Nonalcoholic Fatty Liver Disease Is Exacerbated in High-Fat Diet-Fed Gnotobiotic Mice by Colonization with the Gut Microbiota from Patients with Nonalcoholic Steatohepatitis

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Microbiota Transplantation and Experimental Design
2.3. Clinical Chemistry
2.4. Histopathological Evaluation
2.5. Oil Red O Staining
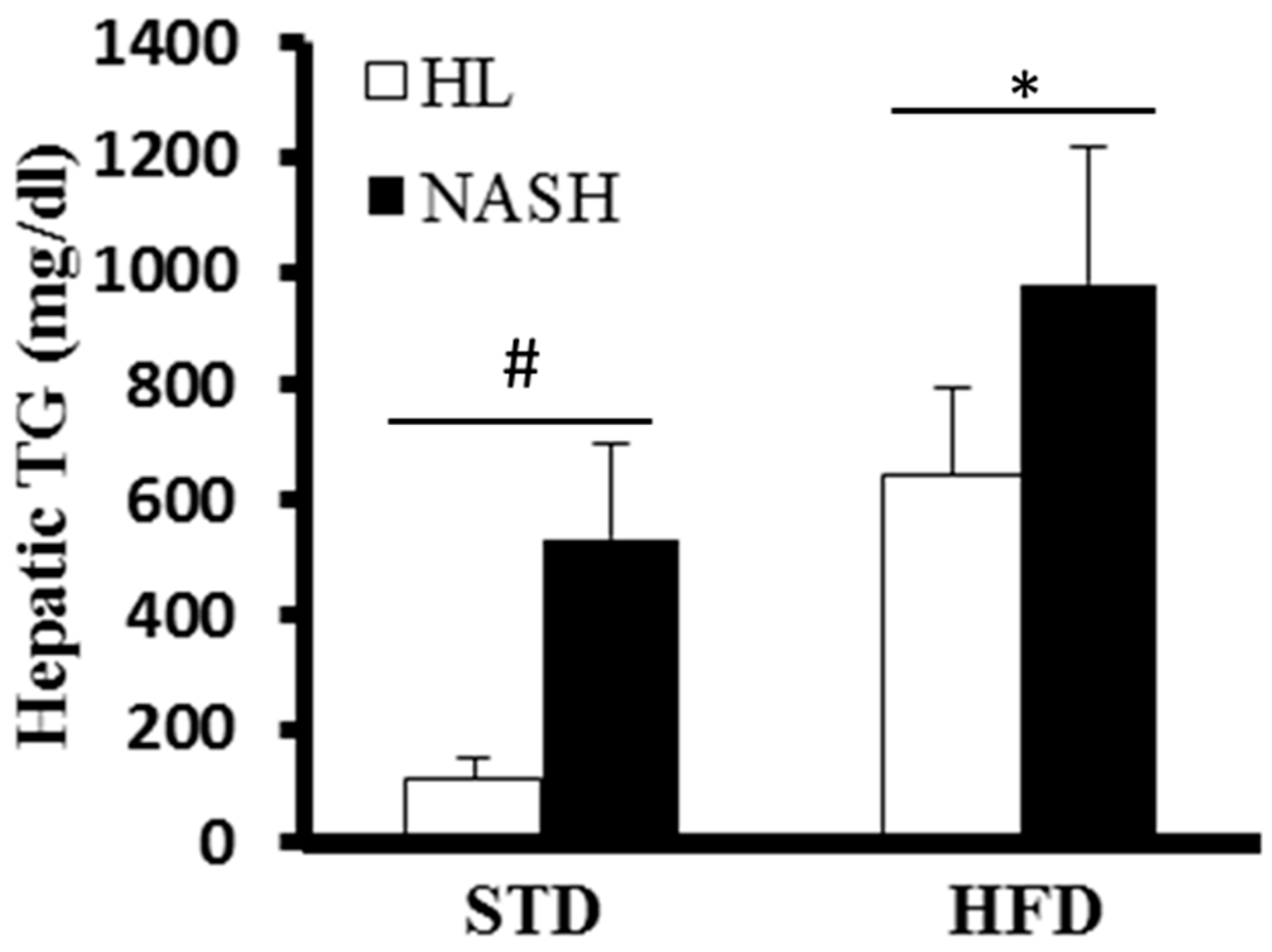
2.6. Liver TGs
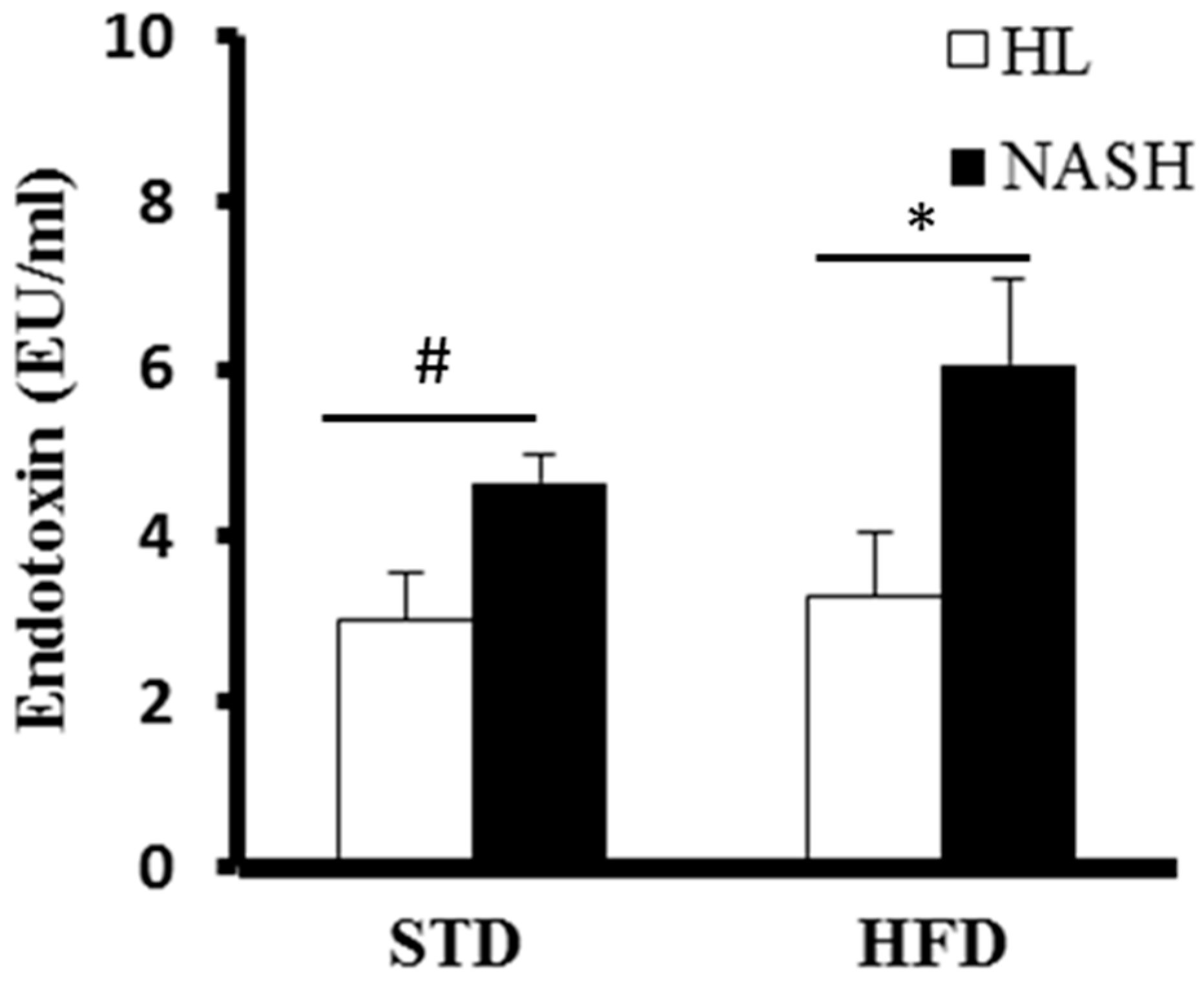
2.7. Limulus Amebocyte Lysate Test
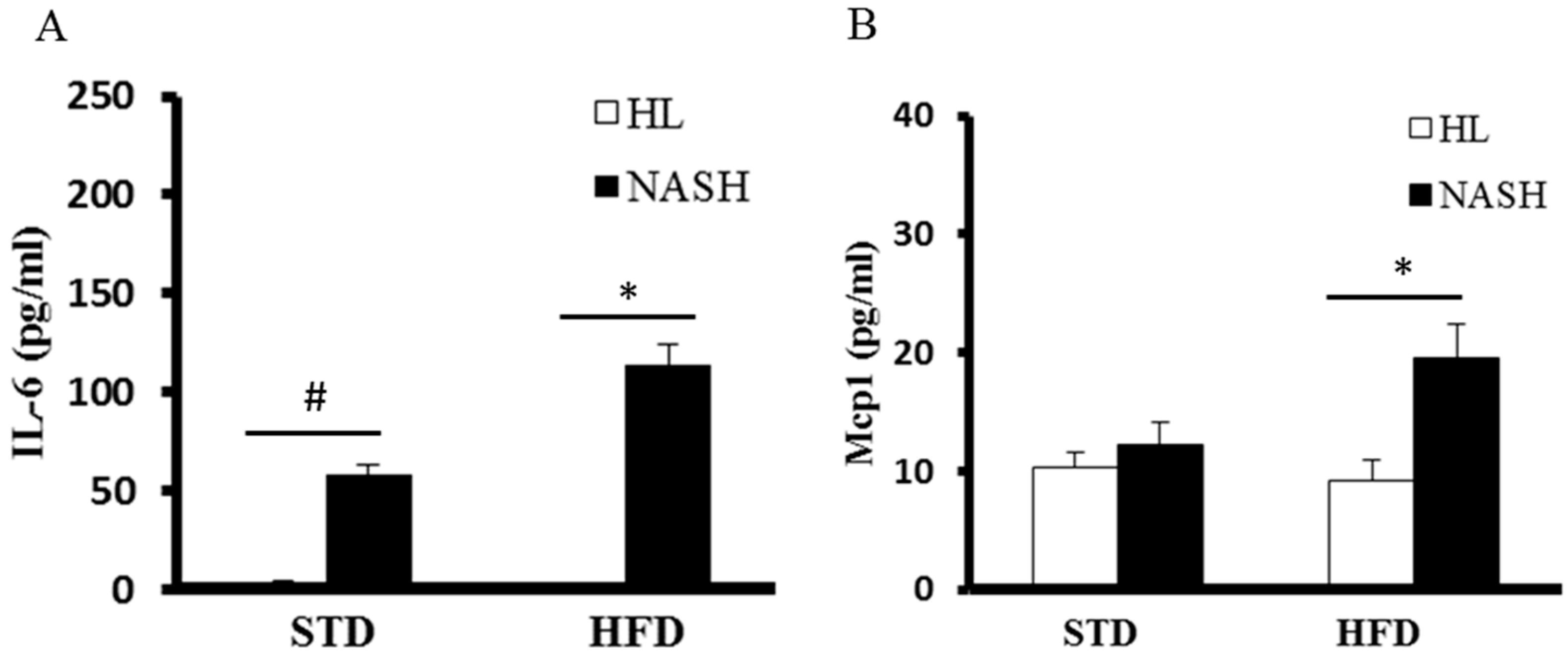
2.8. Serum Levels of Tnf-α, IL-6, Mcp1, and Insulin
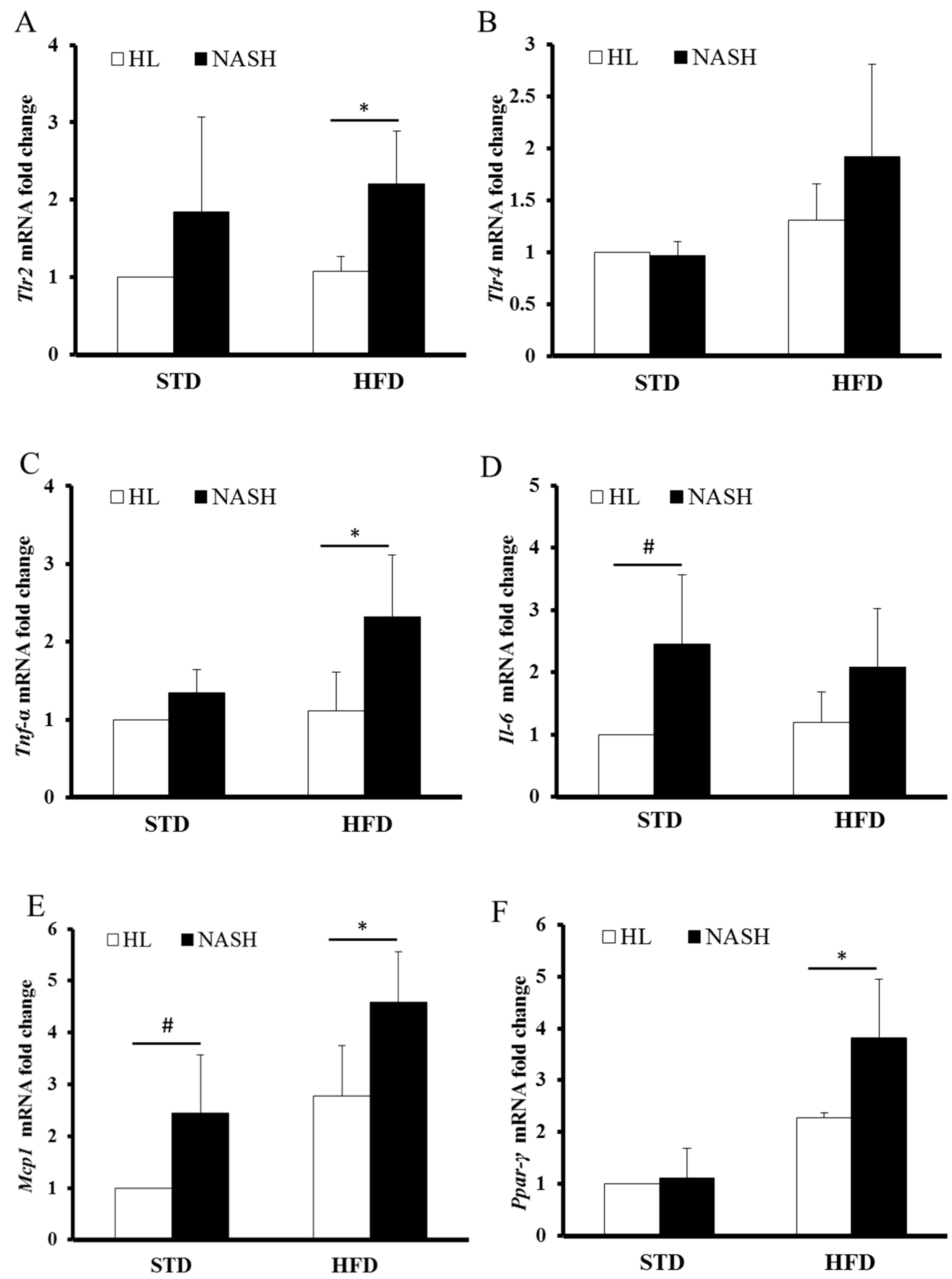
2.9. Quantitative Real-Time Reverse-Transcription PCR (qRT-PCR)
2.10. Gut Bacterial Composition
2.11. Statistical Analysis
3. Results
3.1. Effects of the Gut Microbiota on Body, Liver, and Epididymal Fat Weight in Mice
3.2. Changes in Clinical Biochemistry Parameters as a Result of Colonization by Different Human Gut Microbiota
3.3. Liver of the NASH-HFD Mice Showed a More Advanced Stage of Steatohepatitis
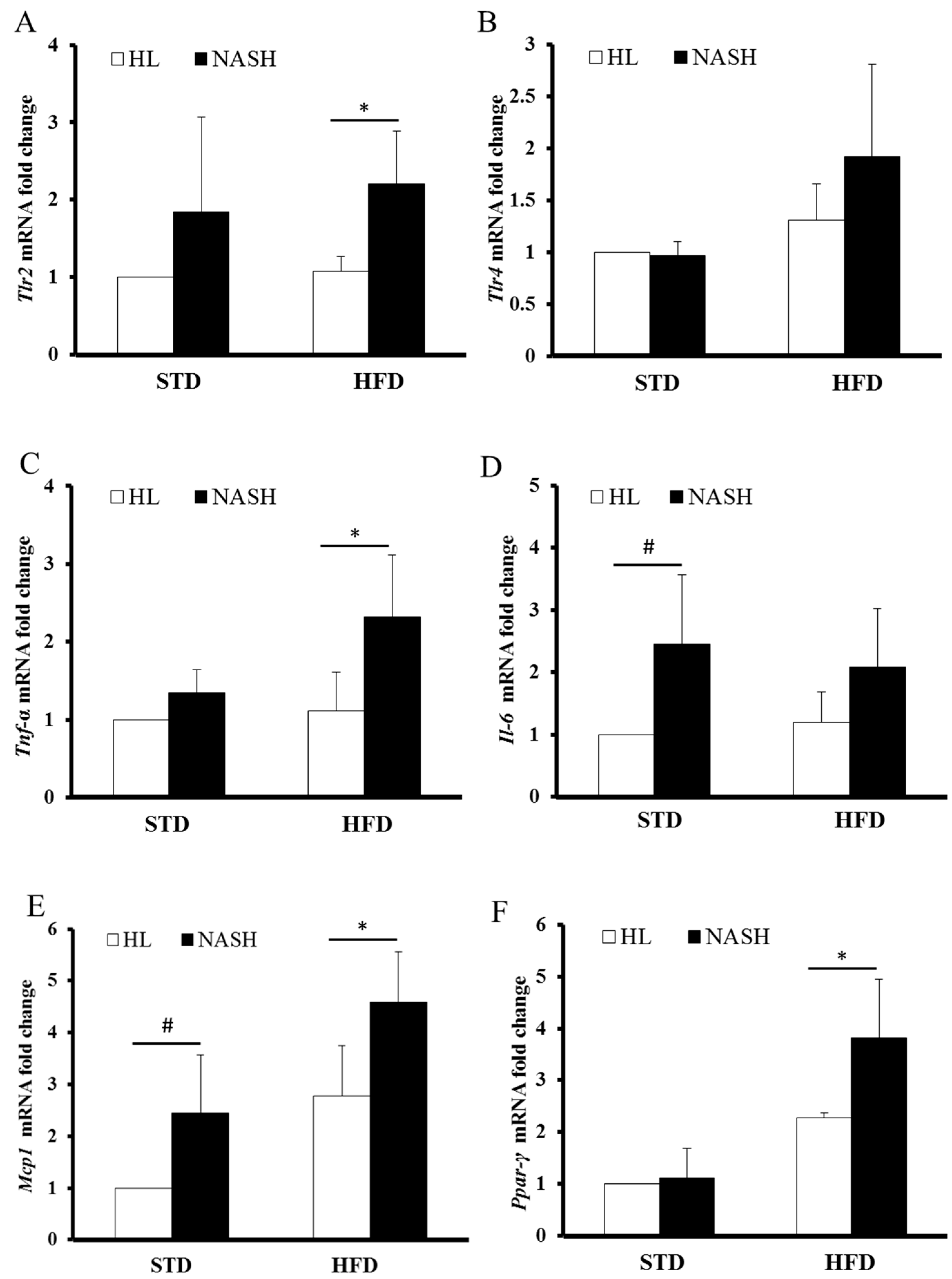
3.4. Serum Levels of Cytokines and Endotoxin Are Significantly Elevated in NASH-HFD Mice
3.5. Analysis of Gene Expression by Real-Time PCR
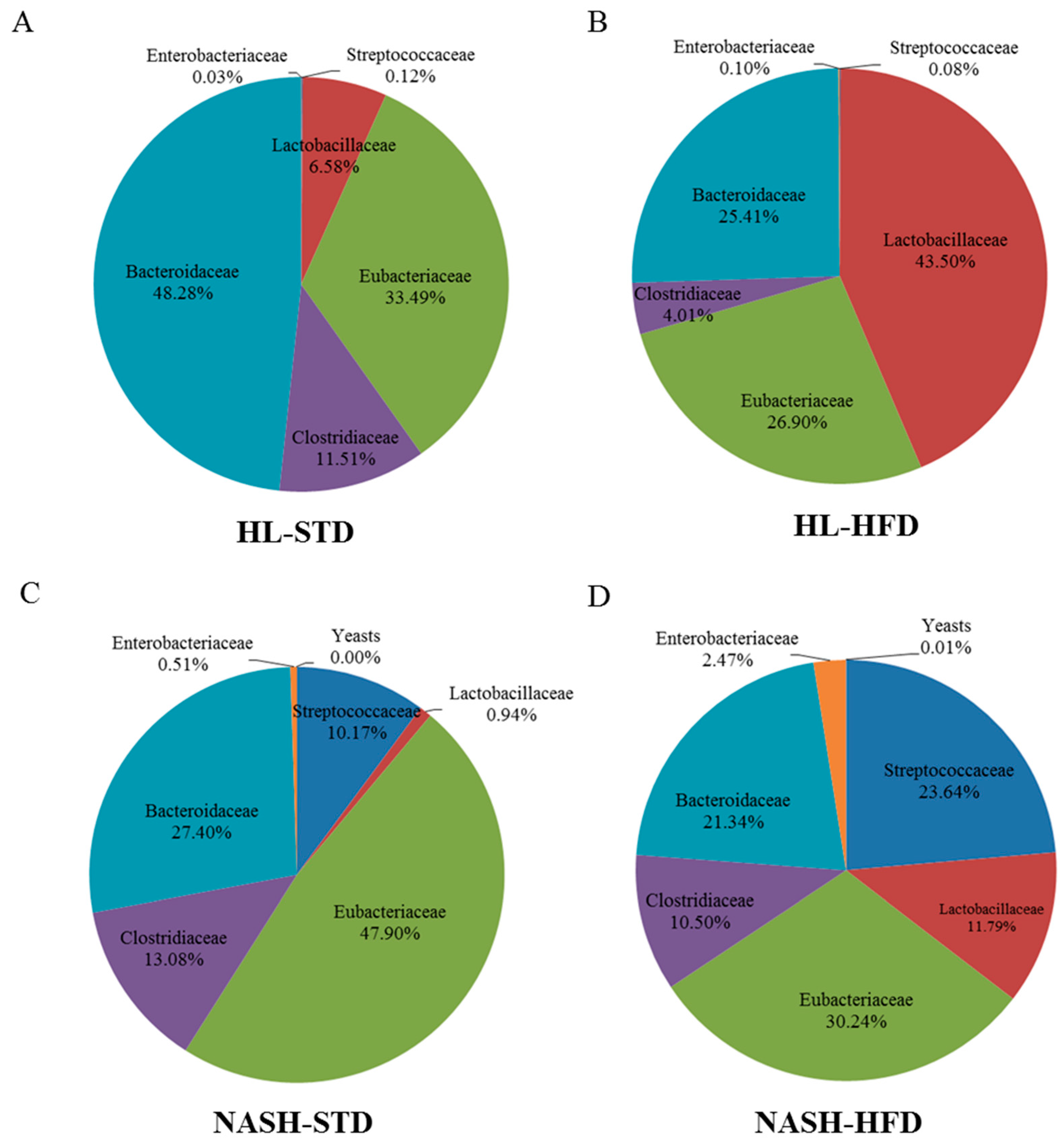
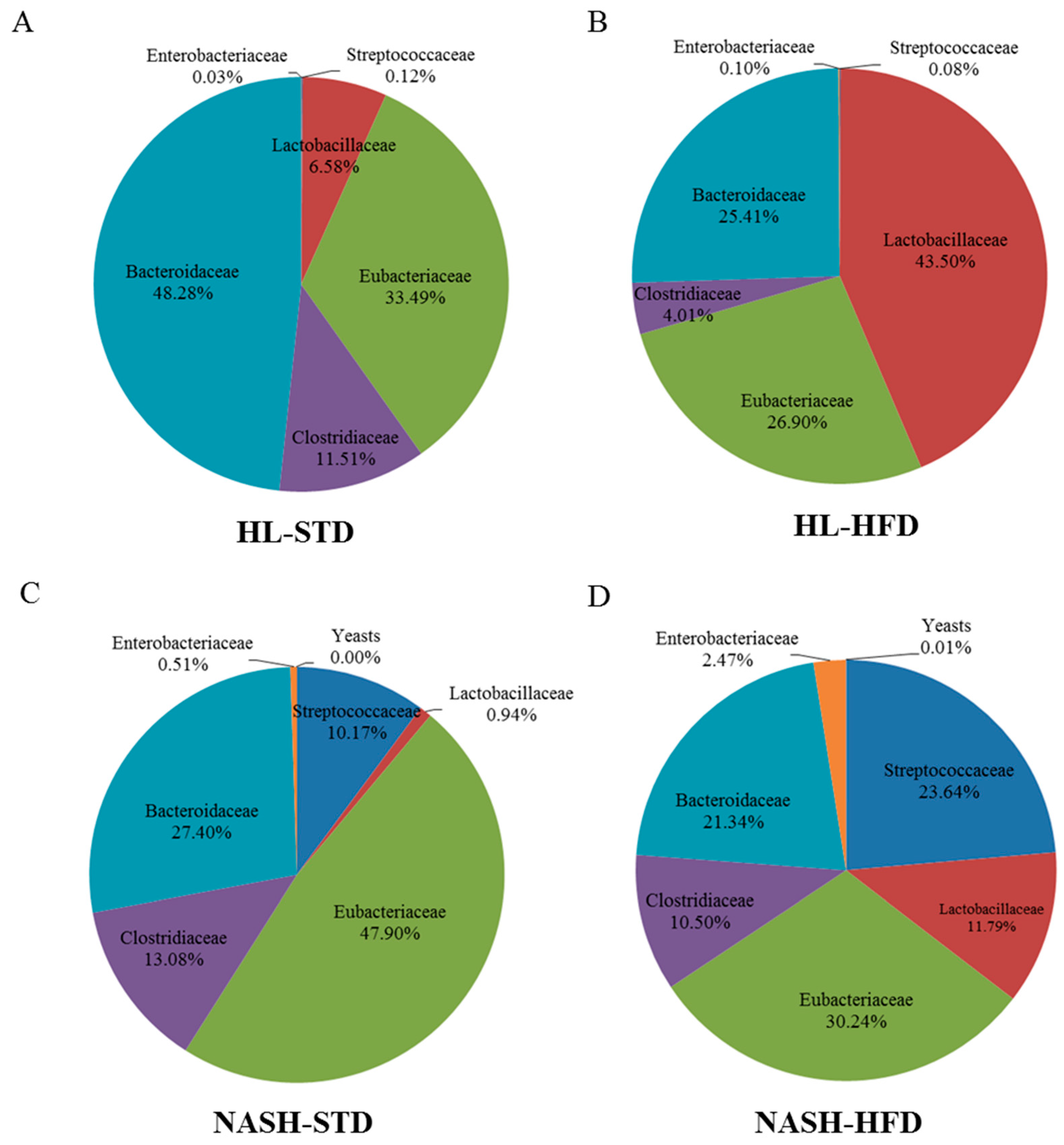
3.6. Analysis of Gut Microbiota Composition
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Adams, L.A.; Lymp, J.F.; St. Sauver, J.; Sanderson, S.O.; Lindor, K.D.; Feldstein, A.; Angulo, P. The natural history of nonalcoholic fatty liver disease: A population-based cohort study. Gastroenterology 2005, 129, 113–121. [Google Scholar] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Toshikuni, N.; Tsutsumi, M.; Arisawa, T. Clinical differences between alcoholic liver disease and nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 8393–8406. [Google Scholar] [PubMed]
- Liang, W.; Menke, A.L.; Driessen, A.; Koek, G.H.; Lindeman, J.H.; Stoop, R.; Havekes, L.M.; Kleemann, R.; van den Hoek, A.M. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014, 9, e115922. [Google Scholar] [CrossRef] [PubMed]
- Ganz, M.; Bukong, T.N.; Csak, T.; Saha, B.; Park, J.K.; Ambade, A.; Kodys, K.; Szabo, G. Progression of non-alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat-cholesterol-sugar diet model in mice. J. Transl. Med. 2015, 13, 193. [Google Scholar] [CrossRef] [PubMed]
- Ellekilde, M.; Selfjord, E.; Larsen, C.S.; Jakesevic, M.; Rune, I.; Tranberg, B.; Vogensen, F.K.; Nielsen, D.S.; Bahl, M.I.; Licht, T.R.; et al. Transfer of gut microbiota from lean and obese mice to antibiotic-treated mice. Sci. Rep. 2014, 4, 5922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory bowel disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef] [PubMed]
- Kelly, L.; Lyon, D.E.; Yoon, S.L.; Horgas, A.L. The microbiome and cancer: Implications for oncology nursing science. Cancer Nurs. 2016, 39, E56–E62. [Google Scholar] [CrossRef] [PubMed]
- Gäbele, E.; Dostert, K.; Hofmann, C.; Wiest, R.; Schölmerich, J.; Hellerbrand, C.; Obermeier, F. DSS induced colitis increases portal LPS levels and enhances hepatic inflammation and fibrogenesis in experimental NASH. J. Hepatol. 2011, 55, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Hudcovic, T.; Mrazek, J.; Kozakova, H.; Srutkova, D.; Schwarzer, M.; Tlaskalova-Hogenova, H.; Kostovcik, M.; Kverka, M. Development of gut inflammation in mice colonized with mucosa-associated bacteria from patients with ulcerative colitis. Gut Pathog. 2015, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Lu, L.; Yu, Y.; Guo, Y.; Wang, Y.; Chang, E.B.; Claud, E.C. Transcriptional modulation of intestinal innate defense/inflammation genes by preterm infant microbiota in a humanized gnotobiotic mouse model. PLoS ONE 2015, 10, e0124504. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The effect of diet on the human gut microbiome: A metagenomic analysis in humanized gnotobiotic mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.L.; Kallstrom, G.; Faith, J.J.; Reyes, A.; Moore, A.; Dantas, G.; Gordon, J.I. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc. Natl. Acad. Sci. USA 2011, 108, 6252–6257. [Google Scholar] [CrossRef] [PubMed]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Mitsuoka, T. Production of gnotobiotic mice with normal physiological functions. I. Selection of useful bacteria from feces of conventional mice. Z. Vers. 1980, 22, 173–178. [Google Scholar]
- Yanabe, M.; Shibuya, M.; Gonda, T.; Asai, H.; Tanaka, T.; Sudou, K.; Narita, T.; Itoh, K. Establishment of specific pathogen-free (SPF) rat colonies using gnotobiotic techniques. Exp. Anim. 2001, 50, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Mitsuoka, T.; Sudo, K.; Suzuki, K. Comparison of fecal flora of mice based upon different strains and different housing conditions. Z. Vers. 1983, 25, 135–146. [Google Scholar]
- Brunt, E.M.; Janney, C.G.; Di Bisceglie, A.M.; Neuschwander-Tetri, B.A.; Bacon, B.R. Nonalcoholic steatohepatitis: A proposal for grading and staging the histological lesions. Am. J. Gastroenterol. 1999, 94, 2467–2474. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Delzenne, N.M. Gut microflora as a target for energy and metabolic homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 729–734. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Wu, E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes 2012, 3, 4–14. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Million, M.; Lagier, J.C.; Yahav, D.; Paul, M. Gut bacterial microbiota and obesity. Clin. Microbiol. Infect. 2013, 19, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Le Roy, T.; Llopis, M.; Lepage, P.; Bruneau, A.; Rabot, S.; Bevilacqua, C.; Martin, P.; Philippe, C.; Walker, F.; Bado, A.; et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut 2013, 62, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.; Tse, C.H.; Lam, T.T.; Wong, G.L.; Chim, A.M.; Chu, W.C.; Yeung, D.K.; Law, P.T.; Kwan, H.S.; Yu, J.; et al. Molecular characterization of the fecal microbiota in patients with nonalcoholic steatohepatitis—A longitudinal study. PLoS ONE 2013, 8, e62885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verma, S.; Jensen, D.; Hart, J.; Mohanty, S.R. Predictive value of ALT levels for non-alcoholic steatohepatitis (NASH) and advanced fibrosis in non-alcoholic fatty liver disease (NAFLD). Liver Int. 2013, 33, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Peverill, W.; Powell, L.W.; Skoien, R. Evolving concepts in the pathogenesis of NASH: Beyond steatosis and inflammation. Int. J. Mol. Sci. 2014, 15, 8591–8638. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.A.; Ng, J.; Lueng, A.; Khajah, M.; Parhar, K.; Li, Y.; Lam, V.; Potentier, M.S.; Ng, K.; Bawa, M.; et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 2011, 17, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Thaissm, C.A.; Zeevi, D.; Dohnalová, L.; Zilberman-Schapira, G.; Mahdi, J.A.; David, E.; Savidor, A.; Korem, T.; Herzig, Y.; et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6. Inflamm. Signal. Cell 2015, 163, 1428–1443. [Google Scholar]
- Sellin, M.E.; Maslowski, K.M.; Maloy, K.J.; Hardt, W.D. Inflammasomes of the intestinal epithelium. Trends Immunol. 2015, 36, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Braunersreuther, V.; Viviani, G.L.; Mach, F.; Montecucco, F. Role of cytokines and chemokines in non-alcoholic fatty liver disease. World J. Gastroenterol. 2012, 18, 727–735. [Google Scholar] [CrossRef] [PubMed]
- Haukeland, J.W.; Damas, J.K.; Konopski, Z.; Løberg, E.M.; Haaland, T.; Goverud, I.; Torjesen, P.A.; Birkeland, K.; Bjøro, K.; Aukrust, P. Systemic inflammation in nonalcoholic fatty liver disease is characterized by elevated levels of CCL2. J. Hepatol. 2006, 44, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Mas, E.; Danjoux, M.; Garcia, V.; Carpentier, S.; Ségui, B.; Levade, T. IL-6 deficiency attenuates murine diet-induced non-alcoholic steatohepatitis. PLoS ONE 2009, 4, e7929. [Google Scholar] [CrossRef] [PubMed]
- Mencin, A.; Kluwe, J.; Schwabe, R.F. Toll-like receptors as targets in chronic liver diseases. Gut 2009, 58, 704–720. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.F.; Fiamoncini, J.; Prist, I.H.; Ariga, S.K.; de Souza, H.P.; de Lima, T.M. Novel role of TLR4 in NAFLD development: Modulation of metabolic enzymes expression. Biochim. Biophys. Acta 2015, 1851, 1353–1359. [Google Scholar] [CrossRef] [PubMed]
- Dapito, D.H.; Mencin, A.; Gwak, G.Y.; Pradere, J.P.; Jang, M.K.; Mederacke, I.; Caviglia, J.M.; Khiabanian, H.; Adeyemi, A.; Bataller, R.; et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012, 21, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Gangarapu, V.; Yıldız, K.; Ince, A.T.; Baysal, B. Role of gut microbiota: Obesity and NAFLD. Turk. J. Gastroenterol. 2014, 25, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ye, D.; Li, F.Y.; Lam, K.S.; Li, H.; Jia, W.; Wang, Y.; Man, K.; Lo, C.M.; Li, X.; Xu, A. Toll-like receptor-4 mediates obesity-induced non-alcoholic steatohepatitis through activation of X-box binding protein-1 in mice. Gut 2012, 61, 1058–1067. [Google Scholar] [CrossRef] [PubMed]
- Caricilli, A.M.; Picardi, P.K.; de Abreu, L.L.; Ueno, M.; Prada, P.O.; Ropelle, E.R.; Hirabara, S.M.; Castoldi, Â.; Vieira, P.; Camara, N.O.; et al. Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol. 2011, 9, e1001212. [Google Scholar] [CrossRef] [PubMed]
- Di Luccia, B.; Crescenzo, R.; Mazzoli, A.; Cigliano, L.; Venditti, P.; Walser, J.C.; Widmer, A.; Baccigalupi, L.; Ricca, E.; Iossa, S. Rescue of fructose-induced metabolic syndrome by antibiotics or faecal transplantation in a rat model of obesity. PLoS ONE 2015, 10, e0134893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prada, P.O.; Zecchin, H.G.; Gasparetti, A.L.; Torsoni, M.A.; Ueno, M.; Hirata, A.E.; Corezola do Amaral, M.E.; Höer, N.F.; Boschero, A.C.; Saad, M.J. Western diet modulates insulin signaling, c-Jun N-terminal kinase activity, and insulin receptor substrate-1ser307 phosphorylation in a tissue-specific fashion. Endocrinology 2005, 146, 1576–1587. [Google Scholar] [CrossRef] [PubMed]
- Membrez, M.; Blancher, F.; Jaquet, M.; Bibiloni, R.; Cani, P.D.; Burcelin, R.G.; Corthesy, I.; Macé, K.; Chou, C.J. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J. 2008, 22, 2416–2426. [Google Scholar] [CrossRef] [PubMed]
- Cryer, P.E. Minireview: Glucagon in the pathogenesis of hypoglycemia and hyperglycemia in diabetes. Endocrinology 2012, 153, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Satoh, H.; Ide, N.; Kagawa, Y.; Maeda, T. Hepatic steatosis with relation to increased expression of peroxisome proliferator-activated receptor-γ in insulin resistant mice. Biol. Pharm. Bull. 2013, 36, 616–623. [Google Scholar] [CrossRef] [PubMed]
- De Minicis, S.; Rychlicki, C.; Agostinelli, L.; Saccomanno, S.; Candelaresi, C.; Trozzi, L.; Mingarelli, E.; Facinelli, B.; Magi, G.; Palmieri, C.; et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 2014, 59, 1738–1749. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.A.; Velazquez, K.T.; Herbert, K.M. Influence of high-fat diet on gut microbiota: A driving force for chronic disease risk. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Sefcíková, Z.; Kmet, V.; Bujnáková, D.; Racek, L.; Mozes, S. Development of gut microflora in obese and lean rats. Folia Microbiol. (Praha) 2010, 55, 373–375. [Google Scholar] [CrossRef] [PubMed]
- Prorok-Hamon, M.; Friswell, M.K.; Alswied, A.; Roberts, C.L.; Song, F.; Flanagan, P.K.; Knight, P.; Codling, C.; Marchesi, J.R.; Winstanley, C.; et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut 2014, 63, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Al-Jashamy, K.; Murad, A.; Zeehaida, M.; Rohaini, M.; Hasnan, J. Prevalence of colorectal cancer associated with Streptococcus bovis among inflammatory bowel and chronic gastrointestinal tract disease patients. Asian Pac. J. Cancer Prev. 2010, 11, 1765–1768. [Google Scholar] [PubMed]
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal microbiome and volatile organic compound metabolome in obese humans with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Wu, N.; Wang, X.; Chi, Y.; Zhang, Y.; Qiu, X.; Hu, Y.; Li, J.; Liu, Y. Dysbiosis gut microbiota associated with inflammation and impaired mucosal immune function in intestine of humans with non-alcoholic fatty liver disease. Sci. Rep. 2015, 5, 8096. [Google Scholar] [CrossRef] [PubMed]
- Lebeer, S.; Vanderleydenm, J.; De Keersmaecker, S.C. Genes and molecules of lactobacilli supporting probiotic action. Microbiol. Mol. Biol. Rev. 2008, 72, 728–764. [Google Scholar] [CrossRef] [PubMed]
- Elshaghabee, F.M.; Bockelmann, W.; Meske, D.; de Vrese, M.; Walte, H.G.; Schrezenmeir, J.; Heller, K.J. Ethanol production by selected intestinal microorganisms and lactic acid bacteria growing under different nutritional conditions. Front. Microbiol. 2016, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.K.; Sheng, L.; Liu, H.X.; Kalanetra, K.M.; Mirsoian, A.; Murphy, W.J.; French, S.W.; Krishnan, V.V.; Mills, D.A.; Wan, Y.Y. Western diet-induced dysbiosis in farnesoid x receptor knockout mice causes persistent hepatic inflammation after antibiotic treatment. Am. J. Pathol. 2017, 187, 1800–1813. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Jena, P.K.; Liu, H.X.; Kalanetra, K.M.; Gonzalez, F.J.; French, S.W.; Krishnan, V.V.; Mills, D.A.; Wan, Y.Y. Gender differences in bile acids and microbiota in relationship with gender dissimilarity in steatosis induced by diet and FXR inactivation. Sci. Rep. 2017, 11, 1748. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | p Values for Two-Way ANOVA | ||||||
|---|---|---|---|---|---|---|---|
| STD | HFD | Main Effect of Microbiota | Main Effect of Diet | Interaction (M × D) | |||
| HL | NASH | HL | NASH | ||||
| Terminal body weight (g) | 30.7 ± 2.8 | 31.6 ± 2.0 | 48.1 ± 3.0 | 48.6 ± 3.0 | 0.532 | <0.0001 | 0.852 |
| Absolute liver weight (g) | 1.4 ± 0.2 | 1.4 ± 0.1 | 2.6 ± 0.3 | 2.4 ± 0.8 | 0.621 | <0.0001 | 0.674 |
| Relative liver weight (percent of terminal body weight) | 4.5 ± 0.3 | 4.3 ± 0.2 | 5.2 ± 0.3 | 4.9 ± 1.5 | 0.394 | 0.049 | 0.747 |
| Absolute epididymal fat weight (g) | 0.9 ± 0.2 | 1.3 ± 0.2 # | 1.9 ± 0.2 | 2.7 ± 0.5 * | <0.0001 | <0.0001 | 0.077 |
| Relative epididymal fat weight (percent of terminal body weight) | 2.8 ± 0.4 | 4.1 ± 0.5 # | 3.9 ± 0.3 | 5.7 ± 1.4 * | <0.0001 | 0.0002 | 0.326 |
| P Values for Two-Way ANOVA | |||||||
|---|---|---|---|---|---|---|---|
| Characteristic | STD | HFD | Main Effect of Microbiota | Main Effect of Diet | Interaction (M × D) | ||
| HL | NASH | HL | NASH | ||||
| ALT (U/L) | 14.7 ± 2.8 | 53.1 ± 25.5 | 138.3 ± 58.7 | 230.7 ± 83.1 * | 0.009 | <0.0001 | 0.248 |
| AST (U/L) | 78.2 ± 14.5 | 161.0 ± 23.9 # | 127.8 ± 30.2 | 218.2 ± 67.0 * | 0.0001 | 0.007 | 0.832 |
| Glucose (mmol/L) | 15.6 ± 0.80 | 16.5 ± 0.87 | 15.7 ± 2.46 | 20.7 ± 0.89 * | 0.017 | 0.078 | 0.085 |
| T-CHO (mmol/L) | 3.67 ± 0.78 | 3.01 ± 0.8 | 6.22 ± 0.32 | 6.48 ± 0.91 | 0.531 | <0.0001 | 0.146 |
| TGs (mmol/L) | 0.63 ± 0.26 | 0.62 ± 0.12 | 0.81 ± 0.21 | 0.98 ± 0.24 | 0.413 | 0.006 | 0.330 |
| HDL-C (mmol/L) | 2.96 ± 0.26 | 2.47 ± 0.62 # | 4.05 ± 0.19 | 3.97 ± 0.37 | 0.087 | <0.0001 | 0.218 |
| NEFAs (mmol/L) | 0.7 ± 0.2 | 0.7 ± 0.2 | 1.4 ± 0.1 | 1.2 ± 0.2 | 0.373 | <0.0001 | 0.135 |
| Insulin (μU/mL) | 74.9 ± 24.1 | 25.4 ± 3.7 # | 171.3 ± 36.1 | 99.9 ± 19.9 * | 0.0001 | <0.0001 | 0.368 |
| HOMA-IR | 52.8 ± 9.3 | 18.4 ± 2.7 # | 119.4 ± 28.1 | 88.9 ± 20.6 | 0.0058 | <0.0001 | 0.8473 |
| Characteristic | STD | HFD | ||
|---|---|---|---|---|
| HL | NASH | HL | NASH | |
| Streptococcaceae | 7.3 ± 0.3 | 9.1 ± 0.4 | 7.1 ± 0.7 | 9.8 ± 0.1 * |
| Lactobacillaceae | 9.1 ± 0.8 | 8.0 ± 0.3 | 9.8 ± 0.2 | 9.5 ± 0.6 |
| Eubacteriaceae | 9.8 ± 0.2 | 9.7 ± 0.4 | 9.6 ± 0.3 | 9.9 ± 0.1 |
| Clostridiaceae | 9.3 ± 0.1 | 9.2 ± 0.6 | 8.8 ± 0.4 | 9.5 ± 0.2 * |
| Bacteroidaceae | 9.6 ± 0.5 | 9.5 ± 0.2 | 9.6 ± 0.6 | 9.8 ± 0.4 |
| Enterobacteriaceae | 6.7 ± 0.4 | 7.8 ± 0.8 | 7.2 ± 0.6 | 8.9 ± 0.9 * |
| Yeasts | 5.4 ± 0.1 # | 6.6 ± 0.1 * | ||
| Total counts | 10.2 ± 0.2 | 10.1 ± 0.2 | 10.2 ± 0.2 | 10.4 ± 0.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, C.-C.; Ching, Y.-H.; Li, Y.-P.; Liu, J.-Y.; Huang, Y.-T.; Huang, Y.-W.; Yang, S.-S.; Huang, W.-C.; Chuang, H.-L. Nonalcoholic Fatty Liver Disease Is Exacerbated in High-Fat Diet-Fed Gnotobiotic Mice by Colonization with the Gut Microbiota from Patients with Nonalcoholic Steatohepatitis. Nutrients 2017, 9, 1220. https://doi.org/10.3390/nu9111220
Chiu C-C, Ching Y-H, Li Y-P, Liu J-Y, Huang Y-T, Huang Y-W, Yang S-S, Huang W-C, Chuang H-L. Nonalcoholic Fatty Liver Disease Is Exacerbated in High-Fat Diet-Fed Gnotobiotic Mice by Colonization with the Gut Microbiota from Patients with Nonalcoholic Steatohepatitis. Nutrients. 2017; 9(11):1220. https://doi.org/10.3390/nu9111220
Chicago/Turabian StyleChiu, Chien-Chao, Yung-Hao Ching, Yen-Peng Li, Ju-Yun Liu, Yen-Te Huang, Yi-Wen Huang, Sien-Sing Yang, Wen-Ching Huang, and Hsiao-Li Chuang. 2017. "Nonalcoholic Fatty Liver Disease Is Exacerbated in High-Fat Diet-Fed Gnotobiotic Mice by Colonization with the Gut Microbiota from Patients with Nonalcoholic Steatohepatitis" Nutrients 9, no. 11: 1220. https://doi.org/10.3390/nu9111220