Modulation of Ras/ERK and Phosphoinositide Signaling by Long-Chain n-3 PUFA in Breast Cancer and Their Potential Complementary Role in Combination with Targeted Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
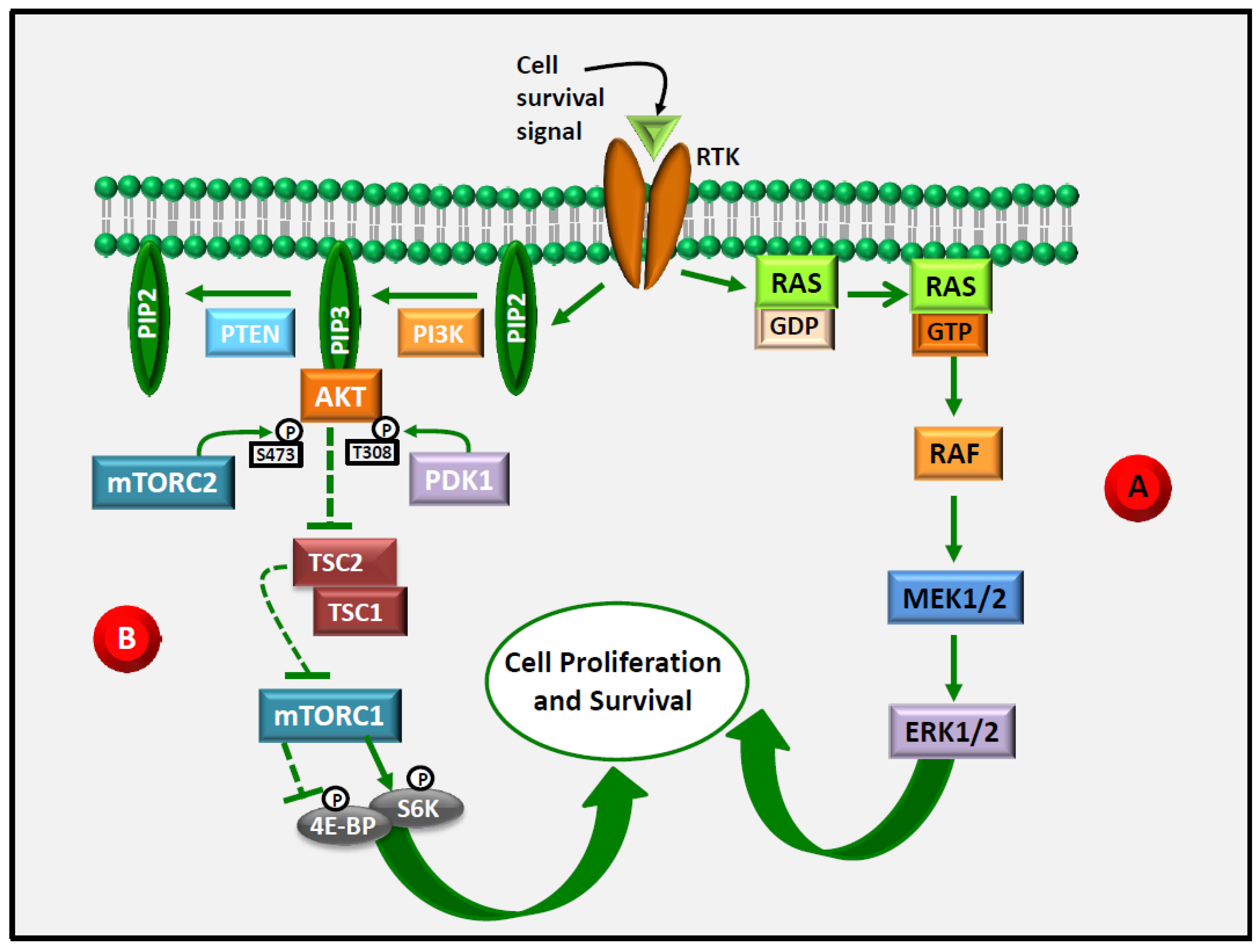
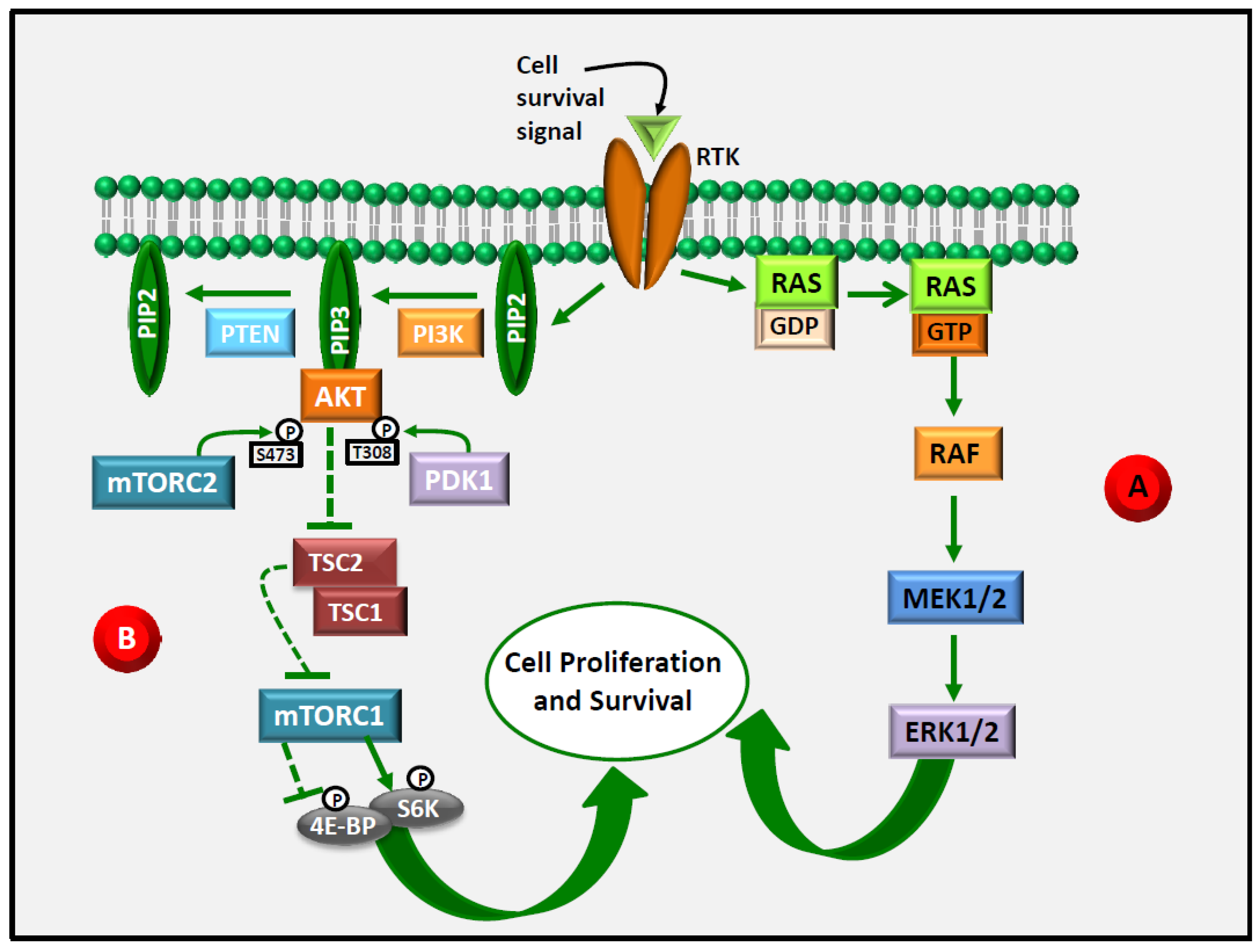
3. ERK1/2 as an Emerging Target in Anti-Cancer Therapy
4. Akt as an Emerging Target in Anti-Cancer Therapy
5. ERK1/2 and Akt Are Central Targets in LCn-3 PUFA Anti-Cancer Action
6. Evidence of an Inhibitory Effect of LCn-3 PUFA on ERK1/2 and/or Akt Phosphorylation/Activation
6.1. In Vitro Research and Preclinical Animal Models
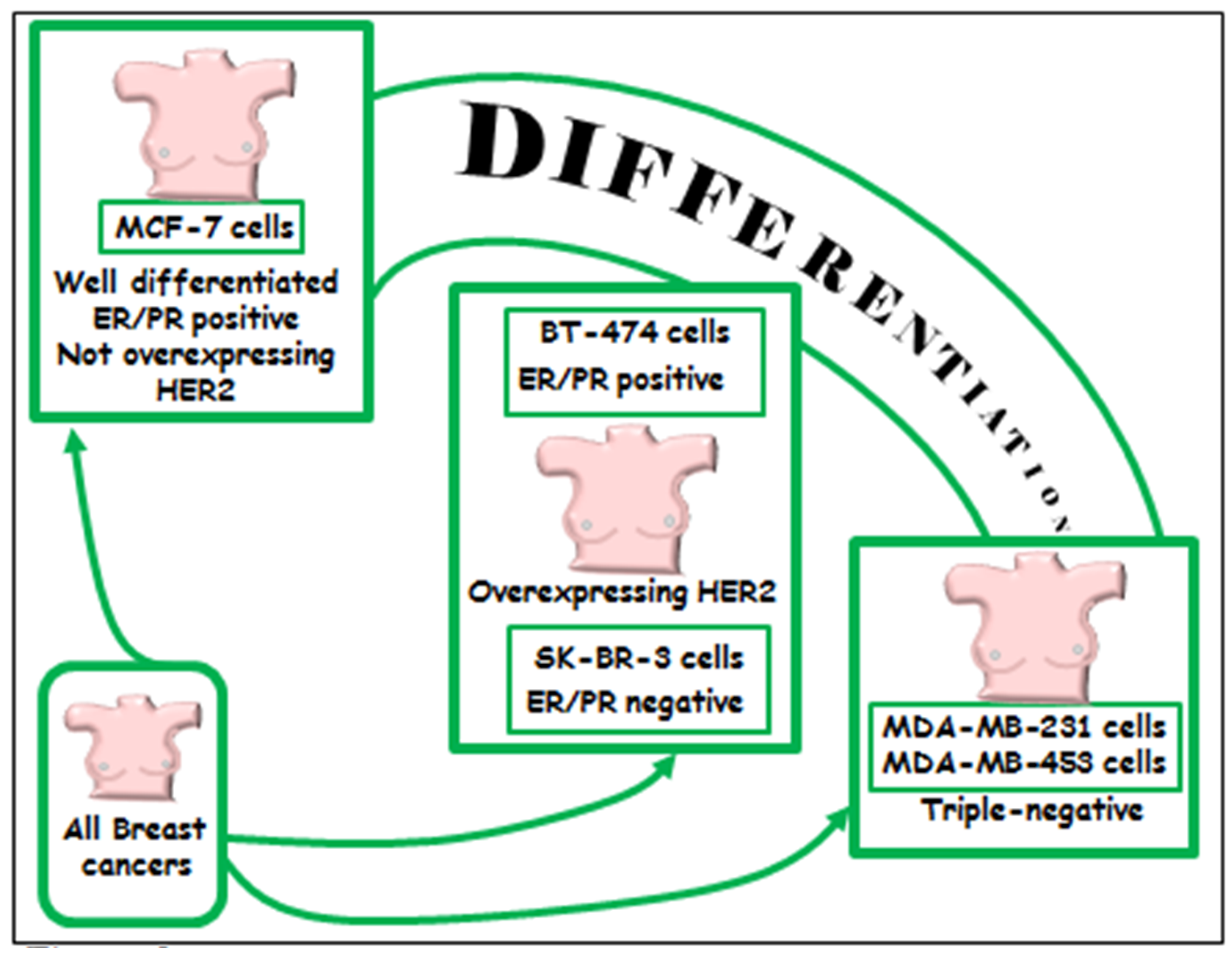
6.1.1. In Vitro Models
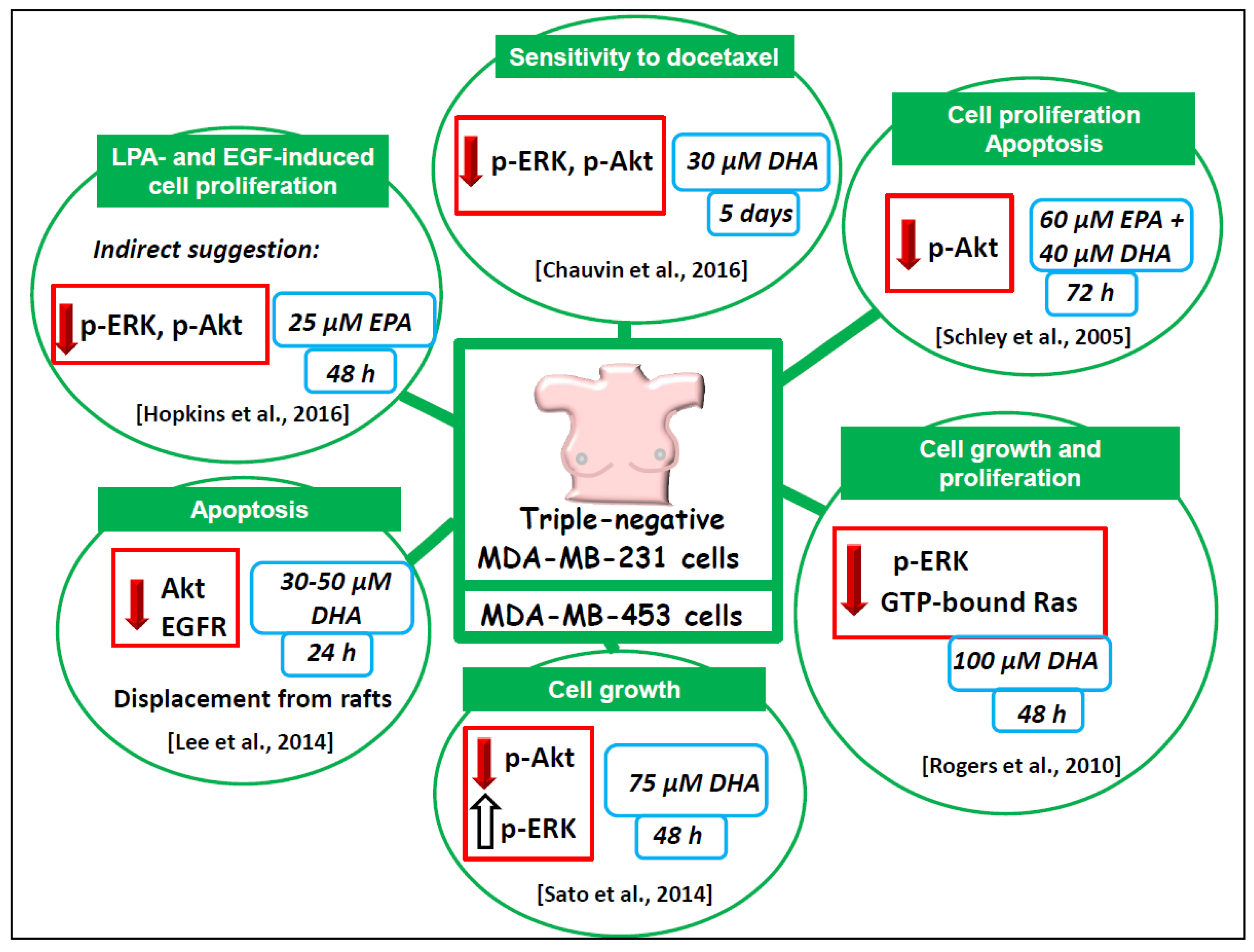
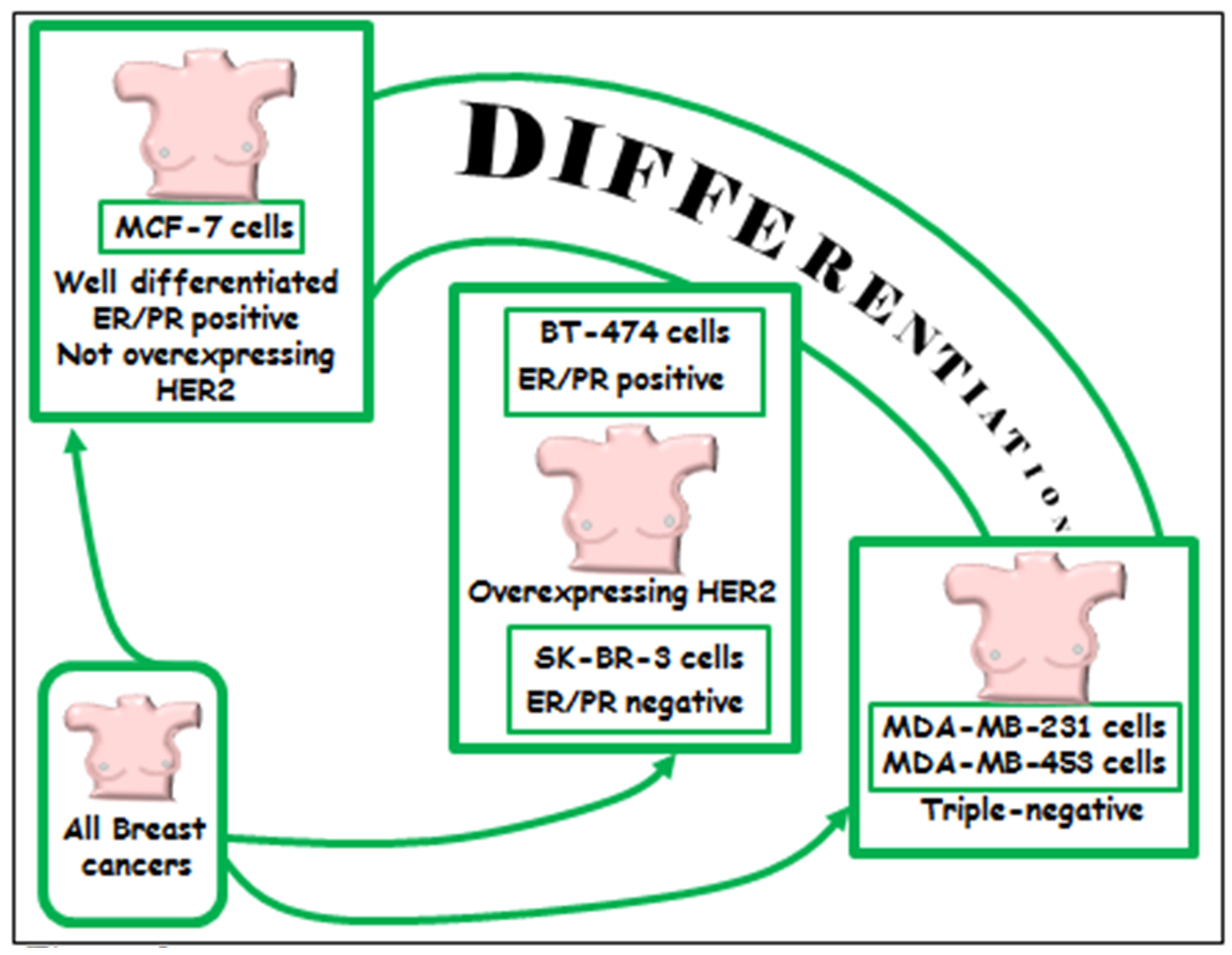
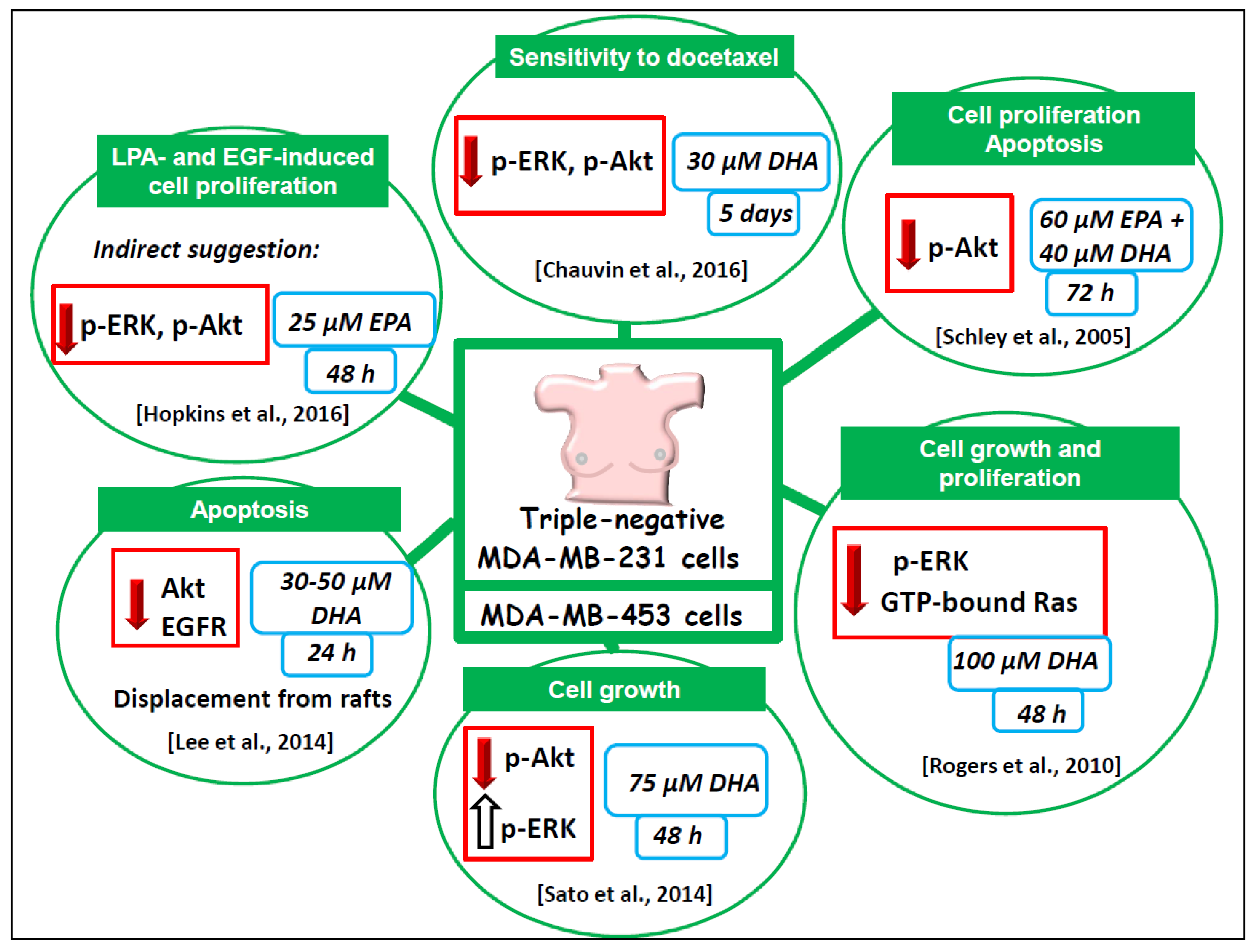
6.1.2. Triple Negative MDA-MB-231 and MDA-MB-453 Breast Cancer Cell Lines
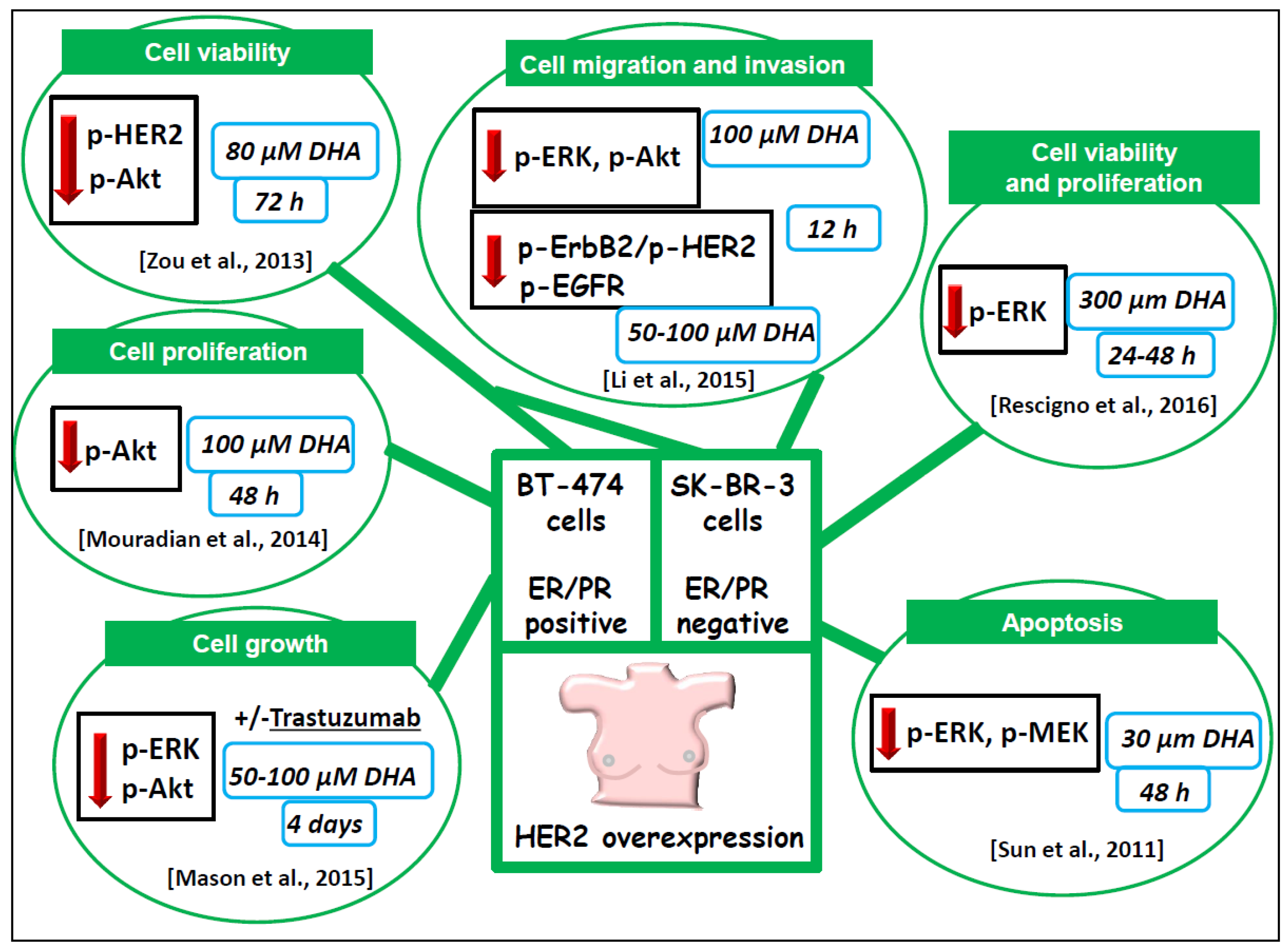
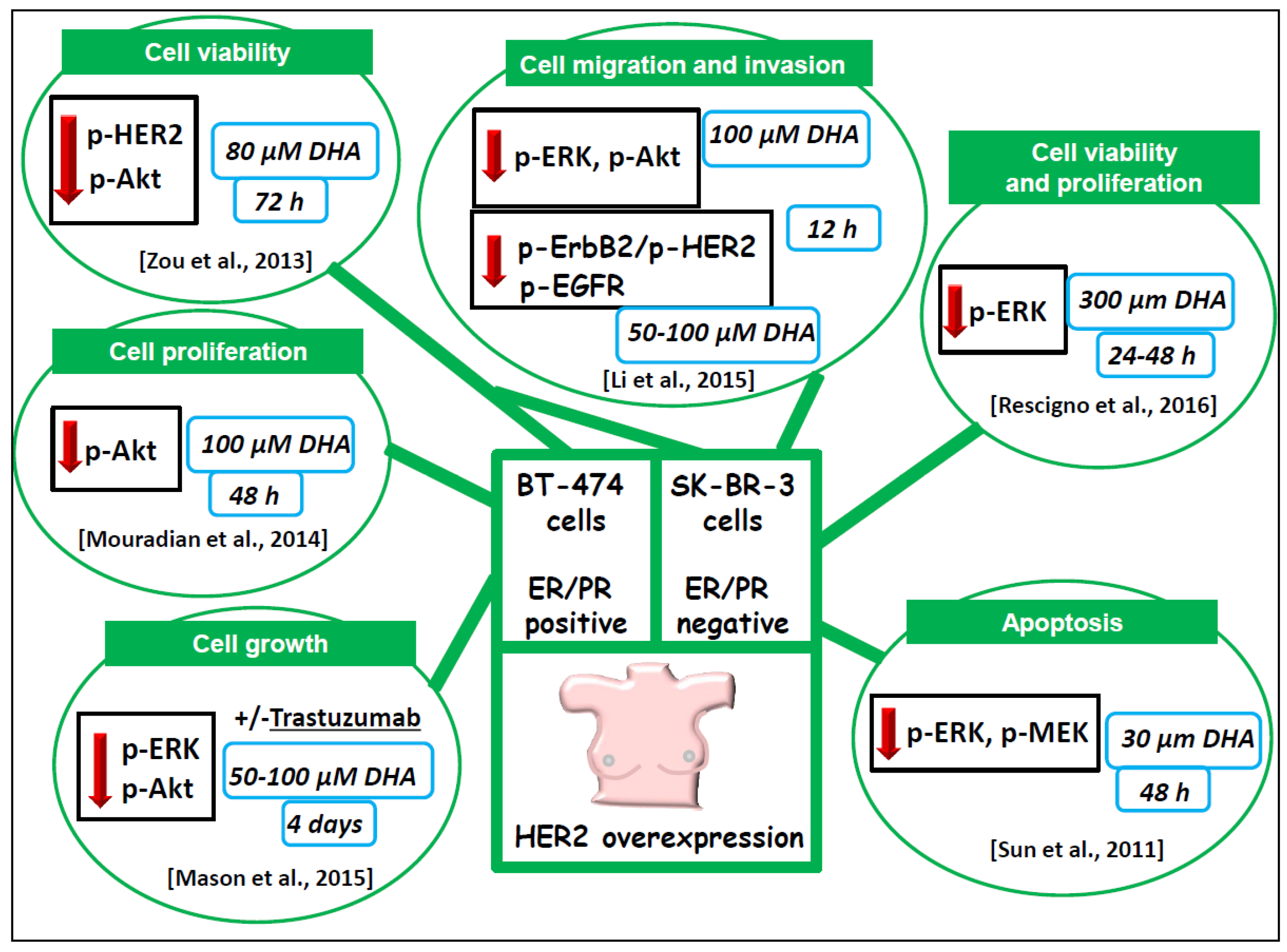
6.1.3. BT-474 and SK-BR-3 Breast Cancer Cells Lines Overexpressing HER2/neu
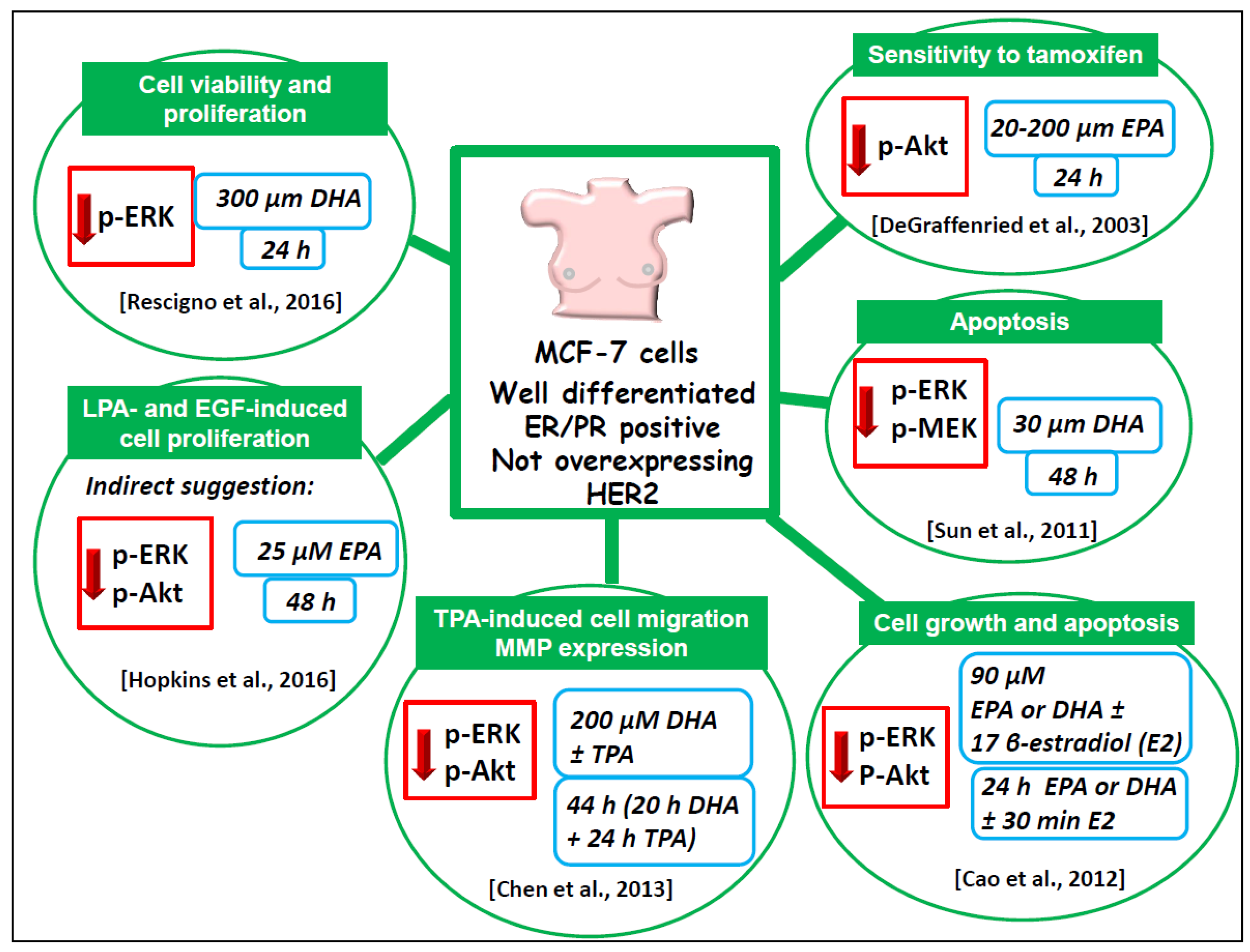
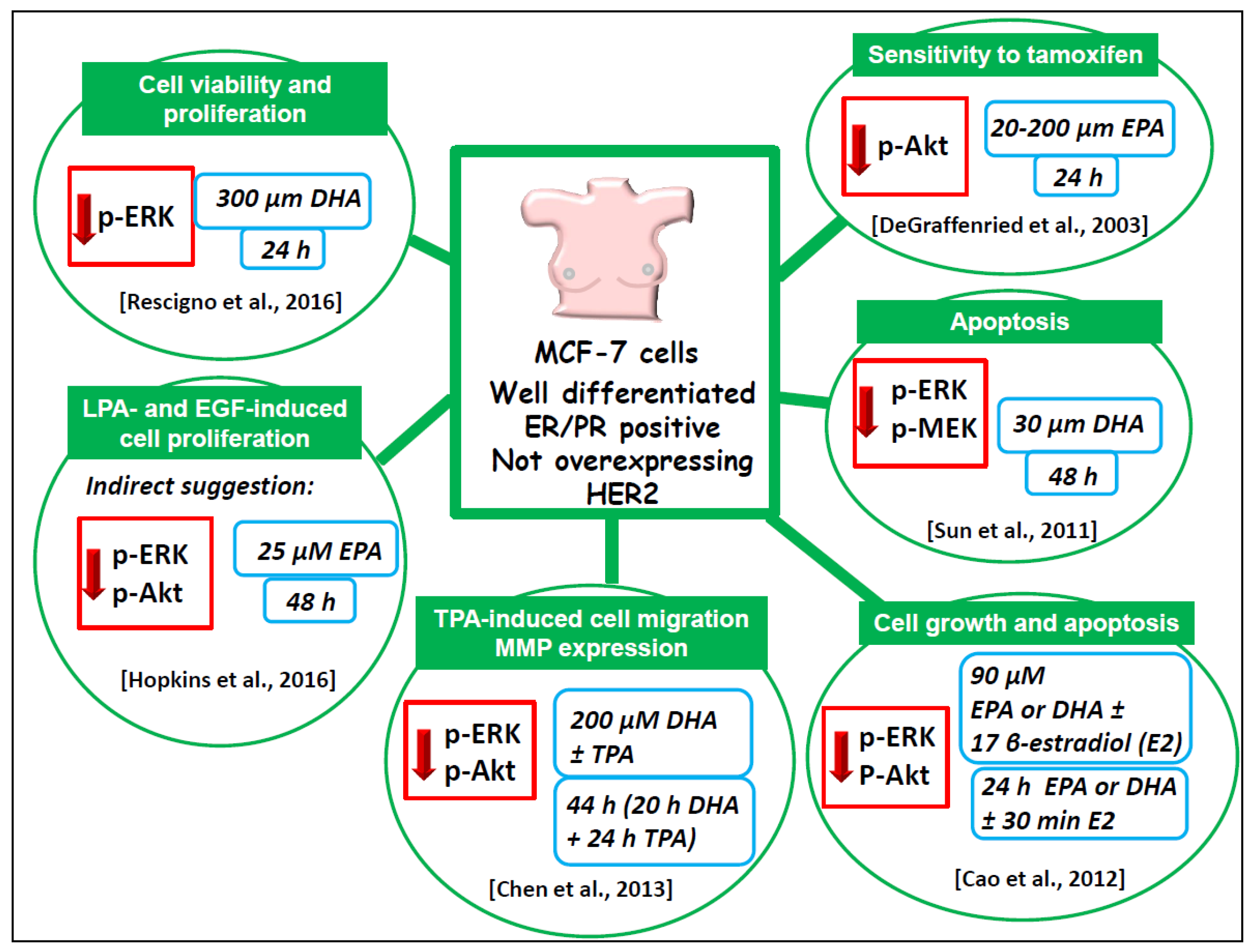
6.1.4. Well-Differentiated MCF-7 Breast Cancer Cell Line
6.1.5. Considerations on the Highly Variable Experimental Conditions Used in the In Vitro Studies
- (1)
- A range of LCn-3 PUFA concentrations should be tested for each single cell line (for most cells usually not higher than 50 µM), as well as variable lengths of incubation, thus trying to avoid those conditions that induce highly cytotoxic effects (i.e., a reduction of viability higher than 10%) for further experiments;
- (2)
- It should not be considered appropriate to justify the use of relatively high concentrations of LCn-3 PUFA only on the basis of their already known physiological serum concentrations in humans in vivo, if these concentrations, when administered in vitro, cause a dramatic reduction of cell viability;
- (3)
- In vitro administration of LCn-3 PUFA to cells, either in the form of FFA or the FA–BSA complex, should be considered equivalent, provided that the doses used are not highly cytotoxic during incubation. However, it is expected that FA–BSA complexes may affect cell processes and/or signaling pathways/molecules more efficiently than comparable concentrations of LCn-3 PUFA in the form FFA.
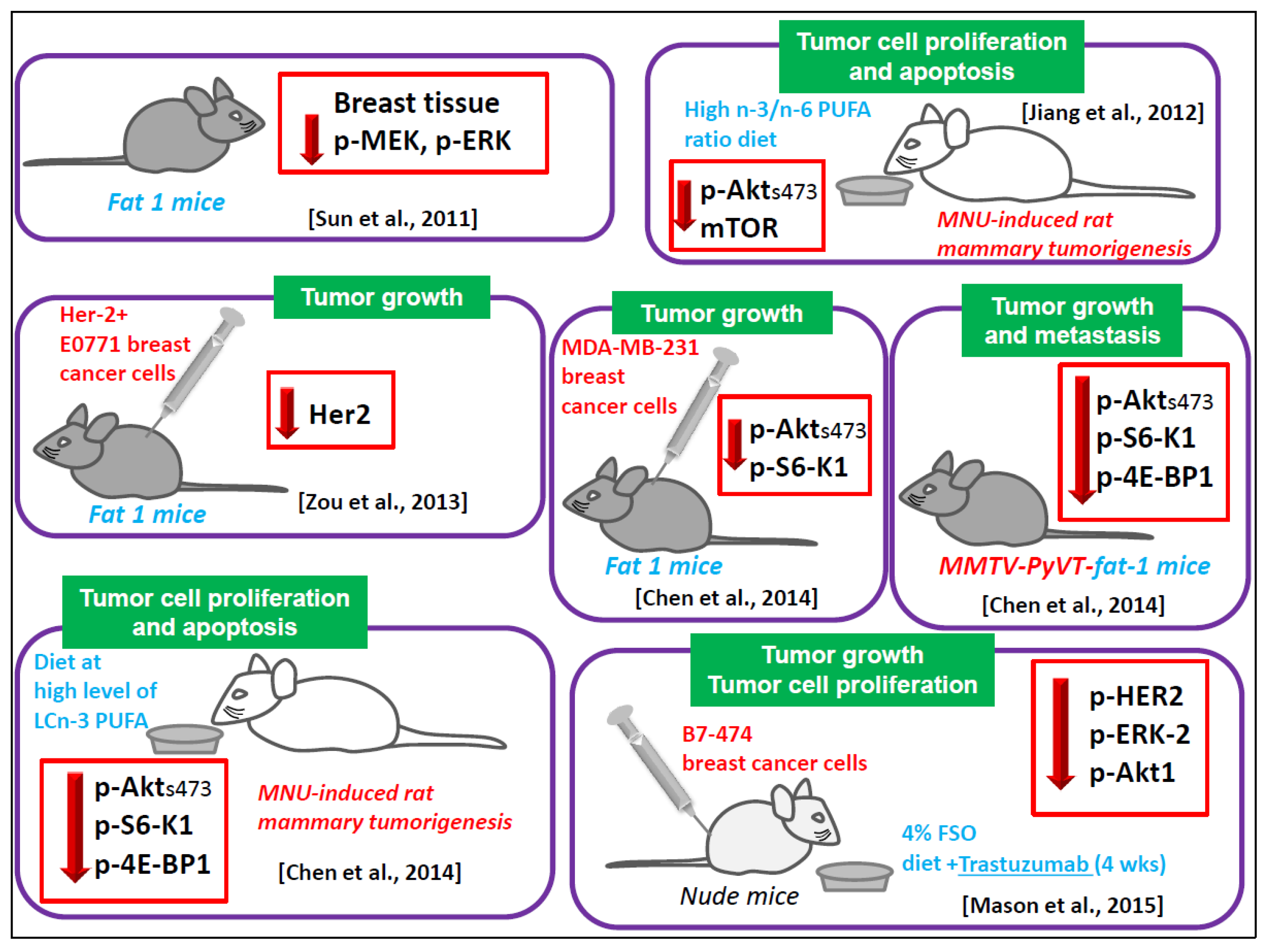
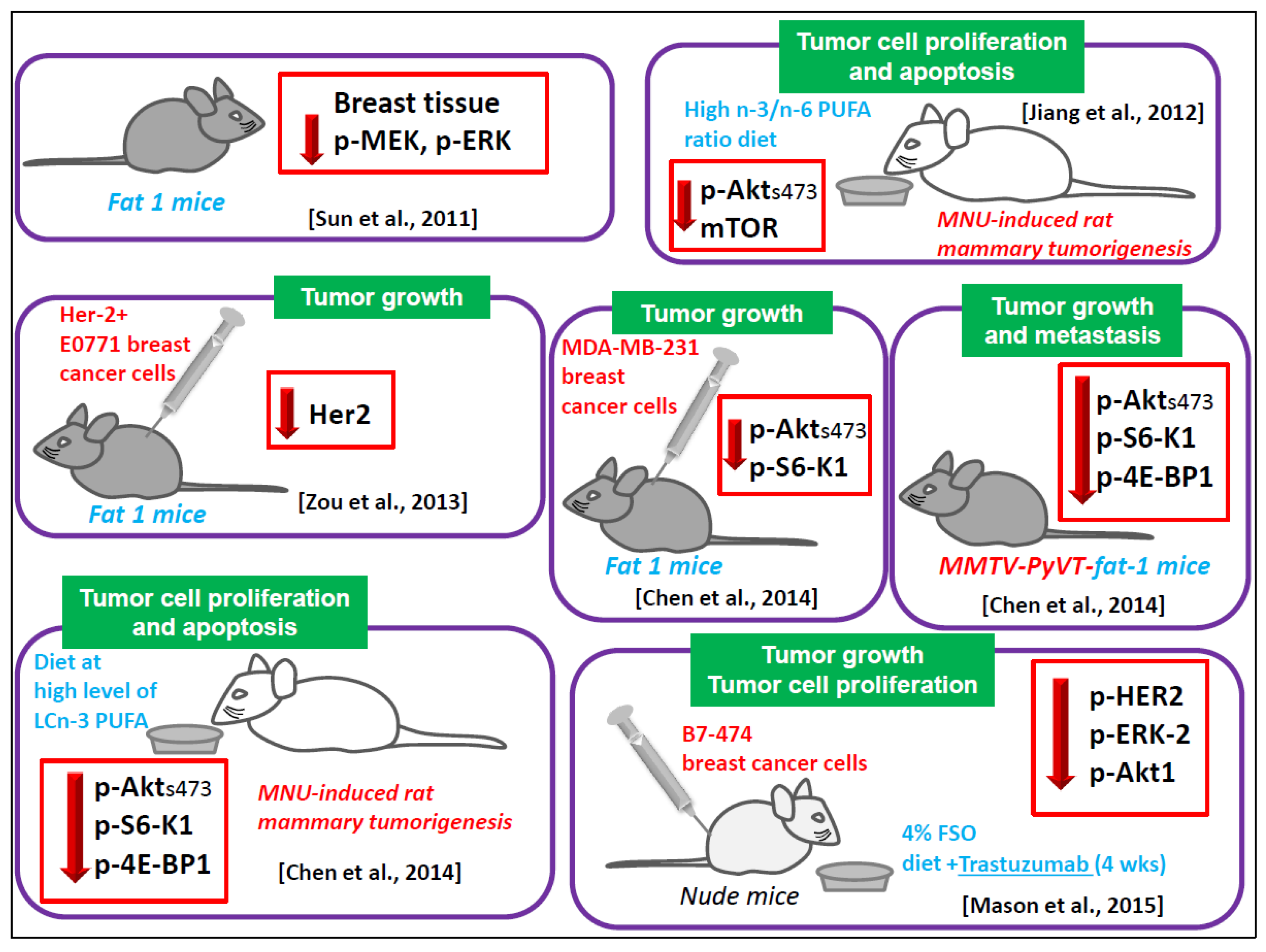
6.1.6. Animal Models
6.1.7. Contrasting Results
6.2. Human Interventional Studies
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moro, K.; Nagahashi, M.; Ramanathan, R.; Takabe, K.; Wakai, T. Resolvins and omega three polyunsaturated fatty acids: Clinical implications in inflammatory diseases and cancer. World J. Clin. Cases 2016, 4, 155–164. [Google Scholar] [CrossRef]
- Gu, Z.; Shan, K.; Chen, H.; Chen, Y.Q. n-3 Polyunsaturated Fatty Acids and their Role in Cancer Chemoprevention. Curr. Pharmacol. Rep. 2015, 1, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhu, J.; Lyu, F.; Panigrahy, D.; Ferrara, K.W.; Hammock, B.; Zhang, G. ω-3 polyunsaturated fatty acids-derived lipid metabolites on angiogenesis, inflammation and cancer. Prostaglandins Other Lipid Mediat. 2014, 113, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, J.A.; Al-Taan, O.; Arshad, A.; Morgan, B.; Metcalfe, M.S.; Dennison, A.R. The multifaceted effects of omega-3 polyunsaturated Fatty acids on the hallmarks of cancer. J. Lipids 2013, 2013, 261247. [Google Scholar] [CrossRef] [PubMed]
- Hardman, W.E. Omega-3 fatty acids to augment cancer therapy. J. Nutr. 2002, 132, 3508S–3512S. [Google Scholar] [PubMed]
- Vara-Messler, M.; Buccellati, C.; Pustina, L.; Folco, G.; Rovati, G.E.; Hoxha, M. A potential role of PUFAs and COXIBs in cancer chemoprevention. Prostaglandins Other Lipid Mediat. 2015, 120, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Fasano, E.; Piccioni, E.; Cittadini, A.R.; Calviello, G. Dietary n-3 polyunsaturated fatty acids and the paradox of their health benefits and potential harmful effects. Chem. Res. Toxicol. 2011, 24, 2093–2105. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Trombino, S.; Oliva, F.; Piccioni, E.; Monego, G.; Resci, F.; Boninsegna, A.; Picci, N.; Ranelletti, F.O.; Calviello, G. Docosahexaenoic acid induces apoptosis in lung cancer cells by increasing MKP-1 and down-regulating p-ERK1/2 and p-p38 expression. Apoptosis 2008, 13, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Wu, J.; Wang, S.; Suburu, J.; Chen, H.; Thomas, M.J.; Shi, L.; Edwards, I.J.; Berquin, I.M.; Chen, Y.Q. Polyunsaturated fatty acids affect the localization and signaling of PIP3/AKT in prostate cancer cells. Carcinogenesis 2013, 34, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, K.; Monsen, V.T.; Hakvåg Pettersen, C.H.; Overland, H.B.; Pettersen, G.; Samdal, H.; Tesfahun, A.N.; Lundemo, A.G.; Bjørkøy, G.; Schønberg, S.A. DHA-induced stress response in human colon cancer cells—Focus on oxidative stress and autophagy. Free Radic. Biol. Med. 2016, 90, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Stillwell, W.; Shaikh, S.R.; Zerouga, M.; Siddiqui, R.; Wassall, S.R. Docosahexaenoic acid affects cell signaling by altering lipid rafts. Reprod. Nutr. Dev. 2005, 45, 559–579. [Google Scholar] [CrossRef] [PubMed]
- Chapkin, R.S.; McMurray, D.N.; Davidson, L.A.; Patil, B.S.; Fan, Y.Y.; Lupton, J.R. Bioactive dietary long-chain fatty acids: Emerging mechanisms of action. Br. J. Nutr. 2008, 100, 1152–1157. [Google Scholar] [CrossRef] [PubMed]
- Ewaschuk, J.B.; Newell, M.; Field, C.J. Docosahexanoic acid improves chemotherapy efficacy by inducing CD95 translocation to lipid rafts in ER(-) breast cancer cells. Lipids 2012, 47, 1019–1030. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Ottes Vasconcelos, R.; Fasano, E.; Calviello, G. How plausible is the use of dietary n-3 PUFA in the adjuvant therapy of cancer? Nutr. Res. Rev. 2016, 29, 102–125. [Google Scholar] [CrossRef] [PubMed]
- Merendino, N.; Costantini, L.; Manzi, L.; Molinari, R.; D’Eliseo, D.; Velotti, F. Dietary ω-3 polyunsaturated fatty acid DHA: A potential adjuvant in the treatment of cancer. Biomed. Res. Int. 2013, 2013, 310186. [Google Scholar] [CrossRef] [PubMed]
- Maione, P.; Gridelli, C.; Troiani, T.; Ciardiello, F. Combining targeted therapies and drugs with multiple targets in the treatment of NSCLC. Oncologist 2006, 11, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Bordoloi, D.; Roy, N.K.; Monisha, J.; Padmavathi, G.; Kunnumakkara, A.B. Multi-targeted agents in cancer cell chemosensitization: What we learnt from curcumin thus far. Recent Pat. Anticancer Drug Discov. 2016, 11, 67–97. [Google Scholar] [CrossRef] [PubMed]
- Monisha, J.; Padmavathi, G.; Roy, N.K.; Deka, A.; Bordoloi, D.; Anip, A.; Kunnumakkara, A.B. NF-κB blockers gifted by mother nature: Prospectives in cancer cell chemosensitization. Curr. Pharm. Des. 2016, 22, 4173–4200. [Google Scholar] [CrossRef] [PubMed]
- Simões, A.E.; Rodrigues, C.M.; Borralho, P.M. The MEK5/ERK5 signalling pathway in cancer: A promising novel therapeutic target. Drug Discov. Today 2016, 21, 1654–1663. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef] [PubMed]
- Uehling, D.E.; Harris, P.A. Recent progress on MAP kinase pathway inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 4047–4056. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.K.; Park, J.I. MEK1/2 Inhibitors: Molecular Activity and Resistance Mechanisms. Semin. Oncol. 2015, 42, 849–862. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Ye, S.; Hu, G.; Lv, M.; Tu, Z.; Zhou, K.; Li, Q. The RAF-MEK-ERK pathway: Targeting ERK to overcome obstacles to effective cancer therapy. Future Med. Chem. 2015, 7, 269–289. [Google Scholar] [CrossRef] [PubMed]
- Strickland, L.R.; Pal, H.C.; Elmets, C.A.; Afaq, F. Targeting drivers of melanoma with synthetic small molecules and phytochemicals. Cancer Lett. 2015, 359, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.K.; Klaire, S.; Kharotia, S.; Wiggins, A.K.; Thompson, L.U. α-linolenic acid and docosahexaenoic acid, alone and combined with trastuzumab, reduce HER2-overexpressing breast cancer cell growth but differentially regulate HER2 signaling pathways. Lipids Health Dis. 2015, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Jeong, S.; Jing, K.; Shin, S.; Kim, S.; Heo, J.Y.; Kweon, G.R.; Park, S.K.; Wu, T.; Park, J.I.; Lim, K. Docosahexaenoic acid induces cell death in human non-small cell lung cancer cells by repressing mTOR via AMPK activation and PI3K/Akt inhibition. Biomed. Res. Int. 2015, 2015, 239764. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Sun, H.; Owens, R.T.; Gu, Z.; Wu, J.; Chen, Y.Q.; O’Flaherty, J.T.; Edwards, I.J. Syndecan-1-dependent suppression of PDK1/Akt/bad signaling by docosahexaenoic acid induces apoptosis in prostate cancer. Neoplasia 2010, 12, 826–836. [Google Scholar] [CrossRef] [PubMed]
- West, K.A.; Castillo, S.S.; Dennis, P.A. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist. Updat. 2002, 5, 234–248. [Google Scholar] [CrossRef]
- Shao, Y.; Aplin, A.E. Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 2010, 70, 6670–6681. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Hugo, W.; Kong, X.; Hong, A.; Koya, R.C.; Moriceau, G.; Chodon, T.; Guo, R.; Johnson, D.B.; Dahlman, K.B.; et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef] [PubMed]
- Mundi, P.S.; Sachdev, J.; McCourt, C.; Kalinsky, K. AKT in cancer: New molecular insights and advances in drug development. Br. J. Clin. Pharmacol. 2016, 82, 943–956. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Bond, P. Regulation of mTORC1 by growth factors, energy status, amino acids and mechanical stimuli at a glance. J. Int. Soc. Sports Nutr. 2016, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Kandel, E.S.; Hay, N. The regulation and activities of the multifunctional serine/threonine kinase AKT/PKB. Exp. Cell Res. 1999, 253, 210–229. [Google Scholar] [CrossRef] [PubMed]
- Nitulescu, G.M.; Margina, D.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Saloustros, E.; Fenga, C.; Spandidos, D.A.; Libra, M.; Tsatsakis, A.M. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 2016, 48, 869–885. [Google Scholar] [CrossRef] [PubMed]
- McCubrey, J.A.; Steelman, L.S.; Abrams, S.L.; Lee, J.T.; Chang, F.; Bertrand, F.E.; Navolanic, P.M.; Terrian, D.M.; Franklin, R.A.; D’Assoro, A.B.; et al. Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv. Enzyme Regul. 2006, 46, 249–279. [Google Scholar] [CrossRef] [PubMed]
- Grabinski, N.; Möllmann, K.; Milde-Langosch, K.; Müller, V.; Schumacher, U.; Brandt, B.; Pantel, K.; Jücker, M. AKT3 regulates ErbB2, ErbB3 and estrogen receptor α expression and contributes to endocrine therapy resistance of ErbB2(+) breast tumor cells from Balb-neuT mice. Cell Signal. 2014, 26, 1021–1029. [Google Scholar] [CrossRef] [PubMed]
- Dienstmann, R.; Rodon, J.; Serra, V.; Tabernero, J. Picking the point of inhibition: A comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol. Cancer Ther. 2014, 13, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, F.; Zoubeidi, A. Dual inhibition of autophagy and the AKT pathway in prostate cancer. Autophagy 2013, 9, 1119–1120. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Tan, A.R.; Im, S.; Villanueva, R.; Valero, V.; Saura, C.; Oliveira, M.; Isakoff, S.J.; Singel, S.M.; Dent, R.A. LOTUS: A randomized, phase II, multicenter, placebo-controlled study of ipatasertib (Ipat, GDC-0068), an inhibitor of Akt, in combination with paclitaxel (Pac) as front-line treatment for patients (pts) with metastatic triple-negative breast cancer (TNBC). J. Clin. Oncol. 2015, 33. [Google Scholar] [CrossRef]
- Frémin, C.; Meloche, S. From basic research to clinical development of MEK1/2 inhibitors for cancer therapy. J. Hematol. Oncol. 2010, 3, 8. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.K.; Fu, M.; Chen, J.; Thompson, L.U. Flaxseed oil enhances the effectiveness of trastuzumab in reducing the growth of HER2-overexpressing human breast tumors (BT-474). J. Nutr. Biochem. 2015, 26, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.K.; Fu, M.H.; Chen, J.; Yu, Z.; Thompson, L.U. Dietary flaxseed-trastuzumab interactive effects on the growth of HER2-overexpressing human breast tumors (BT-474). Nutr. Cancer 2013, 65, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Hudis, C.A. Trastuzumab-mechanism of action and use in clinical practice. N. Engl. J. Med. 2007, 357, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Nikolakopoulou, Z.; Nteliopoulos, G.; Michael-Titus, A.T.; Parkinson, E.K. Omega-3 polyunsaturated fatty acids selectively inhibit growth in neoplastic oral keratinocytes by differentially activating ERK1/2. Carcinogenesis 2013, 34, 2716–2725. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Der, C.J. Targeting the Raf-MEK-ERK mitogenactivated protein kinase cascade for the treatment of cancer. Oncogene 2007, 26, 3291–3310. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Oikonomou, E.; Koustas, E.; Goulielmaki, M.; Pintzas, A. BRAF vs. RAS oncogenes: Are mutations of the same pathway equal? Differential signalling and therapeutic implications. Oncotarget 2014, 5, 11752–11777. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.A.; Beare, D.; Gunasekaran, P.; Leung, K.; Bindal, N.; Boutselakis, H.; Ding, M.; Bamford, S.; Cole, C.; Ward, S.; et al. COSMIC: Exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015, 43, D805–D811. [Google Scholar] [CrossRef] [PubMed]
- Von Lintig, F.C.; Dreilinger, A.D.; Varki, N.M.; Wallace, A.M.; Casteel, D.E.; Boss, G.R. Ras activation in human breast cancer. Breast Cancer Res. Treat. 2000, 62, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Gee, J.M.; Robertson, J.F.; Ellis, I.O.; Nicholson, R.I. Phosphorylation of ERK1/2 mitogen-activated protein kinase is associated with poor response to anti-hormonal therapy and decreased patient survival in clinical breast cancer. Int. J. Cancer 2001, 95, 247–254. [Google Scholar] [CrossRef]
- Eckert, L.B.; Repasky, G.A.; Ulkü, A.S.; McFall, A.; Zhou, H.; Sartor, C.I.; Der, C.J. Involvement of Ras activation in human breast cancer cell signaling, invasion, and anoikis. Cancer Res. 2004, 64, 4585–4592. [Google Scholar] [CrossRef] [PubMed]
- Tsubaki, M.; Satou, T.; Itoh, T.; Imano, M.; Ogaki, M.; Yanae, M.; Nishida, S. Hyperexpression of mitogen-activated protein kinase in human breast cancer. Toxicol. Appl. Pharmacol. 2012, 259, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Schley, P.D.; Jijon, H.B.; Robinson, L.E.; Field, C.J. Mechanisms of omega-3 fatty acid-induced growth inhibition in MDA-MB-231 human breast cancer cells. Breast Cancer Res. Treat. 2005, 92, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Rogers, K.R.; Kikawa, K.D.; Mouradian, M.; Hernandez, K.; McKinnon, K.M.; Ahwah, S.M.; Pardini, R.S. Docosahexaenoic acid alters epidermal growth factor receptor-related signaling by disrupting its lipid raft association. Carcinogenesis 2010, 31, 1523–1530. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, L.; Goupille, C.; Blanc, C.; Pinault, M.; Domingo, I.; Guimaraes, C.; Bougnoux, P.; Chevalier, S.; Mahéo, K. Long chain n-3 polyunsaturated fatty acids increase the efficacy of docetaxel in mammary cancer cells by downregulating Akt and PKCε/δ-induced ERK pathways. Biochim. Biophys. Acta 2016, 1861, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.B.; Park, J.; Kawamoto, J.; Sato, S.; Kurihara, T. Inhibition of constitutive Akt (PKB) phosphorylation by docosahexaenoic acid in the human breast cancer cell line MDA-MB-453. Biochim. Biophys. Acta 2013, 1831, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Yun, U.J.; Koo, K.H.; Sung, J.Y.; Shim, J.; Ye, S.K.; Hong, K.M.; Kim, Y.N. Down-regulation of lipid raft-associated onco-proteins via cholesterol-dependent lipid raft internalization in docosahexaenoic acid-induced apoptosis. Biochim. Biophys. Acta 2014, 1841, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Lasserre, R.; Guo, X.J.; Conchonaud, F.; Hamon, Y.; Hawchar, O.; Bernard, A.M.; Soudja, S.M.; Lenne, P.F.; Rigneault, H.; Olive, D.; et al. Raft nanodomains contribute to Akt/PKB plasma membrane recruitment and activation. Nat. Chem. Biol. 2008, 4, 538–547. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, M.M.; Zhang, Z.; Liu, Z.; Meier, K.E. Eicosopentaneoic acid and other free fatty acid receptor agonists inhibit lysophosphatidic acid- and epidermal growth factor-induced proliferation of human breast cancer cells. J. Clin. Med. 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Mouradian, M.; Kikawa, K.D.; Johnson, E.D.; Beck, K.L.; Pardini, R.S. Key roles for GRB2-associated-binding protein 1, phosphatidylinositol-3-kinase, cyclooxygenase 2, prostaglandin E2 and transforming growth factor alpha in linoleic acid-induced upregulation of lung and breast cancer cell growth. Prostaglandins Leukot. Essent. Fat. Acids 2014, 90, 105–115. [Google Scholar] [CrossRef]
- Li, C.C.; Yao, H.T.; Cheng, F.J.; Hsieh, Y.H.; Lu, C.Y.; Wu, C.C.; Liu, K.L.; Chang, J.W. Docosahexaenoic acid downregulates EGF-induced urokinase plasminogen activator and matrix metalloproteinase 9 expression by inactivating EGFR/ErbB2 signaling in SK-BR3 breast cancer cells. Nutr. Cancer 2015, 67, 771–782. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, T.; Capasso, A.; Tecce, M.F. Effect of docosahexaenoic acid on cell cycle pathways in breast cell lines with different transformation degree. J. Cell. Physiol. 2016, 231, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Hu, Y.; Gu, Z.; Owens, R.T.; Chen, Y.Q.; Edwards, I.J. Omega-3 fatty acids induce apoptosis in human breast cancer cells and mouse mammary tissue through syndecan-1 inhibition of the MEK-Erk pathway. Carcinogenesis 2011, 32, 1518–1524. [Google Scholar] [CrossRef] [PubMed]
- Van der Vusse, G.J. Albumin as fatty acid transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Zou, Z.; Bellenger, S.; Massey, K.A.; Nicolaou, A.; Geissler, A.; Bidu, C.; Bonnotte, B.; Pierre, A.S.; Minville-Walz, M.; Rialland, M.; et al. Inhibition of the HER2 pathway by n-3 polyunsaturated fatty acids prevents breast cancer in fat-1 transgenic mice. J. Lipid Res. 2013, 54, 3453–3463. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Berquin, I.M.; Owens, R.T.; O’Flaherty, J.T.; Edwards, I.J. Peroxisome proliferator-activated receptor gamma-mediated up-regulation of syndecan-1 by n-3 fatty acids promotes apoptosis of human breast cancer cells. Cancer Res. 2008, 68, 2912–2919. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Ma, Z.; Rasenick, M.M.; Yeh, S.; Yu, J. N-3 poly-unsaturated fatty acids shift estrogen signaling to inhibit human breast cancer cell growth. PLoS ONE 2012, 7, e52838. [Google Scholar] [CrossRef]
- Chen, H.W.; Chao, C.Y.; Lin, L.L.; Lu, C.Y.; Liu, K.L.; Lii, C.K.; Li, C.C. Inhibition of matrix metalloproteinase-9 expression by docosahexaenoic acid mediated by heme oxygenase 1 in 12-O-tetradecanoylphorbol- 13-acetate-induced MCF-7 human breast cancer cells. Arch. Toxicol. 2013, 87, 857–869. [Google Scholar] [CrossRef]
- Lin, C.W.; Hou, W.C.; Shen, S.C.; Juan, S.H.; Ko, C.H.; Wang, L.M.; Chen, Y.C. Quercetin inhibition of tumor invasion via suppressing PKC delta/ERK/AP-1-dependent matrix metalloproteinase-9 activation in breast carcinoma cells. Carcinogenesis 2008, 29, 1807–1815. [Google Scholar] [CrossRef] [PubMed]
- Park, S.K.; Hwang, Y.S.; Park, K.K.; Park, H.J.; Seo, J.Y.; Chung, W.Y. Kalopanaxsaponin A inhibits PMA-induced invasion by reducing matrix metalloproteinase-9 via PI3K/Akt- and PKCdelta-mediated signaling in MCF-7 human breast cancer cells. Carcinogenesis 2009, 30, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- DeGraffenried, L.A.; Friedrichs, W.E.; Fulcher, L.; Fernandes, G.; Silva, J.M.; Peralba, J.M.; Hidalgo, M. Eicosapentaenoic acid restores tamoxifen sensitivity in breast cancer cells with high Akt activity. Ann. Oncol. 2003, 14, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Zirpoli, H.; Caputo, M.; Carraturo, A.; Torino, G.; Fazio, A.; Attya, M.; Rastrelli, L.; Tecce, M.F. Selective action of human sera differing in fatty acids and cholesterol content on in vitro gene expression. J. Cell Biochem. 2012, 113, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Van Hees, A.M.; Saris, W.H.; Hul, G.B.; Schaper, N.C.; Timmerman, B.E.; Lovegrove, J.A.; Roche, H.M.; Blaak, E.E. Effects of dietary fat modification on skeletal muscle fatty acid handling in the metabolic syndrome. Int. J. Obes. 2010, 34, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Superko, H.R.; Superko, S.M.; Nasir, K.; Agatston, A.; Garrett, B.C. Omega-3 fatty acid blood levels: Clinical significance and controversy. Circulation 2013, 128, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, R.; Kato, Y.; Imai, T.; Ando, F.; Shimokata, H. Higher serum EPA or DHA, and lower ARA compositions with age independent fatty acid intake in Japanese aged 40 to 79. Lipids 2013, 48, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Folsom, A.R.; Eckfeldt, J.H.; Lewis, L.; Chambless, L.E. Short- and long-term repeatability of fatty acid composition of human plasma phospholipids and cholesterol esters. The Atherosclerosis Risk in Communities (ARIC) Study Investigators. Am. J. Clin. Nutr. 1995, 62, 572–578. [Google Scholar] [PubMed]
- Bønaa, K.H.; Bjerve, K.S.; Nordøy, A. Habitual fish consumption, plasma phospholipid fatty acids, and serum lipids: The Tromsø study. Am. J. Clin. Nutr. 1992, 55, 1126–1134. [Google Scholar] [PubMed]
- Nikkari, T.; Luukkainen, P.; Pietinen, P.; Puska, P. Fatty acid composition of serum lipid fractions in relation to gender and quality of dietary fat. Ann. Med. 1995, 27, 491–498. [Google Scholar] [CrossRef]
- Conquer, J.A.; Holub, B.J. Effect of supplementation with different doses of DHA on the levels of circulating DHA as non-esterified fatty acid in subjects of Asian Indian background. J. Lipid Res. 1998, 39, 286–292. [Google Scholar] [PubMed]
- Fukuzawa, K.; Saitoh, Y.; Akai, K.; Kogure, K.; Ueno, S.; Tokumura, A.; Otagiri, M.; Shibata, A. Antioxidant effect of bovine serum albumin on membrane lipid peroxidation induced by iron chelate and superoxide. Biochim. Biophys. Acta 2005, 1668, 145–155. [Google Scholar] [CrossRef]
- Kanno, S.; Kurauchi, K.; Tomizawa, A.; Yomogida, S.; Ishikawa, M. Albumin modulates docosahexaenoic acid-induced cytotoxicity in human hepatocellular carcinoma cell lines. Toxicol. Lett. 2011, 200, 154–161. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Power, K.A.; Mann, J.; Cheng, A.; Thompson, L.U. Flaxseed alone or in combination with tamoxifen inhibits MCF-7 breast tumor growth in ovariectomized athymic mice with high circulating levels of estrogen. Exp. Biol. Med. 2007, 232, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Saggar, J.K.; Chen, J.; Corey, P.; Thompson, L.U. Dietary flaxseed lignan or oil combined with tamoxifen treatment affects MCF-7 tumor growth through estrogen receptor- and growth factor-signaling pathways. Mol. Nutr. Food Res. 2010, 54, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhang, Y.; Jia, C.; Wang, Y.; Lai, P.; Zhou, X.; Song, Q.; Lin, J.; Ren, Z.; Gao, Q.; et al. mTORC1/2 targeted by n-3 polyunsaturated fatty acids in the prevention of mammary tumorigenesis and tumor progression. Oncogene 2014, 33, 4548–4557. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Zhu, Z.; McGinley, J.N.; El Bayoumy, K.; Manni, A.; Thompson, H.J. Identification of a molecular signature underlying inhibition of mammary carcinoma growth by dietary N-3 fatty acids. Cancer Res. 2012, 72, 3795–3806. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Guo, Y.; Wu, Y.; Zhu, S.; He, Z.; Chen, Y.Q. Omega-3 free fatty acids inhibit tamoxifen-induced cell apoptosis. Biochem. Biophys. Res. Commun. 2015, 459, 294–299. [Google Scholar] [CrossRef] [PubMed]
- Manni, A.; Xu, H.; Washington, S.; Aliaga, C.; Cooper, T.; Richie, J.P.; Bruggeman, R.; Prokopczyk, B.; Calcagnotto, A.; Trushin, N.; et al. The impact of fish oil on the chemopreventive efficacy of tamoxifen against development of N-methyl-N-nitrosourea-induced rat mammary carcinogenesis. Cancer Prev. Res. 2010, 3, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Manni, A.; Richie, J.P., Jr.; Xu, H.; Washington, S.; Aliaga, C.; Bruggeman, R.; Cooper, T.K.; Prokopczyk, B.; Trushin, N.; Calcagnotto, A.; et al. Influence of omega-3 fatty acids on Tamoxifen-induced suppression of rat mammary carcinogenesis. Int. J. Cancer 2014, 134, 1549–1557. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K.; Yang, J.W.; Kim, M.R.; Roh, S.H.; Kim, H.G.; Lee, K.Y.; Jeong, H.G.; Kang, K.W. Increased expression of Nrf2/ARE-dependent anti-oxidant proteins in tamoxifen-resistant breast cancer cells. Free Radic. Biol. Med. 2008, 45, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Jordan, N.J.; Gee, J.M.; Barrow, D.; Wakeling, A.E.; Nicholson, R.I. Increased constitutive activity of PKB/Akt in tamoxifen resistant breast cancer MCF-7 cells. Breast Cancer Res. Treat. 2004, 87, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Fabian, C.J.; Kimler, B.F.; Phillips, T.A.; Box, J.A.; Kreutzjans, A.L.; Carlson, S.E.; Hidaka, B.H.; Metheny, T.; Zalles, C.M.; Mills, G.B.; et al. Modulation of breast cancer risk biomarkers by high-dose omega-3 fatty acids: Phase II pilot study in premenopausal women. Cancer Prev. Res. 2015, 8, 912–921. [Google Scholar] [CrossRef] [PubMed]
- Fabian, C.J.; Kimler, B.F.; Phillips, T.A.; Nydegger, J.L.; Kreutzjans, A.L.; Carlson, S.E.; Hidaka, B.H.; Metheny, T.; Zalles, C.M.; Mills, G.B.; et al. Modulation of breast cancer risk biomarkers by high-dose omega-3 fatty acids: Phase II pilot study in postmenopausal women. Cancer Prev. Res. 2015, 8, 922–931. [Google Scholar] [CrossRef] [PubMed]
- Bougnoux, P.; Hajjaji, N.; Ferrasson, M.N.; Giraudeau, B.; Couet, C.; Le Floch, O. Improving outcome of chemotherapy of metastatic breast cancer by docosahexaenoic acid: A phase II trial. Br. J. Cancer 2009, 101, 1978–1985. [Google Scholar] [CrossRef] [PubMed]
- Aronson, W.J.; Kobayashi, N.; Barnard, R.J.; Henning, S.; Huang, M.; Jardack, P.M.; Liu, B.; Gray, A.; Wan, J.; Konijeti, R.; et al. Phase II prospective randomized trial of a low-fat diet with fish oil supplementation in men undergoing radical prostatectomy. Cancer Prev. Res. 2011, 4, 2062–2071. [Google Scholar] [CrossRef] [PubMed]
- Fasano, E.; Serini, S.; Cittadini, A.; Calviello, G. Long-Chain n-3 PUFA against breast and prostate cancer: Which are the appropriate doses for intervention studies in animals and humans? Crit. Rev. Food Sci. Nutr. 2015. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serini, S.; Calviello, G. Modulation of Ras/ERK and Phosphoinositide Signaling by Long-Chain n-3 PUFA in Breast Cancer and Their Potential Complementary Role in Combination with Targeted Drugs. Nutrients 2017, 9, 185. https://doi.org/10.3390/nu9030185
Serini S, Calviello G. Modulation of Ras/ERK and Phosphoinositide Signaling by Long-Chain n-3 PUFA in Breast Cancer and Their Potential Complementary Role in Combination with Targeted Drugs. Nutrients. 2017; 9(3):185. https://doi.org/10.3390/nu9030185
Chicago/Turabian StyleSerini, Simona, and Gabriella Calviello. 2017. "Modulation of Ras/ERK and Phosphoinositide Signaling by Long-Chain n-3 PUFA in Breast Cancer and Their Potential Complementary Role in Combination with Targeted Drugs" Nutrients 9, no. 3: 185. https://doi.org/10.3390/nu9030185