The Biology of the Cytolethal Distending Toxins

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. CDT Structure and Enzymatic Activity
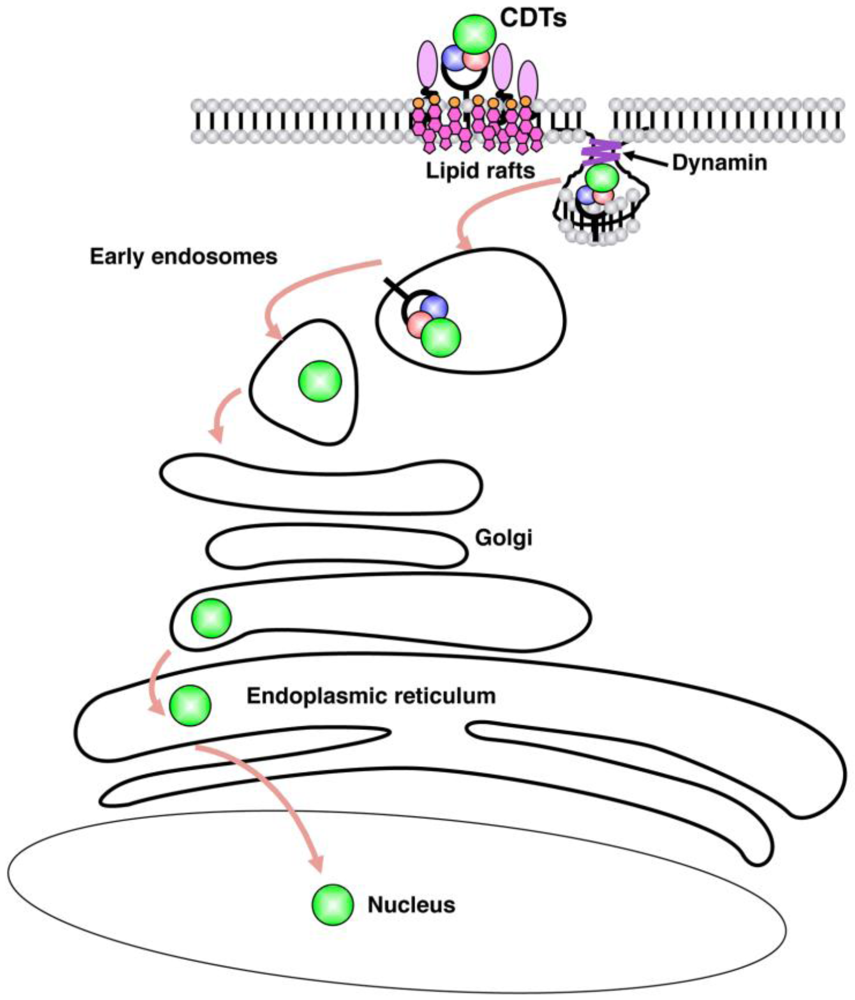
3. Internalization
4. Cellular Responses
4.1. Induction of DNA Damage
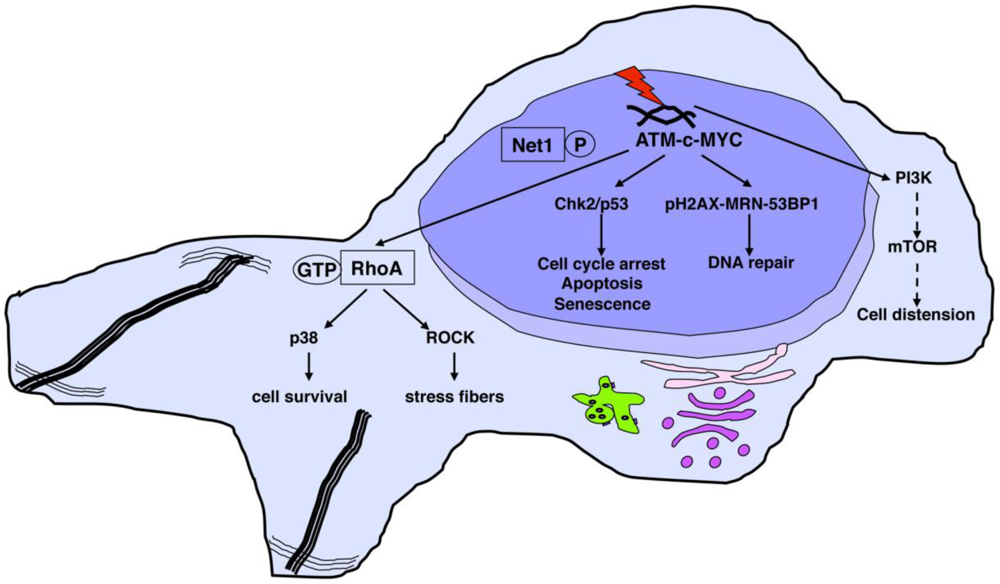
4.2. Activation of DNA Damage Responses
4.3. Survival Signals Activated in Intoxicated Cells
4.4. CDT-Induced Apoptosis
4.5. CDT as Phosphatase
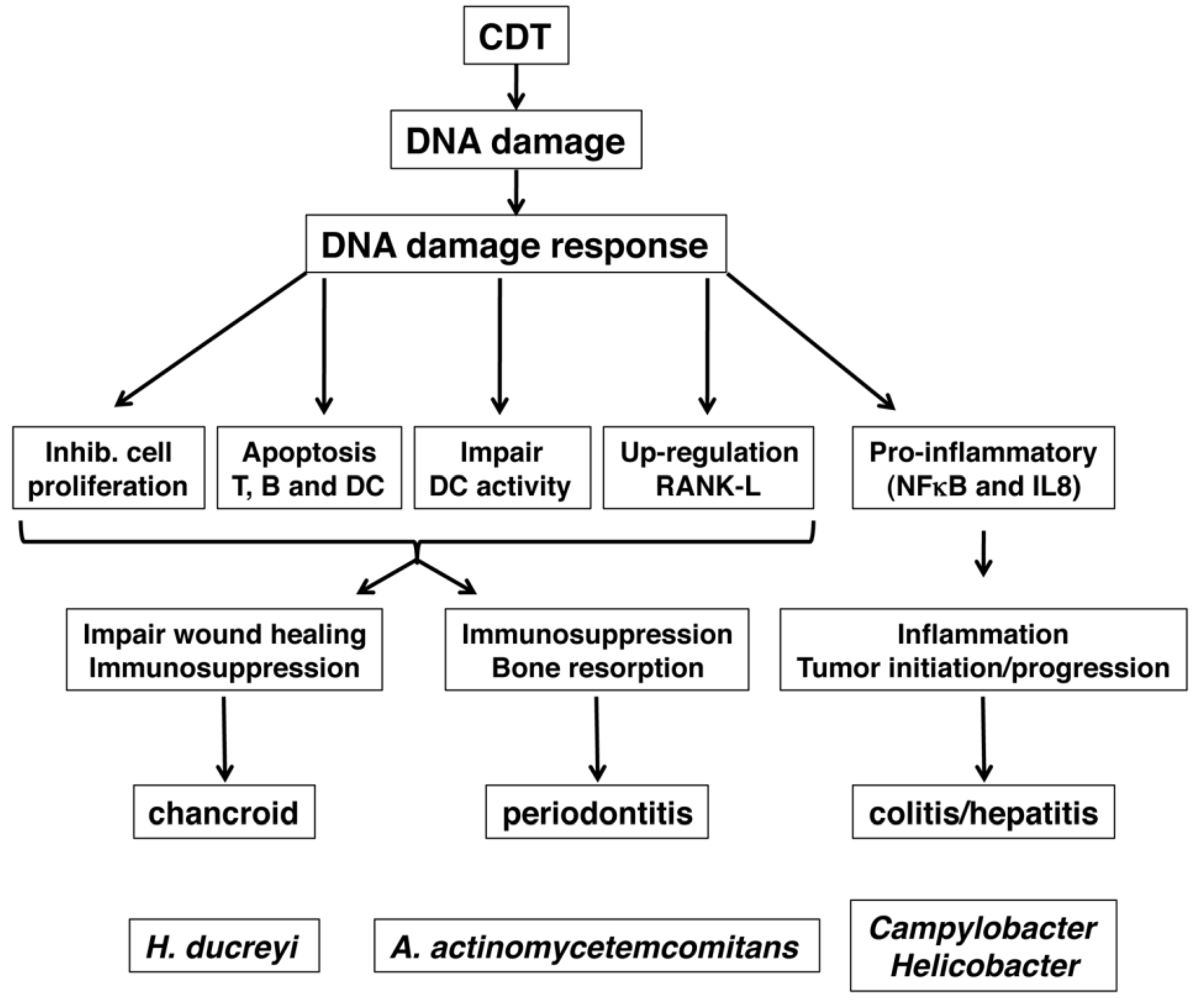
5. Role of CDT as Virulence Factor

5.1. Role of CDT in Colitis and Inflammation-Associated Carcinogenesis
5.2. Chancroid and Wound Healing
5.3. Periodontitis and A. actinomycetemcomitans
6. Conclusions
Acknowledgements
References
- Nougayrede, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar]
- Cortes-Bratti, X.; Frisan, T.; Thelestam, M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon 2001, 39, 1729–1736. [Google Scholar]
- Thelestam, M.; Frisan, T. Cytolethal distending toxins. In The Comprehensive Sourcebook of Bacterial Protein Toxins; Alouf, J., Popoff, M., Eds.; CPL Press: Newbury, UK, 2006; pp. 448–467. [Google Scholar]
- Nesic, D.; Hsu, Y.; Stebbins, C.E. Assembly and function of a bacterial genotoxin. Nature 2004, 429, 429–433. [Google Scholar]
- Elwell, C.A.; Dreyfus, L.A. DNAase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963. [Google Scholar]
- Lara-Tejero, M.; Galan, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000, 290, 354–357. [Google Scholar]
- Hassane, D.C.; Lee, R.B.; Mendenhall, M.D.; Pickett, C.L. Cytolethal distending toxin demonstrates genotoxic activity in a yeast model. Infect. Immun. 2001, 69, 5752–5759. [Google Scholar]
- Guerra, L.; Teter, K.; Lilley, B.N.; Stenerlow, B.; Holmes, R.K.; Ploegh, H.L.; Sandvig, K.; Thelestam, M.; Frisan, T. Cellular internalization of cytolethal distending toxin: A new end to a known pathway. Cell. Microbiol. 2005, 7, 921–934. [Google Scholar]
- Hu, X.; Nesic, D.; Stebbins, C.E. Comparative structure-function analysis of cytolethal distending toxins. Proteins 2006, 62, 421–434. [Google Scholar]
- Ueno, Y.; Ohara, M.; Kawamoto, T.; Fujiwara, T.; Komatsuzawa, H.; Oswald, E.; Sugai, M. Biogenesis of the Actinobacillus actinomycetemcomitans cytolethal distending toxin holotoxin. Infect. Immun. 2006, 74, 3480–3487. [Google Scholar]
- Lee, R.B.; Hassane, D.C.; Cottle, D.L.; Pickett, C.L. Interactions of Campylobacter jejuni cytolethal distending toxin subunits CdtA and CdtC with HeLa cells. Infect. Immun. 2003, 71, 4883–4890. [Google Scholar]
- McSweeney, L.A.; Dreyfus, L.A. Carbohydrate-binding specificity of the Escherichia coli cytolethal distending toxin CdtA-II and CdtC-II subunits. Infect. Immun. 2005, 73, 2051–2060. [Google Scholar]
- Mise, K.; Akifusa, S.; Watarai, S.; Ansai, T.; Nishihara, T.; Takehara, T. Involvement of ganglioside GM3 in G(2)/M cell cycle arrest of human monocytic cells induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect. Immun. 2005, 73, 4846–4852. [Google Scholar]
- Mao, X.; DiRienzo, J.M. Functional studies of the recombinant subunits of a cytolethal distending holotoxin. Cell. Microbiol. 2002, 4, 245–255. [Google Scholar]
- Akifusa, S.; Heywood, W.; Nair, S.P.; Stenbeck, G.; Henderson, B. Mechanism of internalization of the cytolethal distending toxin of Actinobacillus actinomycetemcomitans. Microbiology 2005, 151, 1395–1402. [Google Scholar]
- Frisan, T. 2011; Karolinska Institute, SwedenUnpublished work.
- Boesze-Battaglia, K.; Besack, D.; McKay, T.; Zekavat, A.; Otis, L.; Jordan-Sciutto, K.; Shenker, B.J. Cholesterol-rich membrane microdomains mediate cell cycle arrest induced by Actinobacillus actinomycetemcomitans cytolethal-distending toxin. Cell. Microbiol. 2006, 8, 823–836. [Google Scholar]
- Carette, J.E.; Guimaraes, C.P.; Varadarajan, M.; Park, A.S.; Wuethrich, I.; Godarova, A.; Kotecki, M.; Cochran, B.H.; Spooner, E.; Ploegh, H.L.; Brummelkamp, T.R. Haploid genetic screens in human cells identify host factors used by pathogens. Science 2009, 326, 1231–1235. [Google Scholar]
- Eshraghi, A.; Maldonado-Arocho, F.J.; Gargi, A.; Cardwell, M.M.; Prouty, M.G.; Blanke, S.R.; Bradley, K.A. Cytolethal distending toxin family members are differentially affected by alterations in host glycans and membrane cholesterol. J. Biol. Chem. 2010, 285, 18199–18207. [Google Scholar]
- Parkhill, J.; Dougan, G.; James, K.D.; Thomson, N.R.; Pickard, D.; Wain, J.; Churcher, C.; Mungall, K.L.; Bentley, S.D.; Holden, M.T.; et al. Complete genome sequence of a multiple drug resistant Salmonella enterica serovar Typhi CT18. Nature 2001, 413, 848–852. [Google Scholar]
- Haghjoo, E.; Galan, J.E. Salmonella typhi encodes a functional cytolethal distending toxin that is delivered into host cells by a bacterial-internalization pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 4614–4619. [Google Scholar]
- Spano, S.; Ugalde, J.E.; Galan, J.E. Delivery of a Salmonella typhi exotoxin from a host intracellular compartment. Cell Host Microbe 2008, 3, 30–38. [Google Scholar]
- Cortes-Bratti, X.; Chaves-Olarte, E.; Lagergård, T.; Thelestam, M. Cellular internalization of cytolethal distending toxin from Haemophilus ducreyi. Infect. Immun. 2000, 68, 6903–6911. [Google Scholar]
- Guerra, L.; Nemec, K.N.; Massey, S.; Tatulian, S.A.; Thelestam, M.; Frisan, T.; Teter, K. A novel mode of translocation for cytolethal distending toxin. Biochim. Biophys. Acta 2009, 1793, 489–495. [Google Scholar]
- Nishikubo, S.; Ohara, M.; Ueno, Y.; Ikura, M.; Kurihara, H.; Komatsuzawa, H.; Oswald, E.; Sugai, M. An N-terminal segment of the active component of the bacterial genotoxin cytolethal distending toxin B (CDTB) directs CDTB into the nucleus. J. Biol. Chem. 2003, 278, 50671–50681. [Google Scholar]
- McSweeney, L.A.; Dreyfus, L.A. Nuclear localization of the Escherichia coli cytolethal distending toxin CdtB subunit. Cell. Microbiol. 2004, 6, 447–458. [Google Scholar]
- Lencer, W.I.; Tsai, B. The intracellular voyage of cholera toxin: going retro. Trends Biochem. Sci. 2003, 28, 639–645. [Google Scholar]
- Pickett, C.L.; Whitehouse, C.A. The cytolethal distending toxin family. Trends Microbiol. 1999, 7, 292–297. [Google Scholar]
- Scorza, F.B.; Doro, F.; Rodriguez-Ortega, M.J.; Stella, M.; Liberatori, S.; Taddei, A.R.; Serino, L.; Gomes, M.D.; Nesta, B.; Fontana, M.R.; et al. Proteomics characterization of outer membrane vesicles from the extraintestinal pathogenic Escherichia coli DeltatolR IHE3034 mutant. Mol. Cell. Proteomics 2008, 7, 473–485. [Google Scholar] [PubMed]
- Lindmark, B.; Rompikuntal, P.K.; Vaitkevicius, K.; Song, T.; Mizunoe, Y.; Uhlin, B.E.; Guerry, P.; Wai, S.N. Outer membrane vesicle-mediated release of cytolethal distending toxin (CDT) from Campylobacter jejuni. BMC Microbiol. 2009, 9, 220. [Google Scholar]
- Frisan, T.; Cortes-Bratti, X.; Chaves-Olarte, E.; Stenerlöw, B.; Thelestam, M. The Haemophilus ducreyi cytolethal distending toxin induces DNA double strand breaks and promotes ATM-dependent activation of RhoA. Cell. Microbiol. 2003, 5, 695–707. [Google Scholar]
- Elwell, C.; Chao, K.; Patel, K.; Dreyfus, L. Escherichia coli CdtB mediates cytolethal distending toxin cell cycle arrest. Infect. Immun. 2001, 69, 3418–3422. [Google Scholar]
- Li, L.; Sharipo, A.; Chaves-Olarte, E.; Masucci, M.G.; Levitsky, V.; Thelestam, M.; Frisan, T. The Haemophilus ducreyi cytolethal distending toxin activates sensors of DNA damage and repair complexes in proliferating and non-proliferating cells. Cell. Microbiol. 2002, 4, 87–99. [Google Scholar]
- Smith, J.; Tho, L.M.; Xu, N.; Gillespie, D.A. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv. Cancer Res. 2010, 108, 73–112. [Google Scholar]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar]
- Comayras, C.; Tasca, C.; Peres, S.Y.; Ducommun, B.; Oswald, E.; de Rycke, J. Escherichia coli cytolethal distending toxin blocks the HeLa cell cycle at the G2/M transition by preventing cdc2 protein kinase dephosphorylation and activation. Infect. Immun. 1997, 65, 5088–5095. [Google Scholar]
- Shenker, B.J.; McKay, T.; Datar, S.; Miller, M.; Chowhan, R.; Demuth, D. Actinobacillus actinomycetemcomitans immunosuppressive protein is a member of the family of cytolethal distending toxins capable of causing a G2 arrest in human T cells. J. Immunol. 1999, 162, 4773–4680. [Google Scholar]
- Sert, V.; Cans, C.; Tasca, C.; Bret-Bennis, L.; Oswald, E.; Ducommun, B.; de Rycke, J. The bacterial cytolethal distending toxin (CDT) triggers a G2 cell cycle checkpoint in mammalian cells without preliminary induction of DNA strand breaks. Oncogene 1999, 18, 6296–6304. [Google Scholar]
- Cortes-Bratti, X.; Karlsson, C.; Lagergard, T.; Thelestam, M.; Frisan, T. The Haemophilus ducreyi cytolethal distending toxin induces cell cycle arrest and apoptosis via the DNA damage checkpoint pathways. J. Biol. Chem. 2001, 276, 5296–5302. [Google Scholar]
- Hassane, D.C.; Lee, R.B.; Pickett, C.L. Campylobacter jejuni cytolethal distending toxin promotes DNA repair responses in normal human cells. Infect. Immun. 2003, 71, 541–545. [Google Scholar]
- Yamamoto, K.; Tominaga, K.; Sukedai, M.; Okinaga, T.; Iwanaga, K.; Nishihara, T.; Fukuda, J. Delivery of cytolethal distending toxin B induces cell cycle arrest and apoptosis in gingival squamous cell carcinoma in vitro. Eur. J. Oral. Sci. 2004, 112, 445–451. [Google Scholar]
- Sato, T.; Koseki, T.; Yamato, K.; Saiki, K.; Konishi, K.; Yoshikawa, M.; Ishikawa, I.; Nishihara, T. p53-independent expression of p21(CIP1/WAF1) in plasmacytic cells during G(2) cell cycle arrest induced by Actinobacillus actinomycetemcomitans cytolethal distending toxin. Infect. Immun. 2002, 70, 528–534. [Google Scholar]
- Guerra, L.; Albihn, A.; Tronnersjö, S.; Yan, Q.; Guidi, R.; Stenerlöw, B.; Sterzenbach, T.; Josenhans, C.; Fox, J.G.; Schauer, D.B.; et al. Myc is required for activation of the ATM-dependent checkpoints in response to DNA damage. PLoS One 2010, 5, e8924. [Google Scholar] [PubMed]
- Blazkova, H.; Krejcikova, K.; Moudry, P.; Frisan, T.; Hodny, Z.; Bartek, J. Bacterial Intoxication Evokes Cellular Senescence with Persistent DNA Damage and Cytokine Signaling. J. Cell. Mol. Med. 2010, 14, 357–367. [Google Scholar]
- Kitagawa, T.; Hoshida, H.; Akada, R. Genome-wide analysis of cellular response to bacterial genotoxin CdtB in yeast. Infect. Immun. 2007, 75, 1393–1402. [Google Scholar]
- Kastan, M.B.; Bartek, J. Cell-cycle checkpoints and cancer. Nature 2004, 432, 316–323. [Google Scholar]
- Shiloh, Y. ATM and related protein kinases: safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar]
- Gelfanova, V.; Hansen, E.J.; Spinola, S.M. Cytolethal distending toxin of Haemophilus ducreyi induces apoptotic death of Jurkat T cells. Infect. Immun. 1999, 67, 6394–6402. [Google Scholar]
- Cortes-Bratti, X.; Chaves-Olarte, E.; Lagergård, T.; Thelestam, M. The cytolethal distending toxin from the chancroid bacterium Haemophilus ducreyi induces cell-cycle arrest in the G2 phase. J. Clin. Invest. 1999, 103, 107–115. [Google Scholar]
- Guerra, L.; Carr, H.S.; Richter-Dahlfors, A.; Masucci, M.G.; Thelestam, M.; Frost, J.A.; Frisan, T. A bacterial cytotoxin identifies the RhoA exchange factor Net1 as a key effector in the response to DNA damage. PLoS One 2008, 3, e2254. [Google Scholar]
- Guerra, L.; Guidi, R.; Ilse, S.; Callegari, S.; Sompallae, R.; Pickett, C.L.; Åström, S.; Eisele, F.; Wolf, D.; Sjögren, C.; Masucci, M.G.; Frisan, T. Bacterial genotoxin triggers FEN1-dependent RhoA activation, cytoskeleton remodeling and cell survival. submitted for publication.
- Shenker, B.J.; Hoffmaster, R.H.; Zekavat, A.; Yamaguchi, N.; Lally, E.T.; Demuth, D.R. Induction of apoptosis in human T cells by Actinobacillus actinomycetemcomitans cytolethal distending toxin is a consequence of G2 arrest of the cell cycle. J. Immun. 2001, 167, 435–441. [Google Scholar]
- Ohara, M.; Hayashi, T.; Kusunoki, Y.; Miyauchi, M.; Takata, T.; Sugai, M. Caspase-2 and caspase-7 are involved in cytolethal distending toxin-induced apoptosis in Jurkat and MOLT-4 T-cell lines. Infect. Immun. 2004, 72, 871–879. [Google Scholar]
- Ohara, M.; Hayashi, T.; Kusunoki, Y.; Nakachi, K.; Fujiwara, T.; Komatsuzawa, H.; Sugai, M. Cytolethal distending toxin induces caspase-dependent and -independent cell death in MOLT-4 cells. Infect. Immun. 2008, 76, 4783–4791. [Google Scholar]
- Rabin, S.D.; Flitton, J.G.; Demuth, D.R. Aggregatibacter actinomycetemcomitans cytolethal distending toxin induces apoptosis in nonproliferating macrophages by a phosphatase-independent mechanism. Infect. Immun. 2009, 77, 3161–3169. [Google Scholar]
- Shenker, B.J.; Dlakic, M.; Walker, L.P.; Besack, D.; Jaffe, E.; LaBelle, E.; Boesze-Battaglia, K. A novel mode of action for a microbial-derived immunotoxin: the cytolethal distending toxin subunit B exhibits phosphatidylinositol 3,4,5-triphosphate phosphatase activity. J. Immunol. 2007, 178, 5099–5108. [Google Scholar]
- Matangkasombut, O.; Wattanawaraporn, R.; Tsuruda, K.; Ohara, M.; Sugai, M.; Mongkolsuk, S. Cytolethal distending toxin from Aggregatibacter actinomycetemcomitans induces DNA damage, S/G2 cell cycle arrest, and caspase- independent death in a Saccharomyces cerevisiae model. Infect. Immun. 2010, 78, 783–792. [Google Scholar]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar]
- Karin, M.; Lawrence, T.; Nizet, V. Innate immunity gone awry: linking microbial infections to chronic inflammation and cancer. Cell 2006, 124, 823–835. [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Schistosomes, Liver Flukes and Helicobacter Pylori; World Health Organization: Lyon, France, 1994; 61, pp. 177–240.
- Avenaud, P.; Marais, A.; Monteiro, L.; Le Bail, B.; Bioulac Sage, P.; Balabaud, C.; Megraud, F. Detection of Helicobacter species in the liver of patients with and without primary liver carcinoma. Cancer 2000, 89, 1431–1439. [Google Scholar]
- Nilsson, H.O.; Mulchandani, R.; Tranberg, K.G.; Stenram, U.; Wadstrom, T. Helicobacter species identified in liver from patients with cholangiocarcinoma and hepatocellular carcinoma. Gastroenterology 2001, 120, 323–324. [Google Scholar]
- Purdy, D.; Buswell, C.M.; Hodgson, A.E.; McAlpine, K.; Henderson, I.; Leach, S.A. Characterisation of cytolethal distending toxin (CDT) mutants of Campylobacter jejuni. J. Med. Microbiol. 2000, 49, 473–479. [Google Scholar]
- McAuley, J.L.; Linden, S.K.; Png, C.W.; King, R.M.; Pennington, H.L.; Gendler, S.J.; Florin, T.H.; Hill, G.R.; Korolik, V.; McGuckin, M.A. MUC1 cell surface mucin is a critical element of the mucosal barrier to infection. J. Clin. Invest. 2007, 117, 2313–2324. [Google Scholar]
- Fox, J.G.; Rogers, A.B.; Whary, M.T.; Ge, Z.; Taylor, N.S.; Xu, S.; Horwitz, B.H.; Erdman, S.E. Gastroenteritis in NF-kappaB-deficient mice is produced with wild-type Camplyobacter jejuni but not with C. jejuni lacking cytolethal distending toxin despite persistent colonization with both strains. Infect. Immun. 2004, 72, 1116–1125. [Google Scholar]
- Hickey, T.E.; McVeigh, A.L.; Scott, D.A.; Michielutti, R.E.; Bixby, A.; Carroll, S.A.; Bourgeois, A.L.; Guerry, P. Campylobacter jejunic cytolethal distending toxin mediates release of interleukin-8 from intestinal epithelial cells. Infect. Immun. 2000, 68, 6535–6541. [Google Scholar]
- Chien, C.C.; Taylor, N.S.; Ge, Z.; Schauer, D.B.; Young, V.B.; Fox, J.G. Identification of cdtB homologues and cytolethal distending toxin activity in enterohepatic Helicobacter spp. J. Med. Microbiol. 2000, 49, 525–534. [Google Scholar]
- Ge, Z.; Feng, Y.; Whary, M.T.; Nambiar, P.R.; Xu, S.; Ng, V.; Taylor, N.S.; Fox, J.G. Cytolethal distending toxin is essential for Helicobacter hepaticus colonization in outbred Swiss Webster mice. Infect. Immun. 2005, 73, 3559–3567. [Google Scholar]
- Young, V.B.; Knox, K.A.; Pratt, J.S.; Cortez, J.S.; Mansfield, L.S.; Rogers, A.B.; Fox, J.G.; Schauer, D.B. In vitro and in vivo characterization of Helicobacter hepaticus cytolethal distending toxin mutants. Infect. Immun. 2004, 72, 2521–2527. [Google Scholar]
- Ge, Z.; Rogers, A.B.; Feng, Y.; Lee, A.; Xu, S.; Taylor, N.S.; Fox, J.G. Bacterial cytolethal distending toxin promotes the development of dysplasia in a model of microbially induced hepatocarcinogenesis. Cell. Microbiol. 2007, 9, 2070–2080. [Google Scholar]
- Trees, D.L.; Morse, S.A. Chancroid and Haemophilus ducreyi: An update. Clin. Microbiol. Rev. 1995, 8, 357–375. [Google Scholar]
- Martin, P. Wound healing—aiming for perfect skin regeneration. Science 1997, 276, 75–81. [Google Scholar]
- Park, J.E.; Barbul, A. Understanding the role of immune regulation in wound healing. Am. J. Surg. 2004, 187, 11S–16S. [Google Scholar]
- Wising, C.; Molne, L.; Jonsson, I.M.; Ahlman, K.; Lagergard, T. The cytolethal distending toxin of Haemophilus ducreyi aggravates dermal lesions in a rabbit model of chancroid. Microb. Infect. 2005, 7, 867–874. [Google Scholar]
- Belibasakis, G.N.; Mattsson, A.; Wang, Y.; Chen, C.; Johansson, A. Cell cycle arrest of human gingival fibroblasts and periodontal ligament cells by Actinobacillus actinomycetemcomitans: Involvement of the cytolethal distending toxin. APMIS 2004, 112, 674–685. [Google Scholar]
- Svensson, L.A.; Henning, P.; Lagergard, T. The cytolethal distending toxin of Haemophilus ducreyi inhibits endothelial cell proliferation. Infect. Immun. 2002, 70, 2665–2669. [Google Scholar]
- Svensson, L.; Tarkowski, A.; Thelestam, M.; Lagergård, T. The impact of Haemophilus ducreyi cytolethal distending toxin on cells involved in immune response. Microb. Pathog. 2001, 30, 157–166. [Google Scholar]
- Xu, T.; Lundqvist, A.; Ahmed, H.J.; Eriksson, K.; Yang, Y.; Lagergard, T. Interactions of Haemophilus ducreyi and purified cytolethal distending toxin with human monocyte-derived dendritic cells, macrophages and CD4+ T cells. Microb. Infect. 2004, 6, 1171–1181. [Google Scholar]
- Henderson, B.; Ward, J.M.; Ready, D. Aggregatibacter (Actinobacillus) actinomycetemcomitans: A triple A* periodontopathogen? Periodontol 2000 2010, 54, 78–105. [Google Scholar] [CrossRef] [PubMed]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar]
- Belibasakis, G.; Johansson, A.; Wang, Y.; Chen, C.; Kalfas, S.; Lerner, U.H. The cytolethal distending toxin induces receptor activator of NF-kB ligand expression in human gingival fibroblasts and periodontal ligament cells. Infect. Immun. 2005, 73, 342–351. [Google Scholar]
- Belibasakis, G.N.; Brage, M.; Lagergard, T.; Johansson, A. Cytolethal distending toxin upregulates RANKL expression in Jurkat T-cells. APMIS 2008, 116, 499–506. [Google Scholar]
- Akifusa, S.; Poole, S.; Lewthwaite, J.; Henderson, B.; Nair, S.P. Recombinant Actinobacillus actinomycetemcomitans cytolethal distending toxin proteins are required to interact to inhibit human cell cycle progression and to stimulate human leukocyte cytokine synthesis. Infect. Immun. 2001, 69, 5925–5930. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guerra, L.; Cortes-Bratti, X.; Guidi, R.; Frisan, T. The Biology of the Cytolethal Distending Toxins. Toxins 2011, 3, 172-190. https://doi.org/10.3390/toxins3030172
Guerra L, Cortes-Bratti X, Guidi R, Frisan T. The Biology of the Cytolethal Distending Toxins. Toxins. 2011; 3(3):172-190. https://doi.org/10.3390/toxins3030172
Chicago/Turabian StyleGuerra, Lina, Ximena Cortes-Bratti, Riccardo Guidi, and Teresa Frisan. 2011. "The Biology of the Cytolethal Distending Toxins" Toxins 3, no. 3: 172-190. https://doi.org/10.3390/toxins3030172