Gi/o Protein-Dependent and -Independent Actions of Pertussis Toxin (PTX)

1
Department of Pharmacology, Faculty of Pharmacy, Mahidol University, 447 Sri-Ayudhaya, Rajathevi, Bangkok 10400, Thailand
2
Department of Pharmacology and Toxicology, Graduate School of Pharmaceutical Sciences, Kyushu University, 3-1-1 Maidashi, Higashi-ku, Fukuoka 812-8582, Japan
*
Author to whom correspondence should be addressed.
Toxins 2011, 3(7), 884-899; https://doi.org/10.3390/toxins3070884
Submission received: 13 April 2011
/
Revised: 14 June 2011
/
Accepted: 16 June 2011
/
Published: 15 July 2011
(This article belongs to the Special Issue Novel Properties of Well-Characterized Toxins)
Abstract
: Pertussis toxin (PTX) is a typical A-B toxin. The A-protomer (S1 subunit) exhibits ADP-ribosyltransferase activity. The B-oligomer consists of four subunits (S2 to S5) and binds extracellular molecules that allow the toxin to enter the cells. The A-protomer ADP-ribosylates the α subunits of heterotrimeric Gi/o proteins, resulting in the receptors being uncoupled from the Gi/o proteins. The B-oligomer binds proteins expressed on the cell surface, such as Toll-like receptor 4, and activates an intracellular signal transduction cascade. Thus, PTX modifies cellular responses by at least two different signaling pathways; ADP-ribosylation of the Gαi/o proteins by the A-protomer (Gi/o protein-dependent action) and the interaction of the B-oligomer with cell surface proteins (Gi/o protein-independent action).
Abbreviations
| AC | adenylyl cyclase |
| CHO | Chinese hamster ovary |
| CaMKII | calmodulin kinase II |
| CREB | cAMP response element-binding |
| 8-CPT | 8-(4-chlorophenylthio)-2'-O-methyladenosine-3',5'-cyclic monophosphate |
| eEF2 | eukaryotic elongation factor |
| Epac | exchange protein directly activated by cAMP |
| ERK | extracellular signal-regulated kinase |
| GPCRs | G protein-coupled receptors |
| Gi/oPCRs | Gi/o protein-coupled receptors |
| GPIb | glycoprotein Ib |
| MAPK | mitogen-activated protein kinase |
| NAD+ | nicotinamide adenine dinucleotide |
| PKA | cAMP-dependent protein kinase |
| PTX | Pertussis toxin |
| TLR4 | Toll-like receptor 4 |
| HUVECs | human umbilical vein endothelial cells |
| SAA | serum amyloid A |
| SPC | sphingosylphosphorylcholine |
| TCR | T-cell receptor |
1. Introduction
Bacterial pathogens utilize their toxins to modify or kill host cells. The bacterial ADP-ribosylating toxins are a large family of dangerous and lethal toxins that include pertussis toxin (PTX), cholera toxin, diphtheria toxin, and pseudomonas exotoxin A [1,2]. These toxins are found in a diverse range of bacterial pathogens and are the cytotoxic agents that cause severe infectious diseases including whooping cough, cholera, and diphtheria (Table 1).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ADP-Ribosylating Toxin | Bacterium | Target | Pathological Effect |
|---|---|---|---|
| Pertussis toxin | Bordetella pertussis | Cysteine residue of Gαi subfamily (Gαi, Gαo, and Gαt) except Gαz | Gαi protein-receptor coupling is inhibited, and its signal transduction is blocked. |
| Cholera toxin | Vibrio cholerae | Arginine residue of Gαs subfamily (Gαs and Gαolf) | As GTPase activity of the stimulatory Gαs is inhibited, Gαs protein is permanently activated. |
| Heat-labile enterotoxin | Escherichia coli | Arginine residue of Gαs subfamily (Gαs and Gαolf) | As GTPase activity of stimulatory Gαs is inhibited, the Gαs protein is permanently activated. |
| Diphtheria toxin | Corynebacterium diphtheirae | Diphthamide of eEF2 | Protein synthesis is blocked. |
| Exotoxin A | Pseudomonas aeruginosa | Diphthamide of eEF2 | Protein synthesis is blocked. |
eEF2 = eukaryotic elongation factor 2.
Pertussis toxin (PTX) is the ADP-ribosylating toxin produced by the whooping cough causing bacterium Bordetella pertussis [3]. PTX catalyzes the ADP-ribosylation of the α subunits of the heterotrimeric Gi/o protein family (Gαi, Gαo, and Gαt; except Gαz), thereby preventing the G proteins from interacting with their cognate G protein-coupled receptors (GPCRs) [4]. ADP-ribosylation of the α subunit of heterotrimeric Gi/o proteins (Gαi/o) locks the α subunits into an inactive state (GDP-bound form), thus it is unable to inhibit adenylyl cyclase (AC). This modification of the Gαi/o proteins results in the enhanced accumulation of cAMP, which is one of the mechanisms by which PTX induces the various pathological effects in host cells.
PTX is composed of an A-protomer and B-oligomer. The A-protomer exerts ADP-ribosyltransferase activity on the Gαi/o proteins, leading to inhibition of receptor-G protein coupling [5,6]. The B-oligomer of PTX recognizes and binds carbohydrate-containing receptors that deliver A-protomer into the cytosol [7]. Although many of the effects of PTX are dependent on ADP-ribosylation of the Gαi/o proteins, Gi/o protein-independent effects of PTX have also been reported. For example, interaction of the B-oligomer with receptors on certain eukaryotic cells can mediate biological effects that are independent of the catalytic activity of A-protomer, including enhancement of immune responses [8,9,10], an increase in adenosine A1 receptor density [11], and the activation of tyrosine kinase, mitogen-activated protein kinase (MAPK), and NF-κB [12,13,14]. Moreover, we recently demonstrated a novel function of PTX that induces up-regulation of angiotensin II type 1 receptor independently of ADP-ribosylation of Gαi/o[15]. Thus, PTX can mediate biological effects through at least two signaling pathways; (1) Gi/o protein-dependent pathway through ADP ribosylation of the Gαi/o proteins and (2) Gi/o protein-independent pathway by the binding of B-oligomer to cell surface proteins such as Toll-like receptor 4 (TLR4) [15] but not GPCRs.
In this paper, we review our current understanding of the Gi/o protein-dependent and Gi/o protein-independent pharmacological effects of PTX.
2. Structure of Pertussis Toxin
The PTX molecule is a complex ADP-ribosylating toxin composed of five different subunits: S1, S2, S3, S4 and S5, presented in a ratio of 1:1:1:2:1 and arranged in the A-B architecture [16]. The A-protomer consists of a single S1 subunit that is responsible for ADP-ribosyltransferase activity [17], while the B-oligomer comprises S2, S3, S5, and two S4 subunits [18]. The A-protomer catalyzes the ADP-ribosylation of a cysteine residue in the α subunit of the heterotrimeric Gi/o protein subfamily, whereas the B-oligomer is responsible for binding to specific cell surface receptors and delivering the A-protomer into recipient cells [7].
The crystal structure of PTX [18] revealed that the B-oligomer is composed of 5 noncovalently linked subunits which are organized as a triangular platform around a single catalytic S1 subunit that is on the top of the platform (Figure 1).
Exposing PTX to urea results in dissociation of the A-protomer (S1 subunit) from the B-oligomer and breakdown of the B-oligomer into three moieties: S2-S4 dimer, S3-S4 dimer, and S5 monomer [16]. These results suggest that the B-oligomer consists of two dimers, the S2-S4 and S3-S4 dimers, which are held together by the S5 subunit.
Figure 1.
Pertussis toxin (PTX) structural organization. PTX contains five different subunits that are arranged in a typical A-B structure. The A-protomer contains an enzymatically active S1 subunit that is on the top of B-oligomer. The B-oligomer is composed of two dimers, S2-S4 and S3-S4 dimers, which are held together by the S5 subunit.
Figure 1.
Pertussis toxin (PTX) structural organization. PTX contains five different subunits that are arranged in a typical A-B structure. The A-protomer contains an enzymatically active S1 subunit that is on the top of B-oligomer. The B-oligomer is composed of two dimers, S2-S4 and S3-S4 dimers, which are held together by the S5 subunit.
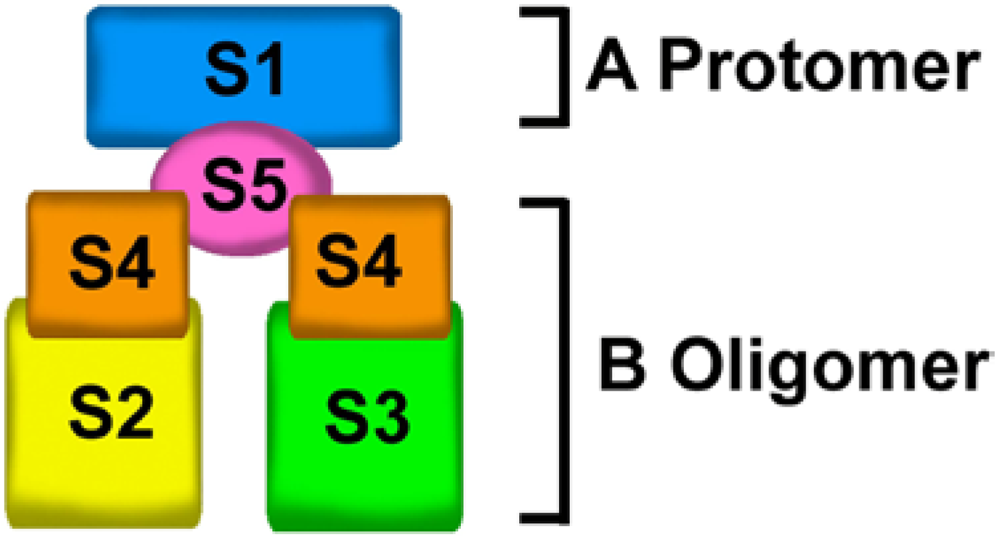
The S2 and S3 subunits of PTX share approximately 75% nucleotide and 70% amino acid homology [19,20]. Despite more than 75% sequence homology, structural and functional studies using site-directed mutagenesis of the S2 and S3 subunits have shown that the S2 subunit mediates binding to nonsialylated glycans, whereas S3 selectively binds sialylated oligosaccharides [21,22].
The S1 subunit of PTX contains regions of sequences homology to the catalytic portion of other ADP-ribosylating toxins such as the cholera toxin, enterotoxin, diphtheria toxin, and exotoxin A [1]. The carboxyl terminus of the S1 subunit is composed of 235 amino acid residues [23]. Previous studies have indicated that the carboxyl terminus is important in the interaction of the S1 subunit with the B-oligomer [24,25]. Residues 195 to 204 are required for optimal ADP-ribosylation of the α subunit of heterotrimeric Gi/o proteins. Residues 205 to 219 are linked to the catalytic region of S1 and are the B-oligomer-binding site of S1 subunit. Residues 220 to 235 are hydrophobic and are important for interaction of the S1 subunit with the B-oligomer [23].
The A protomer contains two cysteine residues at positions 41 and 201, which form disulfide bonds in the native PTX holotoxin. [26]. Reduction of this disulfide bond by dithiothreitol leads to a marked stimulation of S1 catalytic activity, which coincides with a release of the S1 subunit from B-oligomer [27]. Moreover, the presence of ATP leads to the dissociation of the S1 subunit from B-oligomer [28]. This dissociation makes the disulfide bond of the S1 subunit susceptible to cleavage by intracellular reducing compounds.
3. ADP-Ribosylation Mechanism of PTX
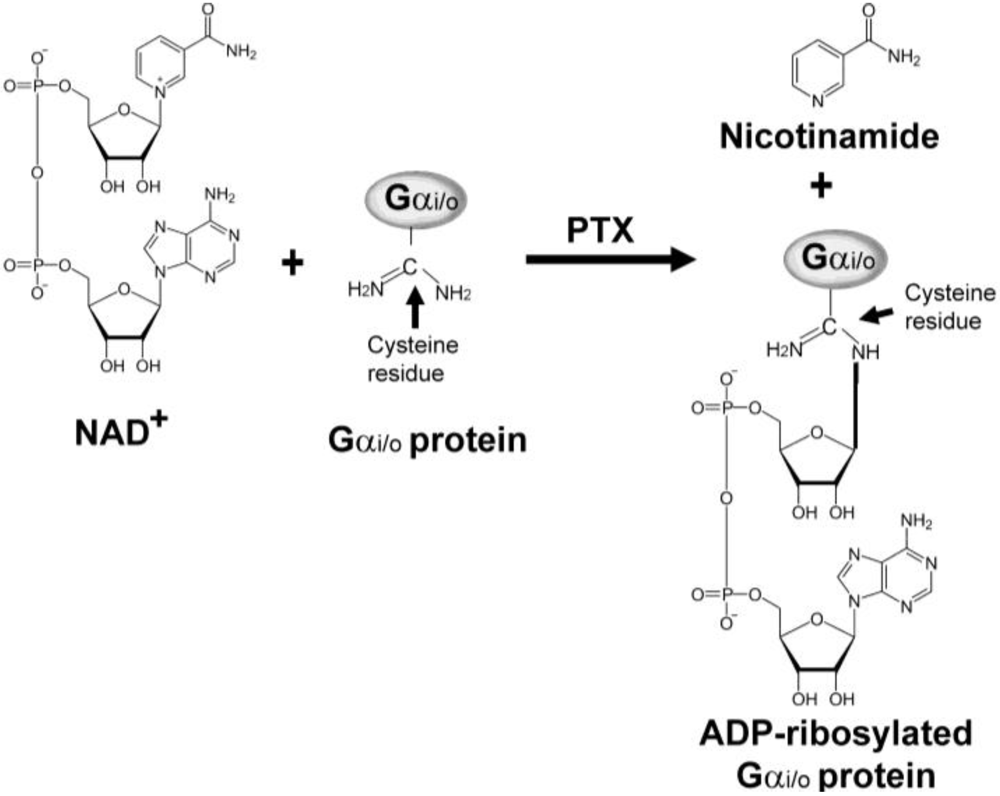
Following attachment of PTX to host cells, S2 and S3 subunits of B-oligomer bind to the exposed sugar residues of glycolipid (gangliosides) on the plasma membrane of host cells. The A-protomer (S1 subunit) penetrates through the membrane and is released from B-oligomer into the cytoplasm. However, the exact molecular events associated with the entry of PTX into host cells are not fully understood. Once inside the cell the A-protomer ribosylates specific target proteins such as the α subunit of heterotrimeric Gi/o proteins (Figure 2) through its ADP-ribosyltransferase activity. ADP-ribosylation is also responsible for the actions of other bacterial ADP-ribosylating toxins, such as cholera toxin, diphtheria toxin, and exotoxin A.
PTX catalyzes the cleavage of the C-N bond between carbon atom of ribose and nitrogen atom of nicotinamide and transfer the ADP-ribosyl moiety from nicotinamide adenine dinucleotide (NAD+) to an acceptor molecule on the target protein (Figure 2). The target proteins for other ribosylating toxins include the eukaryotic elongation factor (eEF2) for diphtheria toxin [29] and arginine residue of the Gαs protein for cholera toxin, which, like the Gαi/o-protein target for PTX, are involved in cell signal transduction [30]. In the case of PTX, ADP-ribosylation of the Gαi/o-proteins prevents the coupling to their cognate GPCRs and consequently disrupts the signal transduction cascade [31,32]. Besides their function as ADP-ribosyl transferases, the ADP-ribosylating toxins also have NAD+ glycohydrolase activity in the absence of an acceptor molecule [17]. However, this activity does not seem to contribute to any effects of PTX in the cell.
The amino acid ADP-ribosylated by PTX is cysteine, which is located four residues from the carboxyl terminus of the α subunits of the Gi/o proteins [33]. The uncoupling of GPCR from the Gαi/o proteins results in disruption of the communication between receptor and the effector molecule AC. Thus, the Gαi/o protein is inactivated and cannot perform its normal function to inhibit AC. In this way it prevents the signal from Gi/oPCRs. Thus, the conversion of ATP to cAMP cannot be halted, resulting in excess intracellular cAMP level and the subsequent disruption of many cellular processes as shown in Figure 3. With the exception of Gαz, all members of the Gαi/o protein family are substrates for PTX.
Figure 2.
Schematic diagram of the ADP-ribosylation of α subunit of heterotrimeric Gi/o protein by pertussis toxin (PTX). PTX catalyzes the cleavage of the C-N bond between a carbon atom of ribose and a nitrogen atom of nicotinamide, and transfers the ADP-ribosyl moiety to an acceptor molecule.
Figure 2.
Schematic diagram of the ADP-ribosylation of α subunit of heterotrimeric Gi/o protein by pertussis toxin (PTX). PTX catalyzes the cleavage of the C-N bond between a carbon atom of ribose and a nitrogen atom of nicotinamide, and transfers the ADP-ribosyl moiety to an acceptor molecule.
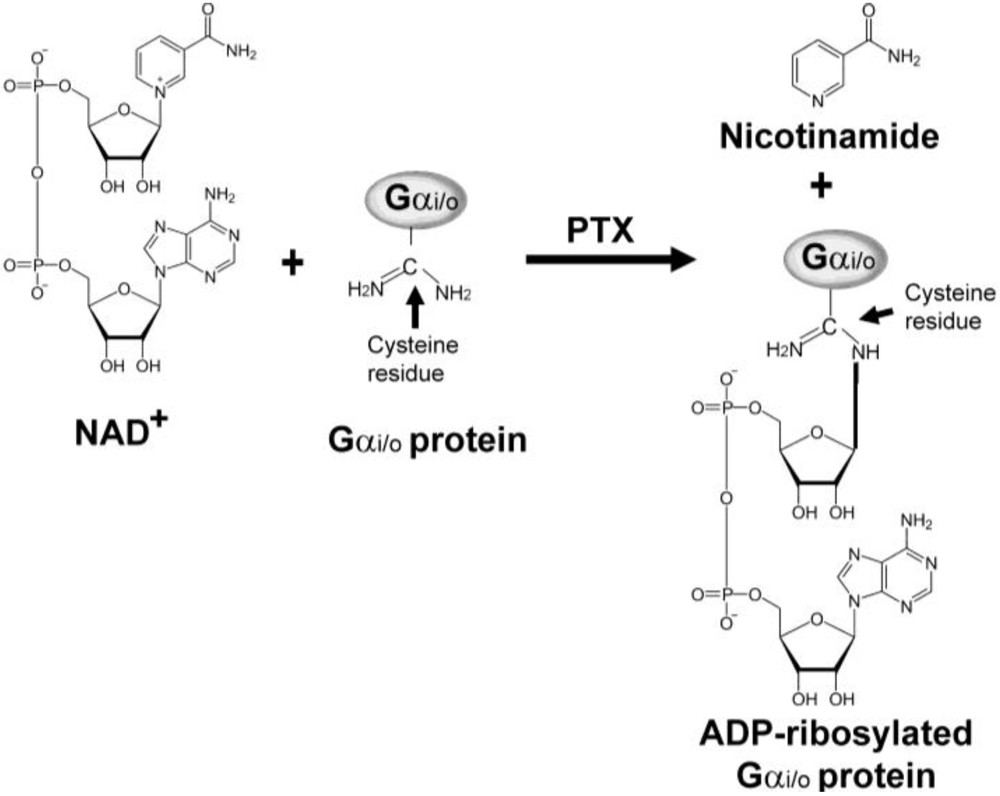
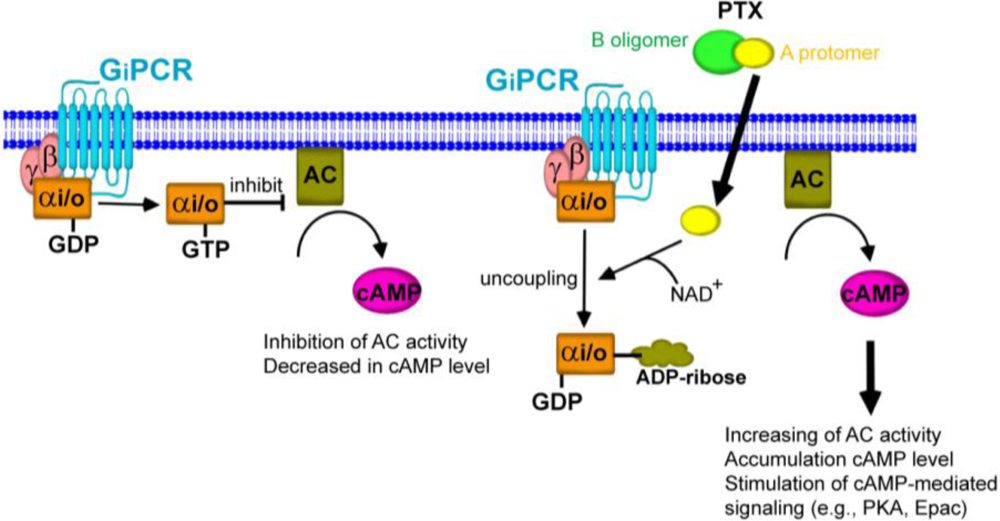
Figure 3.
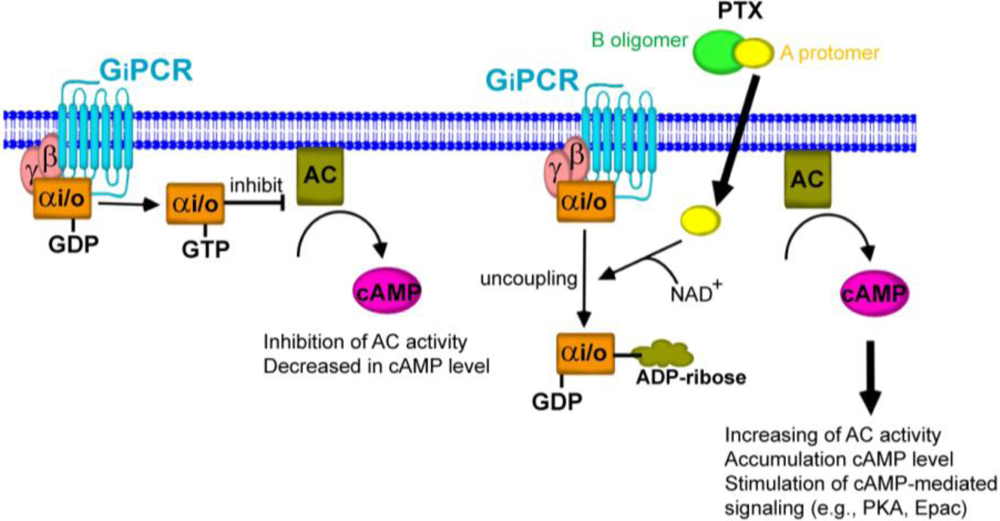
Uncoupling of Gαi/o proteins from their cognate G protein-coupled receptor (GPCR). Activation of GPCRs leads to dissociation of heterotrimeric G protein complex into Gαi/o and βγ subunit. The exchange of GTP from GDP results in activation of the inhibitory G protein (Gαi/o), thereby inhibiting adenylyl cyclase (AC) activity. When the A-protomer of PTX penetrates into the host cells, the Gαi/o is ADP-ribosylated at cysteine residue resulting in inactivation of Gαi/o. The inhibitory effect of Gαi/o on AC activity results in the elevation of intracellular cAMP levels, leading to activation of the cAMP-mediated signaling pathway. This enhanced pathway by PTX is recognized as the Gi/o protein-dependent pathway.
Figure 3.
Uncoupling of Gαi/o proteins from their cognate G protein-coupled receptor (GPCR). Activation of GPCRs leads to dissociation of heterotrimeric G protein complex into Gαi/o and βγ subunit. The exchange of GTP from GDP results in activation of the inhibitory G protein (Gαi/o), thereby inhibiting adenylyl cyclase (AC) activity. When the A-protomer of PTX penetrates into the host cells, the Gαi/o is ADP-ribosylated at cysteine residue resulting in inactivation of Gαi/o. The inhibitory effect of Gαi/o on AC activity results in the elevation of intracellular cAMP levels, leading to activation of the cAMP-mediated signaling pathway. This enhanced pathway by PTX is recognized as the Gi/o protein-dependent pathway.
4. Gi/o Protein-Dependent Actions of PTX
The A-protomer (S1 subunit) of PTX catalyzes the ADP ribosylation of the membrane-bound regulatory Gi/o proteins resulting in inactivation, preventing them from inhibiting AC. As the inhibition of cAMP production by inhibitory GPCRs is only abolished by PTX, PTX treatment results in the enhancement of stimulatory GPCR-induced cAMP production. It is possible that the biological effects of PTX are a result of the disruption of the cAMP-mediated signaling pathway. As the Gi/o proteins are expressed in various tissues, the effects of PTX are observed in almost all cells. Some of the affected cells include the β-cells of pancreatic islets (hence the former name of PTX is islet-activating protein), adipocytes, macrophages, lymphocytes, and other cells that cause paroxysms and neurological disturbance. The ADP-ribosylation of the Gi/o proteins by PTX is irreversible. As a result, restoration of Gi/o function in the cells is dependent on Gi/o protein replacement of ADP-ribosylated Gi/o proteins with new proteins. Unlike cholera toxin, which acts to inhibit GTPase activity of the Gs protein, PTX does not affect basal GTPase activity of the Gi/o proteins, only receptor-stimulated GTPase.
The biological effects of PTX that result from ADP-ribosylation of the Gαi/o proteins are very diverse, some of which can be attributed to the ADP-ribosylation of the α subunits of the heterotrimeric Gi/o protein family, as shown in Table 2.
| α Subunit | Amino Acid | Expression [34,35] | Effect on Effectors | Toxin (Site of Action) |
|---|---|---|---|---|
| αo | 354 | Heart, neurons, neuroendocrine cells | Inhibition of AC activity [36] | PTX at cysteine 351 [1,37,38] |
| Inhibition of Ca2+ channel [39] | ||||
| Activation of K+ channel [40] | ||||
| αi1-αi3 | 354 | Neurons and ubiquitous | Inhibition of AC activity [41] | PTX at cysteine 351 [1,37,38] |
| Inhibition of Ca2+ channel [39,42] | ||||
| Activation of K+ channel [40,43] | ||||
| αz | 355 | Platelets | Inhibition of AC activity [44] | -(cyteine modified by PTX does not present) [46] |
| Inhibition of Ca2+ channel [45] | ||||
| Activation of K+ channel [40] | ||||
| αt | 350 | Rod/cone outer segments | Activation of cGMP-PDE [47] | PTX at cysteine 351 [37,38] |
| CTX at arginine147 [37] | ||||
| αgust | 353 | Taste buds; sweet and/or bitter | Activation of cGMP-PDE [48] | PTX at cysteine 350 [37,38] |
AC: adenylyl cyclase; PDE: phosphodiesterase.
Pancreatic cells show increased insulin secretion upon treatment with PTX [37]. Moreover, PTX inhibits insulin-stimulated autophosphorylation of insulin receptor by inactivation of insulin receptor kinase. Treatment of cells with the catalytically inactive B-oligomer had no effect on insulin receptor kinase activity [49]. These results suggest that PTX can modulate insulin receptor signaling at receptor level. The lipolytic action of PTX has been used as a sensitive assay for the action of toxin, and is caused by PTX-catalyzed ADP ribosylation of the Gαi proteins in adipocytes. Adenosine is produced in vitro as one of the ATP degradation products during preparation of adipocytes from fat pads by the collagenase digestion method, and adenosine inhibits AC via the Gαi[2,50]. As lipolysis is enhanced by the increase in intracellular cAMP, inhibition of adenosine receptor-Gi/o coupling by PTX results in enhanced cAMP accumulation, leading to increased lipolysis.
In vivo administration of PTX induces lymphocytosis, a hallmark of systemic pertussis in children. It also causes hyperinsulinemia and hypoglycemia, as well as modification of histamine sensitization [2]. In addition, PTX, but not a noncatalytic mutant of PTX, inhibits neutrophil migration to rat peritoneal cavities in response to lipopolysaccharide [51]. The increase in skin vascular permeability induced by leukotriene B4 is also inhibited by PTX only, not a catalytically inactive mutant. These results suggest that PTX inhibits neutrophil migration and vascular permeability in vivo and these effects are dependent on the ADP-ribosyltransferase activity of the A-protomer.
Ligand binding to inhibitory GPCRs leads to activation of the Gi/o proteins and then causes the dissociation of heterotrimeric Gi/o proteins into an activated αi/o subunit and a βγ subunit. The activated Gαi/o subunits (GTP-bound form) inhibit AC activity, resulting in the decrease of intracellular cAMP level [30]. This attenuates the cAMP-mediated signaling pathway. Treatment of NG108-15 cells (neuroblastoma-glioma hybrid cells) with PTX attenuates receptor-mediated inhibition of AC, resulting in inhibition of the opiate-stimulated GTPase activity. Moreover, this inhibition effect by PTX requires NAD+, emphasizing that this effect of PTX is mediated by ADP- ribosylation of the Gαi/o proteins. The Gαi/o proteins are ADP-ribosylated by A-protomer, inhibiting the GDP-GTP exchange of the Gi/o proteins by GPCRs. The α subunit is locked in the inactive GDP-bound state resulting in the enhanced increase in cAMP level by stimulatory GPCRs. Thus, Gi/o protein-dependent effects of PTX are mainly mediated by cAMP action. Cyclic AMP is formed from ATP by the action of AC and degraded to AMP by cAMP phosphodiesterase [52]. Most effects of cAMP in animal cells are mediated by the action of cAMP-dependent protein kinase (PKA). The inactive form of PKA is a tetramer consisting of two catalytic and two regulatory subunits. The binding of cAMP to the regulatory subunits leads to their dissociation from the catalytic subunits. The free catalytic subunits are then active and able to phosphorylate serine and threonine residues of their target proteins. The free catalytic subunit of PKA also translocates to the nucleus and phosphorylates the transcriptional factor CREB (cAMP response element-binding), leading to the activation of cAMP-inducible genes [53]. Such regulation of gene expression by cAMP plays important roles in controlling the proliferation, survival, and differentiation of a wide variety of animal cells. Previous studies have identified a family of molecules known as Epac (exchange protein directly activated by cAMP) that directly bind cAMP and exhibit guanine nucleotide exchange factor activity toward Rap1 [54,55]. The binding of cAMP or the selective cAMP analogue 8-CPT (8-(4-chlorophenylthio)-2'-O-methyladenosine-3',5'-cyclic monophosphate) to Epac leads to activation of calmodulin kinase II (CaMKII)-dependent signaling pathways including the release of Ca2+ from sarcoplasmic reticulum [56] and the increased phosphorylation of phospholamban [57]. However, whether the increase in cAMP level by PTX-catalyzed ADP-ribosylation of the Gαi/o proteins activates this Epac-mediated CaMKII signaling pathway remains to be elucidated.
5. Gi/o Protein-Independent Actions of PTX
ADP-ribosylation of the Gαi/o proteins does not account for all responses by the interactions of PTX with host cells. PTX is reported to be a T-cell mitogen [7]. This action can be reproduced by B-oligomer alone suggesting that B-oligomer itself may induce the mitogenic action independent of A-protomer-induced ADP-ribosylation of the Gi/o proteins. Several other Gi/o protein independent actions of PTX have been reported, such as enhancement of immune responses [8,9,10], increase in adenosine A1 receptor density [11], and activation of tyrosine kinase, MAPK, and NF-κB [12,13,14]. Recently, we demonstrated that the B-oligomer of PTX induces Rac activation through a pathway independent of ADP-ribosylation of the Gαi/o proteins [15]. PTX increases IL-1β induction through sequential activation of TLR4, Rac, NADPH oxidase, and NF-κB, which leads to up-regulation of angiotensin II type 1 receptor. Thus, PTX binds to two binding sites; one is TLR4 which activates Rac, and another is the binding site that is required for the entry of PTX into the cells to ADP-ribosylate the Gαi/o[15].
Interestingly, both the S1 mutant of PTX and the B-oligomer completely reproduced several effects of native PTX (holotoxin) action. For example, PTX-induced extracellular signal-regulated kinase (ERK) activation of endothelial cell is entirely independent of ADP-ribosylation of the Gαi/o[58]. However, inactivation of Gαi proteins by PTX inhibits serum amyloid A (SAA)-mediated ERK activation in human umbilical vein endothelial cells (HUVECs) [59]. Preincubation of HUVECs in PTX (100 ng/mL) prior to sphingosylphosphorylcholine (SPC) stimulation markedly reduces the levels of phosphorylated MAPKs including ERK, indicating that Gαi proteins have a role in SPC-induced ERK activity [60]. Thus it is not clear what causes the difference in activation versus inhibition of ERK activity. One possibility could be derived from the differential cellular mechanisms which regulate ERK activity.
Moreover, the B-oligomer alone was as effective as PTX in inducing glucose oxidation in rat adipocytes [7]. It was also reported that the binding of the B-oligomer to eukaryotic cells can alter cellular function independently of ADP-ribosylation. For example, PTX has been shown to bind to glycoprotein Ib (GPIb) on human platelets, leading to subsequent platelet aggregation [61]. The purified B-oligomer also induces mitogenic stimulation of human T cells [62,63], enhances glucose oxidation in adipocytes [64], induces dendritic cell maturation in a TLR4-dependent manner [14], and associates with T-cell receptor complex to initiate signal transduction in T-lymphocytes [65].
Upon prolonged incubation (at least 1 or 2 h) with PTX, the A-protomer is internalized by certain cells and subsequently activated by cleavage of the disulfide bond. The A-protomer then catalyzes the transfer of an ADP-ribose moiety from endogenous NAD+ to a carboxyl terminal cysteine residue in the Gαi/o proteins (Figure 2). A previous study reported that PTX completely ADP-ribosylated the Gαi/o proteins at 10 ng/mL within 3 h in Chinese hamster ovary (CHO) cells [66]. ADP-ribosylation of the Gi/o proteins disrupts their interaction with various inhibitory GPCRs, leading to blockade of certain transmembrane signaling process and eventually cellular intoxication [67]. In addition to its delayed inhibitory effect on the Gi/o proteins, PTX has also been shown to elicit rapid responses (in minutes) in a variety of cell types [64], which may have profound pathological effects as important as its ADP-ribosylation activity. PTX was shown to bind to human platelet GPIb, inducing rapid platelet aggregation and an increase of intracellular Ca2+ level [61,68]. Recent studies also reported that the B-oligomer interacts with extracellular receptors, such as the T-cell receptor (TCR), and activates an intracellular signal transduction pathway by inducing and interacting with the T-cell receptor complex [65,69]. In contrast to the rapid and transient effects caused by B-oligomer, the A-protomer (catalytic S1 subunit) must penetrate into the cytoplasm of host cells to ADP-ribosylate and inactivate the Gαi/o proteins. This process can usually take hours, but permanently modifies G proteins [65,70]. It indicates that PTX-induced responses in T-cells can be divided into two phases: short-term by the B-oligomer, and long-term by the catalytic A-protomer. From these data, it has been concluded that Gi/o protein-dependent effects of PTX through ADP-ribosylation occur more slowly than Gi/o protein-independent effects of PTX through the B-oligomer (Table 3).
PTX detoxified by formaldehyde treatment or genetic modification maintains its protein structure and the immunological properties, but not its enzymatic activity [71,72]. This detoxified PTX is able to activate TLR4 in monocyte derived dendritic cells, a property previously reported for fully active PTX and its B-oligomer [14]. A low dose of detoxified PTX efficiently triggers TLR4 signaling, while a high dose is necessary to activate both TLR4 and TLR2 [73]. This is the first report showing that detoxified PTX also triggers TLR2-mediated signal transduction pathway. As TLR4 plays an essential role in expansion of Th1/Th17 immunity, detoxified PTX may work as an alternative adjuvant to promote Th1/Th17 responses.
Interestingly, a previous study showed that while both PTX and the B-oligomer induce dendritic cell maturation, the induction of cytokines that are produced differ [14]. However, the effect of both PTX and its B-oligomer on dendritic cell maturation was dependent on TLR4 [74]. It is possible that the differential cytokine production of dendritic cells by PTX and the B-oligomer are due to the differences in their utilization of TLR4-mediated intracellular signaling pathways, which may be accounted for by both ADP-ribosylation-dependent and -independent responses.
The toxic effects mediated by catalytic activity of the native PTX (holotoxin) occurs at very low toxin concentrations and appears to alter the function of tissues and organs very distal from the site of bacterial growth. In contrast, effects of PTX mediated by interaction of the B-oligomer with many cell surface proteins require higher doses of the toxin [65]. For example, the B-oligomer of PTX caused concentration-dependent platelet activation, as determined by increasing intracellular Ca2+ concentration and dense granule secretion [61]. In addition to its role in delivering the S1 subunit into the cells, the B-oligomer elicits biological effects such as an increase in intracellular Ca2+, activation of T-lymphocytes [62,75] and platelet aggregation [68]. These B-oligomer-mediated effects may be elicited by the binding of the B-oligomer to a specific receptor and require significantly higher concentrations of toxin than are necessary for the ADP-ribosyltransferase activity of the A-protomer.
Moreover, it appears that most cells would be susceptible to PTX as a result of ADP-ribosylation of the Gαi/o proteins and that PTX holotoxin-mediated effects could occur at many sites in host cells [72]. Recent findings demonstrate that the Gi/o protein-dependent action of PTX (via ADP-ribosylation of Gαi/o) exhibits the differences in signaling pathway when compare to that of Gi/o protein-independent action of PTX (via B-oligomer) as summarized in Table 3.
Table 3.
The differences in characteristics of Gi/o protein-dependent and -independent effects of PTX.
| Gi/o Protein-Dependent Effects of PTX | Gi/o Protein-Independent Effects of PTX | |
|---|---|---|
| Subunit | A-protomer (S1 subunit) | B-oligomer (S2-S3 dimer, S2-S5 dimer, and S5 monomer) |
| Onset of action | Slow | Rapid |
| Concentration of toxin to induce the effects | Low | High |
| Biological effects | Enhance insulin secretion [49] | Induce dendritic cell maturation [14] |
| Inhibit lymphocyte and neutrophil migration [52,76] | Inhibit growth cone guidance [78] | |
| Inhibit enkephalin stimulation of GTPase [77] | Induce myelomonocytic cell adhesion [79] | |
| Inhibit autophosphorylation and activation of insulin receptor kinase [50] | Induce ERK1/2 activation in endothelial cell [59] | |
| Activate platelet aggregation [62,69] | ||
| Activate T lymphocyte [66,80] | ||
| Induce Th1/Th17 immune response through MAPK and IL-10 [74] | ||
| Activate tyrosine kinase signaling [12] | ||
| Inhibit Tat-induced TGF- β production [81] | ||
| Inhibit HIV type 1 replication [82] |
6. Conclusions
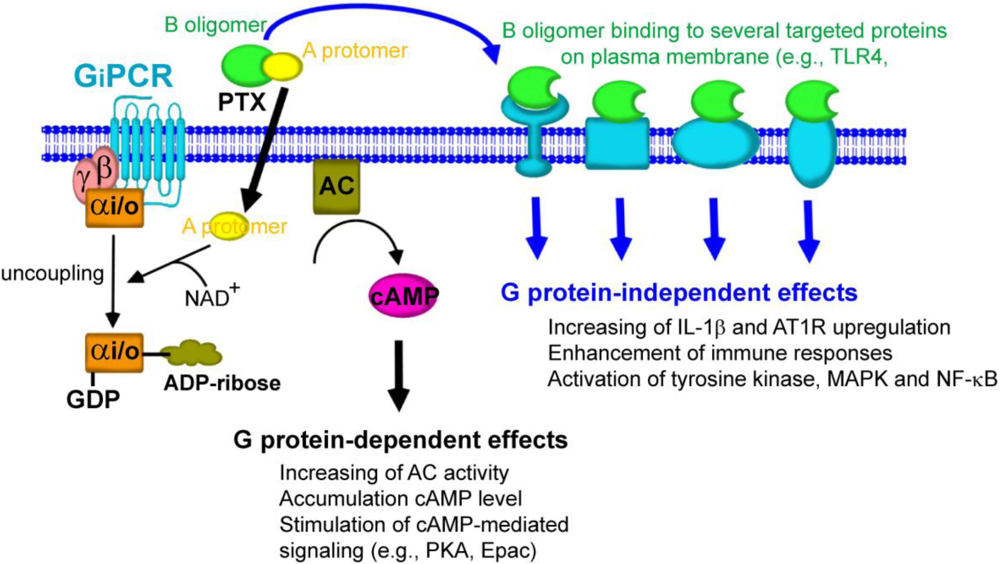
It is clear that PTX exhibits its various effects by eliciting at least two different signaling pathways (Figure 4). One involves the mobilization of the enzymatic A-protomer into host cells leading to the ADP-ribosylation of the α subunit of heterotrimeric Gi/o proteins [72]. This process called Gi/o protein-dependent effect of PTX, has a slow onset (at least 1-2 h) and requires a low concentration of the toxin. A second involves binding of the B-oligomer of PTX to several targeted proteins expressed on the cell surface such as TLR4 [15], GPIb [61], and TCR4 [65] initiating a series of rapid signaling events that require a higher concentration of the toxin. Thus, the B-oligomer of PTX appears to activate Gi/o protein-independent signaling in a diverse array of cell types.
It remains to be determined whether the binding site of PTX for the B-oligomer-mediated responses is the same as that of PTX to enter the cells for the A-protomer to ADP-ribosylate the Gi/o proteins. Another concern generated from these findings is whether the B-oligomer-mediated early transmembrane signaling events will influence the subsequent ADP-ribosylation of the Gαi/o proteins. The various cellular responses of PTX cannot be concluded simply by modification of the Gi/o proteins, as the B-oligomer of PTX can also elicit cellular responses.
In this review, we summarize that PTX can mediate biological responses by at least two different signaling pathways including (1) Gi/o protein-dependent action of PTX through ADP-ribosylation of the Gαi/o proteins and (2) Gi/o protein-independent action of PTX through the binding of the B-oligomer with cell surface specific proteins (Figure 4).
Figure 4.
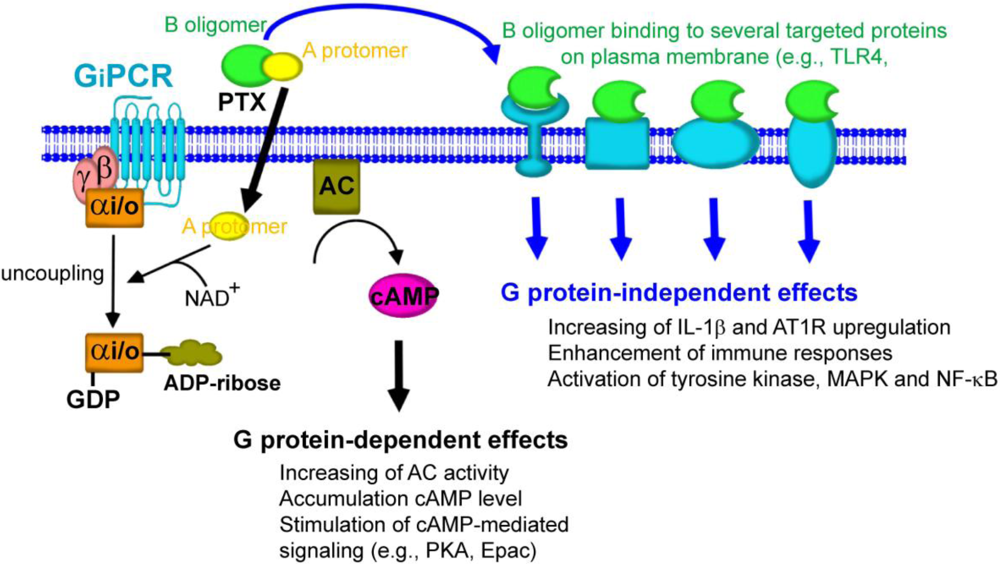
Gi/o protein-dependent and -independent effects of PTX. Following the binding of PTX to host cells, the A-protomer penetrates through the cell membrane. A-protomer is dissociated from B-oligomer and released into the cytoplasm. A-protomer then catalyzes the ADP-ribosylation of Gαi/o that leads to uncoupling of Gαi/o from its cognate inhibitory GPCRs. The inhibitory effect of Gαi/o on AC activity is then halted, resulting in accumulation of cAMP. This action of PTX results in disruption of cellular function through cAMP-mediated signaling pathway (Gi/o protein-dependent effects). In a separate pathway, the B-oligomer binds to and interacts with several targeted proteins on the plasma membrane, leading to the induction of the biological responses that are independent of ADP-ribosylation of Gαi/o protein (Gi/o protein-independent effects).
Figure 4.
Gi/o protein-dependent and -independent effects of PTX. Following the binding of PTX to host cells, the A-protomer penetrates through the cell membrane. A-protomer is dissociated from B-oligomer and released into the cytoplasm. A-protomer then catalyzes the ADP-ribosylation of Gαi/o that leads to uncoupling of Gαi/o from its cognate inhibitory GPCRs. The inhibitory effect of Gαi/o on AC activity is then halted, resulting in accumulation of cAMP. This action of PTX results in disruption of cellular function through cAMP-mediated signaling pathway (Gi/o protein-dependent effects). In a separate pathway, the B-oligomer binds to and interacts with several targeted proteins on the plasma membrane, leading to the induction of the biological responses that are independent of ADP-ribosylation of Gαi/o protein (Gi/o protein-independent effects).
References
- Holbourn, K.P.; Shone, C.C.; Acharya, K.R. A family of killer toxins. Exploring the mechanism of ADP-ribosylating toxins. FEBS J. 2006, 273, 4579–4593. [Google Scholar]
- Locht, C.; Antoine, R. A proposed mechanism of ADP-ribosylation catalyzed by the pertussis toxin S1 subunit. Biochimie 1995, 77, 333–340. [Google Scholar]
- Pittman, M. Pertussis toxin: the cause of the harmful effects and prolonged immunity of whooping cough. A hypothesis. Rev. Infect. Dis. 1979, 1, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Burns, D.L. Subunit structure and enzymic activity of pertussis toxin. Microbiol. Sci. 1988, 5, 285–287. [Google Scholar]
- Katada, T.; Ui, M. ADP ribosylation of the specific membrane protein of C6 cells by islet-activating protein associated with modification of adenylate cyclase activity. J. Biol. Chem. 1982, 257, 7210–7216. [Google Scholar]
- Kurose, H.; Katada, T.; Amano, T.; Ui, M. Specific uncoupling by islet-activating protein, pertussis toxin, of negative signal transduction via alpha-adrenergic, cholinergic, and opiate receptors in neuroblastoma x glioma hybrid cells. J. Biol. Chem. 1983, 258, 4870–4875. [Google Scholar] [PubMed]
- Tamura, M.; Nogimori, K.; Yajima, M.; Ase, K.; Ui, M. A role of the B-oligomer moiety of islet-activating protein, pertussis toxin, in development of the biological effects on intact cells. J. Biol. Chem. 1983, 258, 6756–6761. [Google Scholar]
- Lando, Z.; Teitelbaum, D.; Arnon, R. Induction of experimental allergic encephalomyelitis in genetically resistant strains of mice. Nature 1980, 287, 551–552. [Google Scholar]
- Linthicum, D.S.; Munoz, J.J.; Blaskett, A. Acute experimental autoimmune encephalomyelitis in mice. I. Adjuvant action of Bordetella pertussis is due to vasoactive amine sensitization and increased vascular permeability of the central nervous system. Cell. Immunol. 1982, 73, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Racke, M.K.; Hu, W.; Lovett-Racke, A.E. PTX cruiser: driving autoimmunity via TLR4. Trends Immunol. 2005, 26, 289–291. [Google Scholar]
- Jajoo, S.; Mukherjea, D.; Pingle, S.; Sekino, Y.; Ramkumar, V. Induction of adenosine A1 receptor expression by pertussis toxin via an adenosine 5'-diphosphate ribosylation-independent pathway. J. Pharmacol. Exp. Ther. 2006, 317, 1–10. [Google Scholar]
- Li, H.; Wong, W.S. Pertussis toxin activates tyrosine kinase signaling cascade in myelomonocytic cells: a mechanism for cell adhesion. Biochem. Biophys. Res. Commun. 2001, 283, 1077–1082. [Google Scholar]
- Melien, O.; Sandnes, D.; Johansen, E.J.; Christoffersen, T. Effects of pertussis toxin on extracellular signal-regulated kinase activation in hepatocytes by hormones and receptor-independent agents: evidence suggesting a stimulatory role of G(i) proteins at a level distal to receptor coupling. J. Cell. Physiol. 2000, 184, 27–36. [Google Scholar]
- Wang, Z.Y.; Yang, D.; Chen, Q.; Leifer, C.A.; Segal, D.M.; Su, S.B.; Caspi, R.R.; Howard, Z.O.; Oppenheim, J.J. Induction of dendritic cell maturation by pertussis toxin and its B subunit differentially initiate Toll-like receptor 4-dependent signal transduction pathways. Exp. Hematol. 2006, 34, 1115–1124. [Google Scholar]
- Nishida, M.; Suda, R.; Nagamatsu, Y.; Tanabe, S.; Onohara, N.; Nakaya, M.; Kanaho, Y.; Shibata, T.; Uchida, K.; Sumimoto, H.; et al. Pertussis toxin up-regulates angiotensin type 1 receptors through Toll-like receptor 4-mediated Rac activation. J. Biol. Chem. 2010, 285, 15268–15277. [Google Scholar] [PubMed]
- Tamura, M.; Nogimori, K.; Murai, S.; Yajima, M.; Ito, K.; Katada, T.; Ui, M.; Ishii, S. Subunit structure of islet-activating protein, pertussis toxin, in conformity with the A-B model. Biochemistry 1982, 21, 5516–5522. [Google Scholar] [PubMed]
- Katada, T.; Tamura, M.; Ui, M. The A protomer of islet-activating protein, pertussis toxin, as an active peptide catalyzing ADP-ribosylation of a membrane protein. Arch. Biochem. Biophys. 1983, 224, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Stein, P.E.; Boodhoo, A.; Armstrong, G.D.; Cockle, S.A.; Klein, M.H.; Read, R.J. The crystal structure of pertussis toxin. Structure 1994, 2, 45–57. [Google Scholar]
- Locht, C.; Keith, J.M. Pertussis toxin gene: Nucleotide sequence and genetic organization. Science 1986, 232, 1258–1264. [Google Scholar]
- Nicosia, A.; Perugini, M.; Franzini, C.; Casagli, M.C.; Borri, M.G.; Antoni, G.; Almoni, M.; Neri, P.; Ratti, G.; Rappuoli, R. Cloning and sequencing of the pertussis toxin genes: Operon structure and gene duplication. Proc. Natl. Acad. Sci. USA 1986, 83, 4631–4635. [Google Scholar]
- Saukkonen, K.; Burnette, W.N.; Mar, V.L.; Masure, H.R.; Tuomanen, E.I. Pertussis toxin has eukaryotic-like carbohydrate recognition domains. Proc. Natl. Acad. Sci. USA 1992, 89, 118–122. [Google Scholar]
- van’t Wout, J.; Burnette, W.N.; Mar, V.L.; Rozdzinski, E.; Wright, S.D.; Tuomanen, E.I. Role of carbohydrate recognition domains of pertussis toxin in adherence of Bordetella pertussis to human macrophages. Infect. Immun. 1992, 60, 3303–3308. [Google Scholar]
- Krueger, K.M.; Barbieri, J.T. Assignment of functional domains involved in ADP-ribosylation and B-oligomer binding within the carboxyl terminus of the S1 subunit of pertussis toxin. Infect. Immun. 1994, 62, 2071–2078. [Google Scholar]
- Antoine, R.; Locht, C. Roles of the disulfide bond and the carboxy-terminal region of the S1 subunit in the assembly and biosynthesis of pertussis toxin. Infect. Immun. 1990, 58, 1518–1526. [Google Scholar]
- Burns, D.L.; Hausman, S.Z.; Lindner, W.; Robey, F.A.; Manclark, C.R. Structural characterization of pertussis toxin A subunit. J. Biol. Chem. 1987, 262, 17677–17682. [Google Scholar]
- Sekura, R.D.; Fish, F.; Manclark, C.R.; Meade, B.; Zhang, Y.L. Pertussis toxin. Affinity purification of a new ADP-ribosyltransferase. J. Biol. Chem. 1983, 258, 14647–14651. [Google Scholar] [PubMed]
- Burns, D.L.; Manclark, C.R. Role of cysteine 41 of the A subunit of pertussis toxin. J. Biol. Chem. 1989, 264, 564–568. [Google Scholar]
- Krueger, K.M.; Barbieri, J.T. Molecular characterization of the in vitro activation of pertussis toxin by ATP. J. Biol. Chem. 1993, 268, 12570–12578. [Google Scholar] [PubMed]
- Van Ness, B.G.; Howard, J.B.; Bodley, J.W. ADP-ribosylation of elongation factor 2 by diphtheria toxin. NMR spectra and proposed structures of ribosyl-diphthamide and its hydrolysis products. J. Biol. Chem. 1980, 255, 10710–10716. [Google Scholar] [PubMed]
- Gilman, A.G. G proteins: Transducers of receptor-generated signals. Annu. Rev. Biochem. 1987, 56, 615–649. [Google Scholar]
- Katada, T.I.T.; Takahashi, K.; Nishina, H.; Kanaho, Y. Bacterial Toxins and Virulence Factors in Disease; Dekker, Inc.: New York, NY, USA, 1995; Volume 8, pp. 459–489. [Google Scholar]
- Ui, M. Islet-activating protein, pertussis toxin: A probe for functions of the inhibitory guanine nucleotide regulatory component of adenylate cyclase. Trends Pharmacol. Sci. 1984, 5, 277–279. [Google Scholar]
- West, R.E., Jr.; Moss, J.; Vaughan, M.; Liu, T.; Liu, T.Y. Pertussis toxin-catalyzed ADP-ribosylation of transducin. Cysteine 347 is the ADP-ribose acceptor site. J. Biol. Chem. 1985, 260, 14428–14430. [Google Scholar] [PubMed]
- Fields, T.A.; Casey, P.J. Signalling functions and biochemical properties of pertussis toxin-resistant G-proteins. Biochem. J. 1997, 321, 561–571. [Google Scholar]
- Cabrera-Vera, T.M.; Vanhauwe, J.; Thomas, T.O.; Medkova, M.; Preininger, A.; Mazzoni, M.R.; Hamm, H.E. Insights into G protein structure, function, and regulation. Endocr. Rev. 2003, 24, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.D.; Medzihradsky, F. Go mediates the coupling of the mu opioid receptor to adenylyl cyclase in cloned neural cells and brain. Proc. Natl. Acad. Sci. USA 1993, 90, 4062–6406. [Google Scholar]
- Katada, T.; Ui, M. Slow interaction of islet-activating protein with pancreatic islets during primary culture to cause reversal of alpha-adrenergic inhibition of insulin secretion. J. Biol. Chem. 1980, 255, 9580–9588. [Google Scholar]
- Milligan, G. Techniques used in the identification and analysis of function of pertussis toxin-sensitive guanine nucleotide binding proteins. Biochem. J. 1988, 255, 1–13. [Google Scholar]
- Chen, H.; Lambert, N.A. Inhibition of dendritic calcium influx by activation of G-protein-coupled receptors in the hippocampus. J. Neurophysiol. 1997, 78, 3484–3488. [Google Scholar]
- Yamada, M.; Inanobe, A.; Kurachi, Y. G protein regulation of potassium ion channels. Pharmacol. Rev. 1998, 50, 723–760. [Google Scholar]
- Wittpoth, C.; Scholich, K.; Yigzaw, Y.; Stringfield, T.M.; Patel, T.B. Regions on adenylyl cyclase that are necessary for inhibition of activity by beta gamma and G(ialpha) subunits of heterotrimeric G proteins. Proc. Natl. Acad. Sci. USA 1999, 96, 9551–9556. [Google Scholar]
- Jeong, S.W.; Ikeda, S.R. Effect of G protein heterotrimer composition on coupling of neurotransmitter receptors to N-type Ca(2+) channel modulation in sympathetic neurons. Proc. Natl. Acad. Sci. USA 2000, 97, 907–912. [Google Scholar]
- Chen, C. Gi-3 protein mediates the increase in voltage-gated K+ currents by somatostatin on cultured ovine somatotrophs. Am. J. Physiol. 1998, 275, E278–E284. [Google Scholar]
- Obadiah, J.; Avidor-Reiss, T.; Fishburn, C.S.; Carmon, S.; Bayewitch, M.; Vogel, Z.; Fuchs, S.; Levavi-Sivan, B. Adenylyl cyclase interaction with the D2 dopamine receptor family; differential coupling to Gi, Gz, and Gs. Cell. Mol. Neurobiol. 1999, 19, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.W.; Ikeda, S.R. G protein alpha subunit G alpha z couples neurotransmitter receptors to ion channels in sympathetic neurons. Neuron 1998, 21, 1201–1212. [Google Scholar]
- Tsu, R.C.; Ho, M.K.; Yung, L.Y.; Joshi, S.; Wong, Y.H. Role of amino- and carboxyl-terminal regions of G(alphaZ) in the recognition of Gi-coupled receptors. Mol. Pharmacol. 1997, 52, 38–45. [Google Scholar]
- Rarick, H.M.; Artemyev, N.O.; Hamm, H.E. A site on rod G protein alpha subunit that mediates effector activation. Science 1992, 256, 1031–1033. [Google Scholar]
- Yan, W.; Sunavala, G.; Rosenzweig, S.; Dasso, M.; Brand, J.G.; Spielman, A.I. Bitter taste transduced by PLC-beta(2)-dependent rise in IP(3) and alpha-gustducin-dependent fall in cyclic nucleotides. Am. J. Physiol. Cell Physiol. 2001, 280, C742–C751. [Google Scholar]
- Muller-Wieland, D.; White, M.F.; Behnke, B.; Gebhardt, A.; Neumann, S.; Krone, W.; Kahn, C.R. Pertussis toxin inhibits autophosphorylation and activation of the insulin receptor kinase. Biochem. Biophys. Res. Commun. 1991, 181, 1479–1485. [Google Scholar]
- Ui, M. The Multiple Biological Activities of Pertussis Toxin; John Wiley & Sons, Inc.: London, UK, 1988; pp. 121–145. [Google Scholar]
- Brito, G.A.; Souza, M.H.; Melo-Filho, A.A.; Hewlett, E.L.; Lima, A.A.; Flores, C.A.; Ribeiro, R.A. Role of pertussis toxin A subunit in neutrophil migration and vascular permeability. Infect. Immun. 1997, 65, 1114–1118. [Google Scholar]
- Houslay, M.D.; Milligan, G. Tailoring cAMP-signalling responses through isoform multiplicity. Trends Biochem. Sci. 1997, 22, 217–224. [Google Scholar]
- Montminy, M. Transcriptional regulation by cyclic AMP. Annu. Rev. Biochem. 1997, 66, 807–822. [Google Scholar]
- Bos, J.L. Epac proteins: Multi-purpose cAMP targets. Trends Biochem. Sci. 2006, 31, 680–686. [Google Scholar]
- de Rooij, J.; Zwartkruis, F.J.; Verheijen, M.H.; Cool, R.H.; Nijman, S.M.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar]
- Pereira, L.; Metrich, M.; Fernandez-Velasco, M.; Lucas, A.; Leroy, J.; Perrier, R.; Morel, E.; Fischmeister, R.; Richard, S.; Benitah, J.P.; et al. The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J. Physiol. 2007, 583, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Mangmool, S.; Shukla, A.K.; Rockman, H.A. beta-Arrestin-dependent activation of Ca(2+)/calmodulin kinase II after beta(1)-adrenergic receptor stimulation. J. Cell Biol. 2010, 189, 573–587. [Google Scholar]
- Garcia, J.G.; Wang, P.; Liu, F.; Hershenson, M.B.; Borbiev, T.; Verin, A.D. Pertussis toxin directly activates endothelial cell p42/p44 MAP kinases via a novel signaling pathway. Am. J. Physiol. Cell Physiol. 2001, 280, C1233–C1241. [Google Scholar]
- Lee, H.Y.; Kim, S.D.; Shim, J.W.; Kim, H.J.; Yun, J.; Baek, S.H.; Kim, K.; Bae, Y.S. A pertussis toxin sensitive G-protein-independent pathway is involved in serum amyloid A-induced formyl peptide receptor 2-mediated CCL2 production. Exp. Mol. Med. 2010, 42, 302–309. [Google Scholar]
- Lee, H.Y.; Lee, S.Y.; Kim, S.D.; Shim, J.W.; Kim, H.J.; Jung, Y.S.; Kwon, J.Y.; Baek, S.H.; Chung, J.; Bae, Y.S. Sphingosylphosphorylcholine stimulates CCL2 production from human umbilical vein endothelial cells. J. Immunol. 2011, 186, 4347–4353. [Google Scholar]
- Sindt, K.A.; Hewlett, E.L.; Redpath, G.T.; Rappuoli, R.; Gray, L.S.; Vandenberg, S.R. Pertussis toxin activates platelets through an interaction with platelet glycoprotein Ib. Infect. Immun. 1994, 62, 3108–3114. [Google Scholar]
- Strnad, C.F.; Carchman, R.A. Human T lymphocyte mitogenesis in response to the B oligomer of pertussis toxin is associated with an early elevation in cytosolic calcium concentrations. FEBS Lett. 1987, 225, 16–20. [Google Scholar]
- Thom, R.E.; Casnellie, J.E. Pertussis toxin activates protein kinase C and a tyrosine protein kinase in the human T cell line Jurkat. FEBS Lett. 1989, 244, 181–184. [Google Scholar]
- Wong, W.S.; Rosoff, P.M. Pharmacology of pertussis toxin B-oligomer. Can. J. Physiol. Pharmacol. 1996, 74, 559–564. [Google Scholar]
- Schneider, O.D.; Weiss, A.A.; Miller, W.E. Pertussis toxin utilizes proximal components of the T-cell receptor complex to initiate signal transduction events in T cells. Infect. Immun. 2007, 75, 4040–4049. [Google Scholar]
- Raze, D.; Veithen, A.; Sato, H.; Antoine, R.; Menozzi, F.D.; Locht, C. Genetic exchange of the S2 and S3 subunits in pertussis toxin. Mol. Microbiol. 2006, 60, 1241–1250. [Google Scholar]
- Gierschik, P. ADP-ribosylation of signal-transducing guanine nucleotide-binding proteins by pertussis toxin. Curr. Top. Microbiol. Immunol. 1992, 175, 69–96. [Google Scholar]
- Banga, H.S.; Walker, R.K.; Winberry, L.K.; Rittenhouse, S.E. Pertussis toxin can activate human platelets. Comparative effects of holotoxin and its ADP-ribosylating S1 subunit. J. Biol. Chem. 1987, 262, 14871–14874. [Google Scholar]
- Alfano, M.; Grivel, J.C.; Ghezzi, S.; Corti, D.; Trimarchi, M.; Poli, G.; Margolis, L. Pertussis toxin B-oligomer dissociates T cell activation and HIV replication in CD4 T cells released from infected lymphoid tissue. AIDS 2005, 19, 1007–1014. [Google Scholar]
- Moss, J.; Bruni, P.; Hsia, J.A.; Tsai, S.C.; Watkins, P.A.; Halpern, J.L.; Burns, D.L.; Kanaho, Y.; Chang, P.P.; Hewlett, E.L.; et al. Pertussis toxin-catalyzed ADP-ribosylation: effects on the coupling of inhibitory receptors to the adenylate cyclase system. J. Recept. Res. 1984, 4, 459–474. [Google Scholar]
- Nencioni, L.; Volpini, G.; Peppoloni, S.; Bugnoli, M.; de Magistris, T.; Marsili, I.; Rappuoli, R. Properties of pertussis toxin mutant PT-9K/129G after formaldehyde treatment. Infect. Immun. 1991, 59, 625–630. [Google Scholar]
- Pizza, M.; Bartoloni, A.; Prugnola, A.; Silvestri, S.; Rappuoli, R. Subunit S1 of pertussis toxin: Mapping of the regions essential for ADP-ribosyltransferase activity. Proc. Natl. Acad. Sci. USA 1988, 85, 7521–7525. [Google Scholar]
- Nasso, M.; Fedele, G.; Spensieri, F.; Palazzo, R.; Costantino, P.; Rappuoli, R.; Ausiello, C.M. Genetically detoxified pertussis toxin induces Th1/Th17 immune response through MAPKs and IL-10-dependent mechanisms. J. Immunol. 2009, 183, 1892–1899. [Google Scholar]
- Poltorak, A.; He, X.; Smirnova, I.; Liu, M.Y.; Van Huffel, C.; Du, X.; Birdwell, D.; Alejos, E.; Silva, M.; Galanos, C.; et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science 1998, 282, 2085–2088. [Google Scholar] [PubMed]
- Stewart, S.J.; Prpic, V.; Johns, J.A.; Powers, F.S.; Graber, S.E.; Forbes, J.T.; Exton, J.H. Bacterial toxins affect early events of T lymphocyte activation. J. Clin. Invest. 1989, 83, 234–242. [Google Scholar]
- Spangrude, G.J.; Sacchi, F.; Hill, H.R.; van Epps, D.E.; Daynes, R.A. Inhibition of lymphocyte and neutrophil chemotaxis by pertussis toxin. J. Immunol. 1985, 135, 4135–4143. [Google Scholar]
- Burns, D.L.; Hewlett, E.L.; Moss, J.; Vaughan, M. Pertussis toxin inhibits enkephalin stimulation of GTPase of NG108-15 cells. J. Biol. Chem. 1983, 258, 1435–1438. [Google Scholar]
- Kindt, R.M.; Lander, A.D. Pertussis toxin specifically inhibits growth cone guidance by a mechanism independent of direct G protein inactivation. Neuron 1995, 15, 79–88. [Google Scholar]
- Li, H.; Wong, W.S. Mechanisms of pertussis toxin-induced myelomonocytic cell adhesion: role of CD14 and urokinase receptor. Immunology 2000, 100, 502–509. [Google Scholar]
- Gray, L.S.; Huber, K.S.; Gray, M.C.; Hewlett, E.L.; Engelhard, V.H. Pertussis toxin effects on T lymphocytes are mediated through CD3 and not by pertussis toxin catalyzed modification of a G protein. J. Immunol. 1989, 142, 1631–1638. [Google Scholar]
- Zocchi, M.R.; Contini, P.; Alfano, M.; Poggi, A. Pertussis toxin (PTX) B subunit and the nontoxic PTX mutant PT9K/129G inhibit Tat-induced TGF-beta production by NK cells and TGF-beta-mediated NK cell apoptosis. J. Immunol. 2005, 174, 6054–6061. [Google Scholar]
- Alfano, M.; Pushkarsky, T.; Poli, G.; Bukrinsky, M. The B-oligomer of pertussis toxin inhibits human immunodeficiency virus type 1 replication at multiple stages. J. Virol. 2000, 74, 8767–8770. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Mangmool, S.; Kurose, H. Gi/o Protein-Dependent and -Independent Actions of Pertussis Toxin (PTX). Toxins 2011, 3, 884-899. https://doi.org/10.3390/toxins3070884
AMA Style
Mangmool S, Kurose H. Gi/o Protein-Dependent and -Independent Actions of Pertussis Toxin (PTX). Toxins. 2011; 3(7):884-899. https://doi.org/10.3390/toxins3070884
Chicago/Turabian StyleMangmool, Supachoke, and Hitoshi Kurose. 2011. "Gi/o Protein-Dependent and -Independent Actions of Pertussis Toxin (PTX)" Toxins 3, no. 7: 884-899. https://doi.org/10.3390/toxins3070884