Diversity and Impact of Prokaryotic Toxins on Aquatic Environments: A Review

Abstract
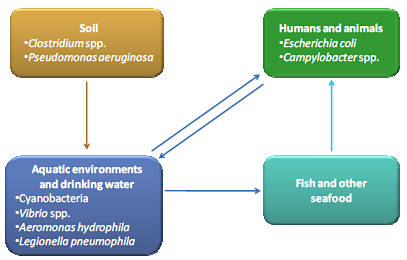
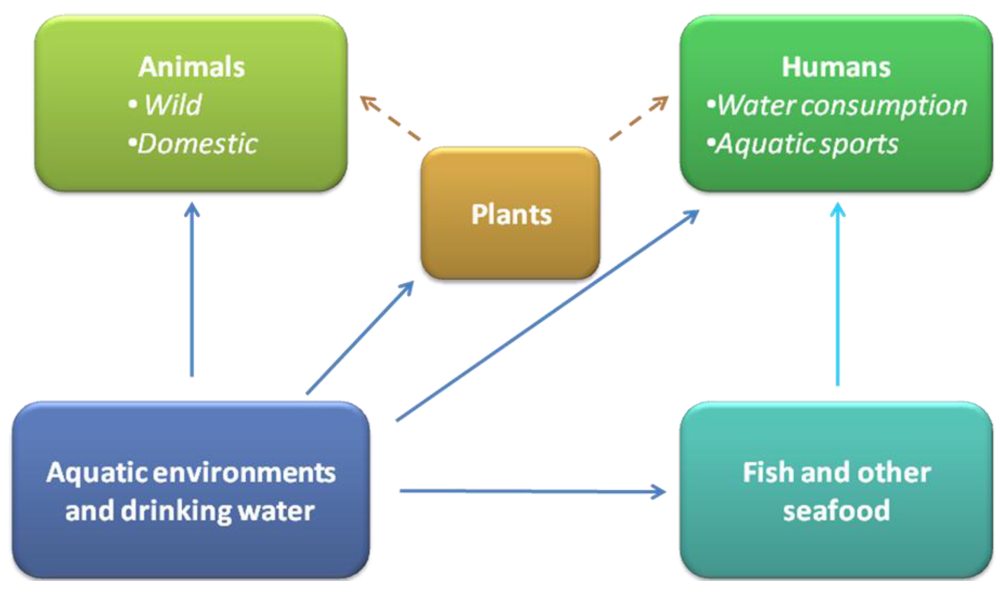
:
1. Introduction
2. Toxins Produced by Cyanobacteria
2.1. Blooms and toxicity
2.2. Importance and impact of the cyanotoxins occurring in aquatic environments
2.3. Hepatotoxins
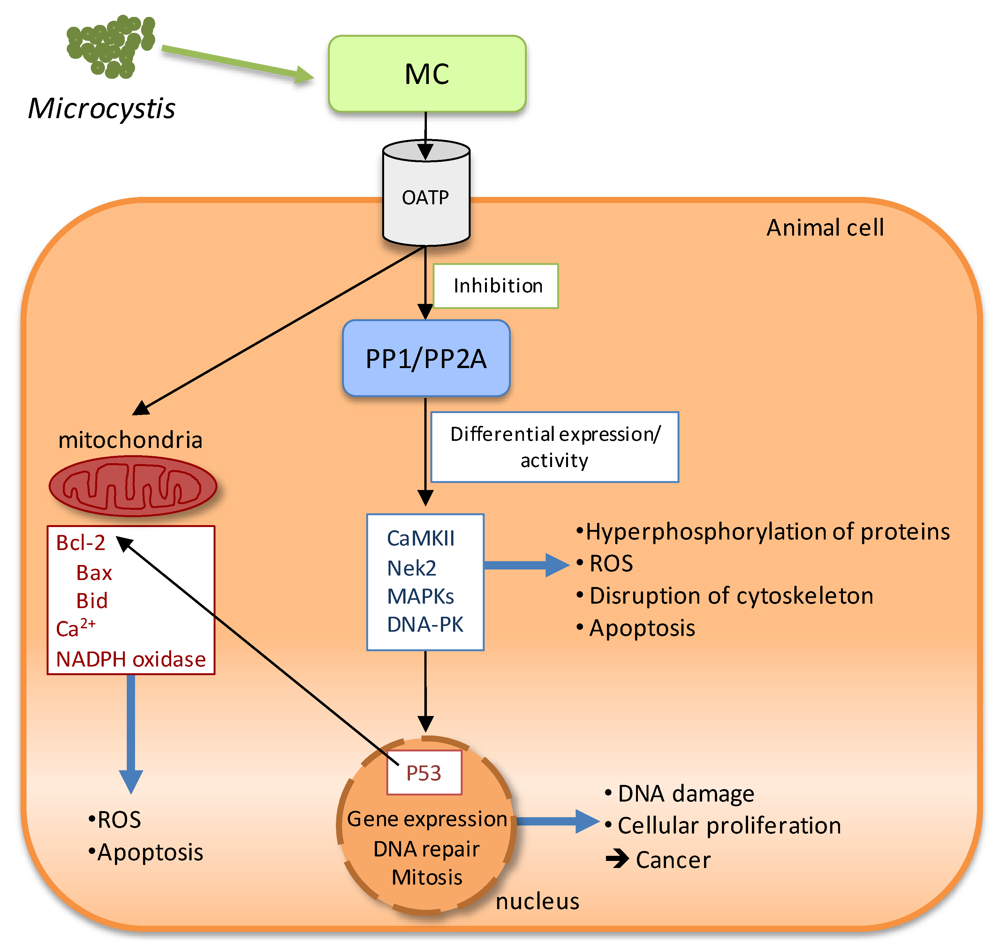
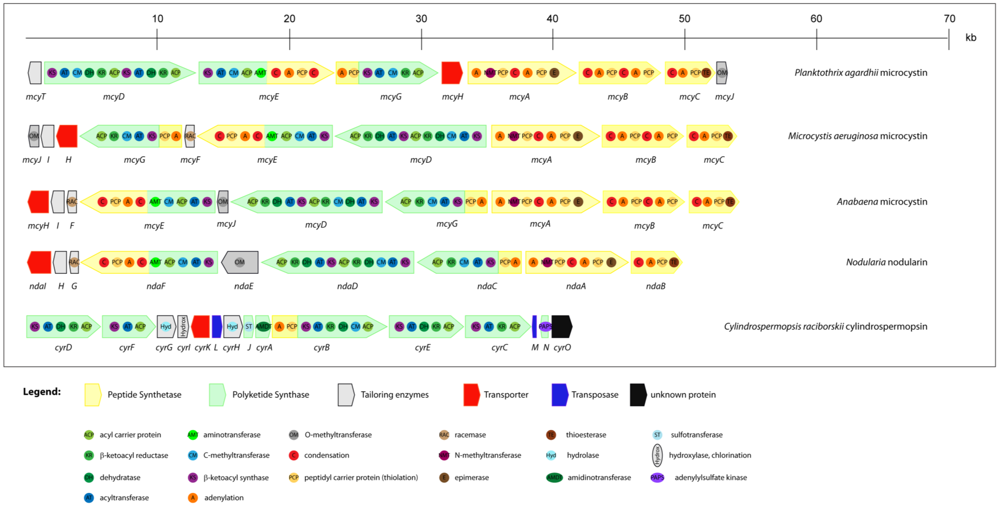
2.3.1. Microcystins

2.3.2. Nodularin
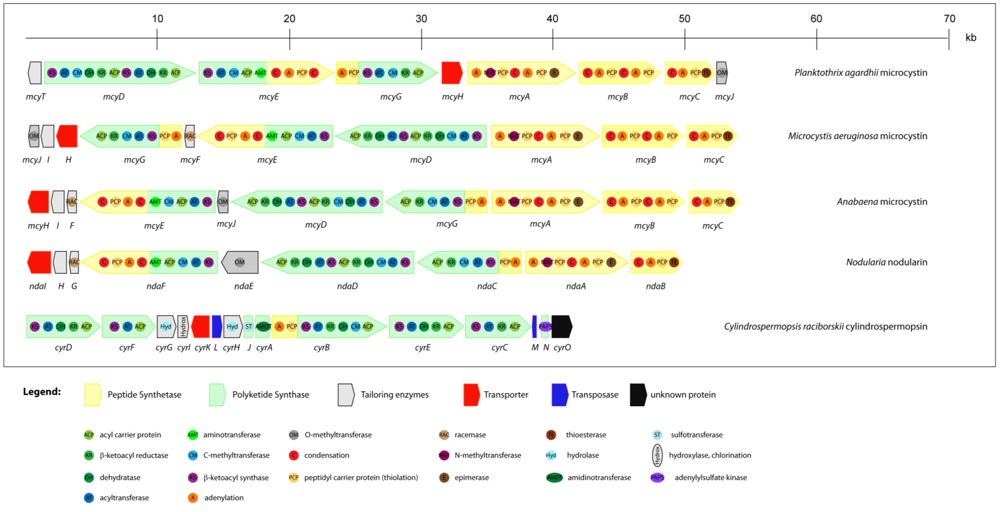
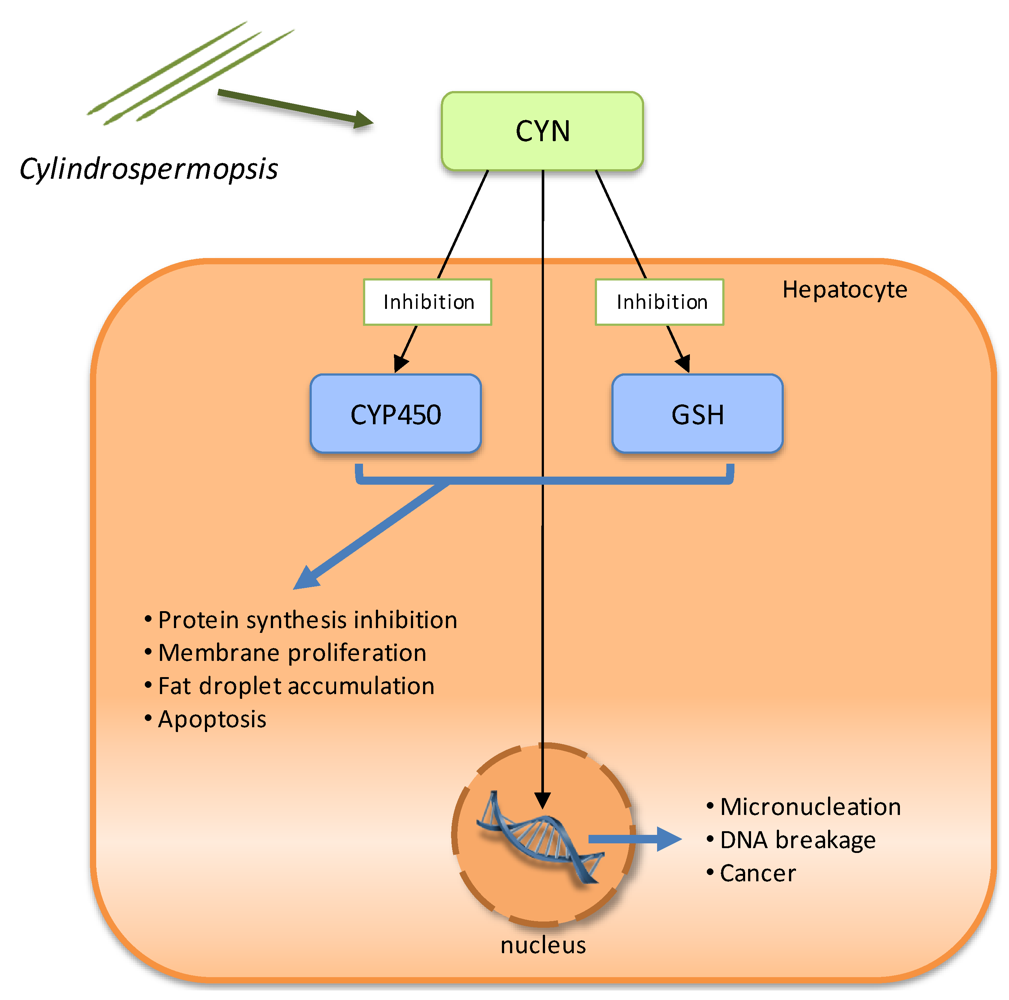
2.4. Cytotoxins: cylindrospermopsin
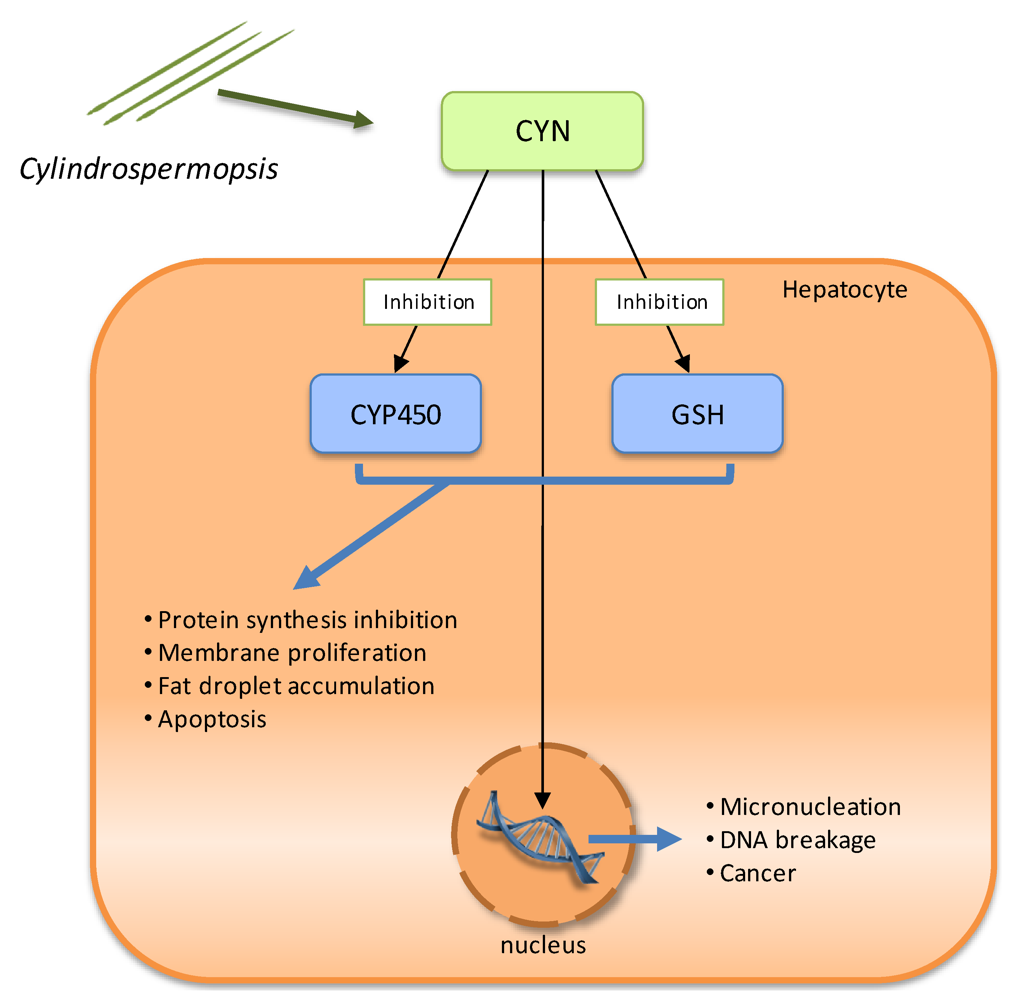
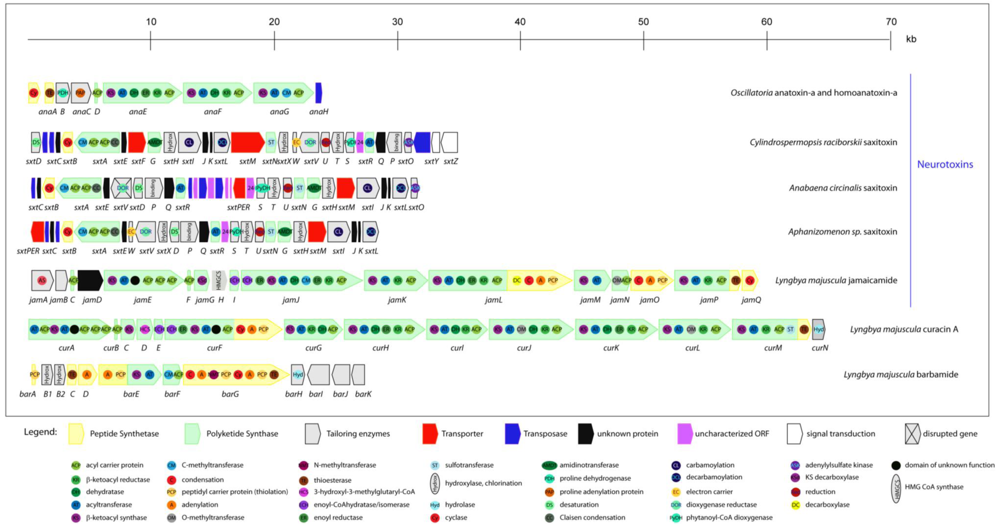
2.5. Neurotoxins
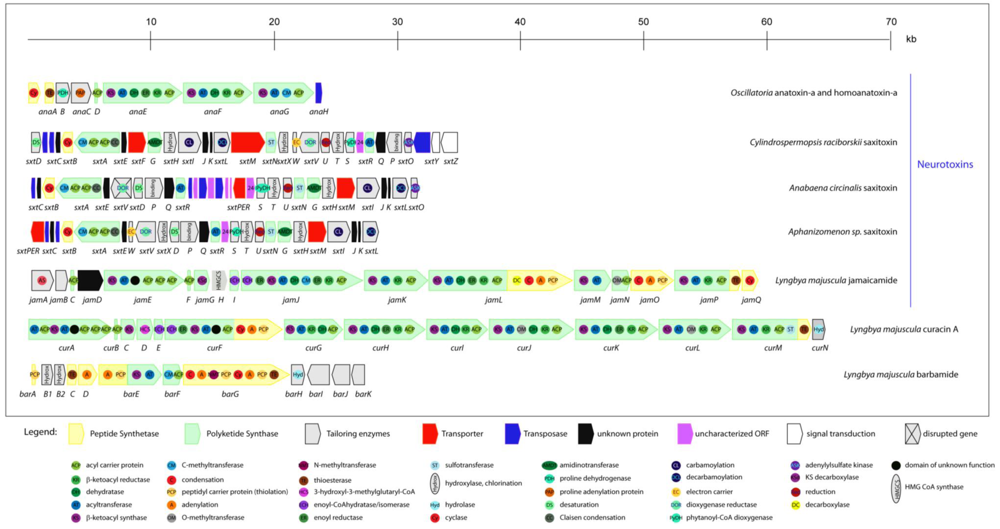
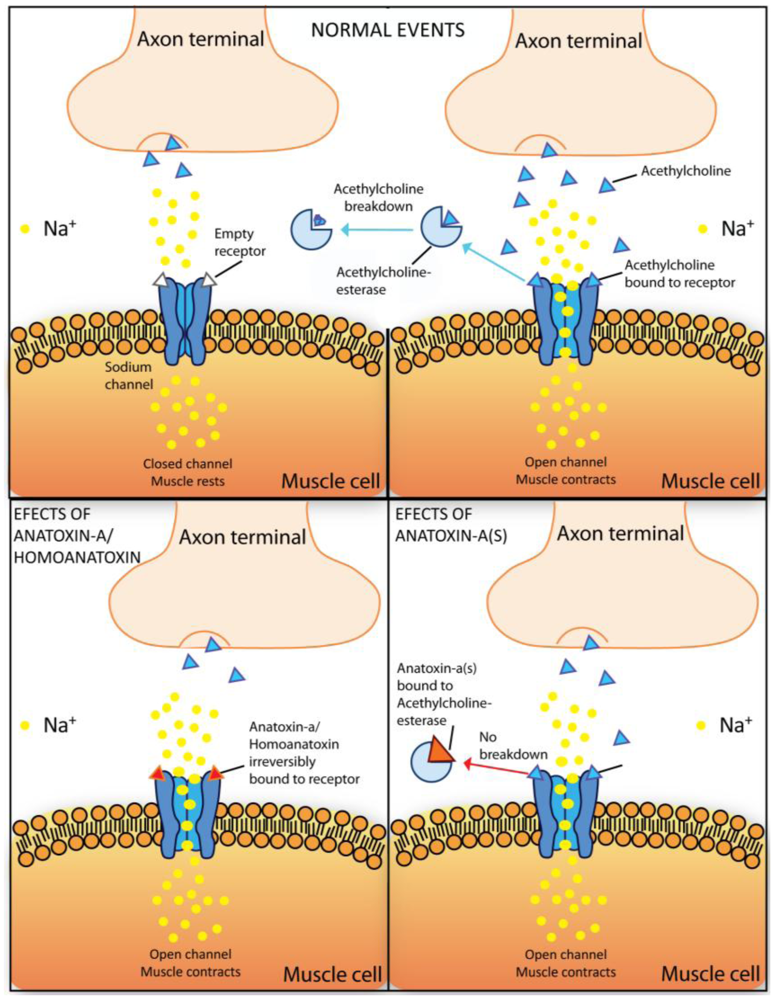
2.5.1. Anatoxin-a and homoanatoxin-a
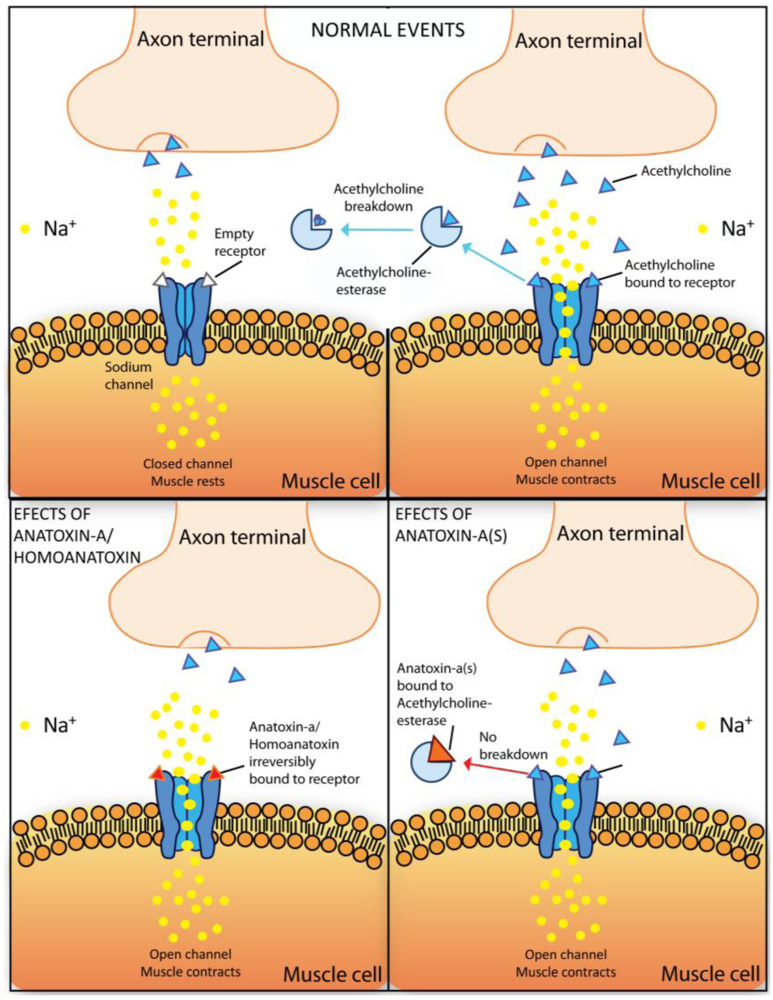
2.5.2. Anatoxin-a(s)
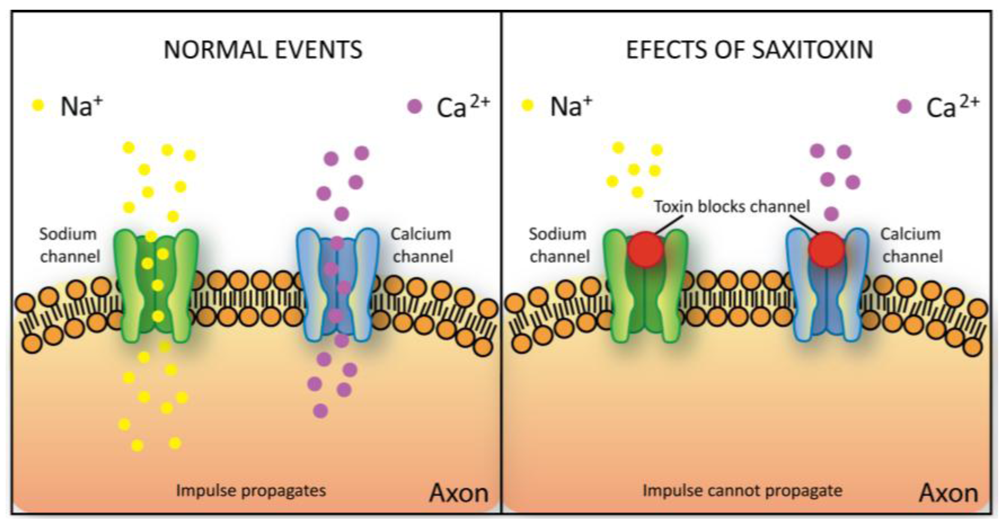
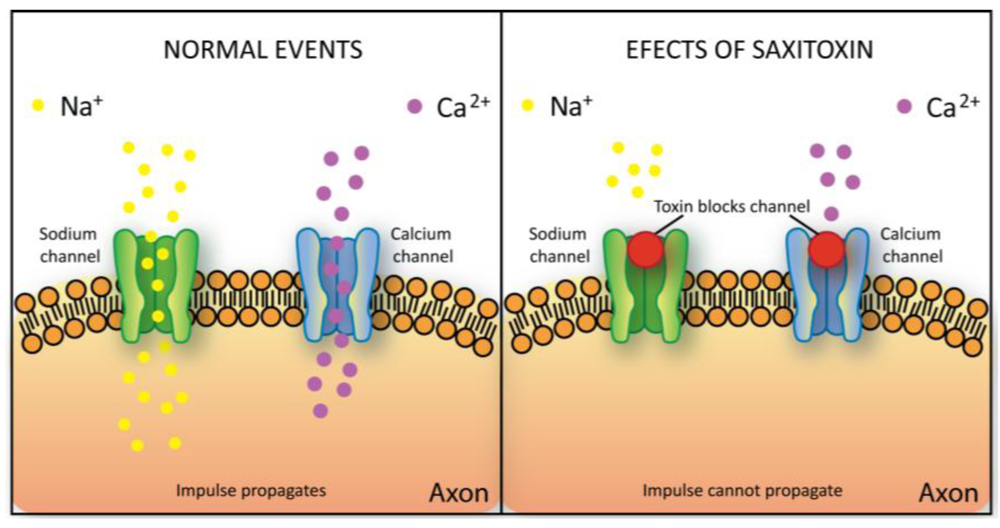
2.5.3. Saxitoxin
2.6. Lipopeptides from marine cyanobacteria
2.6.1. Jamaicamides
2.6.2. Kalkitoxin
2.6.3. Antillatoxins
2.6.4. Curacin A
2.6.5. Barbamide
2.7. Lipopolysaccharides
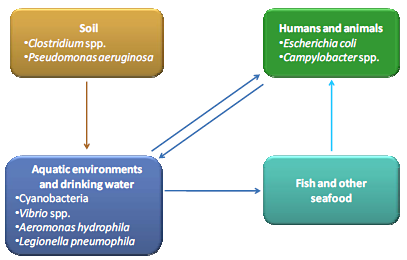
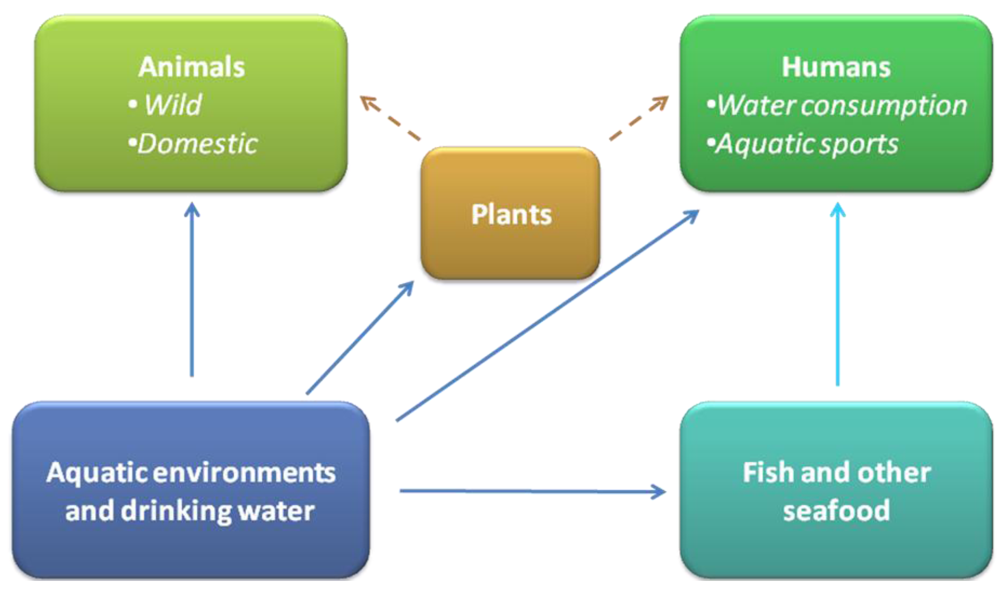
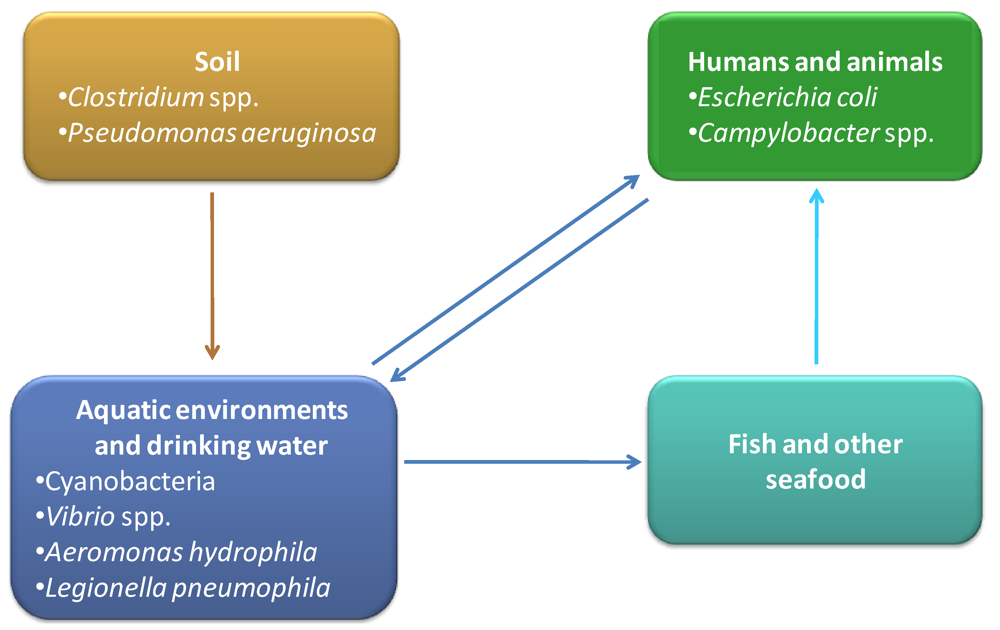
3. Toxins Produced by Other Bacteria in Aquatic Environments
3.1. Vibrio spp.
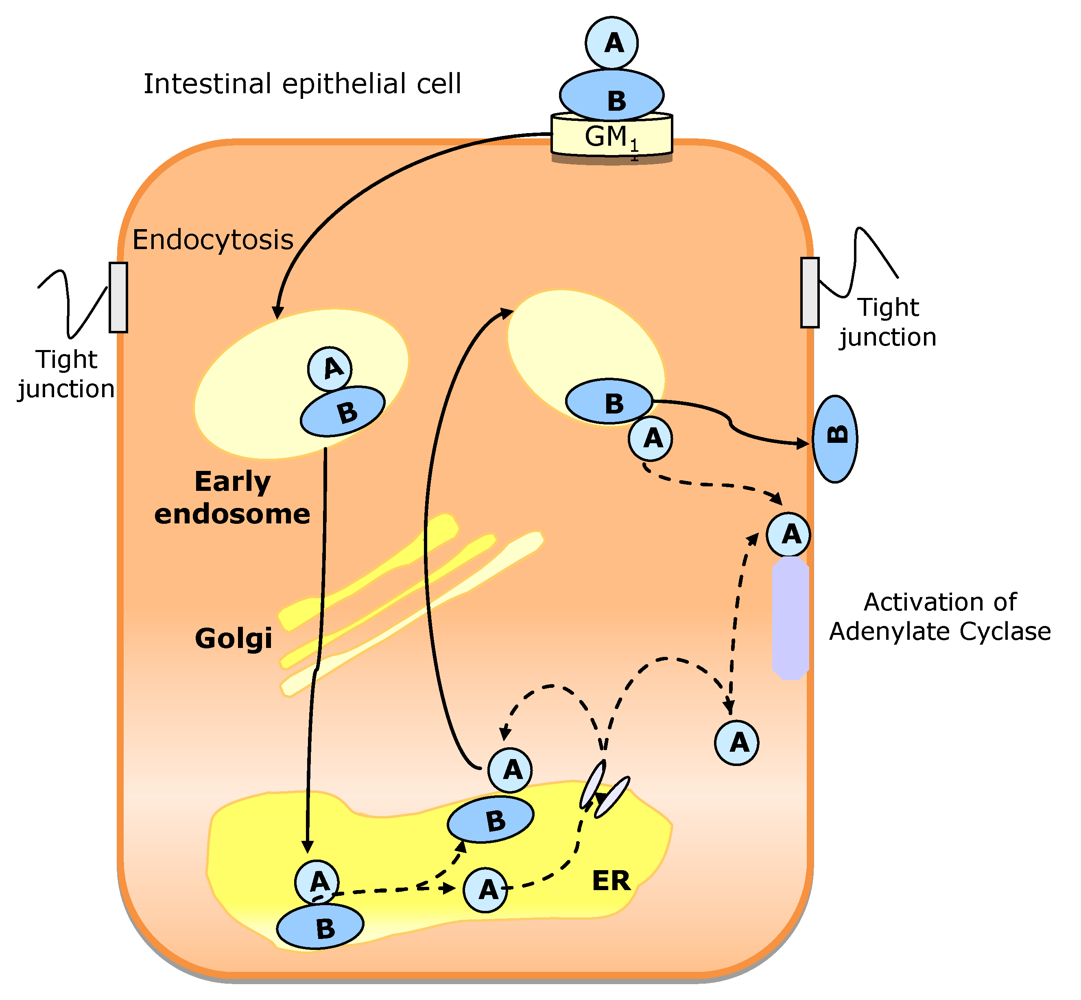
3.1.1. Vibrio cholerae

3.1.2. Vibrio vulnificus
3.2. Aeromonas hydrophila

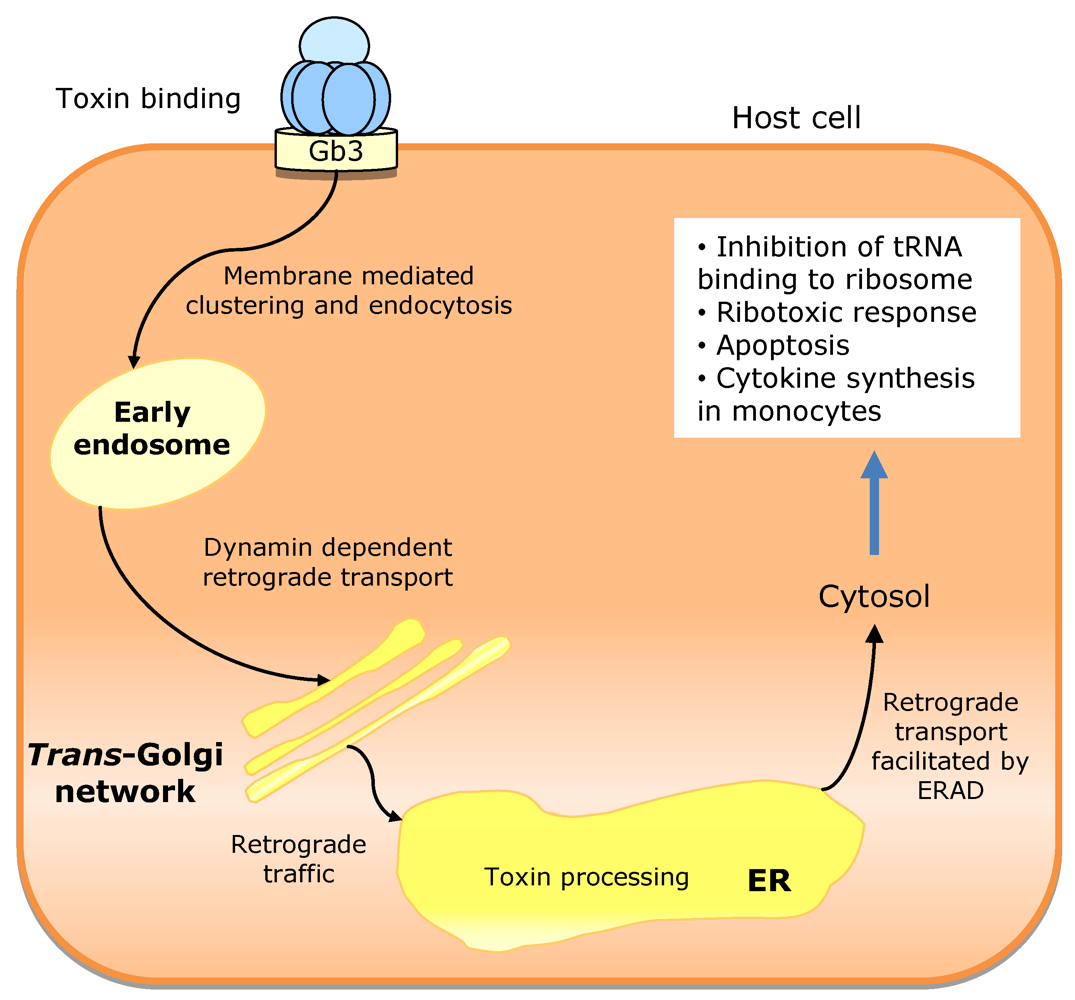
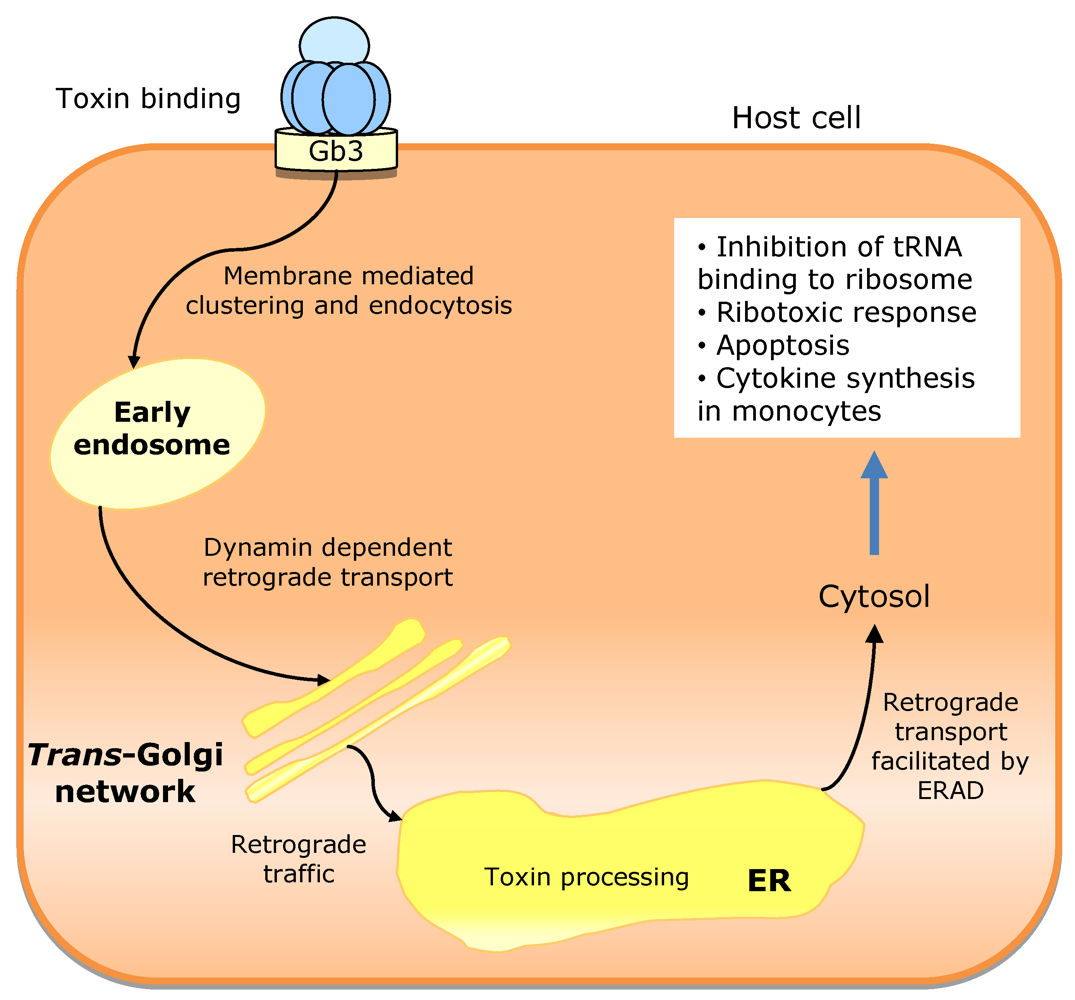
3.3. Escherichia coli
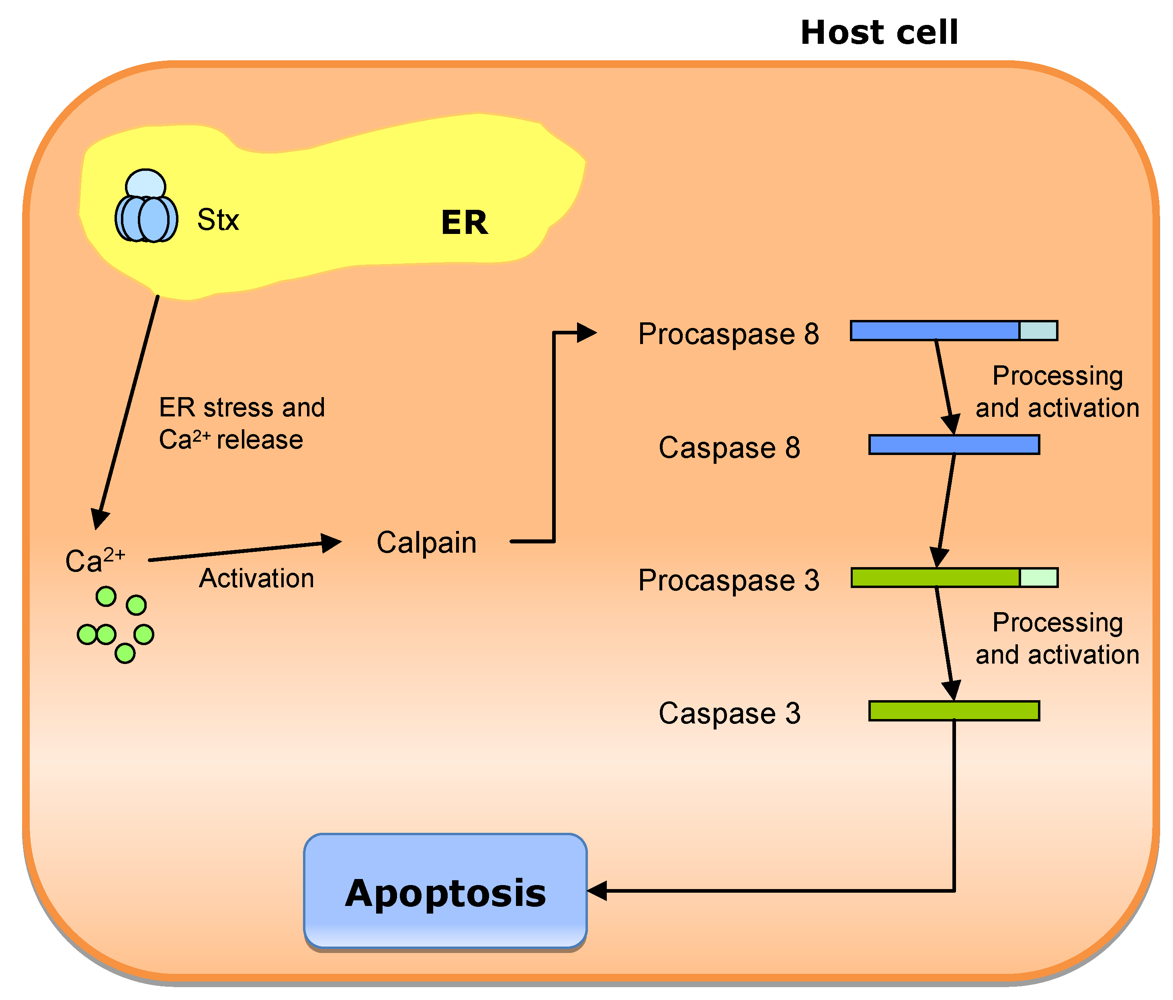
3.4. Legionella pneumophila
3.5. Campylobacter spp.
4. Water Contaminating Toxin Producing Bacteria
4.1. Clostridium spp.
4.1.1. Clostridium botulinum
4.1.2. Clostridium perfringens
4.2. Pseudomonas aeruginosa
5. Final Remarks

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mode of action | Toxin name | Produced by | References |
|---|---|---|---|
| Membrane permeabilizing toxins | Act | A. hydrophila | [179] |
| α-Hemolysin | E. coli | [1] | |
| Bifermentolysin | C. bifermentans | [1] | |
| Botulinolysin | C. botulinum | ||
| Chauveolysin | C. chauvoei | ||
| Histolyticolysin O | C. hystolyticum | ||
| Novyilysin | C. novyi A | ||
| Perfringolysin O | C. perfringens | ||
| Septicolysin O | C. septicum | ||
| Toxins affecting membrane traffic | Botulinum neurotoxin | C. botulinum | [220] |
| Toxins affecting signal transduction | Cholera toxin | V. cholerae | [151] |
| Heat-labile enterotoxin | E. coli | [1] | |
| Toxins affecting protein synthesis | Cholix toxin | V. cholera | [153] |
| Exotoxin A | P. aeruginosa | [237] | |
| Shiga toxin (verotoxin) | E. coli | [187] | |
| Lgt1 | L. pneumophila | [210] | |
| RtxA | V. vulnificus | [165] | |
| RtxA | L. pneumophila | [207] | |
| Toxins inhibiting protein function | Cylindrospermopsin | Cyl. raciborskii | [82,83,84,85,86,87,88,89,90] |
| Umezakia natans | |||
| Aph. ovalisporum | |||
| Raph. curvata | |||
| A. bergii | |||
| Aph. flos-aquae | |||
| Lyngbya wollei | |||
| Microcystins | Microcystis | [27,51] | |
| Planktothrix | |||
| Oscillatoria | |||
| Nostoc | |||
| Anabaena | |||
| Anabaenopsis | |||
| Hapalosiphon | |||
| Snowella | |||
| Woronichinia | |||
| Arthrospira | |||
| Phormidium | |||
| Plectonema | |||
| Pseudoanabaena | |||
| Synechococcus | |||
| Synechocystis | |||
| Nodularins | Nodularia spumigena | [2] | |
| Cytoskeleton-affecting toxins | Anatoxin-a and homoanatoxin-a | Anabaena | [32,59,100,101,102,103,104,105,106] |
| Oscillatoria | |||
| Cylindrospermum | |||
| Microcystis | |||
| Aphanizomenon | |||
| Planktothrix | |||
| C2 toxin | C. botulinum | [246] | |
| Cytotoxic necrotizing factors | E. coli | [1] | |
| DNA damaging | Cytolethal distending toxin | Campylobacter spp. | [213] |
| Voltage-gated ions channel blockers | Saxitonin and gonyautoxins | A. circinalis | [14] |
| Aph. gracile | |||
| C. raciborskii | |||
| L. wollei | |||
| Planktothrix | |||
| Jamaicamides | Lyngbya majuscula | [14] | |
| Kalkitoxin | |||
| Antillatoxin | |||
| Unknown | Lypopolysaccharides (LPS) | E. coli | [39,133] |
| Salmonella spp. | |||
| V. cholera, | |||
| P. aeruginosa | |||
| Cyanobacteria |

References
- Guidebook to Protein Toxins and Their Use in Cell Biology; Rappuoli, R.; Montecucco, C. (Eds.) Sambrook & Tooze Publications at Oxford University Press: New York, NY, USA, 1997.
- Sivonen, K.; Jones, G. Cyanobacterial toxins. In Toxic Cyanobacteria in Water: A Guide to Their Public Health Consequences, Monitoring and Management; Chorus, I., Bartram, J., Eds.; WHO, E & FN Spon: London, UK, 1999; pp. 41–111. Chapter 3. [Google Scholar]
- Wang, D. Neurotoxins from marine dinoflagellates: A brief review. Mar. Drugs 2008, 6, 349–371. [Google Scholar]
- Herrero-Galán, E.; Álvarez-García, E.; Carreras-Sangrà, N.; Lacadena, J.; Alegre-Cebollada, J.; del Pozo, A.M.; Oñaderra, M.; Gavilanes, J.G. Fungal ribotoxins: structure, function and evolution. In Microbial Toxins: Current Research and Future Trends; Proft, T., Ed.; Caister Academic Press: Wymondham, UK, 2009. [Google Scholar]
- Berestetskiy, A.O. A review of fungal phytotoxins: from basic studies to practical use. Appl. Biochem. Microbiol. 2008, 44, 453–465. [Google Scholar]
- Zitzer, A.; Wassenaar, T.; Walev, I.; Bhakdi, S. Potent membrane-permeabilizing and cytocidal action of Vibrio cholerae cytolysin on human intestinal cells. Infect. Immun. 1997, 65, 1293–1298. [Google Scholar]
- Martinez-Arca, S.; Alberts, P.; Zahraoui, A.; Louvard, D.; Galli, T. Role of tetanus neurotoxin insensitive vesicle-associated membrane protein (TI-VAMP) in vesicular transport mediating neurite outgrowth. J. Cell Biol. 2000, 149, 889–900. [Google Scholar]
- Chen, S.; Barbieri, J.T. Engineering botulinum neurotoxin to extend therapeutic intervention. Proc. Natl. Acad. Sci. USA 2009, 106, 9180–9184. [Google Scholar]
- Lencer, W.I.; Strohmeier, G.; Moe, S.; Carlson, S.L.; Constable, C.T.; Madara, J.L. Signal transduction by cholera toxin: processing in vesicular compartments does not require acidification. Am. J. Physiol. Gastrointest. Liver Physiol. 1995, 269, G548–G557. [Google Scholar]
- Cherla, R.P.; Lee, S.-Y.; Mees, P.L.; Tesh, V.L. Shiga toxin 1-induced cytokine production is mediated by MAP kinase pathways and translation initiation factor eIF4E in the macrophage-like THP-1 cell line. J. Leukoc. Biol. 2006, 79, 397–407. [Google Scholar]
- Menestrina, G.; Pederzolli, C.; Forti, S.; Gambale, F. Lipid interaction of Pseudomonas aeruginosa exotoxin A. Acid-triggered permeabilization and aggregation of lipid vesicles. Biophys. J. 1991, 60, 1388–1400. [Google Scholar] [CrossRef] [PubMed]
- Pothoulakis, C.; LaMont, J.T.; Eglow, R.; Gao, N.; Rubins, J.B.; Theoharides, T.C.; Dickey, B.F. Characterization of rabbit lleal receptors for Clostridium difficile toxin A. Evidence for a receptor-coupled G protein. J. Clin. Invest. 1991, 88, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Fiorentini, C.; Arancia, G.; Caprioli, A.; Falbo, V.; Ruggeri, F.M.; Donelli, G. Cytoskeletal changes induced in HEp-2 cells by the cytotoxic necrotizing factor of Escherichia coli. Toxicon 1988, 26, 1047–1056. [Google Scholar]
- Aráoz, R.; Molgó, J.; Tandeau de Marsac, N. Neurotoxic cyanobacterial toxins. Toxicon 2010, 56, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Castenholz, R.W. Species usage, concept, and evolution in the cyanobacteria (blue-green algae). J. Phycol. 1992, 28, 737–745. [Google Scholar]
- Schopf, J.W. The fossil record: tracing the roots of the cyanobacterial lineage. In The Ecology of Cyanobacteria; Whintton, B.A., Potts, M., Eds.; Kluwer: Dordrecht, the Netherlands, 2000; pp. 13–35. [Google Scholar]
- Castenholz, R.W.; Waterbury, J.B. Group I. Cyanobacteria. In Bergey’s Manual of Systematic Bacteriology; Krieg, N.R., Holt, J.G., Eds.; Williams & Wilkins: Baltimore, MD, USA, 1989; Volume 3, pp. 1710–1728. [Google Scholar]
- Falconer, I.R. Cyanobacterial Toxins of Drinking Water Supplies—Cylindrospermopsins and Microcystins; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Sze, P. Prokaryotic algae (Cyanophyta, Prochlorophyta). In A Biology of the Algae, 2nd ed; WCB Publishers: Boston, MA, USA, 1986; pp. 19–34. [Google Scholar]
- Madigan, M.T.; Martinko, J.M.; Parker, J. Brock Biology of Microorganisms, 9th ed; Prentice Hall: Upper Saddle River, NJ, USA, 2000. [Google Scholar]
- Castenholz, R.W. Phylum BX. Cyanobacteria. In Bergey’s Manual of Systematic Bacteriology; Springer: New York, NY, USA, 2001; pp. 473–599. [Google Scholar]
- Mur, L.R.; Skulberg, O.M.; Utkilen, H. Cyanobacteria in the environment. In Toxic Cyanobacteria in Water: A guide to Their Public Health Consequences, Monitoring and Management; Chorus, I., Bartram, J., Eds.; WHO, E & FN Spon: London, UK, 1999; Chapter 2. [Google Scholar]
- Codd, G.A.; Morrison, L.F.; Metcalf, J.S. Cyanobacterial toxins: risk management for health protection. Toxicol. Appl. Pharmacol. 2005, 203, 264–272. [Google Scholar]
- Francis, G. Poisonous Australian lake. Nature 1878, 18, 11–12. [Google Scholar]
- Mahmood, N.A.; Carmichael, W.W.; Pfahler, D. Anticholinesterase poisonings in dogs from a cyanobacterial (blue-green algae) bloom dominated by Anabaena flos-aquae. Am. J. Vet. Res. 1988, 49, 500–503. [Google Scholar]
- Codd, G.A.; Bell, S.G.; Brooks, W.P. Cyanobacterial toxins in water. Water Sci. Technol. 1989, 21, 1–13. [Google Scholar]
- Carmichael, W.W. Cyanobacterial secondary metabolites—the cyanotoxins. J. Appl. Bacteriol. 1992, 72, 445–459. [Google Scholar]
- Negri, A.P.; Jones, G.J.; Hindmarsh, M. Sheep mortality associated with paralytic shellfish poisons from the cyanobacterium Anabaena circinalis. Toxicon 1995, 33, 1321–1329. [Google Scholar]
- Henriksen, P.; Carmichael, W.W.; An, J.; Moestrup, Ø. Detection of an anatoxin-a(s)-like anticholinesterase in natural blooms and cultures of cyanobacteria/blue-green algae from danish lakes and in the stomach contents of poisoned birds. Toxicon 1997, 35, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, H.; Harada, K.-I.; Senma, M.; Ito, Y.; Yasuda, N.; Ushida, S.; Kimura, Y. Possible cause of unnatural mass death of wild birds in a pond in Nishinomiya, Japan: sudden appearance of toxic cyanobacteria. Nat. Toxins 1999, 7, 81–84. [Google Scholar]
- Krienitz, L.; Ballot, A.; Kotut, K.; Wiegand, C.; Putz, S.; Metcalf, J.S.; Codd, G.A.; Pflugmacher, S. Contribution of hot spring cyanobacteria to the mysterious deaths of Lesser Flamingos at Lake Bogoria, Kenya. FEMS Microbiol. Ecol. 2003, 43, 141–148. [Google Scholar]
- Wood, S.A.; Selwood, A.I.; Rueckert, A.; Holland, P.T.; Milne, J.R.; Smith, K.F.; Smits, B.; Watts, L.F.; Cary, C.S. First report of homoanatoxin-a and associated dog neurotoxicosis in New Zealand. Toxicon 2007, 50, 292–301. [Google Scholar]
- Dennison, W.C.; O’Neil, J.M.; Duffy, E.J.; Oliver, P.E.; Shaw, G.R. Blooms of the cyanobacterium Lyngbya majuscula in coastal waters of Queensland, Australia. Bull. Inst. Oceanogr. Monaco 1999, 19, 501–506. [Google Scholar]
- WHO, Guidelines for Safe Recreational Water Environments. Coastal and Freshwaters; World Health Organization: Geneva, Switzerland, 2003; 1, pp. 136–158.
- Teixeira, M.G.L.C.; Costa, M.C.N.; Carvalho, V.L.P.; Pereira, M.S.; Hage, E. Gastroenteritis epidemic in the area of the Itaparica Dam, Bahia, Brazil. Bull. Pan Am. Health Organ. 1993, 27, 244–253. [Google Scholar]
- Carmichael, W.W.; An, J.S.; Azevedo, S.M.F.O.; Lau, S.; Rinehart, K.L.; Jochisen, E.M.; Holmes, C.E.M.; Silva, J.B. Analysis for microcystins involved in outbreak of liver failure and death of humans at a hemodialysis center in Caruaru, Pernambuco, Brazil. In Proceedings of the IV Symposium of the Brazilian Society of Toxinology, São Paulo, Brazil, 6–11 October, 1996.
- Vasconcelos, V.M. Uptake and depuration of the heptapeptide toxin microcystin-LR in Mytilus galloprovincialis. Aquat. Toxicol. 1995, 32, 227–237. [Google Scholar]
- Amorim, A.; Vasconcelos, V. Dynamic of microcystins in the mussel Mytilus galloprovincialis. Toxicon 1999, 37, 1041–1052. [Google Scholar]
- Wiegand, C.; Pflugmacher, S. Ecotoxicological effects of selected cyanobacterial secondary metabolites a short review. Toxicol. Appl. Pharmacol. 2005, 203, 201–218. [Google Scholar]
- Magalhães, V.F.; Marinho, M.M.; Domingos, P.; Oliveira, A.C.; Costa, S.M.; Azevedo, L.O.; Azevedo, S.M.F.O. Microcystins (cyanobacteria hepatotoxins) bioaccumulation in fish and crustaceans from Sepetiba Bay (Brasil, RJ). Toxicon 2003, 42, 289–295. [Google Scholar]
- Vasconcelos, V.; Oliveira, S.; Teles, F.O. Impact of a toxic and a non-toxic strain of Microcystis aeruginosa on the crayfish Procambarus clarkii. Toxicon 2001, 39, 1461–1470. [Google Scholar]
- Saqrane, S.; El ghzali, I.; Ouahid, Y.; El Hassni, M.; El Hadrami, I.; Bouarab, L.; del Campo, F.F.; Oudra, B.; Vasconcelos, V. Phytotoxic effects of cyanobacteria extract on the aquatic plant Lemna gibba: Microcystin accumulation, detoxication and oxidative stress induction. Aquat. Toxicol. 2007, 83, 284–294. [Google Scholar]
- Nogueira, I.C.G.; Pereira, P.; Dias, E.; Pflugmacher, S.; Wiegand, C.; Franca, S.; Vasconcelos, V.M. Accumulation of paralytic shellfish toxins (PST) from the cyanobacterium Aphanizomenon issatschenkoi by the cladoceran Daphnia magna. Toxicon 2004, 44, 773–780. [Google Scholar]
- Negri, A.P.; Jones, G.J. Bioaccumulation of paralytic shellfish poisoning (PSP) toxins from the cyanobacterium Anabaena circinalis by the freshwater mussel Alathyria condola. Toxicon 1995, 33, 667–678. [Google Scholar]
- Pereira, P.; Dias, E.; Franca, S.; Pereira, E.; Carolino, M.; Vasconcelos, V. Accumulation and depuration of cyanobacterial paralytic shellfish toxins by the freshwater mussel Anodonta cygnea. Aquat. Toxicol. 2004, 68, 339–350. [Google Scholar]
- Saker, M.L.; Metcalf, J.S.; Codd, G.A.; Vasconcelos, V.M. Accumulation and depuration of the cyanobacterial toxin cylindrospermopsin in the freshwater mussel Anodonta cygnea. Toxicon 2004, 43, 185–194. [Google Scholar]
- Dittmann, E.; Wiegand, C. Cyanobacterial toxins—occurrence, biosynthesis and impact on human affairs. Mol. Nutr. Food Res. 2006, 50, 7–17. [Google Scholar]
- Stewart, I.; Webb, P.; Schluter, P.; Shaw, G. Recreational and occupational field exposure to freshwater cyanobacteria—a review of anecdotal and case reports, epidemiological studies and the challenges for epidemiologic assessment. Environ. Health 2006, 5, 6. [Google Scholar]
- Okino, T. Heterocycles from Cyanobacteria. Top. Heterocycl. Chem. 2006, 5, 1–19. [Google Scholar]
- Berry, J.P.; Gantar, M.; Perez, M.H.; Berry, G.; Noriega, F.G. Cyanobacterial toxins as allelochemicals with potential applications as algaecides, herbicides and insecticides. Mar. Drugs 2008, 6, 117–146. [Google Scholar]
- Sivonen, K.; Börner, T. Bioactive compounds produced by cyanobacteria. In The Cyanobacteria: Molecular Biology, Genomics and Evolution; Herrero, A., Flores, E., Eds.; Caister Academic Press: Norfolk, UV, USA, 2008; pp. 159–197. [Google Scholar]
- Welker, M.; von Döhren, H. Cyanobacterial peptides—Nature’s own combinatorial biosynthesis. FEMS Microbiol. Rev. 2006, 30, 530–563. [Google Scholar]
- Rapala, J.; Sivonen, K.; Lyra, C.; Niemelä, S.I. Variation of microcystins, cyanobacterial hepatotoxins, in Anabaena spp. as a function of growth stimuli. Appl. Environ. Microbiol. 1997, 63, 2206–2212. [Google Scholar] [PubMed]
- Welker, M.; Brunke, M.; Preussel, K.; Lippert, I.; von Döhren, H. Diversity and distribution of Microcystis (Cyanobacteria) oligopeptide chemotypes from natural communities studied by single-colony mass spectrometry. Microbiology 2004, 150, 1785–1796. [Google Scholar]
- Kameyama, K.; Sugiura, N.; Inamori, Y.; Maekawa, T. Characteristics of microcystin production cell cycle of Microcystis viridis. Environ. Toxicol. 2004, 19, 20–25. [Google Scholar]
- Saker, M.L.; Fastner, J.; Dittmann, E.; Christiansen, G.; Vasconcelos, V.M. Variation between strains of the cyanobacterium Microcystis aeruginosa isolated from a Portuguese river. J. Appl. Microbiol. 2005, 99, 749–757. [Google Scholar]
- Fastner, J.; Heinze, R.; Humpage, A.R.; Mischke, U.; Eaglesham, G.K.; Chorus, I. Cylindrospermopsin occurrence in two German lakes and preliminary assessment of toxicity and toxin production of Cylindrospermopsis raciborskii (cyanobacteria) isolates. Toxicon 2003, 42, 313–321. [Google Scholar]
- Saker, M.L.; Nogueira, I.C.G.; Vasconcelos, V.M.; Neilan, B.A.; Eaglesham, G.H.; Pereira, P. First report and toxicological assessment of the cyanobacterium Cylindrospermopsis raciborskii from Portuguese freshwaters. Ecotoxicol. Environ. Saf. 2003, 55, 243–250. [Google Scholar]
- Park, H.-D.; Watanabe, M.F.; Harada, K.-I.; Nagai, H.; Suzuki, M.; Watanabe, M.; Hayashi, H. Hepatotoxin (microcystin) and neurotoxin (anatoxin-a) contained in natural blooms and strains of cyanobacteria from Japanese freshwaters. Nat. Toxins 1993, 1, 353–360. [Google Scholar]
- Yu, S.; Zhao, N.; Zi, X. The relationship between cyanotoxin (microcystin, MC) in pond-ditch water and primary liver cancer in China. Zhonghua Zhong Liu Za Zhi 2001, 23, 96–99. [Google Scholar]
- Azevedo, S.M.F.O.; Rinehart, K.L.; Eaglesham, G.K.; Lau, S.; Carmichael, W.W.; Jochimsen, E.M.; Shaw, G.R. Human intoxication by microcystins during renal dialysis treatment in Caruaru-Brazil. Toxicolog 2002, 181–182, 441–446. [Google Scholar] [CrossRef]
- Hotto, A.M.; Satchwell, M.F.; Boyer, G.L. Molecular characterization of potential microcystin-producing cyanobacteria in Lake Ontario embayments and nearshore waters. Appl. Environ. Microbiol. 2007, 73, 4570–4578. [Google Scholar]
- Batista, T.; de Sousa, G.; Suput, J.S.; Rahmani, R.; Suput, D. Microcystin-LR causes the collapse of actin filaments in primary human hepatocytes. Aquat. Toxicol. 2003, 65, 85–91. [Google Scholar]
- Campos, A.; Vasconcelos, V. Molecular mechanisms of Microcystin toxicity in animal cells. Int. J. Mol. Sci. 2010, 11, 268–287. [Google Scholar]
- Clark, S.P.; Ryan, T.P.; Searfoss, G.H.; Davis, M.A.; Hooser, S.B. Chronic microcystin exposure induces hepatocyte proliferation with increased expression of mitotic and cyclin-associated genes in P53-deficient mice. Toxicol. Pathol. 2008, 36, 190–203. [Google Scholar]
- Krakstad, C.; Herfindal, L.; Gjertsen, B.T.; Boe, R.; Vintermyr, O.K.; Fladmark, K.E.; Doskeland, S.O. CaM-kinaseII-dependent commitment to microcystin-induced apoptosis is coupled to cell budding, but not to shrinkage or chromatin hypercondensation. Cell Death Differ. 2005, 13, 1191–1202. [Google Scholar]
- Li, M.; Satinover, D.L.; Brautigan, D.L. Phosphorylation and functions of inhibitor-2 family of proteins. Biochemistry 2007, 46, 2380–2389. [Google Scholar]
- Gehringer, M.M. Microcystin-LR and okadaic acid-induced cellular effects: a dualistic response. FEBS Lett. 2004, 557, 1–8. [Google Scholar]
- Guzman, R.E.; Solter, P.F.; Runnegar, M.T. Inhibition of nuclear protein phosphatase activity in mouse hepatocytes by the cyanobacterial toxin microcystin-LR. Toxicon 2003, 41, 773–781. [Google Scholar]
- Weng, D.; Lu, Y.; Wei, Y.; Liu, Y.; Shen, P. The role of ROS in microcystin-LR-induced hepatocyte apoptosis and liver injury in mice. Toxicology 2007, 232, 15–23. [Google Scholar]
- Tillett, D.; Dittmann, E.; Erhard, M.; von Döhren, H.; Börner, T.; Neilan, B.A. Structural organization of microcystin biosynthesis in Microcystis aeruginosa PCC 7806: an integrated peptide-polyketide synthetase system. Chem. Biol. 2000, 7, 753–764. [Google Scholar]
- Christiansen, G.; Fastner, J.; Erhard, M.; Börner, T.; Dittmann, E. Microcystin biosynthesis in Planktothrix: genes, evolution, and manipulation. J. Bacteriol. 2003, 185, 564–572. [Google Scholar]
- Rouhiainen, L.; Vakkilainen, T.; Siemer, B.L.; Buikema, W.; Haselkorn, R.; Sivonen, K. Genes coding for hepatotoxic heptapeptides (microcystins) in the cyanobacterium Anabaena strain 90. Appl. Environ. Microbiol. 2004, 70, 686–692. [Google Scholar]
- Kurmayer, R.; Christiansen, G. The genetic basis of toxin production in Cyanobacteria. Freshwater Rev. 2009, 2, 31–50. [Google Scholar]
- Pearson, L.A.; Hisbergues, M.; Börner, T.; Dittmann, E.; Neilan, B.A. Inactivation of an ABC transporter gene, mcyH, results in loss of microcystin production in the cyanobacterium Microcystis aeruginosa PCC 7806. Appl. Environ. Microbiol. 2004, 70, 6370–6378. [Google Scholar]
- Shi, L.; Carmichael, W.W.; Miller, I. Immuno-gold localization of hepatotoxins in cyanobacterial cells. Arch. Microbiol. 1995, 163, 7–15. [Google Scholar]
- Young, F.M.; Thomson, C.; Metcalf, J.S.; Lucocq, J.M.; Codd, G.A. Immunogold localisation of microcystins in cryosectioned cells of Microcystis. J. Struct. Biol. 2005, 151, 208–214. [Google Scholar]
- Kaebernick, M.; Neilan, B.; Borner, T.; Dittmann, E. Light and the transcriptional response of the microcystin biosynthesis gene cluster. Appl. Environ. Microbiol. 2000, 66, 3387–3392. [Google Scholar]
- Bagu, J.R.; Sykes, B.D.; Craig, M.M.; Holmes, C.F.B. A molecular basis for different interactions of marine toxins with protein phosphatase-1. J. Biol. Chem. 1997, 272, 5087–5097. [Google Scholar]
- Moffitt, M.C.; Neilan, B.A. Characterization of the Nodularin synthetase gene cluster and proposed theory of the evolution of cyanobacterial hepatotoxins. Appl. Environ. Microbiol. 2004, 70, 6353–6362. [Google Scholar]
- Pearson, L.A.; Moffitt, M.C.; Ginn, H.P.; Neilan, B.A. The molecular genetics and regulation of cyanobacterial peptide hepatotoxin biosynthesis. Crit. Rev. Toxicol. 2008, 38, 847–856. [Google Scholar]
- Hawkins, P.R.; Runnegar, M.T.C.; Jackson, A.R.B.; Falconer, I.R. Severe hepatotoxicity caused by the tropical cyanobacterium (blue-green Alga) Cylindrospermopsis raciborskii (Woloszynska) Seenaya and Subba Raju isolated from a domestic water supply reservoir. Appl. Environ. Microbiol. 1985, 50, 1292–1295. [Google Scholar]
- Ohtani, I.; Moore, R.E.; Runnegar, M.T.C. Cylindrospermopsin: A potent hepatotoxin from the blue-green alga Cylindrospermopsis raciborskii. J. Am. Chem. Soc. 1992, 114, 7941–7942. [Google Scholar]
- Li, R.; Carmichael, W.W.; Brittain, S.; Eaglesham, G.K.; Shaw, G.R.; Mahakhant, A.; Noparatnaraporn, N.; Yongmanitchai, W.; Kaya, K.; Watanabe, M.M. Isolation and identification of the cyanotoxin cylindrospermopsin and deoxy-cylindrospermopsin from a Thailand strain of Cylindrospermopsis raciborskii (Cyanobacteria). Toxicon 2001, 39, 973–980. [Google Scholar]
- Harada, K.-I.; Ohtani, I.; Iwamoto, K.; Suzuki, M.; Watanabe, M.F.; Watanabe, M.; Terao, K. Isolation of cylindrospermopsin from a cyanobacterium Umezakia natans and its screening method. Toxicon 1994, 32, 73–84. [Google Scholar]
- Banker, R.; Carmeli, S.; Hadas, O.; Teltsch, B.; Porat, R.; Skenik, A. Identification of cylindrospermopsin in the cyanobacterium Aphanizomenon ovalisporum (Cyanophyceae) isolated from lake Kinneret, Israel. J. Phycol. 1997, 33, 613–616. [Google Scholar]
- Li, R.; Carmichael, W.W.; Brittain, S.; Eaglesham, G.K.; Shaw, G.R.; Yongding, L.; Watanabe, M.M. First report of the cyanotoxins cylindrospermopsin and deoxycylindrospermopsin from Raphidiopsis curvata (cyanobacteria). J. Phycol. 2001, 37, 1121–1126. [Google Scholar]
- Schembri, M.A.; Neilan, B.A.; Saint, C.P. Identification of genes implicated in toxin production in the cyanobacterium Cylindrospermopsis raciborskii. Environ. Toxicol. 2001, 16, 413–421. [Google Scholar]
- Preuβel, K.; Stüken, A.; Wiedner, C.; Chorus, I.; Fastner, J. First report on cylindrospermopsin producing Aphanizomenon flos-aquae (Cyanobacteria) isolated from two German lakes. Toxicon 2006, 47, 156–162. [Google Scholar]
- Seifert, M.; McGregor, G.; Eaglesham, G.; Wickramasinghe, W.; Shaw, G. First evidence for the production of cylindrospermopsin and deoxy-cylindrospermopsin by the freshwater benthic cyanobacterium, Lyngbya wollei (Farlow ex Gomont) Speziale and Dyck. Harmful Algae 2007, 6, 73–80. [Google Scholar]
- Terao, K.; Ohmori, S.; Igarashi, K.; Ohtani, I.; Watanabe, M.F.; Harada, K.I.; Ito, E.; Watanabe, M. Electron microscopic studies on experimental poisoning in mice induced by cylindrospermopsin isolated from blue-green alga Umezakia natans. Toxicon 1994, 32, 833–843. [Google Scholar]
- Harada, K.-I.; Kimura, Y.; Ogawa, K.; Suzuki, M.; Dahlem, A.M.; Beasley, V.R.; Carmichael, W.W. A new procedure for the analysis and purification of naturally occurring anatoxin-a from the blue-green alga Anabaena flos-aquae. Toxicon 1989, 27, 1289–1296. [Google Scholar]
- Falconer, I.R.; Hardy, S.J.; Humpage, A.R.; Froscio, S.M.; Tozer, G.J.; Hawkins, P.R. Hepatic and renal toxicity of the blue-green alga (cyanobacterium) Cylindrospermopsis raciborskii in male Swiss albino mice. Environ. Toxicol. 1999, 14, 143–150. [Google Scholar]
- Mihali, T.K.; Kellmann, R.; Muenchhoff, J.; Barrow, K.D.; Neilan, B.A. Characterization of the gene cluster responsible for cylindrospermopsin biosynthesis. Appl. Environ. Microbiol. 2008, 74, 716–722. [Google Scholar]
- Falconer, I.R.; Humpage, A.R. Preliminary evidence for in vivo tumour initiation by oral administration of extracts of the blue-green alga Cylindrospermopsis raciborskii containing the toxin cylindrospermopsin. Environ. Toxicol. 2001, 16, 192–195. [Google Scholar]
- Runnegar, M.T.; Kong, S.-M.; Zhong, Y.-Z.; Lu, S.C. Inhibition of reduced glutathione synthesis by cyanobacterial alkaloid cylindrospermopsin in cultured rat hepatocytes. Biochem. Pharmacol. 1995, 49, 219–225. [Google Scholar]
- Shen, X.; Lam, P.K.S.; Shaw, G.R.; Wickramasinghe, W. Genotoxicity investigation of a cyanobacterial toxin, cylindrospermopsin. Toxicon 2002, 40, 1499–1501. [Google Scholar]
- Shalev-Alon, G.; Sukenik, A.; Livnah, O.; Schwarz, R.; Kaplan, A. A novel gene encoding amidinotransferase in the cylindrospermopsin producing cyanobacterium Aphanizomenon ovalisporum. FEMS Microbiol. Lett. 2002, 209, 87–91. [Google Scholar]
- Kellmann, R.; Mills, T.; Neilan, B. Functional modeling and phylogenetic distribution of putative cylindrospermopsin biosynthesis enzymes. J. Mol. Evol. 2006, 62, 267–280. [Google Scholar]
- Sivonen, K.; Himberg, K.; Luukkainen, R.; Niemelä, S.I.; Poon, G.K.; Codd, G.A. Preliminary characterization of neurotoxic cyanobacteria blooms and strains from Finland. Toxic. Assess. 1989, 4, 339–352. [Google Scholar]
- Namikoshi, M.; Murakami, T.; Watanabe, M.F.; Oda, T.; Yamada, J.; Tsujimura, S.; Nagai, H.; Oishi, S. Simultaneous production of homoanatoxin-a, anatoxin-a, and a new non-toxic 4-hydroxyhomoanatoxin-a by the cyanobacterium Raphidiopsis mediterranea Skuja. Toxicon 2003, 42, 533–538. [Google Scholar]
- Viaggiu, E.; Melchiorre, S.; Volpi, F.; Corcia, A.D.; Mancini, R.; Garibaldi, L.; Crichigno, G.; Bruno, M. Anatoxin-a toxin in the cyanobacterium Planktothrix rubescens from a fishing pond in northern Italy. Environ. Toxicol. 2004, 19, 191–197. [Google Scholar]
- Selwood, A.I.; Holland, P.T.; Wood, S.A.; Smith, K.F.; McNabb, P.S. Production of Anatoxin-a and a novel biosynthetic precursor by the cyanobacterium Aphanizomenon issatschenkoi. Environ. Sci. Technol. 2006, 41, 506–510. [Google Scholar]
- Skulberg, O.M.; Skulberg, R.; Carmichael, W.W.; Andersen, R.A.; Matsunaga, S.; Moore, R.E. Investigations of a neurotoxic oscillatorialean strain (Cyanophyceae) and its toxin. Isolation and characterization of homoanatoxin-a. Environ. Toxicol. Chem. 1992, 11, 321–329. [Google Scholar] [CrossRef]
- Furey, A.; Crowley, J.; Shuilleabhain, A.N.; Skulberg, O.M.; James, K.J. The first identification of the rare cyanobacterial toxin, homoanatoxin-a, in Ireland. Toxicon 2003, 41, 297–303. [Google Scholar]
- Aráoz, R.; Nghiem, H.-O.; Rippka, R.; Palibroda, N.; de Marsac, N.T.; Herdman, M. Neurotoxins in axenic oscillatorian cyanobacteria: coexistence of anatoxin-a and homoanatoxin-a determined by ligand-binding assay and GC/MS. Microbiology 2005, 151, 1263–1273. [Google Scholar]
- Cadel-Six, S.; Iteman, I.; Peyraud-Thomas, C.; Mann, S.; Ploux, O.; Mejean, A. Identification of a polyketide synthase coding sequence specific for anatoxin-a-producing Oscillatoria cyanobacteria. Appl. Environ. Microbiol. 2009, 75, 4909–4912. [Google Scholar]
- Méjean, A.; Mann, S.; Maldiney, T.; Vassiliadis, G.; Lequin, O.; Ploux, O. Evidence that biosynthesis of the neurotoxic alkaloids anatoxin-a and homoanatoxin-a in the cyanobacterium Oscillatoria PCC 6506 occurs on a modular polyketide synthase initiated by L-proline. J. Am. Chem. Soc. 2009, 131, 7512–7513. [Google Scholar]
- Mahmood, N.A.; Carmichael, W.W. The pharmacology of anatoxin-a(s), a neurotoxin produced by the freshwater cyanobacterium Anabaena flos-aquae NRC 525-17. Toxicon 1986, 24, 425–434. [Google Scholar]
- Matsunaga, S.; Moore, R.E.; Niemczura, W.P.; Carmichael, W.W. Anatoxin-a(s), a potent anticholinesterase from Anabaena flos-aquae. J. Am. Chem. Soc. 1989, 111, 8021–8023. [Google Scholar]
- Cook, W.O.; Beasley, V.R.; Dahlem, A.M.; Dellinger, J.A.; Harlin, K.S.; Carmichael, W.W. Comparison of effects of anatoxin-a(s) and paraoxon, physostigmine and pyridostigmine on mouse brain cholinesterase activity. Toxicon 1988, 26, 750–753. [Google Scholar]
- Pita, R.; Anadón, A.; Martínez-Larrañaga, M.R. Neurotoxins with anticholinesterase activity and their possible use as warfare agents. Med. Clin. (Barc.) 2003, 121, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Kellmann, R.; Neilan, B.A. Biochemical characterization of paralytic shellfish toxin biosynthesis in vitro. J. Phycol. 2007, 43, 497–508. [Google Scholar]
- Mihali, T.K.; Kellmann, R.; Neilan, B.A. Characterisation of the paralytic shellfish toxin biosynthesis gene clusters in Anabaena circinalis AWQC131C and Aphanizomenon sp. NH-5. BMC Biochem. 2009, 10, 8. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.Y. Structure-activity relations of tetrodotoxin, saxitoxin, and analogues. Ann. N. Y. Acad. Sci. 1986, 479, 52–67. [Google Scholar]
- Chang, Z.; Sitachitta, N.; Rossi, J.V.; Roberts, M.A.; Flatt, P.M.; Jia, J.; Sherman, D.H.; Gerwick, W.H. Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2004, 67, 1356–1367. [Google Scholar]
- Tamplin, M.L. A bacterial source of tetrodotoxins and saxitoxins. In Marine Toxins: Origin, Structure, and Molecular Pharmacology; Hall, S., Strichartz, G., Eds.; American Chemical Society: Washington, DC, USA, 1990; Volume 418, pp. 78–86. [Google Scholar]
- Llewellyn, L.E. Saxitoxin, a toxic marine natural product that targets a multitude of receptors. Nat. Prod. Rep. 2006, 23, 200–222. [Google Scholar]
- Toxins: Potential chemical weapons from living organisms. Available online: http://www.opcw.org/about-chemical-weapons/types-of-chemical-agent/toxins/ (accessed on 24 June 2010).
- Kellmann, R.; Mihali, T.K.; Jeon, Y.J.; Pickford, R.; Pomati, F.; Neilan, B.A. Biosynthetic intermediate analysis and functional homology reveal a saxitoxin gene cluster in cyanobacteria. Appl. Environ. Microbiol. 2008, 74, 4044–4053. [Google Scholar]
- Edwards, D.J.; Marquez, B.L.; Nogle, L.M.; McPhail, K.; Goeger, D.E.; Roberts, M.A.; Gerwick, W.H. Structure and biosynthesis of the jamaicamides, new mixed polyketide-peptide neurotoxins from the marine cyanobacterium Lyngbya majuscula. Chem. Biol. 2004, 11, 817–833. [Google Scholar]
- LePage, K.T.; Goeger, D.; Yokokawa, F.; Asano, T.; Shioiri, T.; Gerwick, W.H.; Murray, T.F. The neurotoxic lipopeptide kalkitoxin interacts with voltage-sensitive sodium channels in cerebellar granule neurons. Toxicol. Lett. 2005, 158, 133–139. [Google Scholar]
- Wu, M.; Okino, T.; Nogle, L.M.; Marquez, B.L.; Williamson, R.T.; Sitachitta, N.; Berman, F.W.; Murray, T.F.; McGough, K.; Jacobs, R.; Colsen, K.; Asano, T.; Yokokawa, F.; Shioiri, T.; Gerwick, W.H. Structure, synthesis, and biological properties of Kalkitoxin, a novel neurotoxin from the marine cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 2000, 122, 12041–12042. [Google Scholar]
- Orjala, J.; Nagle, D.G.; Hsu, V.; Gerwick, W.H. Antillatoxin: an exceptionally ichthyotoxic cyclic lipopeptide from the tropical cyanobacterium Lyngbya majuscula. J. Am. Chem. Soc. 1995, 117, 8281–8282. [Google Scholar]
- Li, W.I.; Berman, F.W.; Okino, T.; Yokokawa, F.; Shioiri, T.; Gerwick, W.H.; Murray, T.F. Antillatoxin is a marine cyanobacterial toxin that potently activates voltage-gated sodium channels. Proc. Natl. Acad. Sci. USA 2001, 98, 7599–7604. [Google Scholar]
- Berman, F.W.; Gerwick, W.H.; Murray, T.F. Antillatoxin and kalkitoxin, ichthyotoxins from the tropical cyanobacterium Lyngbya majuscula, induce distinct temporal patterns of NMDA receptor-mediated neurotoxicity. Toxicon 1999, 37, 1645–1648. [Google Scholar]
- Calabresi, P.; Chabner, B. Chemotherapy of neoplastic diseases. In The Pharmacological Basis of Therapeutics, 9th; Hardman, J.G., Limbird, L.E., Molinoff, P.B., Ruddin, R.W., Guilman, A.G., Eds.; Pergamon Press: New York, NY, USA, 1996; pp. 1225–1287. [Google Scholar]
- Chang, Z.; Sitachitta, N.; Rossi, J.V.; Roberts, M.A.; Flatt, P.M.; Jia, J.; Sherman, D.H.; Gerwick, W.H. Biosynthetic pathway and gene cluster analysis of curacin A, an antitubulin natural product from the tropical marine cyanobacterium Lyngbya majuscula. J. Nat. Prod. 2004, 67, 1356–1367. [Google Scholar]
- Orjala, J.; Gerwick , W.H. Barbamide, a chlorinated metabolite with molluscicidal activity from the Caribbean cyanobacterium Lyngbya majuscula. J. Nat. Prod. 1996, 59, 427–430. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.; Flatt, P.; Gerwick, W.H.; Nguyen, V.-A.; Willis, C.L.; Sherman, D.H. The barbamide biosynthetic gene cluster: A novel cyanobacterial system of mixed polyketide synthase (PKS)-non-ribosomal peptide synthetase (NRPS) origin involving an unusual trichloroleucyl starter unit. Gene 2002, 296, 235–247. [Google Scholar]
- Doekel, S.; Marahiel, M.A. Biosynthesis of natural products on modular peptide synthetases. Metab. Eng. 2001, 3, 64–77. [Google Scholar]
- Jenke-Kodama, H.; Sandmann, A.; Muller, R.; Dittmann, E. Evolutionary implications of bacterial polyketide synthases. Mol. Biol. Evol. 2005, 22, 2027–2039. [Google Scholar]
- Choi, S.H.; Kim, S.G. Lipopolysaccharide inhibition of rat hepatic microsomal epoxide hydrolase and glutathione S-transferase gene expression irrespective of nuclear factor-[κ]B activation. Biochem. Pharmacol. 1998, 56, 1427–1436. [Google Scholar]
- Biscardi, D.; Castaldo, A.; Gualillo, O.; Fusco, R. The occurrence of Aeromonas hydrophila strains in Italian mineral and thermal waters. Sci. Total Environ. 2002, 292, 255–263. [Google Scholar]
- Holt, J.G.; Krieg, N.R.; Sneath, P.H.A.; Staley, J.T.; Williams, S.T. Bergey’s Manual of Determinative Bacteriology, 9th ed; Williams & Wilkins: Baltimore, MD, USA, 1994. [Google Scholar]
- Barbieri, E.; Falzano, L.; Fiorentini, C.; Pianetti, A.; Baffone, W.; Fabbri, A.; Matarrese, P.; Casiere, A.; Katouli, M.; Kuhn, I.; et al. Occurrence, diversity, and pathogenicity of halophilic Vibrio spp. and non-O1 Vibrio cholerae from estuarine waters along the Italian Adriatic coast. Appl. Environ. Microbiol. 1999, 65, 2748–2753. [Google Scholar] [PubMed]
- Heidelberg, J.F.; Heidelberg, K.B.; Colwell, R.R. Seasonality of Chesapeake Bay bacterioplankton species. Appl. Environ. Microbiol. 2002, 68, 5488–5497. [Google Scholar]
- Ortigosa, M.; Esteve, C.; Pujalte, M.J. Vibrio species in seawater and mussels: abundance and numerical taxonomy. Syst. Appl. Microbiol. 1989, 12, 316–325. [Google Scholar]
- Ortigosa, M.; Garay, E.; Pujalte, M.J. Numerical taxonomy of Vibrionaceae isolated from oysters and seawater along an annual cycle. Syst. Appl. Microbiol. 1994, 17, 216–225. [Google Scholar]
- Arias, C.R.; Garay, E.; Aznar, R. Nested PCR method for rapid and sensitive detection of Vibrio vulnificus in fish, sediments, and water. Appl. Environ. Microbiol. 1995, 61, 3476–3478. [Google Scholar]
- Grisez, L.; Reyniers, J.; Verdonck, L.; Swings, J.; Ollevier, F. Dominant intestinal microflora of sea bream and sea bass larvae from two hatcheries, during larval development. Aquaculture 1997, 155, 387–399. [Google Scholar] [CrossRef]
- Bauer, A.; Østensvik, Ø.; Florvåg, M.; Ørmen, Ø.; Rørvik, L.M. Occurrence of Vibrio parahaemolyticus, V. cholerae, and V. vulnificus in Norwegian blue mussels (Mytilus edulis). Appl. Environ. Microbiol. 2006, 72, 3058–3061. [Google Scholar] [CrossRef] [PubMed]
- Roque, A.; Lopez-Joven, C.; Lacuesta, B.; Elandaloussi, L.; Wagley, S., Furones; Ruiz-Zarzuela, I.; de Blas, I.; Rangdale, R.; Gomez-Gil, B. Detection and identification of tdh- and trh-positive Vibrio parahaemolyticus Strains from four species of cultured bivalve molluscs on the Spanish Mediterranean coast. Appl. Environ. Microbiol. 2009, 75, 7574–7577. [Google Scholar] [PubMed]
- Gomez-Gil, B.; Roque, A.; Turnbull, J.F.; Tron-Mayen, L. Species of Vibrio isolated from hepatopancreas, haemolymph and digestive tract of a population of healthy juvenile Penaeus vannamei. Aquaculture 1998, 163, 1–9. [Google Scholar]
- Vandenberghe, J.; Li, Y.; Verdonck, L.; Sorgeloos, P.; Xu, H.S.; Swings, J. Vibrios associated with Penaeus chinensis (Crustacea: Decapoda) larvae in Chinese shrimp hatcheries. Aquaculture 1998, 169, 121–132. [Google Scholar]
- Wai, S.N.; Mizunoe, Y.; Yoshida, S. How Vibrio cholerae survive during starvation. FEMS Microbiol. Lett. 1999, 180, 123–131. [Google Scholar]
- Lightner, D.V.; Redman, R.M. Shrimp diseases and currentdiagnostic methods. Aquaculture 1998, 164, 201–220. [Google Scholar]
- Bergh, O.; Nilsen, F.; Samuelsen, O.B. Diseases, prophylaxis and treatment of the Atlantic halibut Hippoglossus hippoglossus: a review. Dis. Aquat. Org. 2001, 48, 57–74. [Google Scholar]
- Diggles, B.K.; Carson, J.; Hine, P.M.; Hickman, R.W.; Tait, M.J. Vibrio species associated with mortalities in hatchery-reared turbot (Colistium nudipinnis) and brill (C. guntheri) in New Zealand. Aquaculture 2000, 183, 1–12. [Google Scholar] [CrossRef]
- Hansen, G.H.; Olafsen, J.A. Bacterial interactions in early life stages of marine cold water fish. Microb. Ecol. 1999, 38, 1–26. [Google Scholar]
- Matson, J.S.; Withey, J.H.; DiRita, V.J. Regulatory networks controlling Vibrio cholerae virulence gene expression. Infect. Immun. 2007, 75, 5542–5549. [Google Scholar]
- Thompson, F.L.; Tetsuya, I.; Swings, J. Biodiversity of Vibrios. Microbiol. Mol. Biol. Rev. 2004, 68, 403–431. [Google Scholar]
- Jørgensen, A.R.; Purdy, R.E.; Fieldhouse, J.; Kimber, M.S.; Bartlett, D.H.; Merrill, A.R. Cholix toxin, a novel ADP-ribosylating factor from Vibrio cholerae. J. Biol. Chem. 2008, 283, 10671–10678. [Google Scholar]
- Davis, B.M.; Moyer, K.E.; Boyd, E.F.; Waldor, M.K. CTX prophages in classical biotype Vibrio cholerae: functional phage genes but dysfunctional phage genomes. J. Bacteriol. 2000, 182, 6992–6998. [Google Scholar]
- Higgins, D.; Nazareno, E.; DiRita, V.J. The virulence gene activator ToxT from Vibrio cholerae is a member of the AraC family of transcriptional activators. J. Bacteriol. 1992, 174, 6974–6980. [Google Scholar]
- Schuhmacher, D.A.; Klose, K.E. Environmental signals modulate ToxT-dependent virulence factor expression in Vibrio cholerae. J. Bacteriol. 1999, 181, 1508–1514. [Google Scholar]
- Prouty, M.G.; Osorio, C.R.; Klose, K.E. Characterization of functional domains of the Vibrio cholerae virulence regulator ToxT. Mol. Microbiol. 2005, 58, 1143–1156. [Google Scholar]
- Withey, J.H.; DiRita, V.J. The toxbox: specific DNA sequence requirements for activation of Vibrio cholerae virulence genes by ToxT. Mol. Microbiol. 2006, 59, 1779–1789. [Google Scholar]
- Yu, R.R.; DiRita, V.J. Regulation of gene expression in Vibrio cholerae by ToxT involves both antirepression and RNA polymerase stimulation. Mol. Microbiol. 2002, 43, 119–134. [Google Scholar]
- Lee, S.H.; Hava, D.L.; Waldor, M.K.; Camilli, A. Regulation and temporal expression patterns of Vibrio cholerae virulence genes during infection. Cell 1999, 99, 625–634. [Google Scholar]
- Lencer, W.I. Microbes and microbial toxins: paradigms for microbial-mucosal interactions V. Cholera: invasion of the intestinal epithelial barrier by a stably folded protein toxin. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 280, 781–786. [Google Scholar]
- Lencer, W.I.; Hirst, T.R.; Holmes, R.K. Membrane traffic and cellular uptake of cholera toxin. Biochim. Biophys. Acta 1999, 1450, 177–190. [Google Scholar]
- Sixma, T.K.; Pronk, S.E.; Kalk, H.H.; Wartna, E.S.; van Zanten, B.A.M.; Witholt, B.; Hol, W.G.J. Crystal structure of a cholera toxin-related heat-labile enterotoxin from E. coli. Nature 1991, 351, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Fasano, A. Cellular microbiology: can we learn cell physiology from microorganisms? Am. J. Physiol. Cell Physiol. 1999, 276, 765–776. [Google Scholar]
- Satchell, K.J.F. MARTX, multifuntional autoprocessing repeats-in-toxin toxins. Infect. Immun. 2007, 75, 5079–5084. [Google Scholar]
- Jorgensen, R.; Merrill, A.R.; Andersen, G.R. The life and death of translation elongation factor 2. Biochem. Soc. Trans. 2006, 34, 1–6. [Google Scholar]
- DePaola, A.; Capers, G.M.; Alexander, D. Densities of Vibrio vulnificus in the intestines of fish from the U.S. Gulf Coast. Appl. Environ. Microbiol. 1994, 60, 984–988. [Google Scholar] [PubMed]
- do Nascimento, S.M.; Fernandes Vieira, R.H.; Theophilo, G.N.; Rodrigues, P.D.; Vieira, G.H. Vibrio vulnificus as a health hazard for shrimp consumers. Rev. Inst. Med. Trop. Sao Paulo 2001, 43, 263–266. [Google Scholar]
- Baffone, W.; Tarsi, R.; Pane, L.; Campana, R.; Repetto, B.; Mariottini, G.L.; Pruzzo, C. Detection of free-living and plankton-bound vibrios in coastal waters of the Adriatic Sea (Italy) and study of their pathogenicity associated properties. Environ. Microbiol. 2006, 8, 1299–1305. [Google Scholar]
- Jones, K.J.; Oliver, J.D. Vibrio vulnificus: disease and pathogenesis. Infect. Immun. 2009, 77, 1723–1733. [Google Scholar]
- Chuang, Y.C.; Yuan, C.Y.; Liu, C.Y.; Lan, C.K.; Huang, A.H. Vibrio vulnificus infection in Taiwan: report of 28 cases and review of clinical manifestations and treatment. Clin. Infect. Dis. 1992, 15, 271–276. [Google Scholar]
- Oliver, J.D. Wound infections caused by Vibrio vulnificus and other marine bacteria. Epidemiol. Infect. 2005, 133, 383–391. [Google Scholar]
- Gray, L.D.; Kreger, A.S. Purification and characterization of an extracellular cytolysin produced by Vibrio vulnificus. Infect. Immun. 1985, 48, 62–72. [Google Scholar]
- Kothary, M.H.; Kreger, A.S. Purification and characterization of an elastolytic protease of Vibrio vulnificus. J. Gen. Microbiol. 1987, 133, 1783–1791. [Google Scholar]
- Shao, C.P.; Hor, L.I. Metalloprotease is not essential for Vibrio vulnificus virulence in mice. Infect. Immun. 2000, 68, 3569–3573. [Google Scholar]
- Kim, Y.R.; Lee, S.E.; Kook, H.; Yeom, J.A.; Na, H.S.; Kim, S.Y.; Chung, S.S.; Choy, H.E.; Rhee, J.H. Vibrio vulnificus RTX toxin kills host cells only after contact of the bacteria with host cells. Cell. Microbiol. 2008, 10, 848–862. [Google Scholar] [CrossRef] [PubMed]
- Gulig, P.A.; Bourdage, K.L.; Starks, A.M. Molecular pathogenesis of Vibrio vulnificus. J. Microbiol. 2005, 43, 118–131. [Google Scholar]
- Lee, B.C.; Lee, J.H.; Kim, M.W.; Kim, B.S.; Oh, M.H.; Kim, K.S.; Kim, T.S.; Choi, S.H. Vibrio vulnificus rtxE is important for virulence and its expression is induced by exposure to host cells. Infect. Immun. 2008, 76, 1509–1517. [Google Scholar]
- Chopra, A.K. Hyper production, purification, and mechanism of action of the cytotoxic enterotoxin produced by Aeromonas hydrophila. Infect. Immun. 1997, 65, 4299–4308. [Google Scholar]
- Yamada, S.; Matsushita, S.; Dejsirilet, S.; Kudoh, Y. Incidence and clinical symptoms of Aeromonas-associated traveller’s diarrhoea in Tokyo. Epidemiol. Infect. 1997, 119, 121–126. [Google Scholar]
- Sha, J.; Kozlova, E.V.; Chopra, K. Role of various enterotoxins in Aeromonas hyrophila-induced gastroenteritis: generation of enterotoxin gene-deficient mutants and evaluation of their enterotoxin activity. Infect. Immun. 2002, 70, 1924–1935. [Google Scholar]
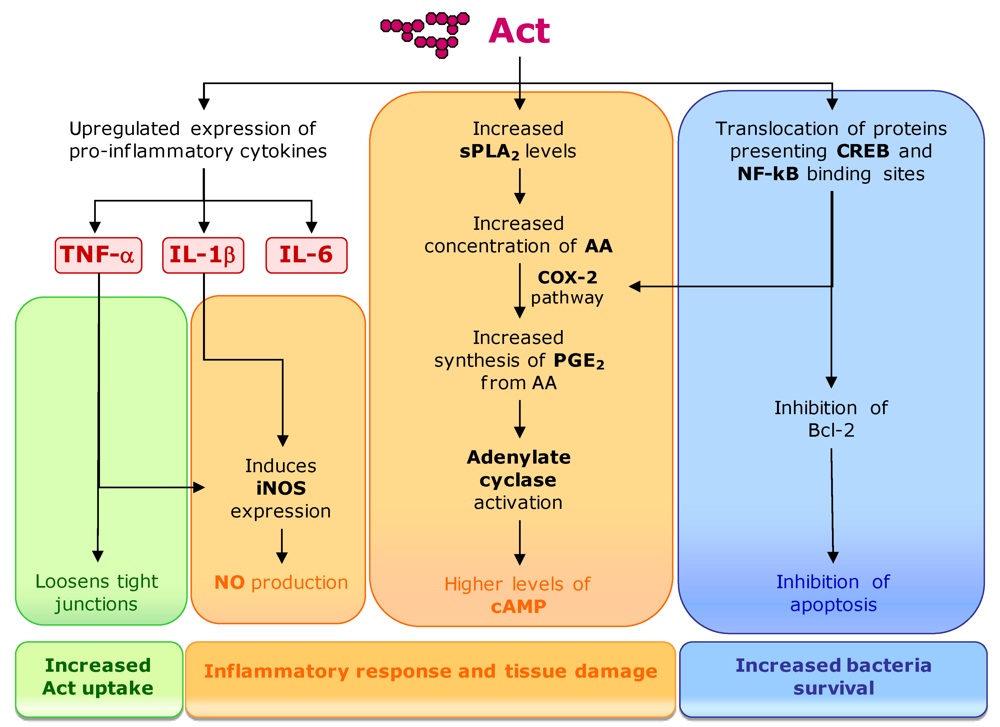
- Chopra, A.K.; Xu, X.J.; Ribardo, D.; Gonzalez, M.; Kuhl, K.; Peterson, J.W.; Houston, C.W. The cytotoxic enterotoxin of Aeromonas hydrophila induces proinflammatory cytokine production and activates arachidonic acid metabolism in macrophages. Infect. Immun. 2000, 68, 2808–2818. [Google Scholar]
- Shenkar, R.; Abraham, E. Mechanisms of lung neutrophil activation after hemorrhage or endotoxemia: roles of reactive oxygen intermediates, NF-kB, and cyclic AMP response element binding protein. J. Immunol. 1999, 163, 954–962. [Google Scholar]
- Howard, S.P.; Garland, W.J.; Green, M.J.; Buckley, J.T. Nucleotide sequence of the gene for the hole-forming toxin aerolysin of Aeromonas hydrophila. J. Bacteriol. 1987, 169, 2869–2871. [Google Scholar]
- Ashok, K.C.; Houston, C.W.; Kurosky, A. Genetic variation in related cytolytic toxins produced by different species of Aeromonas. FEMS Microbiol. Lett. 1991, 78, 231–238. [Google Scholar]
- Gyles, C.L. Shiga toxin-producing Escherichia coli: an overview. J. Anim. Sci. 2007, 85, E45–E62. [Google Scholar]
- Kaper, J.B.; Nataro, J.P.; Mobley, H.L. Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2004, 2, 123–140. [Google Scholar]
- Johannes, L.; Romer, W. Shiga toxins-from cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar]
- Herold, S.; Karch, H.; Schmidt, H. Shiga toxinencoding bacteriophages–genomes in motion. Int. J. Med. Microbiol. 2004, 294, 115–121. [Google Scholar]
- Zhang, W.; Bielaszewska, M.; Kuczius, T.; Karch, H. Identification, characterization, and distribution of a Shiga toxin 1 gene variant (stx(1c)) in Escherichia coli strains isolated from humans. J. Clin. Microbiol. 2002, 40, 1441–1446. [Google Scholar]
- Waddell, T.; Cohen, A.; Lingwood, C.A. Induction of verotoxin sensitivity in receptor-deficient cell lines using the receptor glycolipid globotriosylceramide. Proc. Natl. Acad. Sci. USA 1990, 87, 7898–7901. [Google Scholar]
- Soltyk, A.M.; Mackenzie, C.R.; Wolski, V.M.; Hirama, T.; Kitov, P.I.; Bundle, D.R.; Brunton, J.L. A mutational analysis of the globotriaosylceramide-binding sites of verotoxin VT1. J. Biol. Chem. 2002, 277, 5351–5359. [Google Scholar] [PubMed]
- Kiarash, A.; Boyd, B.; Lingwood, C.A. Glycosphingolipid receptor function is modified by fatty acid content. Verotoxin 1 and verotoxin 2c preferentially recognize different globotriaosyl ceramide fatty acid homologues. J. Biol. Chem. 1994, 269, 11138–11146. [Google Scholar] [PubMed]
- Sandvig, K.; Spilsberg, B.; Lauvrak, S.U.; Torgersen, M.L.; Iversen, T.G.; van Deurs, B. Pathways followed by protein toxins into cells. Int. J. Med. Microbiol. 2004, 293, 483–490. [Google Scholar]
- Lauvrak, S.U.; Torgersen, M.L.; Sandvig, K. Efficient endosome-to-Golgi transport of Shiga toxin is dependent on dynamin and clathrin. J. Cell Sci. 2004, 117, 2321–2331. [Google Scholar]
- Popoff, V.; Mardones, G.A.; Tenza, D.; Rojas, R.; Lamaze, C.; Bonifacino, J.S.; Raposo, G.; Johannes, L. The retromer complex and clathrin define an early endosomal retrograde exit site. J. Cell Sci. 2007, 120, 2022–2031. [Google Scholar]
- Endo, Y. Site of action of a Vero toxin (VT2) from Escherichia coli O157:H7 and of Shiga toxin on eukaryotic ribosomes. RNA N-glycosidase activity of the toxins. Eur. J. Biochem. 1988, 171, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Chen, S.L.; Magun, B.E. Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the α-sarcin/ricin loop in the 28S rRNA. Mol. Cell Biol. 1997, 17, 3373–3381. [Google Scholar]
- Foster, G.H.; Tesh, V.L. Shiga toxin 1-induced activation of c-Jun NH2-terminal kinase and p38 in the human monocytic cell line THP-1: possible involvement in the production of TNF-α. J. Leukoc. Biol. 2002, 71, 107–114. [Google Scholar]
- Yamasaki, C.; Natori, Y.; Zeng, X.T.; Ohmura, M.; Yamasaki, S.; Takeda, Y.; Natori, Y. Induction of cytokines in a human colon epithelial cell line by Shiga toxin 1 (Stx1) and Stx2 but not by non-toxic mutant Stx1 which lacks N-glycosidase activity. FEBS Lett. 1999, 442, 231–234. [Google Scholar]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003, 71, 1497–1504. [Google Scholar]
- Lee, S.Y.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell. Microbiol. 2008, 10, 10770–10780. [Google Scholar]
- Suh, J.K.; Hovde, C.J.; Robertus, J.D. Shiga toxin attacks bacterial ribosomes as effectively as eucaryotic ribosomes. Biochemistry 1998, 37, 9394–9398. [Google Scholar]
- Brieland, J.; McClain, M.; Heath, L.; Chrisp, C.; Huffnagle, G.; LeGendre, M.; Hurley, M.; Fantone, J.; Engleberg, C. Coinoculation with Hartmannella vermiformis enhances replicative Legionella pneumophila lung infection in a murine model of Legionnaires’ disease. Infect. Immun. 1996, 64, 2449–2456. [Google Scholar]
- Cirillo, S.L.G.; Littman, L.Y.M.; Samrakandi, M.M.; Cirillo, J.D. Role of the Legionella pneumophila rtxA gene in amoebae. Microbiology 2002, 148, 1667–1677. [Google Scholar] [PubMed]
- Horwitz, M.A. Formation of a novel phagosome by the Legionnaires’ disease bacterium (Legionella pneumophila) in human monocytes. J. Exp. Med. 1983, 158, 1319–1331. [Google Scholar]
- Samrakandi, M.M.; Cirillo, S.L.; Ridenour, D.A.; Bermudez, L.E.; Cirillo, J.D. Genetic and phenotypic differences between Legionella pneumophila strains. J. Clin. Microbiol. 2002, 40, 1352–1362. [Google Scholar]
- Ambagala, T.C.; Ambagala, A.P.N.; Srikumaran, S. The leukotoxin of Pasteurella haemolytica binds to b2 integrins on bovine leukocytes. FEMS Microbiol. Lett. 1999, 179, 161–167. [Google Scholar]
- D’Auria, G.; Jimenez, N.; Peris-Bondia, F.; Pelaz, C.; Latorre, A.; Moya, A. Virulence factor rtx in Legionella pneumophila, evidence suggesting it is a modular multifunctional protein. BMC Genomics 2008, 9, 14. [Google Scholar] [CrossRef] [PubMed]
- Belyi, Y.; Niggeweg, R.; Opitz, B.; Vogelsgesang, M.; Hippenstiel, S.; Wilm, M.; Aktories, K. Legionella pneumophila glucosyltransferase inhibits host elongation factor 1A. Proc. Natl. Acad. Sci. USA 2006, 103, 16953–16958. [Google Scholar]
- Nachamkin, I. Campylobacter jejuni. In Food Microbiology, Fundamentals and Frontiers, 2nd; Doyle, M.P., Beuchat, L.R., Montville, T.J., Eds.; ASM Press: Washington, DC, USA, 2001. [Google Scholar]
- Skirrow, M.B.; Blaser, M.J. Clinical aspects of Campylobacter infection. In Campylobacter, 2nd; Nachamkin, I., Blaser, M.J., Eds.; ASM Press: Washington, DC, USA, 2000; pp. 69–88. [Google Scholar]
- Whitehouse, C.A.; Balbo, P.B.; Pesci, E.C.; Cottle, D.L.; Mirabito, P.M.; Pickett, C.L. Campylobacter jejuni cytolethal distending toxin causes a G2-phase cell cycle block. Infect. Immun. 1998, 66, 1934–1940. [Google Scholar]
- Okuda, J.; Kurazono, H.; Takeda, Y. Distribution of the cytolethal distending toxin A gene (cdtA) among species of Shigella and Vibrio, and cloning and sequencing of the cdt gene from Shigella dysenteriae. Microb. Pathog. 1995, 18, 167–172. [Google Scholar]
- Young, V.B.; Knox, K.A.; Schauer, D.B. Cytolethal distending toxin sequence and activity in the enterohepatic pathogen Helicobacter hepaticus. Infect. Immun. 2000, 68, 184–191. [Google Scholar]
- Zheng, J.; Meng, J.; Zhao, S.; Singh, R.; Song, W. Campylobacter-induced interleukin-8 secretion in polarized human intestinal epithelial cells requires Campylobacter-secreted cytolethal distending toxin- and Toll-like receptor-mediated activation of NF-kB. Infect. Immun. 2008, 76, 4498–4508. [Google Scholar]
- Tejero, L.M.; Galan, J.E. A bacterial toxin that controls cell cycle progression as a deoxyribonuclease I-like protein. Science 2000, 290, 354–357. [Google Scholar]
- Tejero, L.M.; Galan, J.E. CdtA, CdtB, and CdtC form a tripartite complex required for cytolethal distending toxin. Infect. Immun. 2001, 69, 4358–4365. [Google Scholar]
- Long, S.C.; Tauscher, T. Watershed issues associated with Clostridium botulinum: A literature review. J. Water Health 2006, 04.3, 277–288. [Google Scholar]
- DasGupta, B.R.; Sugiyama, H. A common subunit structure in Clostridium botulinum type A, B and E toxins. Biochem. Biophys. Res. Commun. 1972, 48, 108–112. [Google Scholar]
- Tsuzuki, K.; Kimura, K.; Fujii, N.; Yokosawa, N.; Indoh, T.; Murakami, T.; Oguma, K. Cloning and complete nucleotide sequence of the gene for the main component of hemagglutinin produced by Clostridium botulinum type C. Infect. Immun. 1990, 58, 3173–3177. [Google Scholar]
- Tsuzuki, K.; Kimura, K.; Fujii, N.; Yokosawa, N.; Oguma, K. The complete nucleotide sequence of the gene coding for the nontoxic-nonhemagglutinin component of Clostridium botulinum type C progenitor toxin. Biochem. Biophys. Res. Commun. 1992, 183, 1273–1279. [Google Scholar]
- Pellizzari, R.; Guidi-Rontani, C.; Vitale, G.; Mock, M.; Montecucco, C. Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFN gamma-induced release of NO and TNFalpha. FEBS Lett. 1999, 462, 199–204. [Google Scholar]
- Montecucco, C.; Schiavo, G.; Rossetto, O. The mechanism of action of tetanus and botulinum neurotoxins. Arch. Toxicol. Suppl. 1996, 18, 342–354. [Google Scholar]
- Montecucco, C.; Schiavo, G. Mechanism of action of tetanus and botulinum neurotoxins. Mol. Microbiol. 1994, 13, 1–8. [Google Scholar]
- East, F.A.K.; Collins, M.D. Conserved structure of genes encoding components of botulinum neurotoxin complex M and the sequence of the gene coding for the nontoxic component in nonproteolytic Clostridium botulinum type. Curr. Microbiol. 1994, 29, 69–77. [Google Scholar]
- Binz, T.; Kurazono, H.; Wille, M.; Frevert, J.; Wernars, K.; Niemann, H. The complete sequence of botulinum neurotoxin type A and comparison with other clostridial neurotoxins. J. Biol. Chem. 1990, 265, 9153–9158. [Google Scholar]
- Hauser, D.; Eklund, M.W.; Boquet, P.; Popoff, M.R. Organization of the botulinum neurotoxin C1 gene and its associated non-toxic protein genes in Clostridium botulinum C 468. Mol. Gen. Genet. 1994, 243, 631–640. [Google Scholar]
- Zhou, Y.; Sugiyama, H.; Nakano, H.; Johnson, E.A. The genes for the Clostridium botulinum type G toxin complex are on a plasmid. Infect. Immun. 1995, 63, 2087–2091. [Google Scholar]
- Songer, J.G.; Meer, R.M. Genotyping of Clostridium perfringens by polymerase chain reaction is a useful adjunct to diagnosis of clostridial enteric disease in animals. Anaerobe 1996, 2, 197–203. [Google Scholar]
- Hunter, S.E.; Brown, J.E.; Oyston, P.C.; Sakurai, J.; Titball, R.W. Molecular genetic analysis of beta-toxin of Clostridium perfringens reveals sequence homology with alpha-toxin, gamma-toxin, and leukocidin of Staphylococcus aureus. Infect. Immun. 1993, 61, 3958–3965. [Google Scholar] [PubMed]
- Czeczulin, J.R.; Hanna, P.C.; McClane, B.A. Cloning, nucleotidesequencing, and expression of the Clostridium perfringens enterotoxin gene in Escherichia coli. Infect. Immun. 1993, 61, 3429–3439. [Google Scholar]
- Brynestad, S.; Iwanejko, L.A.; Stewart, G.S.A.B.; Granum, P.E. A complex array of Hpr consensus DNA recognition sequences proximal to the enterotoxin gene in Clostridium perfringens type A. Microbiology 1994, 140, 97–104. [Google Scholar]
- Anzai, Y.K.H.; Park, J.Y.; Wakabayashi, H. Phylogenetic affiliation of the pseudomonads based on 16S rRNA sequence. Int. J. Syst. Evol. Microbiol. 2000, 50, 1563–1589. [Google Scholar]
- Hassett, D.; Cuppoletti, J.; Trapnell, B.; Lymar, S.; Rowe, J.; Yoon, S.; Hilliard, G.; Parvatiyar, K.; Kamani, M.; Wozniak, D.; Hwang, S.; McDermott, T.; Ochsner, U. Anaerobic metabolism and quorum sensing by Pseudomonas aeruginosa biofilms in chronically infected cystic fibrosis airways: rethinking antibiotic treatment strategies and drug targets. Adv. Drug. Deliv. Rev. 2002, 54, 1425–1443. [Google Scholar]
- Van Eldere, J. Multicentre surveillance of Pseudomonas aeruginosa susceptibility patterns in nosocomial infections. J. Antimicrob. Chemother. 2003, 51, 347–352. [Google Scholar]
- Wolf, P.; Elsasser-Beile, Ú. Pseudomonas exotoxin A: From virulence actor toanti-cancera gent. Int. J. Med. Microbiol. 2009, 299, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Domenighini, M.; Rappuoli, R. Three conserved consensus sequences identify the NAD-binding site of ADP-ribosylating enzymes, expressed by eukaryotes, bacteria and T-even bacteriophages. Microbiology 1996, 21, 667–674. [Google Scholar]
- Siegall, C.B.; Chaudhary, V.K.; FitzGerald, D.J.; Pastan, I. Functional analysis of domains II, Ib, and III of Pseudomonas exotoxin. J. Biol. Chem. 1989, 264, 14256–14261. [Google Scholar]
- Ogata, M.; Fryling, C.M.; Pastan, I.; FitzGerald, D.J. Cell-mediated cleavage of Pseudomonas exotoxin between Arg279 and Gly280 generates the enzymatically active fragment which translocates to the cytosol. J. Biol. Chem. 1992, 267, 25396–25401. [Google Scholar]
- Lombardi, D.; Soldati, T.; Riederer, M.A.; Goda, Y.; Zerial, M.; Pfeffer, S.R. Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J. 1993, 12, 677–682. [Google Scholar]
- Jackson, M.E.; Simpson, J.C.; Girod, A.; Pepperkok, R.; Roberts, L.M.; Lord, J.M. The KDEL retrieval system is exploited by Pseudomonas exotoxin A, but not by Shiga-like toxin-1, during retrograde transport from the Golgi complex to the endoplasmic reticulum. J. Cell Sci. 1999, 112, 467–475. [Google Scholar] [PubMed]
- Duris, J.W.; Haack, S.K.; Fogarty, L.R. Gene and antigen markers of Shiga-toxin producing E. coli from Michigan and Indiana river water: Occurrence and relation to recreational water quality criteria. J. Environ. Qual. 2009, 38, 1878–1886. [Google Scholar] [CrossRef] [PubMed]
- Belkin, S.; Colwell, R.J. Oceans and Health: Pathogens in the Marine Environment; Springer Science+Business Media: New York, NY, USA, 2005. [Google Scholar]
- Janda, J.M.; Abbott, S.L. The genus Aeromonas: taxonomy, pathogenicity, and infection. Clin. Microbiol. Rev. 2010, 23, 35–73. [Google Scholar]
- Barth, H.; Aktories, K.; Popoff, M.R.; Stiles, B.G. Binary Bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol. Mol. Biol. Rev. 2004, 68, 373–402. [Google Scholar]
- Glenn, G.M.; Francis, D.H.; Danielsen, E.M. Toxin-mediated effects on the innate mucosal defenses: implications for enteric vaccines. Infect. Immun. 2009, 77, 5206–5215. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Valério, E.; Chaves, S.; Tenreiro, R. Diversity and Impact of Prokaryotic Toxins on Aquatic Environments: A Review. Toxins 2010, 2, 2359-2410. https://doi.org/10.3390/toxins2102359
Valério E, Chaves S, Tenreiro R. Diversity and Impact of Prokaryotic Toxins on Aquatic Environments: A Review. Toxins. 2010; 2(10):2359-2410. https://doi.org/10.3390/toxins2102359
Chicago/Turabian StyleValério, Elisabete, Sandra Chaves, and Rogério Tenreiro. 2010. "Diversity and Impact of Prokaryotic Toxins on Aquatic Environments: A Review" Toxins 2, no. 10: 2359-2410. https://doi.org/10.3390/toxins2102359