol. Rev. 1993, 131, 43–59. [Google Scholar] [CrossRef]
- Tiedemann, R.E.; Fraser, J.D. Cross-linking of MHC class II molecules by staphylococcal enterotoxin A is essential for antigen-presenting cell and T cell activation. J. Immunol. 1996, 157, 3958–3966. [Google Scholar]
- Carlsson, R.; Fischer, H.; Sjogren, H.O. Binding of staphylococcal enterotoxin A to accessory cells is a requirement for its ability to activate human T cells. J. Immunol. 1998, 140, 2484–2488. [Google Scholar]
- Anderson, M.R.; Tary-Lehmann, M. Staphylococcal enterotoxin-B-induced lethal shock in mice is T-cell-dependent, but disease susceptibility is defined by the non-T-cell compartment. Clin. Immunol. 2001, 98, 85–94. [Google Scholar] [CrossRef]
- Fleischer, B.; Schrezenmeier, H. T cell stimulation by staphylococcal enterotoxins. Clonally variable response and requirement for major histocompatibility complex class II molecules on accessory or target cells. J. Exp. Med. 1988, 167, 1697–1707. [Google Scholar] [CrossRef]
- Linsley, P.S.; Ledbetter, J.A. The role of the CD28 receptor during T cell responses to antigen. Ann. Rev. Immunol. 1993, 11, 191–212. [Google Scholar] [CrossRef]
- Krakauer, T. Co-stimulatory receptors for the superantigen staphyloccoccal enterotoxin B on human vascular endothelial cells and T cells. J. Leukco. Biol. 1994, 56, 458–463. [Google Scholar]
- Cemerski, S.; Shaw, A. Immune synapses in T-cell activation. Curr. Opin. Immunol. 2006, 18, 298–304. [Google Scholar]
- Fraser, J.; Newton, M.; Weiss, A. CD28 and T-cell antigen receptor signal transduction coordinately regulates interleukin 2 gene expression in response to superantigen stimulation. J. Exp. Med. 1992, 175, 1131–1134. [Google Scholar] [CrossRef]
- Boise, L.H.; Minn, A.J.; Noel, P.J.; June, C.H.; Accavitti, M.A.; Lindsten, T.; Thompson, C.B. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-xl. Immunity 1995, 3, 87–98. [Google Scholar] [CrossRef]
- Weiss, A.T. Lymphocyte Activation. In Fundamental Immunology, 4th; Paul, W., Ed.; Lippincott-Raven: Philadelphia, PA, USA, 1998; pp. 411–447. [Google Scholar]
- Van Leeuwen, J.E.; Samelson, L.E. T cell-antigen receptor signal transduction. Curr. Opin. Immunol. 1999, 11, 242–248. [Google Scholar]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Isakov, N.; Altman, A. PKC-theta-mediated signal delivery from the TCR/CD28 surface receptors. Front. Immunol. 2012, 3, 273–284. [Google Scholar]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NFκB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef]
- Deane, J.A.; Fruman, D.A. Phosphoinositide 3-kinase: Diverse roles in immune cell activation. Annu. Rev. Immunol. 2004, 22, 563–598. [Google Scholar] [CrossRef]
- Park, S.G.; Schulze-Luehrman, J.; Hayden, M.S.; Hashimoto, N.; Ogawa, W; Kasuga, M.; Ghosh, S.P. Phosphoinositide-dependent kinase 1 integrates T cell receptor and CD28 co-receptor signaling to effect NFκB induction and T cell activation. Nat. Immunol. 2009, 10, 158–166. [Google Scholar] [CrossRef]
- Cartwright, N.G.; Kashyap, A.K.; Schaefer, B.C. An active kinase domain is required for retention of PKCθ at the immunological synapse. Mol. Biol. Cell 2011, 22, 3491–3497. [Google Scholar] [CrossRef]
- Carling, D.; Thornton, C.; Woods, A.; Sanders, M.J. AMP-activated protein kinase: New regulation, new roles? Biochem. J. 2012, 445, 11–27. [Google Scholar] [CrossRef]
- Chatila, T.; Wood, N.; Parsonnet, J.; Geha, R.S. Toxic shock syndrome toxin-1 induces inositol phospholipid turnover, protein kinase C translocation, and calcium mobilization in human T cell. J. Immunol. 1988, 140, 1250–1255. [Google Scholar]
- Scholl, P.R.; Trede, N.; Chatila, T.A.; Geha, R.S. Role of protein tyrosine phosphorylation in monokine induction by the staphylococcal superantigen toxic shock syndrome toxin-1. J. Immunol. 1992, 148, 2237–2241. [Google Scholar]
- Bueno, C.; Lemke, C.D.; Criado, G.; Baroja, M.L.; Ferguson, S.S.; Rahman, A.K.; Tsoukas, C.D.; McCormick, J.K.; Madrenas, J. Bacterial superantigens bypass Lck-dependent T cell receptor signaling by activating a Galpha11-dependent, PLC-beta-mediated pathway. Immunity 2006, 25, 67–78. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PBK signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Memmott, R.M.; Dennis, P.A. Akt-dependent and independent mechanisms of mTOR regulation in cancer. Cell. Signal. 2009, 21, 656–664. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Janus kinases in immune cell signaling. Immunol. Rev. 2009, 228, 273–287. [Google Scholar] [CrossRef]
- Hulme, M.A.; Wasserfall, C.H.; Atkinson, M.A.; Brusko, T.M. Centrol role for interleukin-2 in type 1 diabetes. Diabetes 2012, 61, 14–22. [Google Scholar] [CrossRef]
- Thomson, A.W.; Turnquist, H.R.; Raimondi, G. Immunoregulatory functions of mTOR inhibition. Nat. Rev. Immunol. 2009, 9, 324–337. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. cell. Sci. 2009, 122, 3389–3394. [Google Scholar]
- Abraham, R.T.; Wiederrecht, O.J. Immunopharmacology of rapamycin. Ann. Rev. Immunol. 1996, 14, 483–510. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef]
- Yang, O.; Guan, K.L. Expanding mTOR signaling. Cell Res. 2007, 17, 666–681. [Google Scholar] [CrossRef]
- Krakauer, T.; Buckley, M.; Issaq, H.J.; Fox, S.D. Rapamycin protects mice from staphylococcal enterotoxin B-induced toxic shock and blocks cytokine release in vitro and in vivo. Antimicrob. Agents Chemother. 2010, 54, 1125–1131. [Google Scholar] [CrossRef]
- Sims, J.E.; Smith, D.E. The IL-1 family: Regulators of immunity. Nat. Rev. Immunol. 2010, 10, 89–102. [Google Scholar]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NFκB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Mele, T.; Madrenas, J. TLR2 signalling: At the crossroads of commensalism, invasive infections and toxic shock syndrome by Staphylococcus aureus. Int. J. Biochem. Cell. Biol. 2010, 42, 1066–1071. [Google Scholar] [CrossRef]
- Keystone, E.C.; Ware, C.F. Tumor necrosis factor and anti-tumor necrosis factor therapies. J. Rheumatol. 2010, 85, 27–39. [Google Scholar]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef]
- Murray, P.J. The JAK-STAT signaling pathway: Input and output integration. J. Immunol. 2007, 178, 2623. [Google Scholar]
- Ramana, C.V.; Gil, M.P.; Schreiber, R.D.; Stark, G.R. Stat-1-dependent and -independent pathways in IFN-dependent signaling. Trends Immunol. 2002, 23, 96–101. [Google Scholar] [CrossRef]
- Lu, J.; Philpott, D.J.; Saunders, P.R.; Perdue, M.H.; Yang, P.C.; McKay, D.M. Epithelial ion transport and barrier abnormalities evoked by superantigen-activated immune cells are inhibited by interleukin-10 but not interleukin-4. J. Pharmacol. Exp. Ther. 1998, 287, 128–136. [Google Scholar]
- Matthys, P.; Mitera, T.; Heremans, H.; van Damme, J.; Billiau, A. Anti-gamma interferon and anti-interleukin-6 antibodies affect staphylococcal enterotoxin B-induced weight loss, hypoglycemia, and cytokine release in D-galactosamine-sensitized and unsensitized mice. Infect. Immun. 1995, 63, 1158–1164. [Google Scholar]
- Malek, T.R.; Castro, I. Interleukin-2 receptor signaling: At the interface between tolerance and immunity. Immunity 2010, 33, 153–165. [Google Scholar] [CrossRef]
- Liu, D.; Zienkiewicz, J.; DiGiandomenico, A.; Hawiger, J. Suppression of acute lung inflammation by intracellular peptide delivery of a nuclear import inhibitor. Mol. Ther. 2009, 17, 796–802. [Google Scholar] [CrossRef]
- Huzella, L.M.; Buckley, M.J.; Alves, D.A.; Stiles, B.G.; Krakauer, T. Central roles for IL-2 and MCP-1 following intranasal exposure to SEB: A new mouse model. Vet. Res. Sci. 2009, 86, 241–247. [Google Scholar] [CrossRef]
- Khan, A.A.; Priya, S.; Saha, B. IL-2 regulates SEB induced toxic shock syndrome in BALB/c mice. PLoS One 2009, 4, e8473. [Google Scholar]
- Wang, X.; Lupardus, P.; LaPorte, S.L.; Garcia, K.C. Structural biology of shared cytokine receptors. Annu. Rev. Immunol. 2009, 27, 27–60. [Google Scholar]
- Sadik, C.D.; Kim, N.D.; Luster, A.D. Neutrophils cascading their way to inflammation. Trends Immunol. 2011, 32, 452–460. [Google Scholar] [CrossRef]
- Zlotnik, A.; Yoshie, D. The chemokine superfamily revisited. Immunity 2012, 36, 705–716. [Google Scholar] [CrossRef]
- Krakauer, T.; Buckley, M.; Huzella, L.M.; Alves, D. Critical timing, location and duration of glucocorticoid administration rescues mice from superantigen-induced shock and attenuates lung injury. Int. Immunopharmacol. 2009, 9, 1168–1174. [Google Scholar] [CrossRef]
- Darenberg, J.; Soderquist, B.; Normark, B.H.; Norrby-Teglund, A. Differences in potency of intravenous polyspecific immunoglobulin G against streptococcal and staphylococcal superantigens: Implications for therapy of toxic shock syndrome. Clin. Infect. Dis. 2004, 38, 836–842. [Google Scholar] [CrossRef]
- Tilahun, M.E.; Rajagopalan, G.; Shah-Mahoney, N.; Lawlor, R.G.; Tilahun, A.Y.; Xie, C.; Natarajan, K.; Margulies, D.H.; Ratner, D.I.; Osborne, B.A.; Goldsby, R.A. Potent neutralization of staphylococcal enterotoxin B by synergistic action of chimeric antibodies. Infect. Immun. 2010, 78, 2801–2811. [Google Scholar] [CrossRef]
- Larkin, E.A.; Stiles, B.G.; Ulrich, R.G. Inhibition of toxic shock by human monoclonal antibodies against staphylococcal enterotoxin B. PLoS One 2010, 5, e13253. [Google Scholar]
- Varshney, A.K.; Wang, X.; Cook, E.; Dutta, K.; Scharff, M.D.; Goger, M.J.; Fries, B.C. Generation, characterization, and epitope mapping of neutralizing and protective monoclonal antibodies against staphylococcal enterotoxin B-induced lethal shock. J. Biol. Chem. 2011, 286, 9737–9747. [Google Scholar]
- Bavari, S.; Dyas, B.; Ulrich, R.G. Superantigen vaccines: A comparative study of genetically attenuated receptor-binding mutants of staphylococcal enterotoxin A. J. Infect. Dis. 1996, 174, 338–345. [Google Scholar] [CrossRef]
- Grumann, D.; Ruotsalainen, E.; Kolata, J.; Kuusela, P.; Jarvinen, A.; Kontinen, V.P.; Broker, B.M.; Holtfreter, S. Characterization of infecting strains and superantigen-neutralizing antibodies in Staphylococcus aureus bacteremia. Clin. Vaccine Immunol. 2001, 18, 487–493. [Google Scholar]
- Holtfreter, S.; Roschack, K.; Eichler, P.; Eske, K.; Holtfreter, B.; Kohler, C.; Engelmann, S.; Hecker, M.; Greinacher, A.; Broker, B.M. Staphylococcus aureus carriers neutralize superantigens by antibodies specific for their colonizing strain: A potential explanation for their improved prognosis in severe sepsis. J. Infect. Dis. 2006, 193, 1275–1278. [Google Scholar] [CrossRef]
- Arad, G.; Levy, R.; Hillman, D.; Kaempfer, R. Superantigen antagonist protects against lethal shock and defines a new domain for T-cell activation. Nat. Med. 2000, 6, 414–421. [Google Scholar] [CrossRef]
- Visvanathan, K.; Charles, A.; Bannan, J.; Pugach, P.; Kashfi, K.; Zabriskie, J.B. Inhibition of bacterial superantigens by peptides and antibodies. Infect. Immun. 2001, 69, 875–884. [Google Scholar] [CrossRef]
- Rajagopalan, G.; Sen, M.M.; David, C.S. In vitro and in vivo evaluation of staphylococcal superantigen peptide antagonists. Infect. Immun. 2004, 72, 6733–6737. [Google Scholar] [CrossRef]
- Geller-Hong, E.; Möllhoff, M.; Shiflett, P.R.; Gupta, G. Design of chimeric receptor mimics with different TcRVβ isoforms: Type-specific inhibition of superantigen pathogenesis. J. Biol. Chem. 2004, 279, 5676–5684. [Google Scholar]
- Wang, N.; Mattis, D.M.; Sundberg, E.J.; Schlievert, P.M.; Kranz, D.M. A single, engineered protein therapeutic agent neutralizes exotoxins from both Staphylococcus aureus and Streptococcus pyogenes. Clin. Vacc. Immunol. 2010, 17, 1781–1789. [Google Scholar] [CrossRef]
- Saha, B.; Jaklic, B.; Harlan, D.M.; Gray, G.S.; June, C.H.; Abe, R. Toxic shock syndrome toxin-1 induced death is prevented by CTLA4Ig. J. Immunol. 1996, 157, 3869–3875. [Google Scholar]
- DeGrasse, J.A. A single-stranded DNA apttamer that selectively binds to Staphhylococcus aureeus enterotoxin B. PLos One 2012, 7, e33410. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-κb, a key role in inflammatory diseases. J. Clin. Invest. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Liu, D.; Liu, X.Y.; Robinson, D.; Burnett, C.; Jackson, C.; Seele, L.; Veach, R.A.; Downs, S.; Collins, R.D.; Ballard, R.W.; Hawiger, J. Suppression of staphylococcal enterotoxin B-induced toxicity by a nuclear import inhibitor. J. Biol. Chem. 2004, 279, 19239–19246. [Google Scholar]
- Sprung, C.L.; Goodman, S.; Weiss, Y.G. Steriod therapy of septic shock. Crit. Care Clin. 2009, 25, 825–834. [Google Scholar] [CrossRef]
- Krakauer, T. Differential inhibitory effects of interleukin-10, interleukin-4, and dexamethasone on staphylococcal enterotoxin-induced cytokine production and T cell activation. J. Leukoc. Biol. 1995, 57, 450–454. [Google Scholar]
- Krakauer, T.; Buckley, M. Dexamethasone attenuates staphylococcal enterotoxin B-induced hypothermic response and protects mice from superantigen-induced toxic shock. Antimicrob. Agents Chemother. 2006, 50, 391–395. [Google Scholar] [CrossRef]
- Tilahun, A.Y.; Theuer, J.E.; Patel, R.; David, C.S.; Rajagopalan, G. Detrimental effect of the proteasome inhibitor, bortezomib in bacterial superantigen- and lipopolysaccharide-induced systemic inflammation. Mol. Ther. 2010, 18, 1143–1154. [Google Scholar] [CrossRef]
- Krakauer, T. Comparative potency of green tea and red wine polyphenols in attenuating staphylococcal superantigen-induced immune responses. Am. J. Biomed. Sci. 2012. [Google Scholar]
- Watson, J.L.; Vicario, M.; Wang, A.; Moreto, M.; McKay, D.M. Immune cell activation and subsequent epithelial dysfunction by staphylococcal enterotoxin B is attenuated by the green tea polyphenol (−)− epigallocatechin gallate. Cell. Immunol. 2005, 237, 7–16. [Google Scholar] [CrossRef]
- Rieder, S.A.; Nagarkatti, P.; Nagarkatti, M. Identification of multiple anti-inflammatory pathways triggered by resveratrol leading to amelioration of staphylococcal enterotoxin B-induced lung inflammation. Brit. J. Pharmacol. 2012, 167, 1244–1258. [Google Scholar] [CrossRef]
- Kissner, T.L.; Ruthel, G.; Alam, S.; Mann, E.; Ajami, D.; Rebek, M.; Larkin, E.; Fernandez, S.; Ulrich, R.G.; Ping, S.; et al. Therapeutic inhibition of pro-inflammatory signaling and toxicity to Staphylococcal enterotoxin B by a synthetic dimeric BB-loop mimetic of MyD88. PLos One 2012, 7, e40773. [Google Scholar]
- Kissner, T.L.; Moisan, L.; Mann, E.; Alam, S.; Ruthel, G.; Ulrich, R.G.; Rebek, M.; Rebek, J., Jr.; Saikh, K.U. A small molecule that mimics the BB-loop in the Toll interleukin-1 (IL-1) receptor domain of MyD88 attenuates staphylococcal enterotoxin B-induced pro-inflammatory cytokine production and toxicity in mice. J. Biol. Chem. 2011, 286, 31385–31396. [Google Scholar]
- See, R.H.; Chow, A.W. Staphylococcal toxic shock syndrome toxin 1-induced tumor necrosis factor alpha and interleukin-1β secretion by human peripheral blood monocytes and T lymphocytes is differentially suppressed by protein kinase inhibitors. Infect. Immun. 1992, 60, 3456–3459. [Google Scholar]
- Krakauer, T. Suppression of endotoxin- and staphylococcal exotoxin-induced cytokines and chemokines by a phospholipase C inhibitor in human peripheral blood mononuclear cells. Clin. Diagn. Lab. Immunol. 2001, 8, 449–453. [Google Scholar]
- Tschaikowsky, K.J.; Schmidt, J.; Meisner, M. Modulation of mouse endotoxin shock by inhibition of phosphatidylcholine-specific phospholipase C. J. Pharmacol. Exp. Therap. 1999, 285, 800–804. [Google Scholar]
- Jo, D.; Liu, D.; Yao, S.; Collins, R.D.; Hawiger, J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat. Med. 2005, 11, 892–898. [Google Scholar] [CrossRef]
- Komisar, J.L.; Weng, C.F.; Oyejide, A.; Hunt, R.E.; Briscoe, C.; Tseng, J. Cellular and cytokine responses in the circulation and tissue reactionsin the lung of rhesus monkeys (Macaca mulatta) pretreated with cyclosporine A and challenged with staphylococcal enterotoxin B. Toxicol. Pathol. 2001, 29, 369. [Google Scholar] [CrossRef]
- Tilahun, A.Y.; Karau, M.J.; Clark, C.R.; Patel, R.; Rajagopalan, G. The impact of tacrolimus on the immunopathogenesis of with staphylococcal enterotoxin-induced systemic inflammatory response syndrome and pneumonia. Microbes Infect. 2012, 14, 528–536. [Google Scholar] [CrossRef]
- Krakauer, T.; Buckley, B. Intranasal rapamycin rescues mice from staphylococcal enterotoxin-induced shock. Toxins 2012, 4, 718–728. [Google Scholar] [CrossRef]
- Krakauer, T.; Buckley, M. The potency of anti-oxidants in attenuating superantigen-induced proinflammatory cytokines correlates with inactivation of NFκB. Immunopharmacol. Immunotoxic. 2008, 30, 163–179. [Google Scholar] [CrossRef]
- Bean, A.G.; Freiberg, R.A.; Andrade, S.; Menon, S.; Zlotnik, A. Interleukin 10 protects mice against staphylococcal enterotoxin B-induced lethal shock. Infect. Immun. 1993, 61, 4937–4939. [Google Scholar]
- Florquin, S.; Amraoui, Z.; Abramowicz, D.; Goldman, M. Systemic release and protective role of IL-10 in staphylococcal enterotoxin B-induced shock in mice. J. Immunol. 1994, 153, 2618–2623. [Google Scholar]
- LeClaire, R.D.; Kell, W.; Bavari, S.; Smith, T.; Hunt, R.E. Protective effects of niacinamide in staphylococcal enterotoxin B induced toxicity. Toxicology 1996, 107, 69–81. [Google Scholar] [CrossRef]
- Krakauer, T.; Buckley, M. Doxycycline is anti-inflammatory and inhibits staphylococcal exotoxin-induced cytokines and chemokines. Antimicrob. Agents Chemother. 2003, 47, 3630–3633. [Google Scholar] [CrossRef]
- Krakauer, T.; Stiles, B.G. Pentoxifylline inhibits staphylococcal superantigen induced toxic shock and cytokine release. Clin. Diagn. Lab. Immunol. 1999, 6, 594–598. [Google Scholar]
- Krakauer, T. Caspase inhibitors attenuate superantigen-induced inflammatory cytokines, chemokines and T-cell proliferation. Clin. Diagn. Lab. Immunol. 2004, 11, 621–624. [Google Scholar]
- Takei, Y.; Kunikata, T.; Aga, M.; Inoue, S.; Ushio, S.; Iwaki, K.; Ikeda, M.; Kurimoto, M. Tryptanthrin inhibits interferon-γ production by Peyer’s patch lymphocytes derived from mice that had been orally administered staphylococcal enterotoxin. Biol. Pharm. Bull. 2003, 26, 365–367. [Google Scholar] [CrossRef]
- Krakauer, T.; Li, B.; Young, H. The flavonoid baicalin inhibits staphylococcal superantigen-induced inflammatory cytokine and chemokine production in human peripheral blood mononuclear cells. FEBS Lett. 2001, 500, 50–55. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Krakauer, T. PI3K/Akt/mTOR, a Pathway Less Recognized for Staphylococcal Superantigen-Induced Toxicity. Toxins 2012, 4, 1343-1366. https://doi.org/10.3390/toxins4111343
Krakauer T. PI3K/Akt/mTOR, a Pathway Less Recognized for Staphylococcal Superantigen-Induced Toxicity. Toxins. 2012; 4(11):1343-1366. https://doi.org/10.3390/toxins4111343
Chicago/Turabian StyleKrakauer, Teresa. 2012. "PI3K/Akt/mTOR, a Pathway Less Recognized for Staphylococcal Superantigen-Induced Toxicity" Toxins 4, no. 11: 1343-1366. https://doi.org/10.3390/toxins4111343