Novel Cytotoxic Vectors Based on Adeno-Associated Virus

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Primary Murine Breast Cancer Cells
2.3. pAAVTetO2 Plasmid Backbone
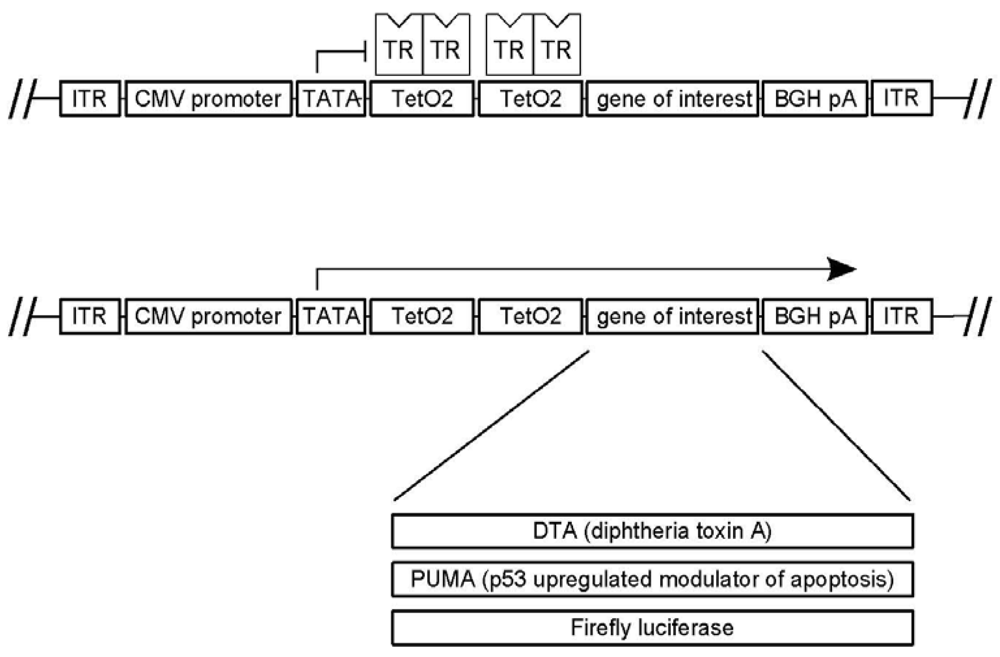
2.4. pAAVTetO2-Based Transgene Constructs
2.5. Production of Recombinant rAAV Vectors
2.6. Titration of AAV Libraries and Recombinant AAV Vectors
2.7. Luciferase Gene Transduction
2.8. Flow Cytometry
2.9. Viability/toxicity Assay
2.10. Statistics
3. Results
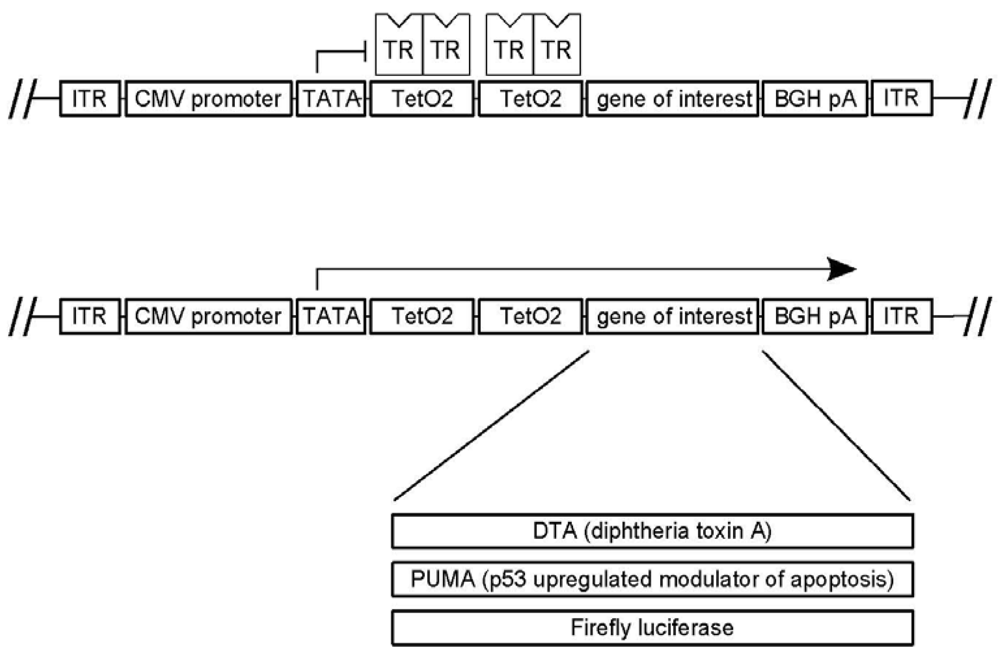
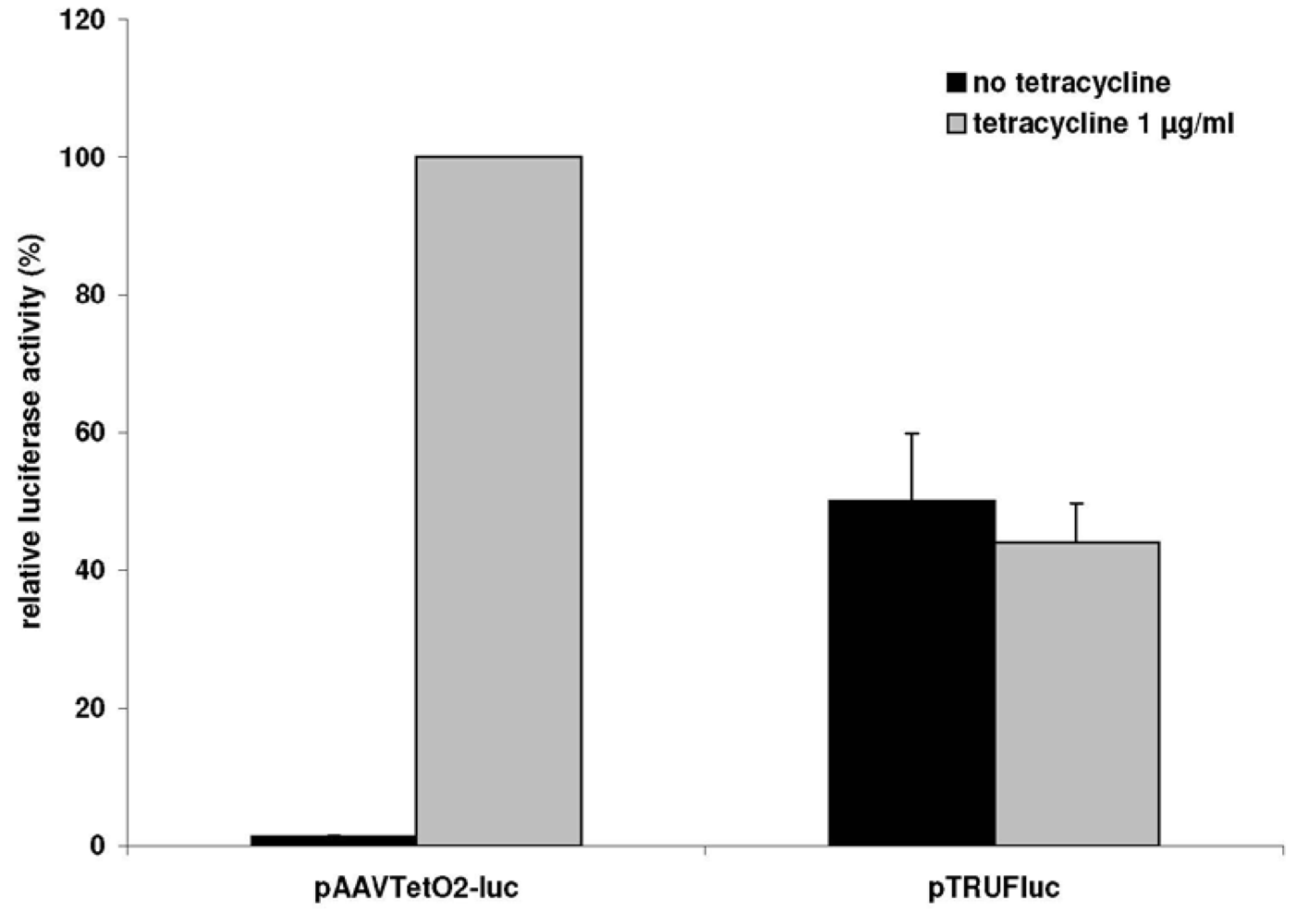
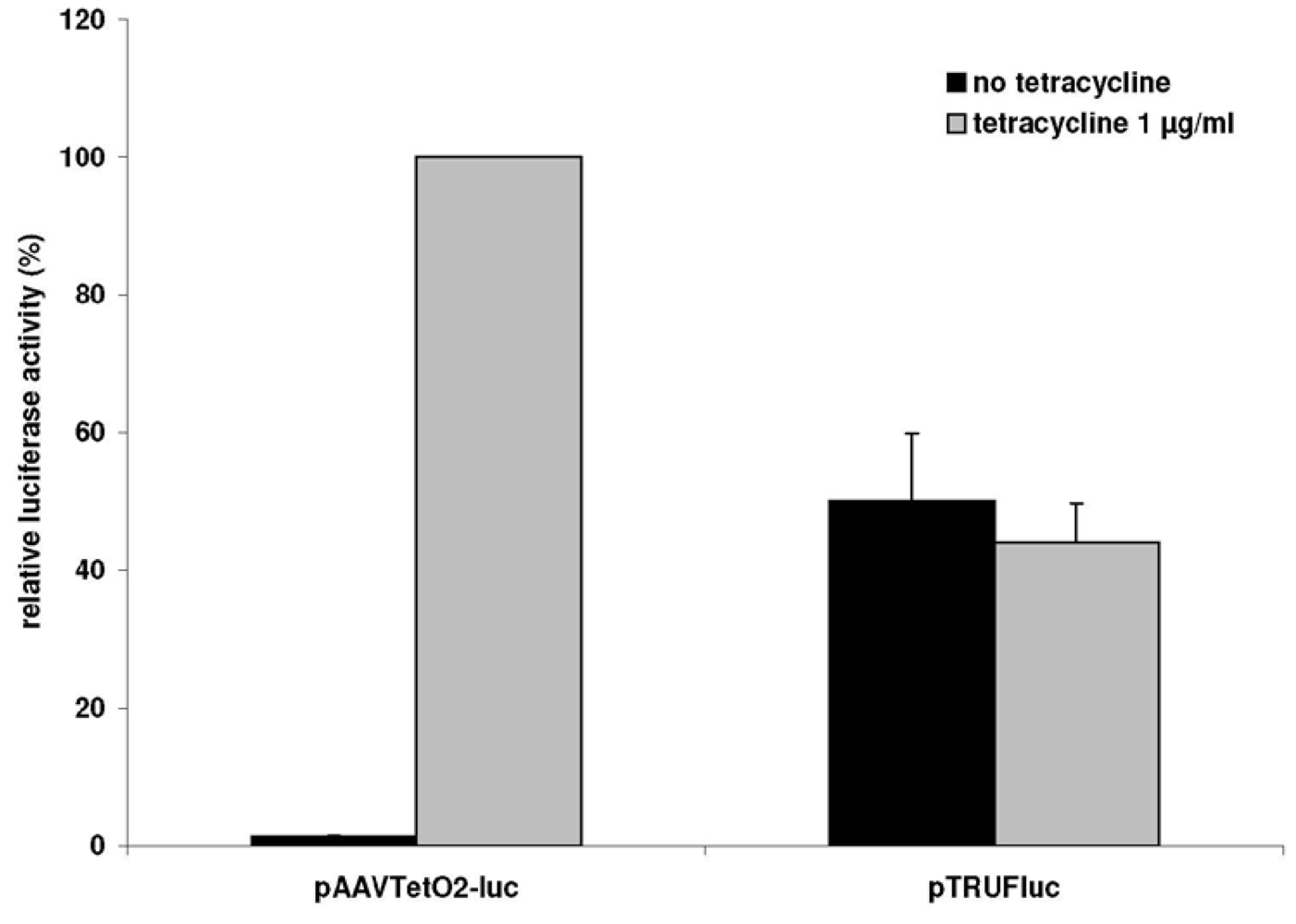
3.1. A Novel Tet-Regulated Plasmid for Production of Toxic AAV Vectors
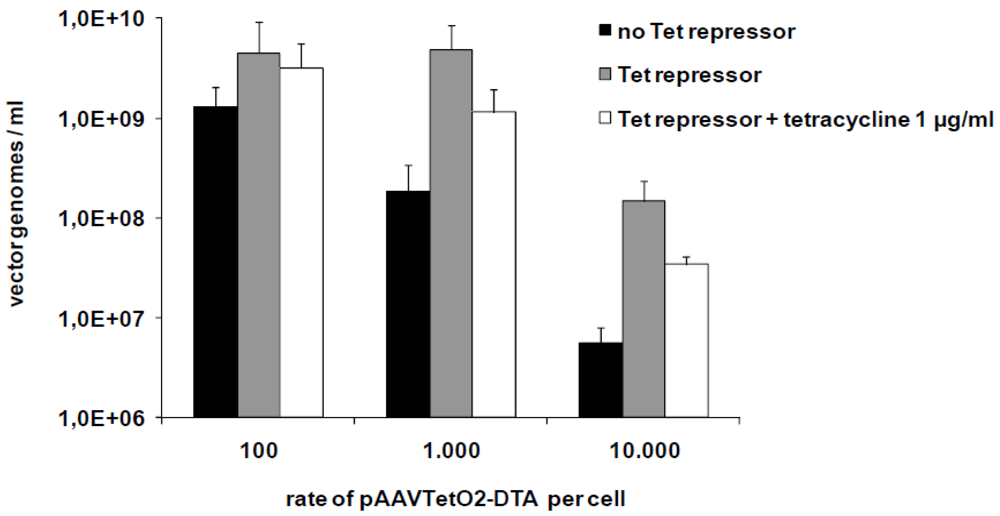
3.2. Production of Toxic Vectors
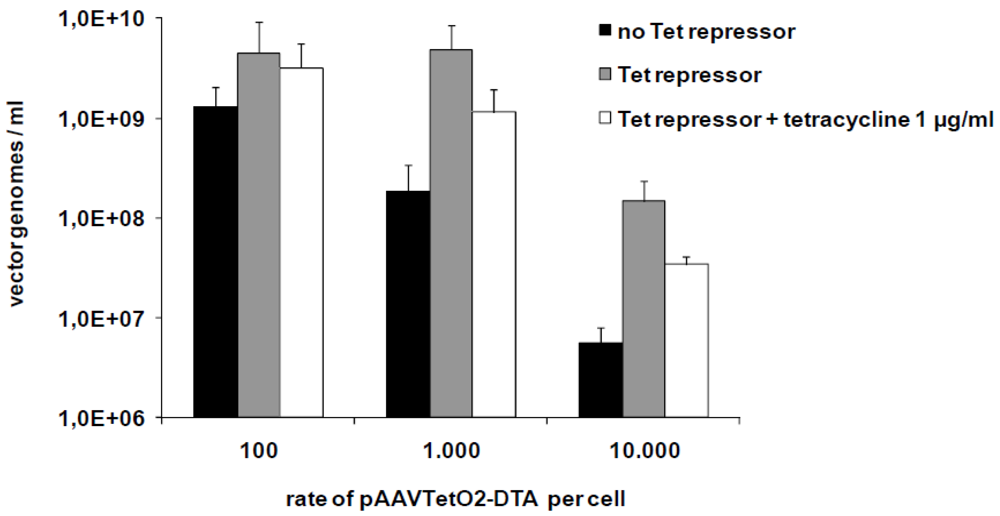
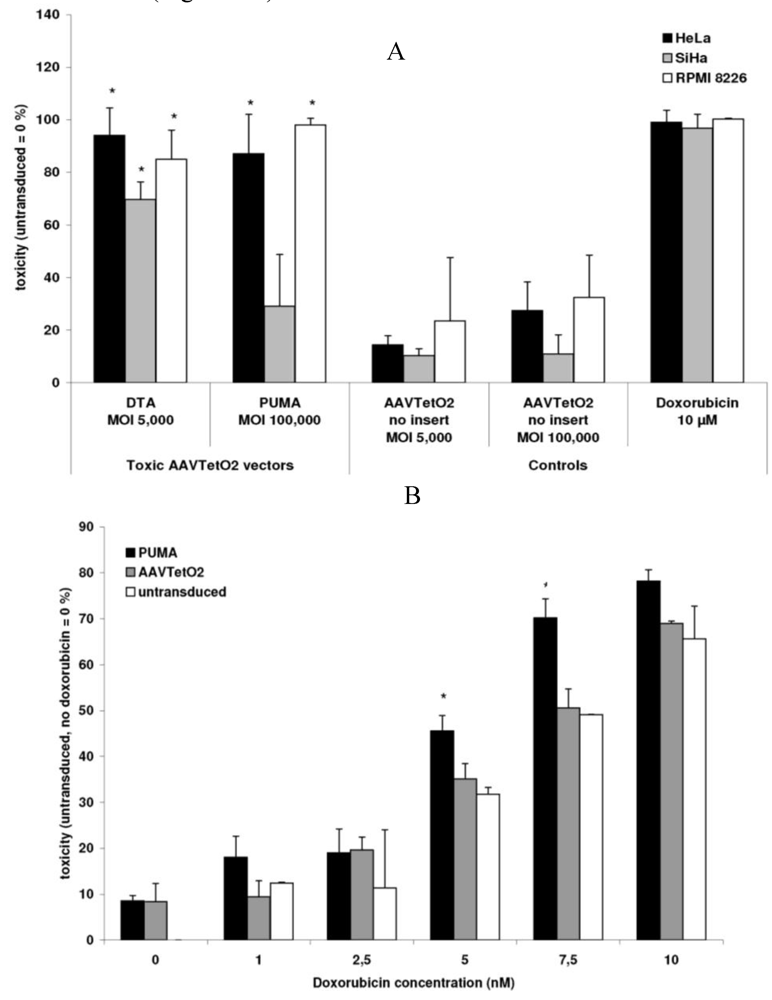
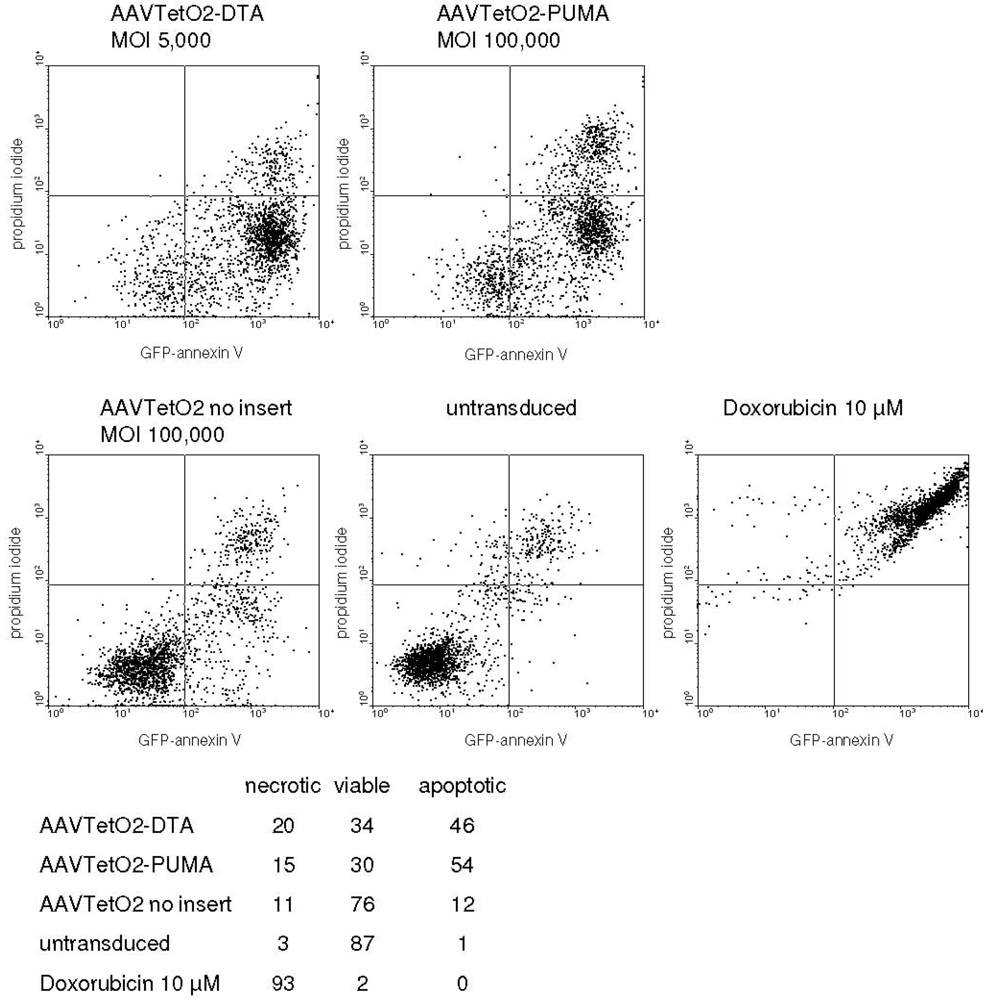
3.3. Evaluation of Novel Toxic Vectors
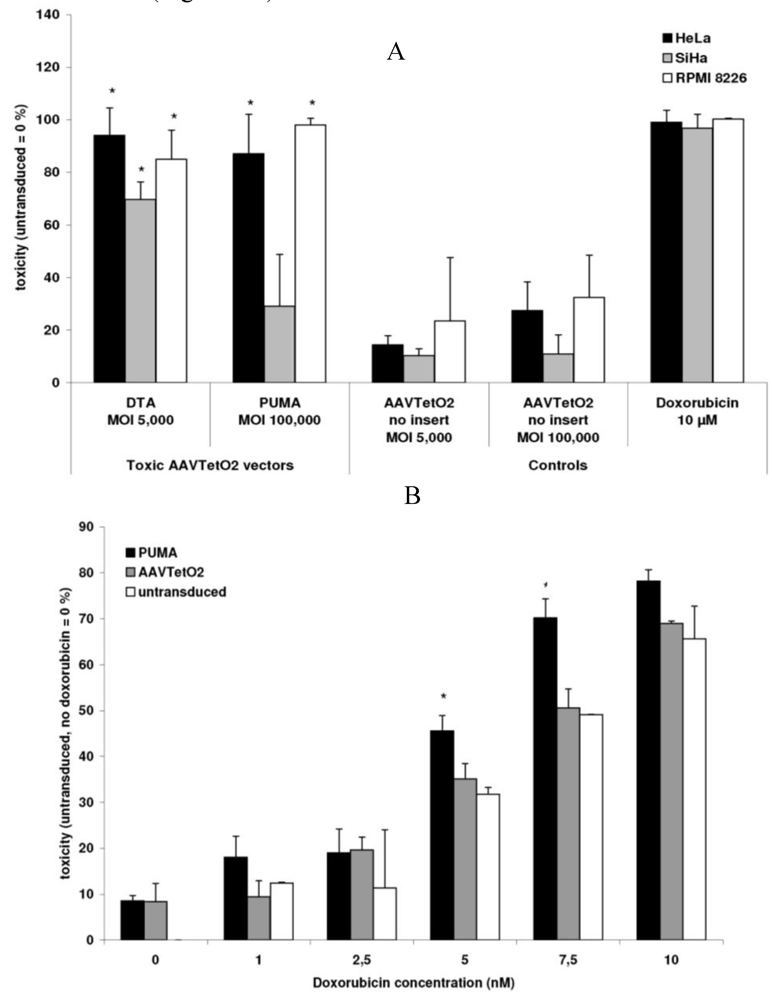
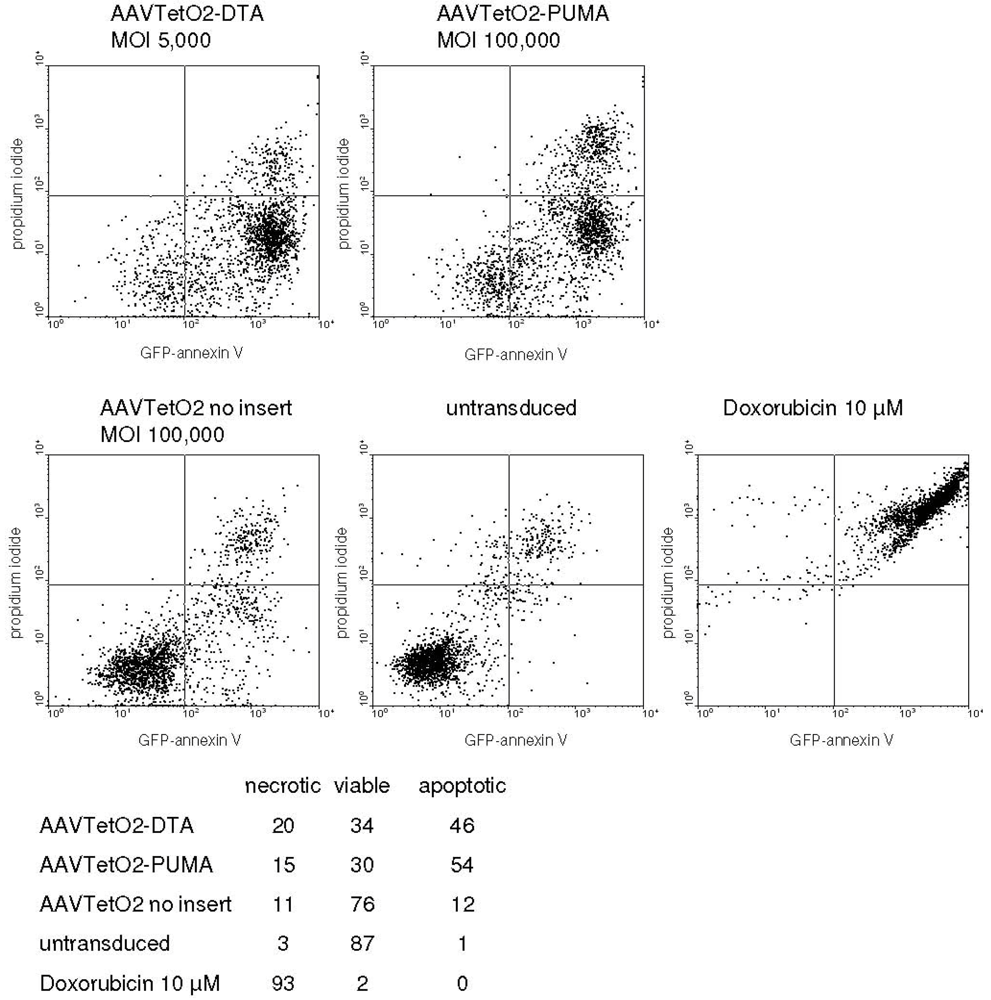
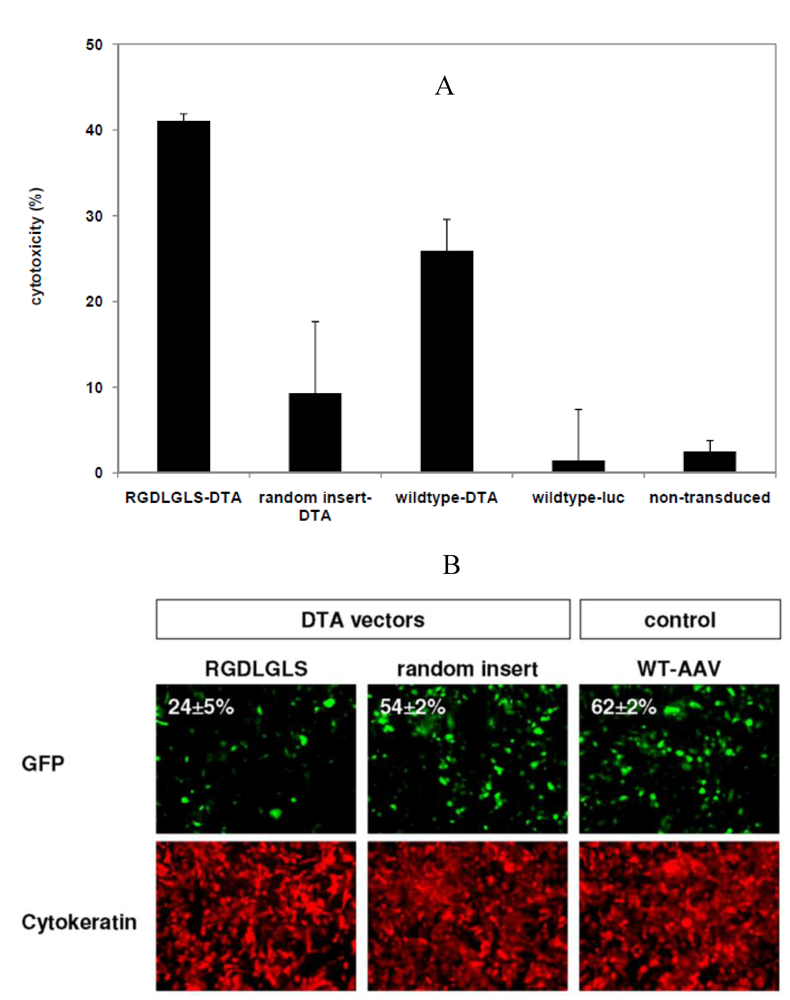
3.4. Targeted Delivery of the DTA Vector to PymT Cells
4. Discussion
5. Conclusions
Acknowledgements
Appendix
References
- Carter, B.J. Adeno-associated virus vectors in clinical trials. Hum. Gene Ther. 2005, 16, 541–550. [Google Scholar]
- Zaiss, A.K.; Muruve, D.A. Immune responses to adeno-associated virus vectors. Curr. Gene Ther. 2005, 5, 323–331. [Google Scholar]
- Bell, P.; Wang, L.; Lebherz, C.; Flieder, D.B.; Bove, M.S.; Wu, D.; Gao, G.P.; Wilson, J.M.; Wivel, N.A. No evidence for tumorigenesis of AAV vectors in a large-scale study in mice. Mol. Ther. 2005, 12, 299–306. [Google Scholar]
- McCarty, D.M.; Young, S.M., Jr.; Samulski, R.J. Integration of adeno-associated virus (AAV) and recombinant AAV vectors. Annu. Rev. Genet. 2004, 38, 819–845. [Google Scholar]
- Michelfelder, S.; Trepel, M. Adeno-associated viral vectors and their redirection to cell-type specific receptors. Adv. Genet. 2009, 67, 29–60. [Google Scholar]
- Nicklin, S.A.; Baker, A.H. Tropism-modified adenoviral and adeno-associated viral vectors for gene therapy. Curr. Gene Ther. 2002, 2, 273–293. [Google Scholar]
- Wu, Z.; Asokan, A.; Samulski, R.J. Adeno-associated virus serotypes: Vector toolkit for human gene therapy. Mol.Ther. 2006, 14, 316–327. [Google Scholar]
- Girod, A.; Ried, M.; Wobus, C.; Lahm, H.; Leike, K.; Kleinschmidt, J.; Deleage, G.; Hallek, M. Genetic capsid modifications allow efficient re-targeting of adeno-associated virus type 2. Nat. Med. 1999, 5, 1052–1056. [Google Scholar]
- Grifman, M.; Trepel, M.; Speece, P.; Gilbert, L.B.; Arap, W.; Pasqualini, R.; Weitzman, M.D. Incorporation of tumor-targeting peptides into recombinant adeno-associated virus capsids. Mol. Ther. 2001, 3, 964–975. [Google Scholar]
- Shi, W.; Bartlett, J.S. RGD inclusion in VP3 provides adeno-associated virus type 2 (AAV2)-based vectors with a heparan sulfate-independent cell entry mechanism. Mol. Ther. 2003, 7, 515–525. [Google Scholar]
- Bartlett, J.S.; Kleinschmidt, J.; Boucher, R.C.; Samulski, R.J. Targeted adeno-associated virus vector transduction of nonpermissive cells mediated by a bispecific F(ab'gamma)2 antibody. Nat. Biotechnol. 1999, 17, 181–186. [Google Scholar]
- Ponnazhagan, S.; Mahendra, G.; Kumar, S.; Thompson, J.A.; Castillas, M., Jr. Conjugate-based targeting of recombinant adeno-associated virus type 2 vectors by using avidin-linked ligands. J. Virol. 2002, 76, 12900–12907. [Google Scholar]
- Trepel, M.G.; Weitzman, M.D.; Pasqualini, R. Molecular adaptors for vascular-targeted adenoviral gene delivery. Hum. Gene Ther. 2000, 11, 1971–1981. [Google Scholar]
- Müller, O.J.; Kaul, F.; Weitzman, M.D.; Pasqualini, R.; Arap, W.; Kleinschmidt, J.A.; Trepel, M. Random peptide libraries displayed on adeno-associated virus to select for targeted gene therapy vectors. Nat. Biotechnol. 2003, 21, 1040–1046. [Google Scholar]
- Waterkamp, D.A.; Muller, O.J.; Ying, Y.; Trepel, M.; Kleinschmidt, J.A. Isolation of targeted AAV2 vectors from novel virus display libraries. J. Gene Med. 2006, 8, 1307–1319. [Google Scholar]
- Michelfelder, S.; Lee, M.K.; deLima-Hahn, E.; Wilmes, T.; Kaul, F.; Muller, O.; Kleinschmidt, J.A.; Trepel, M. Vectors selected from adeno-associated viral display peptide libraries for leukemia cell-targeted cytotoxic gene therapy. Exp. Hematol. 2007, 35, 1766–1776. [Google Scholar]
- Michelfelder, S.; Kohlschutter, J.; Skorupa, A.; Pfennings, S.; Muller, O.; Kleinschmidt, J.A.; Trepel, M. Successful expansion but not complete restriction of tropism of adeno-associated virus by in vivo biopanning of random virus display peptide libraries. PLoS One 2009, 4, e5122. [Google Scholar]
- Ying, Y.; Muller, O.J.; Goehringer, C.; Leuchs, B.; Trepel, M.; Katus, H.A.; Kleinschmidt, J.A. Heart-targeted adeno-associated viral vectors selected by in vivo biopanning of a random viral display peptide library. Gene Ther. 2010, 17, 980–990. [Google Scholar]
- Li, C.; Bowles, D.E.; van Dyke, T.; Samulski, R.J. Adeno-associated virus vectors: Potential applications for cancer gene therapy. Canc. Gene Ther. 2005, 12, 913–925. [Google Scholar]
- Su, H.; Chang, J.C.; Xu, S.M.; Kan, Y.W. Selective killing of AFP-positive hepatocellular carcinoma cells by adeno-associated virus transfer of the herpes simplex virus thymidine kinase gene. Hum. Gene Ther. 1996, 7, 463–470. [Google Scholar]
- Hajitou, A.; Trepel, M.; Lilley, C.E.; Soghomonyan, S.; Alauddin, M.M.; Marini, F.C., 3rd; Restel, B.H.; Ozawa, M.G.; Moya, C.A.; Rangel, R.; Sun, Y.; Zaoui, K.; Schmidt, M.; von Kalle, C.; Weitzman, M.D.; Gelovani, J.G.; Pasqualini, R.; Arap, W. A hybrid vector for ligand-directed tumor targeting and molecular imaging. Cell 2006, 125, 385–398. [Google Scholar]
- Trepel, M.; Stoneham, C.A.; Eleftherohorinou, H.; Mazarakis, N.D.; Pasqualini, R.; Arap, W.; Hajitou, A. A heterotypic bystander effect for tumor cell killing after adeno-associated virus/phage-mediated, vascular-targeted suicide gene transfer. Mol. Canc. Ther. 2009, 8, 2383–2391. [Google Scholar]
- Wang, Y.; Huang, F.; Cai, H.; Zhong, S.; Liu, X.; Tan, W.S. Potent antitumor effect of TRAIL mediated by a novel adeno-associated viral vector targeting to telomerase activity for human hepatocellular carcinoma. J. Gene Med. 2008, 10, 518–526. [Google Scholar]
- Zhang, Y.; Ma, H.; Zhang, J.; Liu, S.; Liu, Y.; Zheng, D. AAV-mediated TRAIL gene expression driven by hTERT promoter suppressed human hepatocellular carcinoma growth in mice. Life Sci. 2008, 82, 1154–1161. [Google Scholar]
- Nettelbeck, D.M.; Jerome, V.; Muller, R. A strategy for enhancing the transcriptional activity of weak cell type-specific promoters. Gene Ther. 1998, 5, 1656–1664. [Google Scholar]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar]
- Choe, S.; Bennett, M.J.; Fujii, G.; Curmi, P.M.; Kantardjieff, K.A.; Collier, R.J.; Eisenberg, D. The crystal structure of diphtheria toxin. Nature 1992, 357, 216–222. [Google Scholar]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell. 2001, 7, 683–694. [Google Scholar]
- Guy, C.T.; Cardiff, R.D.; Muller, W.J. Induction of mammary tumors by expression of polyomavirus middle T oncogene: a transgenic mouse model for metastatic disease. Mol. Cell. Biol. 1992, 12, 954–961. [Google Scholar]
- Grimm, D.; Kern, A.; Pawlita, M.; Ferrari, F.; Samulski, R.; Kleinschmidt, J. Titration of AAV-2 particles via a novel capsid ELISA: packaging of genomes can limit production of recombinant AAV-2. Gene Ther. 1999, 6, 1322–1330. [Google Scholar]
- Lee, E.J.; Jameson, J.L. Cell-specific Cre-mediated activation of the diphtheria toxin gene in pituitary tumor cells: Potential for cytotoxic gene therapy. Hum. Gene Ther. 2002, 13, 533–542. [Google Scholar]
- Xiao, X.; Li, J.; Samulski, R.J. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. J. Virol. 1998, 72, 2224–2232. [Google Scholar]
- Hermens, W.T.; ter Brake, O.; Dijkhuizen, P.A.; Sonnemans, M.A.; Grimm, D.; Kleinschmidt, J.A.; Verhaagen, J. Purification of recombinant adeno-associated virus by iodixanol gradient ultracentrifugation allows rapid and reproducible preparation of vector stocks for gene transfer in the nervous system. Hum. Gene Ther. 1999, 10, 1885–1891. [Google Scholar]
- Slater, T.F.; Sawyer, B.; Straeuli, U. Studies on Succinate-Tetrazolium Reductase Systems.Iii. Points of Coupling of Four Different Tetrazolium Salts. Biochim. Biophys. Acta 1963, 77, 383–393. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Method. 1983, 65, 55–63. [Google Scholar]
- Wang, H.; Qian, H.; Yu, J.; Zhang, X.; Zhang, L.; Fu, M.; Liang, X.; Zhan, Q.; Lin, C. Administration of PUMA adenovirus increases the sensitivity of esophageal cancer cells to anticancer drugs. Canc. Biol. Ther. 2006, 5, 380–385. [Google Scholar]
- Yu, J.; Yue, W.; Wu, B.; Zhang, L. PUMA sensitizes lung cancer cells to chemotherapeutic agents and irradiation. Clin. Canc. Res. 2006, 12, 2928–2936. [Google Scholar]
- Freeman, S.M.; Abboud, C.N.; Whartenby, K.A.; Packman, C.H.; Koeplin, D.S.; Moolten, F.L.; Abraham, G.N. The "bystander effect": Tumor regression when a fraction of the tumor mass is genetically modified. Canc. Res. 1993, 53, 5274–5283. [Google Scholar]
- Massuda, E.S.; Dunphy, E.J.; Redman, R.A.; Schreiber, J.J.; Nauta, L.E.; Barr, F.G.; Maxwell, I.H.; Cripe, T.P. Regulated expression of the diphtheria toxin A chain by a tumor-specific chimeric transcription factor results in selective toxicity for alveolar rhabdomyosarcoma cells. Proc. Natl. Acad. Sci. USA 1997, 94, 14701–14706. [Google Scholar]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kohlschütter, J.; Michelfelder, S.; Trepel, M. Novel Cytotoxic Vectors Based on Adeno-Associated Virus. Toxins 2010, 2, 2754-2768. https://doi.org/10.3390/toxins2122754
Kohlschütter J, Michelfelder S, Trepel M. Novel Cytotoxic Vectors Based on Adeno-Associated Virus. Toxins. 2010; 2(12):2754-2768. https://doi.org/10.3390/toxins2122754
Chicago/Turabian StyleKohlschütter, Johannes, Stefan Michelfelder, and Martin Trepel. 2010. "Novel Cytotoxic Vectors Based on Adeno-Associated Virus" Toxins 2, no. 12: 2754-2768. https://doi.org/10.3390/toxins2122754