Identification of Small Molecule Inhibitors of Clostridium perfringens ε-Toxin Cytotoxicity Using a Cell-Based High-Throughput Screen

Abstract
:
1. Introduction
2. Materials and Methods
2.1. Cell culture
2.2. Recombinant ε-toxin expression and purification
2.3. Compound library
2.4. High-throughput screen for ε-toxin inhibitors
2.5. Secondary screens
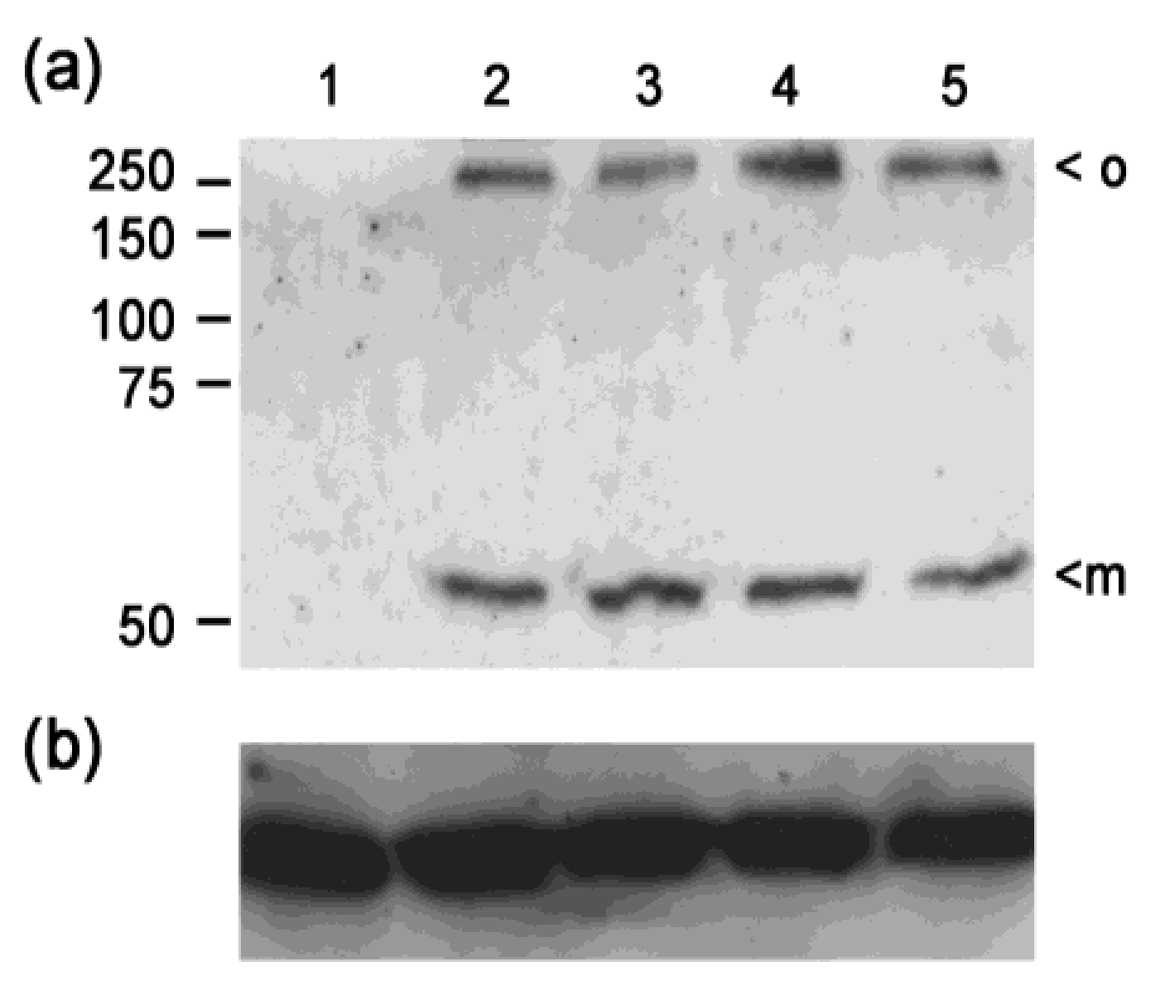
2.6. Immunoblotting
2.7. Propidium iodide influx
2.8. Select agent
3. Results and Discussion
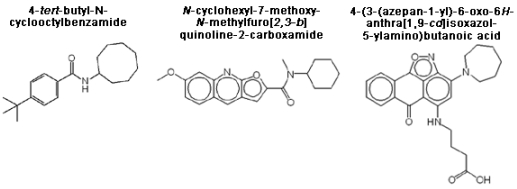
3.1. Assay validation

3.2. High-throughput screen
3.3. Secondary screens
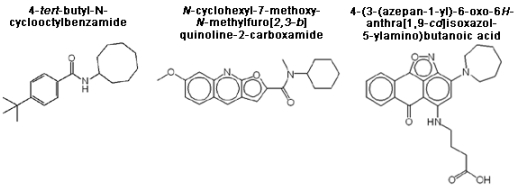
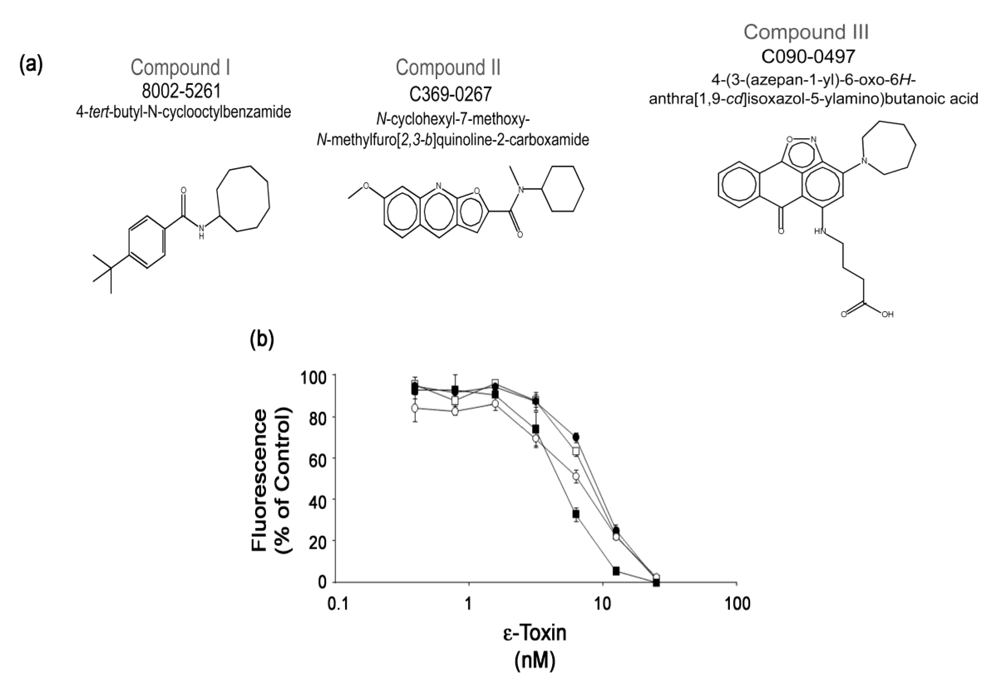
3.4. Analysis of confirmed hits


3.5. Structure-activity relationships

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | CT50 (25 μM) a | P < 0.05 (25 μM) b | CT50 (50 μM) a | P < 0.05 (50 μM) b |
|---|---|---|---|---|
 | 8.1 | Yes | 10.1 | Yes |
 | 7.2 | Yes | 9.1 | Yes |
 | 7.4 | Yes | 8.5 | Yes |
 | 6.1 | Yes | 7.2 | Yes |
 | 5.9 | Yes | 6.7 | Yes |
 | 5.8 | Yes | 6.6 | Yes |
 | 4.7 | No | 5.7 | No |
 | 5.1 | No | 5.7 | No |
| No compound | 4.4 | - | 4.8 | - |
4. Conclusions
Acknowledgements
References
- Gill, D.M. Bacterial toxins: A table of lethal amounts. Microbiol. Rev. 1982, 46, 86–94. [Google Scholar]
- Minami, J.; Katayama, S.; Matsushita, O.; Matsushita, C.; Okabe, A. Lambda-toxin of Clostridium perfringens activates the precursor of epsilon-toxin by releasing its N- and C-terminal peptides. Microbiol. Immunol. 1997, 41, 527–535. [Google Scholar]
- Gleeson-White, M.H.; Bullen, J.J. Clostridium welchii epsilon toxin in the intestinal contents of man. Lancet 1955, 268, 384–385. [Google Scholar]
- Kohn, J.; Warrack, G.H. Recovery of clostridium welchii type d from man. Lancet 1955, 268, 385. [Google Scholar]
- Smith, L.D.S.; Williams, B.L. The Pathogenic Anaerobic Bacteria, 3rd; Charles, C., Ed.; Thomas: Springfield, IL, USA, 1984. [Google Scholar]
- Miller, C.; Florman, S.; Kim-Schluger, L.; Lento, P.; De La Garza, J.; Wu, J.; Xie, B.; Zhang, W.; Bottone, E.; Zhang, D.; Schwartz, M. Fulminant and fatal gas gangrene of the stomach in a healthy live liver donor. Liver Transpl. 2004, 10, 1315–1319. [Google Scholar]
- Morinaga, G.; Nakamura, T.; Yoshizawa, J.; Nishida, S. Isolation of Clostridium perfringens type D from a case of gas gangrene. J. Bacteriol. 1965, 90, 826. [Google Scholar]
- Soler-Jover, A.; Blasi, J.; de Aranda, I.G.; Navarro, P.; Gibert, M.; Popoff, M.R.; Martin-Satue, M. Effect of epsilon toxin-GFP on MDCK cells and renal tubules in vivo. J. Histochem. Cytochem. 2004, 52, 931–942. [Google Scholar]
- Soler-Jover, A.; Dorca, J.; Popoff, M.R.; Gibert, M.; Saura, J.; Tusell, J.M.; Serratosa, J.; Blasi, J.; Martin-Satue, M. Distribution of Clostridium perfringens epsilon toxin in the brains of acutely intoxicated mice and its effect upon glial cells. Toxicon 2007, 50, 530–540. [Google Scholar]
- Tamai, E.; Ishida, T.; Miyata, S.; Matsushita, O.; Suda, H.; Kobayashi, S.; Sonobe, H.; Okabe, A. Accumulation of Clostridium perfringens epsilon-toxin in the mouse kidney and its possible biological significance. Infect. Immun. 2003, 71, 5371–5375. [Google Scholar]
- Miyata, S.; Matsushita, O.; Minami, J.; Katayama, S.; Shimamoto, S.; Okabe, A. Cleavage of a C-terminal peptide is essential for heptamerization of Clostridium perfringens epsilon-toxin in the synaptosomal membrane. J. Biol. Chem. 2001, 276, 13778–13783. [Google Scholar]
- Miyata, S.; Minami, J.; Tamai, E.; Matsushita, O.; Shimamoto, S.; Okabe, A. Clostridium perfringens epsilon-toxin forms a heptameric pore within the detergent-insoluble microdomains of madin-darby canine kidney cells and rat synaptosomes. J. Biol. Chem. 2002, 277, 39463–39468. [Google Scholar]
- Petit, L.; Gibert, M.; Gillet, D.; Laurent-Winter, C.; Boquet, P.; Popoff, M.R. Clostridium perfringens epsilon-toxin acts on MDCK cells by forming a large membrane complex. J. Bacteriol. 1997, 179, 6480–6487. [Google Scholar]
- Petit, L.; Maier, E.; Gibert, M.; Popoff, M.R.; Benz, R. Clostridium perfringens epsilon toxin induces a rapid change of cell membrane permeability to ions and forms channels in artificial lipid bilayers. J. Biol. Chem. 2001, 276, 15736–15740. [Google Scholar]
- Petit, L.; Gibert, M.; Gourch, A.; Bens, M.; Vandewalle, A.; Popoff, M.R. Clostridium perfringens epsilon toxin rapidly decreases membrane barrier permeability of polarized MDCK cells. Cell. Microbiol. 2003, 5, 155–164. [Google Scholar]
- McClain, M.S.; Cover, T.L. Functional analysis of neutralizing antibodies against Clostridium perfringens epsilon-toxin. Infect. Immun. 2007, 75, 1785–1793. [Google Scholar]
- Nagahama, M.; Ochi, S.; Sakurai, J. Assembly of Clostridium perfringens epsilon-toxin on MDCK cell membrane. J. Nat. Toxins 1998, 7, 291–302. [Google Scholar]
- Lindsay, C.D. Assessment of aspects of the toxicity of Clostridium perfringens epsilon-toxin using the MDCK cell line. Hum. Exp. Toxicol. 1996, 15, 904–908. [Google Scholar]
- Odendaal, M.W.; Visser, J.J.; Botha, W.J.; Prinsloo, H. The passive protection of lambs against Clostridium perfringens type D with semi-purified hyperimmune serum. Onderstepoort J. Vet. Res. 1988, 55, 47–50. [Google Scholar]
- Uzal, F.A.; Bodero, D.A.; Kelly, W.R.; Nielsen, K. Variability of serum antibody responses of goat kids to a commercial Clostridium perfringens epsilon toxoid vaccine. Vet. Rec. 1998, 143, 472–474. [Google Scholar]
- Aiello, S.E. Merck Veterinary Manual, 8th ed; Merck and Co., Inc.: Whitehouse Station, NJ, USA, 2003. [Google Scholar]
- Pelish, T.M.; McClain, M.S. Dominant-negative inhibitors of the Clostridium perfringens epsilon-toxin. J. Biol. Chem. 2009, 284, 29446–29453. [Google Scholar]
- Hauer, P.J.; Clough, N.E. Development of monoclonal antibodies suitable for use in antigen quantification potency tests for clostridial veterinary vaccines. Dev. Biol. Stand. 1999, 101, 85–94. [Google Scholar]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar]
- Payne, D.W.; Williamson, E.D.; Havard, H.; Modi, N.; Brown, J. Evaluation of a new cytotoxicity assay for Clostridium perfringens type D epsilon toxin. FEMS Microbiol. Lett. 1994, 116, 161–167. [Google Scholar]
- Makarenkov, V.; Kevorkov, D.; Zentilli, P.; Gagarin, A.; Malo, N.; Nadon, R. Hts-corrector: Software for the statistical analysis and correction of experimental high-throughput screening data. Bioinformatics 2006, 22, 1408–1409. [Google Scholar]
- Tukey, J.W. A survey of sampling from contaminated distributions. In Contributions to Probability and Statistics; Olkin, I., Ed.; Stanford University Press: Stanford, CA, USA, 1960; pp. 448–485. [Google Scholar]
- Malo, N.; Hanley, J.A.; Cerquozzi, S.; Pelletier, J.; Nadon, R. Statistical practice in high-throughput screening data analysis. Nat. Biotechnol. 2006, 24, 167–175. [Google Scholar]
- Brideau, C.; Gunter, B.; Pikounis, B.; Liaw, A. Improved statistical methods for hit selection in high-throughput screening. J. Biomol. Screen. 2003, 8, 634–647. [Google Scholar]
- Katz, D.; Ito, E.; Lau, K.S.; Mocanu, J.D.; Bastianutto, C.; Schimmer, A.D.; Liu, F.F. Increased efficiency for performing colony formation assays in 96-well plates: Novel applications to combination therapies and high-throughput screening. Biotechniques 2008, 44, ix–xiv. [Google Scholar]
- Weiss, J.; Burhenne, J.; Riedel, K.D.; Haefeli, W.E. Poor solubility limiting significance of in-vitro studies with HIV protease inhibitors. AIDS 2002, 16, 674–676. [Google Scholar]
- Weiss, J.; Haefeli, W.E. Evaluation of inhibitory potencies for compounds inhibiting P-glycoprotein but without maximum effects: F2 values. Drug Metab. Dispos. 2006, 34, 203–207. [Google Scholar]
- Dorca-Arevalo, J.; Soler-Jover, A.; Gibert, M.; Popoff, M.R.; Martin-Satue, M.; Blasi, J. Binding of varepsilon-toxin from Clostridium perfringens in the nervous system. Vet. Microbiol. 2008, 131, 14–25. [Google Scholar]
- Casadevall, A. Passive antibody administration (immediate immunity) as a specific defense against biological weapons. Emerg. Infect. Dis. 2002, 8, 833–841. [Google Scholar]
- Lee, Y.S.; Bergson, P.; He, W.S.; Mrksich, M.; Tang, W.J. Discovery of a small molecule that inhibits the interaction of anthrax edema factor with its cellular activator, calmodulin. Chem. Biol. 2004, 11, 1139–1146. [Google Scholar]
- Soelaiman, S.; Wei, B.Q.; Bergson, P.; Lee, Y.S.; Shen, Y.; Mrksich, M.; Shoichet, B.K.; Tang, W.J. Structure-based inhibitor discovery against adenylyl cyclase toxins from pathogenic bacteria that cause anthrax and whooping cough. J. Biol. Chem. 2003, 278, 25990–25997. [Google Scholar]
- Panchal, R.G.; Hermone, A.R.; Nguyen, T.L.; Wong, T.Y.; Schwarzenbacher, R.; Schmidt, J.; Lane, D.; McGrath, C.; Turk, B.E.; Burnett, J.; Aman, M.J.; Little, S.; Sausville, E.A.; Zaharevitz, D.W.; Cantley, L.C.; Liddington, R.C.; Gussio, R.; Bavari, S. Identification of small molecule inhibitors of anthrax lethal factor. Nat. Struct. Mol. Biol. 2004, 11, 67–72. [Google Scholar]
- Burnett, J.C.; Schmidt, J.J.; Stafford, R.G.; Panchal, R.G.; Nguyen, T.L.; Hermone, A.R.; Vennerstrom, J.L.; McGrath, C.F.; Lane, D.J.; Sausville, E.A. Novel small molecule inhibitors of botulinum neurotoxin a metalloprotease activity. Biochem. Biophys. Res. Commun. 2003, 310, 84–93. [Google Scholar]
- Zhu, P.J.; Hobson, J.P.; Southall, N.; Qiu, C.; Thomas, C.J.; Lu, J.; Inglese, J.; Zheng, W.; Leppla, S.H.; Bugge, T.H.; Austin, C.P.; Liu, S. Quantitative high-throughput screening identifies inhibitors of anthrax-induced cell death. Bioorg. Med. Chem. 2009, 17, 5139–5145. [Google Scholar]
- Saenz, J.B.; Doggett, T.A.; Haslam, D.B. Identification and characterization of small molecules that inhibit intracellular toxin transport. Infect. Immun. 2007, 75, 4552–4561. [Google Scholar]
- Wahome, P.G.; Bai, Y.; Neal, L.M.; Robertus, J.D.; Mantis, N.J. Identification of small-molecule inhibitors of ricin and shiga toxin using a cell-based high-throughput screen. Toxicon 2010. [Google Scholar]
- Ma, T.; Thiagarajah, J.R.; Yang, H.; Sonawane, N.D.; Folli, C.; Galietta, L.J.; Verkman, A.S. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J. Clin. Invest. 2002, 110, 1651–1658. [Google Scholar]
- Peinado, J.R.; Kacprzak, M.M.; Leppla, S.H.; Lindberg, I. Cross-inhibition between furin and lethal factor inhibitors. Biochem. Biophys. Res. Commun. 2004, 321, 601–605. [Google Scholar]
- Cole, A.R.; Gibert, M.; Popoff, M.; Moss, D.S.; Titball, R.W.; Basak, A.K. Clostridium perfringens epsilon-toxin shows structural similarity to the pore-forming toxin aerolysin. Nat. Struct. Mol. Biol. 2004, 11, 797–798. [Google Scholar]
- Parker, M.W.; Buckley, J.T.; Postma, J.P.; Tucker, A.D.; Leonard, K.; Pattus, F.; Tsernoglou, D. Structure of the aeromonas toxin proaerolysin in its water-soluble and membrane-channel states. Nature 1994, 367, 292–295. [Google Scholar]
- Wheeler, D.L.; Barrett, T.; Benson, D.A.; Bryant, S.H.; Canese, K.; Chetvernin, V.; Church, D.M.; DiCuccio, M.; Edgar, R.; Federhen, S.; Geer, L.Y.; Helmberg, W.; Kapustin, Y.; Kenton, D.L.; Khovayko, O.; Lipman, D.J.; Madden, T.L.; Maglott, D.R.; Ostell, J.; Pruitt, K.D.; Schuler, G.D.; Schriml, L.M.; Sequeira, E.; Sherry, S.T.; Sirotkin, K.; Souvorov, A.; Starchenko, G.; Suzek, T.O.; Tatusov, R.; Tatusova, T.A.; Wagner, L.; Yaschenko, E. Database resources of the national center for biotechnology information. Nucleic Acids Res. 2006, 34, D173–D180. [Google Scholar]
- Seiler, K.P.; George, G.A.; Happ, M.P.; Bodycombe, N.E.; Carrinski, H.A.; Norton, S.; Brudz, S.; Sullivan, J.P.; Muhlich, J.; Serrano, M.; Ferraiolo, P.; Tolliday, N.J.; Schreiber, S.L.; Clemons, P.A. Chembank: A small-molecule screening and cheminformatics resource database. Nucleic Acids Res. 2008, 36, D351–D359. [Google Scholar]
- Wunberg, T.; Hendrix, M.; Hillisch, A.; Lobell, M.; Meier, H.; Schmeck, C.; Wild, H.; Hinzen, B. Improving the hit-to-lead process: Data-driven assessment of drug-like and lead-like screening hits. Drug Discov. Today 2006, 11, 175–180. [Google Scholar]
- Keseru, G.M.; Makara, G.M. Hit discovery and hit-to-lead approaches. Drug Discov. Today 2006, 11, 741–748. [Google Scholar]
| Compound | z-score |
|---|---|
 | 34.0 |
 | 28.1 |
 | 21.3 |
 | 6.6 |
 | 5.1 |
 | 3.8 |
 | 3.8 |
 | 3.6 |
 | 2.1 |
 | 2.1 |
 | 2.0 |
 | 1.5 |
 | 1.3 |
 | 1.3 |
 | 1.1 |
 | 0.9 |
 | 0.6 |
 | 0.5 |
 | 0.4 |
 | 0.3 |
 | 0.2 |
 | 0.2 |
 | 0.1 |
 | 0.0 |
 | 0.0 |
 | 0.0 |
 | 0.0 |
 | -0.2 |
 | -0.2 |
 | -0.3 |
 | -0.3 |
 | -0.4 |
 | -0.4 |
 | -0.4 |
 | -0.6 |
 | -0.6 |
 | -0.8 |
 | -0.8 |
 | -0.9 |
 | -1.1 |
 | -1.5 |
 | -1.6 |
 | -1.8 |
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lewis, M.; Weaver, C.D.; McClain, M.S. Identification of Small Molecule Inhibitors of Clostridium perfringens ε-Toxin Cytotoxicity Using a Cell-Based High-Throughput Screen. Toxins 2010, 2, 1825-1847. https://doi.org/10.3390/toxins2071825
Lewis M, Weaver CD, McClain MS. Identification of Small Molecule Inhibitors of Clostridium perfringens ε-Toxin Cytotoxicity Using a Cell-Based High-Throughput Screen. Toxins. 2010; 2(7):1825-1847. https://doi.org/10.3390/toxins2071825
Chicago/Turabian StyleLewis, Michelle, Charles David Weaver, and Mark S. McClain. 2010. "Identification of Small Molecule Inhibitors of Clostridium perfringens ε-Toxin Cytotoxicity Using a Cell-Based High-Throughput Screen" Toxins 2, no. 7: 1825-1847. https://doi.org/10.3390/toxins2071825