Precision Nutrition and Omega-3 Polyunsaturated Fatty Acids: A Case for Personalized Supplementation Approaches for the Prevention and Management of Human Diseases

Abstract
:1. Introduction
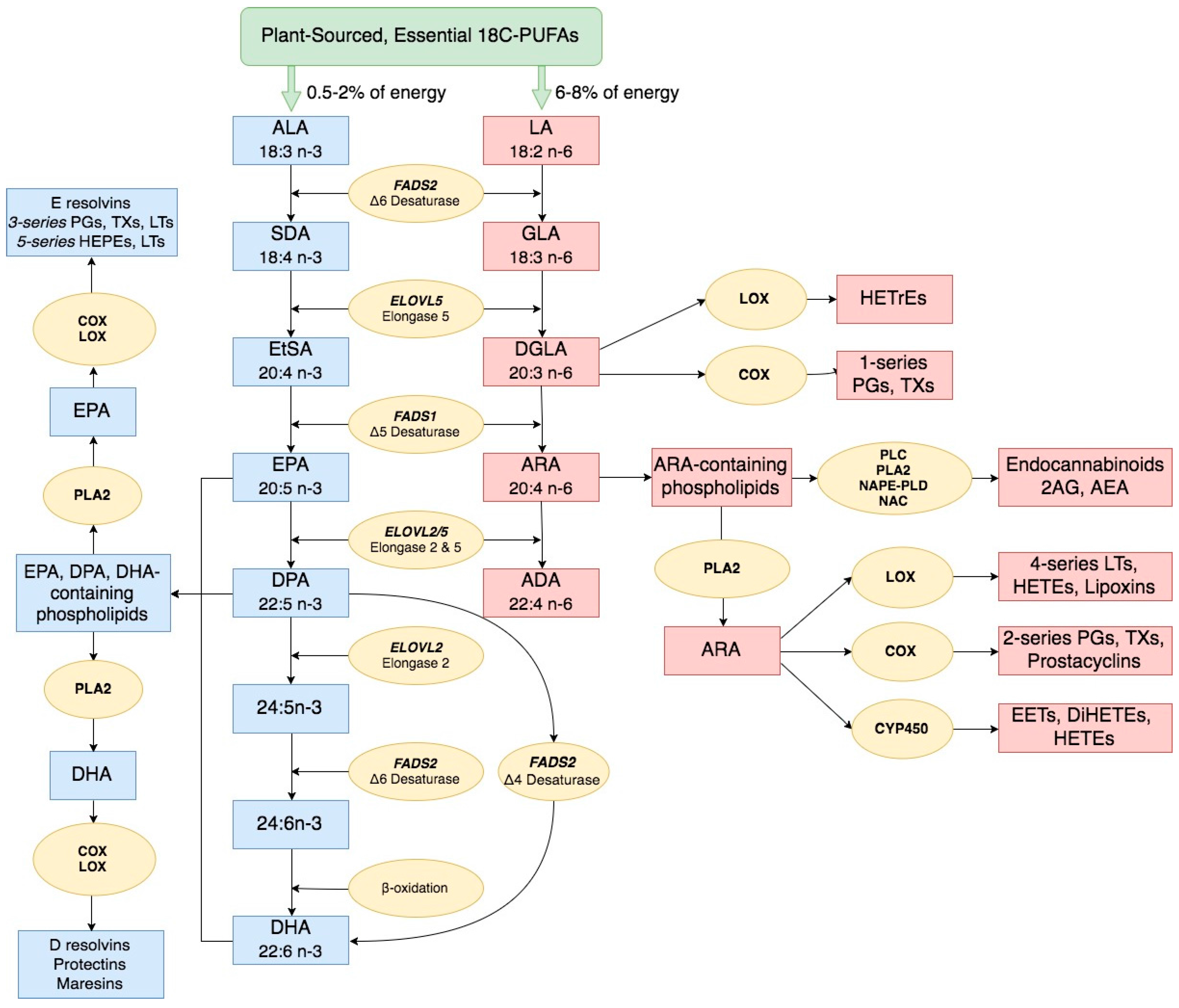
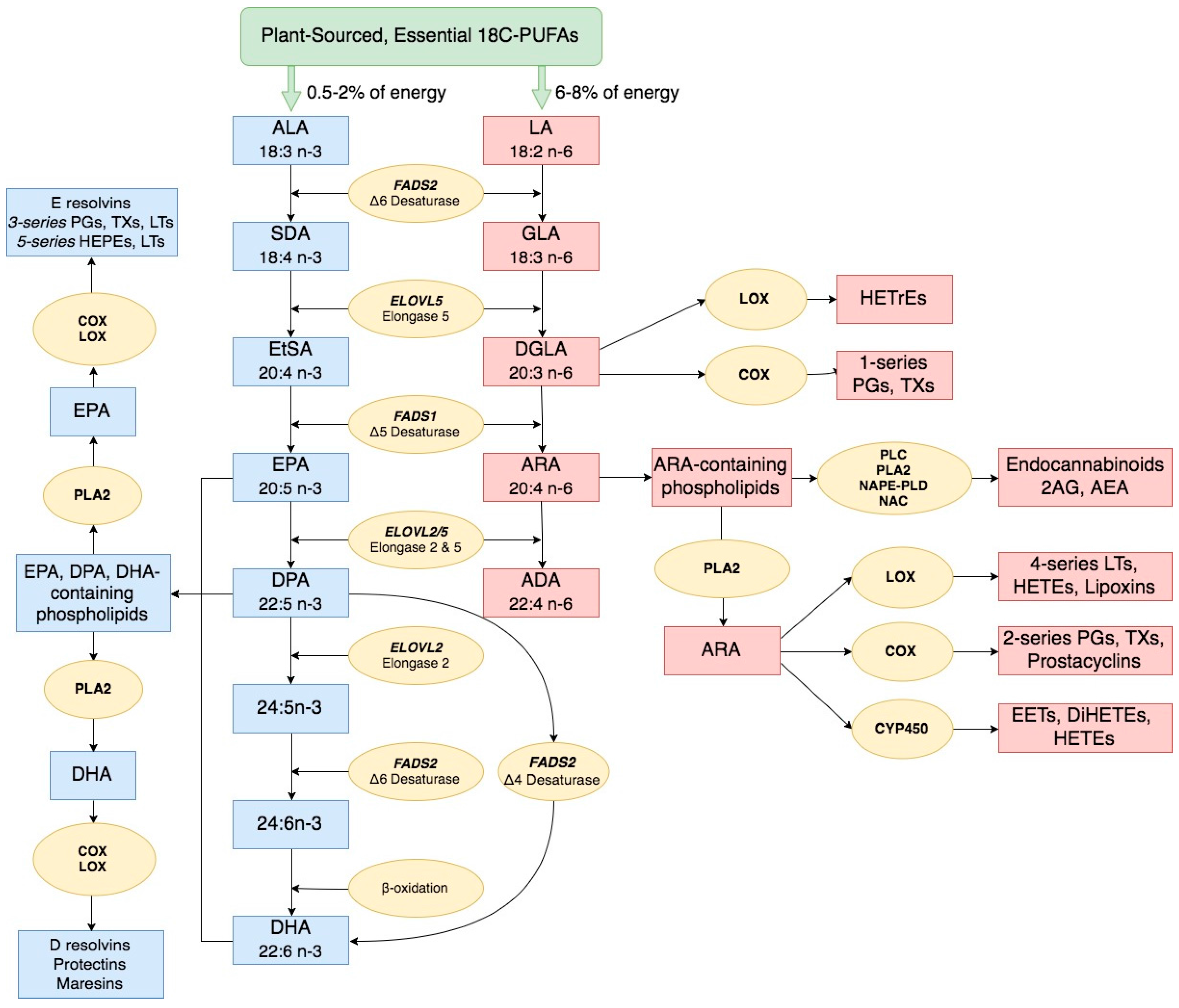
2. Long Chain Polyunsaturated Fatty Acid Biosynthesis and Biological Activities
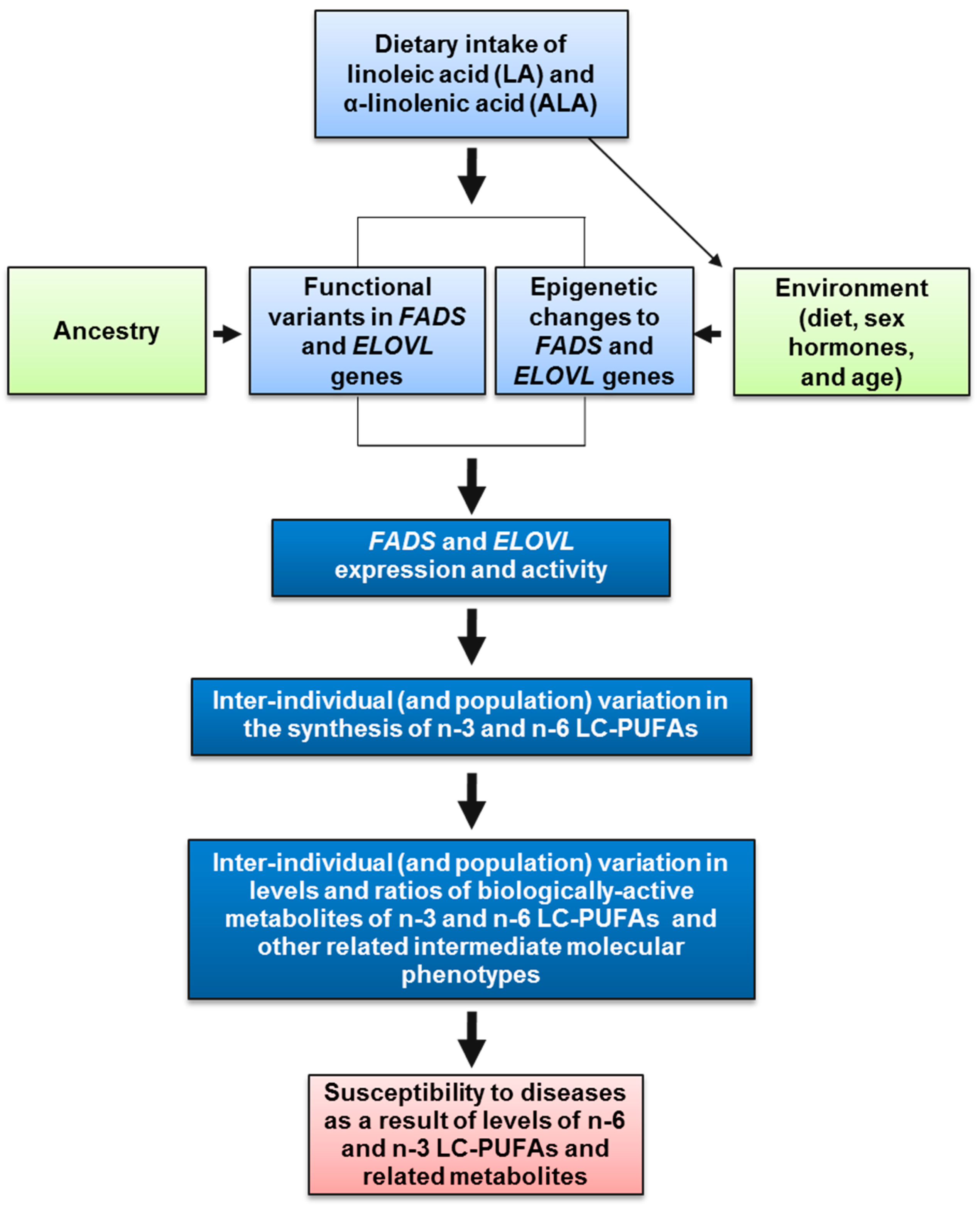
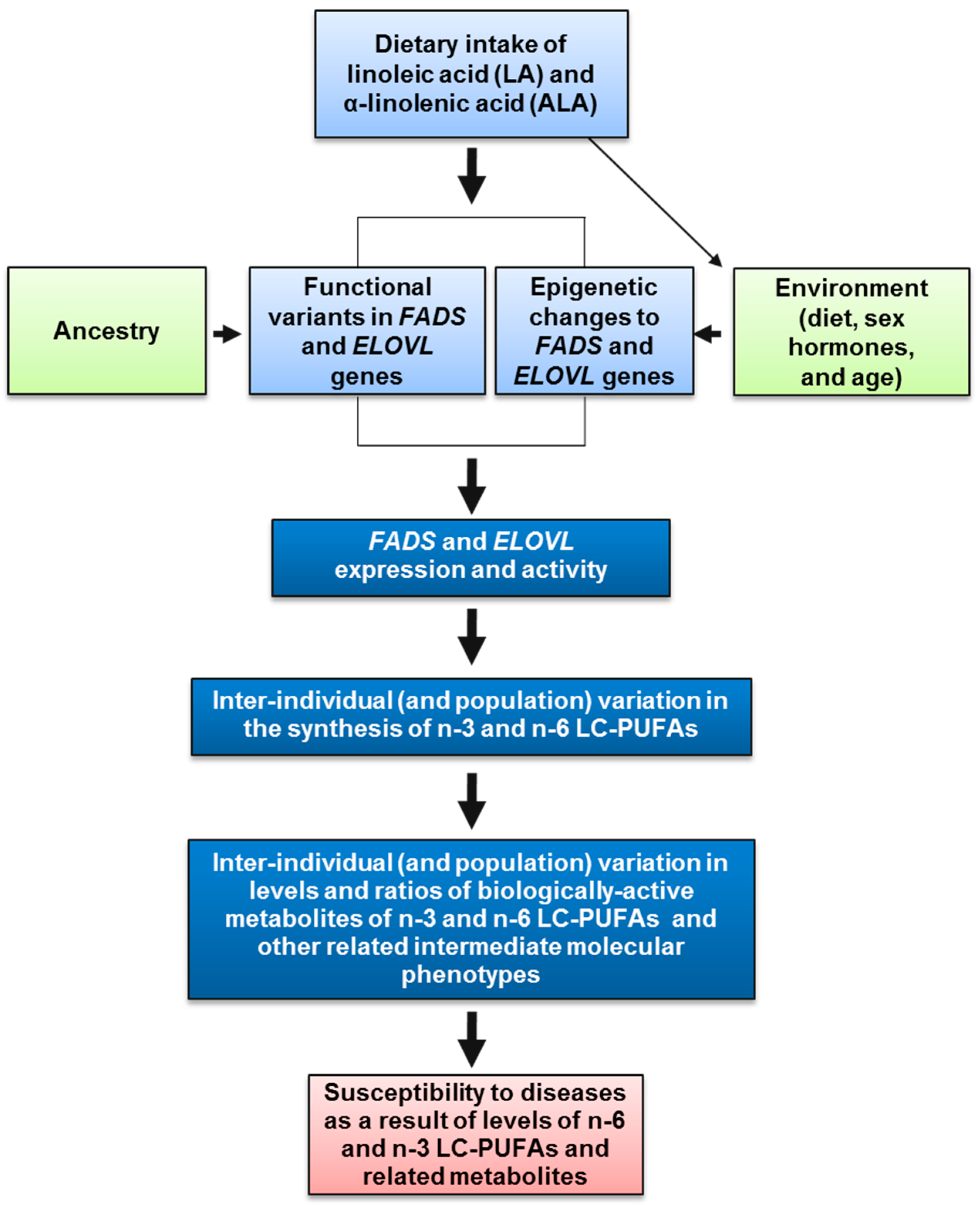
3. Impact of Dietary Linoleic Acid and α-Linolenic Acid Levels on n-3 LC-PUFA Biosynthesis
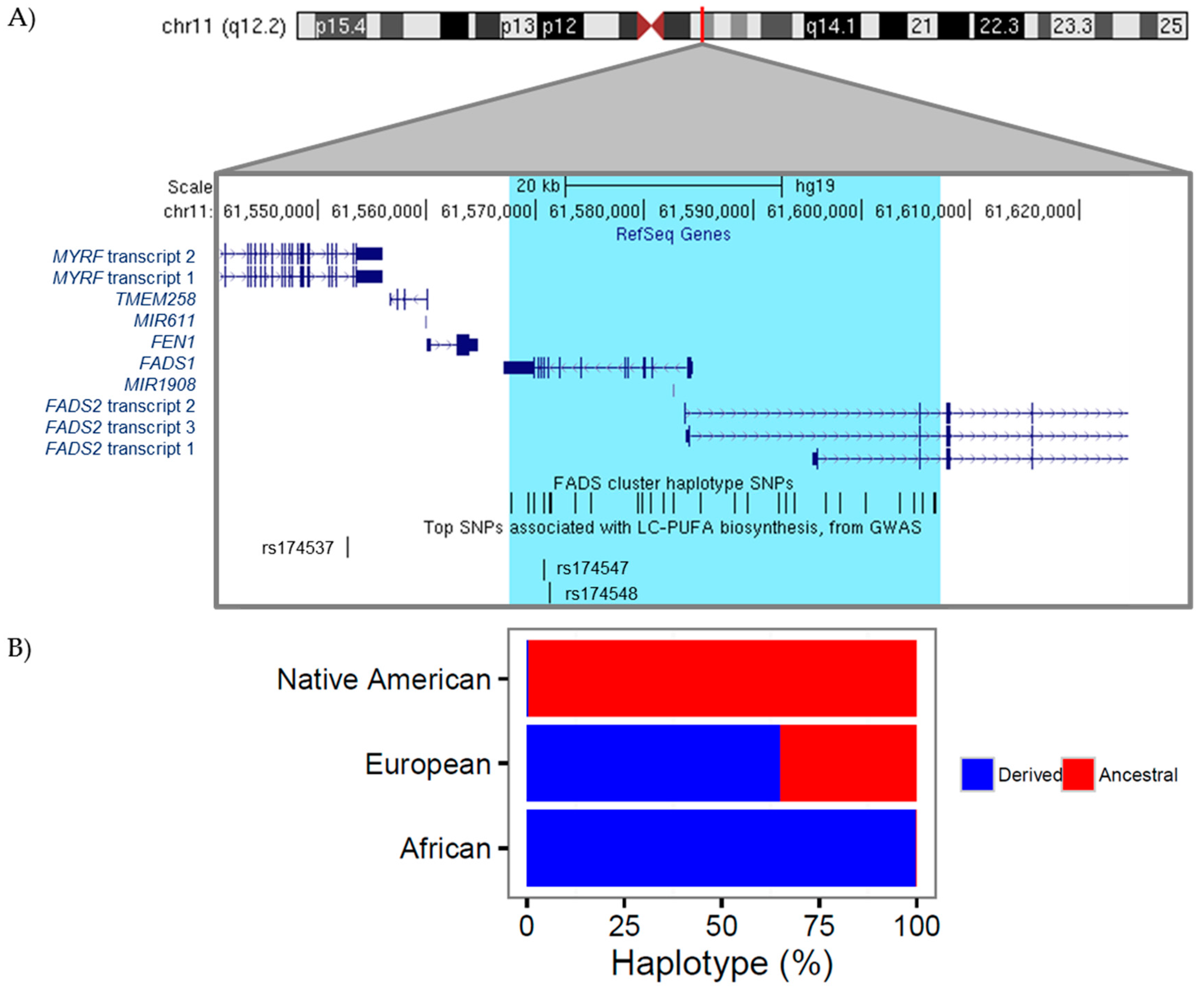
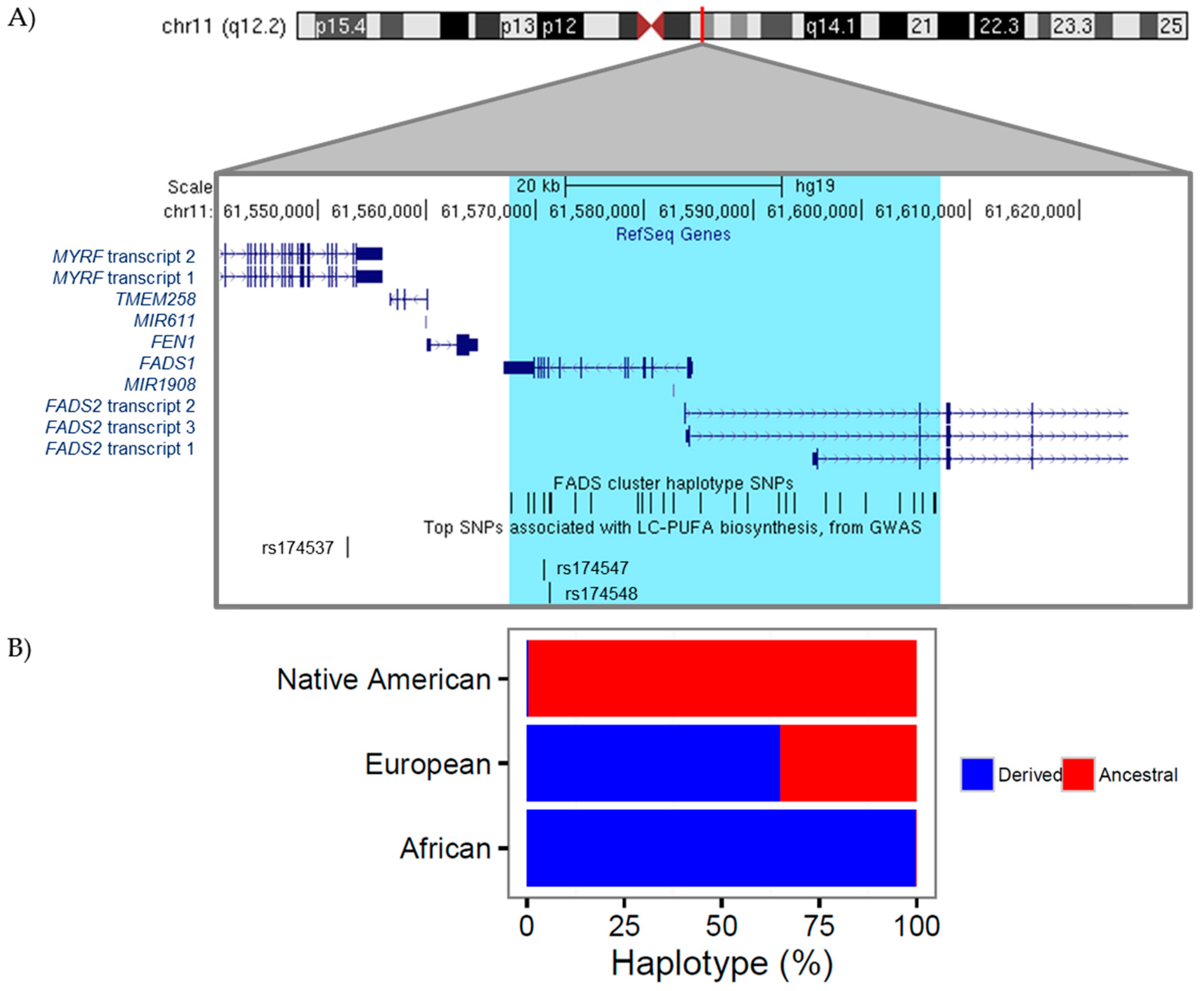
4. The Genetics and Evolution of LC-PUFA Biosynthesis
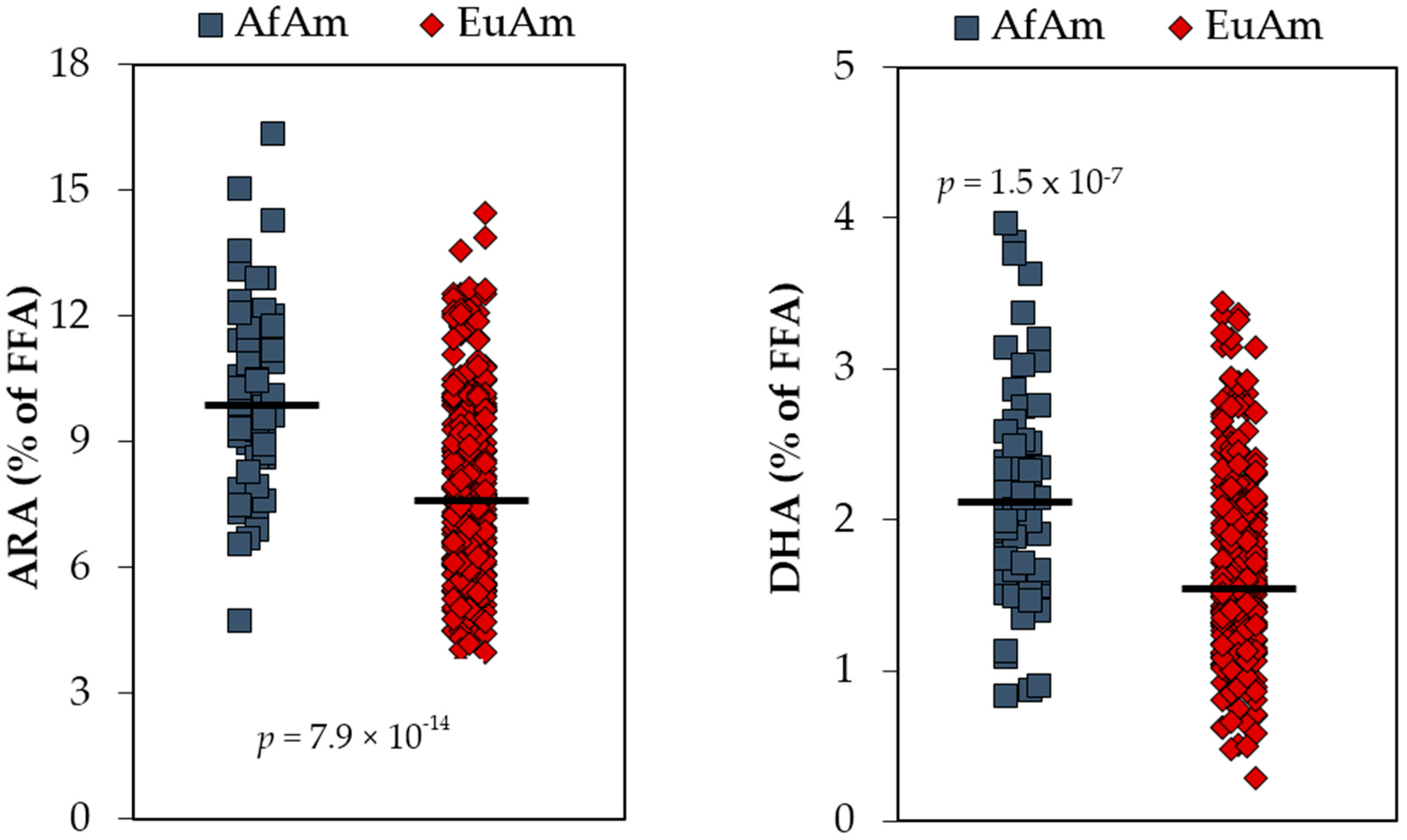
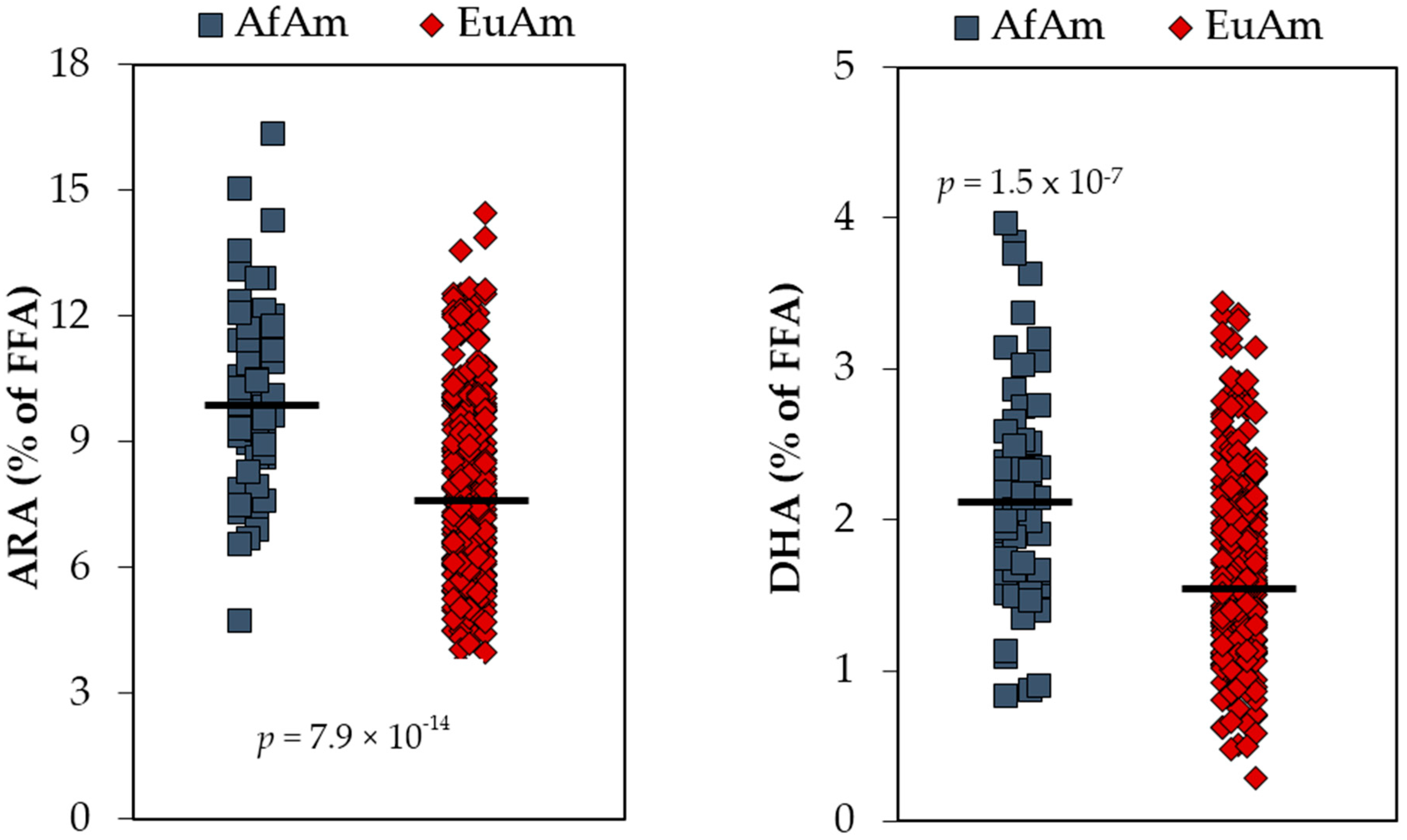
5. Anatomy of n-6 LC-PUFA Excesses and n-3 LC-PUFA Deficiencies
6. Implications of Non-Uniform n-3 LC-PUFA Biosynthesis on the Efficacy of n-3 LC-PUFA Supplementation Trials
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Blasbalg, T.L.; Hibbeln, J.R.; Ramsden, C.E.; Majchrzak, S.F.; Rawlings, R.R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am. J. Clin. Nutr. 2011, 93, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Chilton, F.H.; Murphy, R.C.; Wilson, B.A.; Sergeant, S.; Ainsworth, H.; Seeds, M.C.; Mathias, R.A. Diet-gene interactions and pufa metabolism: A potential contributor to health disparities and human diseases. Nutrients 2014, 6, 1993–2022. [Google Scholar] [CrossRef] [PubMed]
- Cordain, L.; Eaton, S.B.; Sebastian, A.; Mann, N.; Lindeberg, S.; Watkins, B.A.; O’Keefe, J.H.; Brand-Miller, J. Origins and evolution of the Western diet: Health implications for the 21st century. Am. J. Clin. Nutr. 2005, 81, 341–354. [Google Scholar] [PubMed]
- Popkin, B.M. Global nutrition dynamics: The world is shifting rapidly toward a diet linked with noncommunicable diseases. Am. J. Clin. Nutr. 2006, 84, 289–298. [Google Scholar] [PubMed]
- Ferrante, A.W., Jr. Obesity-induced inflammation: A metabolic dialogue in the language of inflammation. J. Intern. Med. 2007, 262, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Hamminga, E.A.; van der Lely, A.J.; Neumann, H.A.; Thio, H.B. Chronic inflammation in psoriasis and obesity: Implications for therapy. Med. Hypotheses 2006, 67, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Forsythe, L.K.; Wallace, J.M.; Livingstone, M.B. Obesity and inflammation: The effects of weight loss. Nutr. Res. Rev. 2008, 21, 117–133. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, X.-M.T.; Lane, J.; Smith, B.R.; Nguyen, N.T. Changes in inflammatory biomarkers across weight classes in a representative US population: A link between obesity and inflammation. J. Gastrointest. Surg. 2009, 13, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Calle, E.E.; Thun, M.J. Obesity and cancer. Oncogene 2004, 23, 6365–6378. [Google Scholar] [CrossRef] [PubMed]
- Beuther, D.A.; Weiss, S.T.; Sutherland, E.R. Obesity and asthma. Am. J. Respir. Crit. Care Med. 2006, 174, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Naderali, E.K.; Ratcliffe, S.H.; Dale, M.C. Obesity and Alzheimer’s disease: A link between body weight and cognitive function in old age. Am. J. Alzheimers Dis. Other Demen. 2009, 24, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Leveille, S.G.; Wee, C.C.; Iezzoni, L.I. Trends in obesity and arthritis among baby boomers and their predecessors, 1971–2002. Am. J. Public Health 2005, 95, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Serini, S.; Calviello, G. Reduction of oxidative/nitrosative stress in brain and its involvement in the neuroprotective effect of n-3 pufa in Alzheimer’s disease. Curr. Alzheimer Res. 2016, 13, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Han, S.B.; Nam, S.Y.; Oh, K.W.; Hong, J.T. Inflammation and Alzheimer’s disease. Arch. Pharmcal Res. 2010, 33, 1539–1556. [Google Scholar] [CrossRef] [PubMed]
- Sankar, P.; Cho, M.K.; Condit, C.M.; Hunt, L.M.; Koenig, B.; Marshall, P.; Lee, S.S.; Spicer, P. Genetic research and health disparities. JAMA 2004, 291, 2985–2989. [Google Scholar] [CrossRef] [PubMed]
- Sankar, P.; Cho, M.K.; Mountain, J. Race and ethnicity in genetic research. Am. J. Med. Genet. 2007, 143A, 961–970. [Google Scholar] [CrossRef] [PubMed]
- Mensah, G.A.; Mokdad, A.H.; Ford, E.S.; Greenlund, K.J.; Croft, J.B. State of disparities in cardiovascular health in the United States. Circulation 2005, 111, 1233–1241. [Google Scholar] [CrossRef] [PubMed]
- Krieger, N.; Chen, J.T.; Waterman, P.D.; Soobader, M.J.; Subramanian, S.V.; Carson, R. Geocoding and monitoring of US socioeconomic inequalities in mortality and cancer incidence: Does the choice of area-based measure and geographic level matter? The Public Health Disparities Geocoding Project. Am. J. Epidemiol. 2002, 156, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Braveman, P. Health disparities and health equity: Concepts and measurement. Annu. Rev. Public Health 2006, 27, 167–194. [Google Scholar] [CrossRef] [PubMed]
- Kuzawa, C.W.; Sweet, E. Epigenetics and the embodiment of race: Developmental origins of US racial disparities in cardiovascular health. Am. J. Hum. Biol. 2009, 21, 2–15. [Google Scholar] [CrossRef] [PubMed]
- De Toro-Martin, J.; Arsenault, B.J.; Despres, J.P.; Vohl, M.C. Precision Nutrition: A review of personalized nutritional approaches for the prevention and management of metabolic syndrome. Nutrients 2017, 9, 913. [Google Scholar] [CrossRef] [PubMed]
- American Heart Association & American Stroke Association. 50 Years of American Heart Association Dietary Fats Recommendations. 2015. Available online: http://www.heart.org/idc/groups/heart-public/@wcm/@fc/documents/downloadable/ucm_475005.pdf (accessed on 9 February 2017).
- Miller, M.; Stone, N.J.; Ballantyne, C.; Bittner, V.; Criqui, M.H.; Ginsberg, H.N.; Goldberg, A.C.; Howard, W.J.; Jacobson, M.S.; Kris-Etherton, P.M.; et al. Triglycerides and cardiovascular disease: A scientific statement from the American Heart Association. Circulation 2011, 123, 2292–2333. [Google Scholar] [CrossRef] [PubMed]
- Nutrient Content of the U.S. Food Supply, 1909-99: A Summary Report. U.S. Department of Agriculture, Center for Nutrition Policy and Promotion 2002, Home Economics Research Report No. 55. Available online: https://www.cnpp.usda.gov/sites/default/files/nutrient_content_of_the_us_food_supply/FoodSupply1909-1999Report.pdf (accessed on 9 February 2017).
- Eaton, S.B. Humans, lipids and evolution. Lipids 1992, 27, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Hibbeln, J.R.; Nieminen, L.R.; Blasbalg, T.L.; Riggs, J.A.; Lands, W.E. Healthy intakes of n-3 and n-6 fatty acids: Estimations considering worldwide diversity. Am. J. Clin. Nutr. 2006, 83, 1483S–1493S. [Google Scholar] [PubMed]
- Simopoulos, A.P. Importance of the omega-6/omega-3 balance in health and disease: Evolutionary aspects of diet. World Rev. Nutr. Diet. 2011, 102, 10–21. [Google Scholar] [PubMed]
- Burr, G.O.; Burr, M.M. A new deficiency disease produced by the rigid exclusion of fat from the diet. J. Biol. Chem. 1929, 82, 345–367. [Google Scholar] [CrossRef]
- Burr, G.O.; Burr, M.M. On the nature and role of the fatty acids essential in nutrition. J. Biol. Chem. 1930, 86, 587–621. [Google Scholar] [CrossRef]
- Holman, R.T. Essential fatty acids. Nutr. Rev. 1958, 16, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Holman, R.T. The slow discovery of the importance of omega 3 essential fatty acids in human health. J. Nutr. 1998, 128, 427S–433S. [Google Scholar] [PubMed]
- Sprecher, H. Biochemistry of essential fatty acids. Prog. Lipid Res. 1981, 20, 13–22. [Google Scholar] [CrossRef]
- Sprecher, H.; Luthria, D.L.; Mohammed, B.S.; Baykousheva, S.P. Reevaluation of the pathways for the biosynthesis of polyunsaturated fatty acids. J. Lipid Res. 1995, 36, 2471–2477. [Google Scholar] [PubMed]
- Sprecher, H.; Chen, Q. Polyunsaturated fatty acid biosynthesis: A microsomal-peroxisomal process. Prostaglandins Leukot. Essent. Fat. Acids 1999, 60, 317–321. [Google Scholar] [CrossRef]
- Park, W.J.; Kothapalli, K.S.; Lawrence, P.; Tyburczy, C.; Brenna, J.T. An alternate pathway to long-chain polyunsaturates: The FADS2 gene product Delta8-desaturates 20:2n-6 and 20:3n-3. J. Lipid Res. 2009, 50, 1195–1202. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.A.; Pani, V.; Chilton, F.H. Genetic variants in the FADS gene: Implications for dietary recommendations for fatty acid intake. Curr. Nutr. Rep. 2014, 3, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Park, H.G.; Park, W.J.; Kothapalli, K.S.; Brenna, J.T. The fatty acid desaturase 2 (FADS2) gene product catalyzes Delta4 desaturation to yield n-3 docosahexaenoic acid and n-6 docosapentaenoic acid in human cells. FASEB J. 2015, 29, 3911–3919. [Google Scholar] [CrossRef] [PubMed]
- Spector, A.A. Plasma free fatty acid and lipoproteins as sources of polyunsaturated fatty acid for the brain. J. Mol. Neurosci. 2001, 16, 159–165. [Google Scholar] [CrossRef]
- Lands, W.E.; Hart, P. Metabolism of glycerolipids.VI. Specificities of acyl coenzyme A: Phospolipid acyltransferases. J. Biol. Chem. 1965, 240, 1905–1911. [Google Scholar] [PubMed]
- Gibson, R.A.; Muhlhausler, B.; Makrides, M. Conversion of linoleic acid and alpha-linolenic acid to long-chain polyunsaturated fatty acids (LCPUFAs), with a focus on pregnancy, lactation and the first 2 years of life. Matern. Child Nutr. 2011, 7 (Suppl. 2), 17–26. [Google Scholar] [CrossRef] [PubMed]
- McNamara, R.K.; Carlson, S.E. Role of omega-3 fatty acids in brain development and function: Potential implications for the pathogenesis and prevention of psychopathology. Prostaglandins Leukot. Essent. Fat. Acids 2006, 75, 329–349. [Google Scholar] [CrossRef] [PubMed]
- McCann, J.C.; Ames, B.N. Is docosahexaenoic acid, an n-3 long-chain polyunsaturated fatty acid, required for development of normal brain function? An overview of evidence from cognitive and behavioral tests in humans and animals. Am. J. Clin. Nutr. 2005, 82, 281–295. [Google Scholar] [PubMed]
- Weiser, M.J.; Butt, C.M.; Mohajeri, M.H. Docosahexaenoic acid and cognition throughout the lifespan. Nutrients 2016, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Hibbeln, J.R.; Davis, J.M.; Steer, C.; Emmett, P.; Rogers, I.; Williams, C.; Golding, J. Maternal seafood consumption in pregnancy and neurodevelopmental outcomes in childhood (ALSPAC study): An observational cohort study. Lancet 2007, 369, 578–585. [Google Scholar] [CrossRef]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science 1999, 285, 1276–1279. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.L. The eicosanoids and their biochemical mechanisms of action. Biochem. J. 1989, 259, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N.; Chiang, N.; Van Dyke, T.E. Resolving inflammation: Dual anti-inflammatory and pro-resolution lipid mediators. Nat. Rev. Immunol. 2008, 8, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Di Marzoa, V.M.D.; Bisogno, T.; De Petrocellis, L. Endocannabinoids: Endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998, 22, 521–528. [Google Scholar] [CrossRef]
- Buczynski, M.W.; Dumlao, D.S.; Dennis, E.A. Thematic Review Series: Proteomics. An integrated omics analysis of eicosanoid biology. J. Lipid Res. 2009, 50, 1015–1038. [Google Scholar] [CrossRef] [PubMed]
- Chilton, F.H.; Fonteh, A.N.; Surette, M.E.; Triggiani, M.; Winkler, J.D. Control of arachidonate levels within inflammatory cells. Biochim. Biophys. Acta 1996, 1299, 1–15. [Google Scholar] [CrossRef]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed]
- Haeggstrom, J.Z.; Funk, C.D. Lipoxygenase and leukotriene pathways: Biochemistry, biology, and roles in disease. Chem. Rev. 2011, 111, 5866–5898. [Google Scholar] [CrossRef] [PubMed]
- Lands, W.E.; Libelt, B.; Morris, A.; Kramer, N.C.; Prewitt, T.E.; Bowen, P.; Schmeisser, D.; Davidson, M.H.; Burns, J.H. Maintenance of lower proportions of (n-6) eicosanoid precursors in phospholipids of human plasma in response to added dietary (n-3) fatty acids. Biochim. Biophys. Acta 1992, 1180, 147–162. [Google Scholar] [CrossRef]
- James, M.J.; Gibson, R.A.; Cleland, L.G. Dietary polyunsaturated fatty acids and inflammatory mediator production. Am. J. Clin. Nutr. 2000, 71, 343S–348S. [Google Scholar] [PubMed]
- Lands, B. A critique of paradoxes in current advice on dietary lipids. Prog. Lipid Res. 2008, 47, 77–106. [Google Scholar] [CrossRef] [PubMed]
- Lands, B. Omega-3 PUFAs lower the propensity for arachidonic acid cascade overreactions. Biomed. Res. Int. 2015, 2015, 285135. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, G.; Ecker, J. The opposing effects of n-3 and n-6 fatty acids. Prog. Lipid Res. 2008, 47, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Novel pro-resolving lipid mediators in inflammation are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Cianci, E.; Simiele, F.; Recchiuti, A. Lipoxins and aspirin-triggered lipoxins in resolution of inflammation. Eur. J. Pharmacol. 2015, 760, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urban, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; et al. Hardwiring the brain: Endocannabinoids shape neuronal connectivity. Science 2007, 316, 1212–1216. [Google Scholar] [CrossRef] [PubMed]
- Matias, I.; Di Marzo, V. Endocannabinoids and the control of energy balance. Trends Endocrinol. Metab. 2007, 18, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Batkai, S.; Pacher, P.; Osei-Hyiaman, D.; Radaeva, S.; Liu, J.; Harvey-White, J.; Offertaler, L.; Mackie, K.; Rudd, M.A.; Bukoski, R.D.; et al. Endocannabinoids acting at cannabinoid-1 receptors regulate cardiovascular function in hypertension. Circulation 2004, 110, 1996–2002. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.; Cascio, M.G.; Rotondo, D.; Pertwee, R.G.; Heys, S.D.; Wahle, K.W. Cannabinoids and omega-3/6 endocannabinoids as cell death and anticancer modulators. Prog. Lipid Res. 2013, 52, 80–109. [Google Scholar] [CrossRef] [PubMed]
- Brown, I.; Cascio, M.G.; Wahle, K.W.; Smoum, R.; Mechoulam, R.; Ross, R.A.; Pertwee, R.G.; Heys, S.D. Cannabinoid receptor-dependent and -independent anti-proliferative effects of omega-3 ethanolamides in androgen receptor-positive and -negative prostate cancer cell lines. Carcinogenesis 2010, 31, 1584–1591. [Google Scholar] [CrossRef] [PubMed]
- McDougle, D.R.; Watson, J.E.; Abdeen, A.A.; Adili, R.; Caputo, M.P.; Krapf, J.E.; Johnson, R.W.; Kilian, K.A.; Holinstat, M.; Das, A. Anti-inflammatory omega-3 endocannabinoid epoxides. Proc. Natl. Acad. Sci. USA 2017, 114, E6034–E6043. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Castillo, P.E.; Manzoni, O.J.; Tonini, R. Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology 2017, 124, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.C.; Glass, M. The cannabinoid CB2 receptor as a target for inflammation-dependent neurodegeneration. Curr. Neuropharmacol. 2007, 5, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Di Marzo, V. Endocannabinoids in nervous system health and disease: The big picture in a nutshell. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 3193–3200. [Google Scholar] [CrossRef] [PubMed]
- Sloan, M.E.; Gowin, J.L.; Ramchandani, V.A.; Hurd, Y.L.; Le Foll, B. The endocannabinoid system as a target for addiction treatment: Trials and tribulations. Neuropharmacology 2017, 124, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Murillo-Rodriguez, E.; Poot-Ake, A.; Arias-Carrion, O.; Pacheco-Pantoja, E.; Fuente-Ortegon Ade, L.; Arankowsky-Sandoval, G. The emerging role of the endocannabinoid system in the sleep-wake cycle modulation. Cent. Nerv. Syst. Agents Med. Chem. 2011, 11, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Malek, N.; Starowicz, K. Dual-acting compounds targeting endocannabinoid and endovanilloid systems-a novel treatment option for chronic pain management. Front. Pharmacol. 2016, 7, 257. [Google Scholar] [CrossRef] [PubMed]
- Lau, B.K.; Cota, D.; Cristino, L.; Borgland, S.L. Endocannabinoid modulation of homeostatic and non-homeostatic feeding circuits. Neuropharmacology 2017, 124, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Kruk-Slomka, M.; Dzik, A.; Budzynska, B.; Biala, G. Endocannabinoid system: The direct and indirect involvement in the memory and learning processes-a short review. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Hedlund, P.; Gratzke, C. The endocannabinoid system—A target for the treatment of LUTS? Nat. Rev. Urol. 2016, 13, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Giaginis, C.; Lakiotaki, E.; Korkolopoulou, P.; Konstantopoulos, K.; Patsouris, E.; Theocharis, S. Endocannabinoid system: A promising therapeutic target for the treatment of haematological malignancies? Curr. Med. Chem. 2016, 23, 2350–2362. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, B.M.; Costa, M.A.; Almada, M.; Correia-da-Silva, G.; Teixeira, N.A. Endogenous cannabinoids revisited: A biochemistry perspective. Prostaglandins Other Lipid Mediat. 2013, 102–103, 13–30. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: Cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chem. Rev. 2011, 111, 5899–5921. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.E.; Hibbeln, J.R.; Majchrzak, S.F.; Davis, J.M. n-6 fatty acid-specific and mixed polyunsaturate dietary interventions have different effects on CHD risk: A meta-analysis of randomised controlled trials. Br. J. Nutr. 2010, 104, 1586–1600. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, C.E.; Zamora, D.; Majchrzak-Hong, S.; Faurot, K.R.; Broste, S.K.; Frantz, R.P.; Davis, J.M.; Ringel, A.; Suchindran, C.M.; Hibbeln, J.R. Re-evaluation of the traditional diet-heart hypothesis: Analysis of recovered data from Minnesota Coronary Experiment (1968-73). BMJ 2016, 353, i1246. [Google Scholar] [CrossRef] [PubMed]
- Lands, B. Dietary omega-3 and omega-6 fatty acids compete in producing tissue compositions and tissue responses. Mil. Med. 2014, 179, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.A.; King, D.J.; Zibrik, D.; Innis, S.M. Decreasing linoleic acid with constant alpha-linolenic acid in dietary fats increases (n-3) eicosapentaenoic acid in plasma phospholipids in healthy men. J. Nutr. 2007, 137, 945–952. [Google Scholar] [PubMed]
- Wood, K.E.; Mantzioris, E.; Gibson, R.A.; Ramsden, C.E.; Muhlhausler, B.S. The effect of modifying dietary LA and ALA intakes on omega-3 long chain polyunsaturated fatty acid (n-3 LCPUFA) status in human adults: A systematic review and commentary. Prostaglandins Leukot. Essent. Fat. Acids 2015, 95, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Macintosh, B.A.; Ramsden, C.E.; Faurot, K.R.; Zamora, D.; Mangan, M.; Hibbeln, J.R.; Mann, J.D. Low-n-6 and low-n-6 plus high-n-3 diets for use in clinical research. Br. J. Nutr. 2013, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, H. Dietary fatty acids—The N-6/N-3 balance and chronic elderly diseases. Excess linoleic acid and relative N-3 deficiency syndrome seen in Japan. Prog. Lipid Res. 1996, 35, 409–457. [Google Scholar] [CrossRef]
- Cleland, L.G.; James, M.J.; Neumann, M.A.; D’Angelo, M.; Gibson, R.A. Linoleate inhibits EPA incorporation from dietary fish-oil supplements in human subjects. Am. J. Clin. Nutr. 1992, 55, 395–399. [Google Scholar] [PubMed]
- Blank, C.; Neumann, M.A.; Makrides, M.; Gibson, R.A. Optimizing DHA levels in piglets by lowering the linoleic acid to alpha-linolenic acid ratio. J. Lipid Res. 2002, 43, 1537–1543. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.E.; Lau, A.; Mantzioris, E.; Gibson, R.A.; Ramsden, C.E.; Muhlhausler, B.S. A low omega-6 polyunsaturated fatty acid (n-6 PUFA) diet increases omega-3 (n-3) long chain PUFA status in plasma phospholipids in humans. Prostaglandins Leukot. Essent. Fat. Acids 2014, 90, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Brenna, J.T.; Salem, N., Jr.; Sinclair, A.J.; Cunnane, S.C.; International Society for the Study of Fatty, Acids and Lipids, ISSFAL. alpha-Linolenic acid supplementation and conversion to n-3 long-chain polyunsaturated fatty acids in humans. Prostaglandins Leukot. Essent. Fat. Acids 2009, 80, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Arterburn, L.M.; Hall, E.B.; Oken, H. Distribution, interconversion, and dose response of n-3 fatty acids in humans. Am. J. Clin. Nutr. 2006, 83, 1467S–1476S. [Google Scholar] [PubMed]
- Goyens, P.L.; Spilker, M.E.; Zock, P.L.; Katan, M.B.; Mensink, R.P. Conversion of alpha-linolenic acid in humans is influenced by the absolute amounts of alpha-linolenic acid and linoleic acid in the diet and not by their ratio. Am. J. Clin. Nutr. 2006, 84, 44–53. [Google Scholar] [PubMed]
- Schaeffer, L.; Gohlke, H.; Muller, M.; Heid, I.M.; Palmer, L.J.; Kompauer, I.; Demmelmair, H.; Illig, T.; Koletzko, B.; Heinrich, J. Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum. Mol. Genet. 2006, 15, 1745–1756. [Google Scholar] [CrossRef] [PubMed]
- Malerba, G.; Schaeffer, L.; Xumerle, L.; Klopp, N.; Trabetti, E.; Biscuola, M.; Cavallari, U.; Galavotti, R.; Martinelli, N.; Guarini, P.; et al. SNPs of the FADS gene cluster are associated with polyunsaturated fatty acids in a cohort of patients with cardiovascular disease. Lipids 2008, 43, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Innis, S.M. Genetic variants of the FADS1 FADS2 gene cluster are associated with altered (n-6) and (n-3) essential fatty acids in plasma and erythrocyte phospholipids in women during pregnancy and in breast milk during lactation. J. Nutr. 2008, 138, 2222–2228. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Shen, J.; Abecasis, G.R.; Kisialiou, A.; Ordovas, J.M.; Guralnik, J.M.; Singleton, A.; Bandinelli, S.; Cherubini, A.; Arnett, D.; et al. Genome-wide association study of plasma polyunsaturated fatty acids in the InCHIANTI Study. PLoS Genet. 2009, 5, e1000338. [Google Scholar] [CrossRef] [PubMed]
- Rzehak, P.; Heinrich, J.; Klopp, N.; Schaeffer, L.; Hoff, S.; Wolfram, G.; Illig, T.; Linseisen, J. Evidence for an association between genetic variants of the fatty acid desaturase 1 fatty acid desaturase 2 (FADS1 FADS2) gene cluster and the fatty acid composition of erythrocyte membranes. Br. J. Nutr. 2009, 101, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Innis, S.M. Association of fatty acid desaturase gene polymorphisms with blood lipid essential fatty acids and perinatal depression among Canadian women: A pilot study. J. Nutrigenet. Nutrigenom. 2009, 2, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.A.; Vergara, C.; Gao, L.; Rafaels, N.; Hand, T.; Campbell, M.; Bickel, C.; Ivester, P.; Sergeant, S.; Barnes, K.C.; et al. FADS genetic variants and omega-6 polyunsaturated fatty acid metabolism in a homogeneous island population. J. Lipid Res. 2010, 51, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Bokor, S.; Dumont, J.; Spinneker, A.; Gonzalez-Gross, M.; Nova, E.; Widhalm, K.; Moschonis, G.; Stehle, P.; Amouyel, P.; De, H.S.; et al. Single nucleotide polymorphisms in the FADS gene cluster are associated with delta-5 and delta-6 desaturase activities estimated by serum fatty acid ratios. J. Lipid Res. 2010, 51, 2325–2333. [Google Scholar] [CrossRef] [PubMed]
- Rzehak, P.; Thijs, C.; Standl, M.; Mommers, M.; Glaser, C.; Jansen, E.; Klopp, N.; Koppelman, G.H.; Singmann, P.; Postma, D.S.; et al. Variants of the FADS1 FADS2 gene cluster, blood levels of polyunsaturated fatty acids and eczema in children within the first 2 years of life. PLoS ONE 2010, 5, e13261. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.A.; Sergeant, S.; Ruczinski, I.; Torgerson, D.G.; Hugenschmidt, C.E.; Kubala, M.; Vaidya, D.; Suktitipat, B.; Ziegler, J.T.; Ivester, P.; et al. The impact of FADS genetic variants on omega6 polyunsaturated fatty acid metabolism in African Americans. BMC Genet. 2011, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Lemaitre, R.N.; Tanaka, T.; Tang, W.; Manichaikul, A.; Foy, M.; Kabagambe, E.K.; Nettleton, J.A.; King, I.B.; Weng, L.C.; Bhattacharya, S.; et al. Genetic loci associated with plasma phospholipid n-3 fatty acids: A meta-analysis of genome-wide association studies from the CHARGE Consortium. PLoS Genet. 2011, 7, e1002193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morales, E.; Bustamante, M.; Gonzalez, J.R.; Guxens, M.; Torrent, M.; Mendez, M.; Garcia-Esteban, R.; Julvez, J.; Forns, J.; Vrijheid, M.; et al. Genetic variants of the FADS gene cluster and ELOVL gene family, colostrums LC-PUFA levels, breastfeeding, and child cognition. PLoS ONE 2011, 6, e17181. [Google Scholar] [CrossRef] [PubMed]
- Lattka, E.; Rzehak, P.; Szabo, E.; Jakobik, V.; Weck, M.; Weyermann, M.; Grallert, H.; Rothenbacher, D.; Heinrich, J.; Brenner, H.; et al. Genetic variants in the FADS gene cluster are associated with arachidonic acid concentrations of human breast milk at 1.5 and 6 mo postpartum and influence the course of milk dodecanoic, tetracosenoic, and trans-9-octadecenoic acid concentrations over the duration of lactation. Am. J. Clin. Nutr. 2011, 93, 382–391. [Google Scholar] [PubMed]
- Koletzko, B.; Lattka, E.; Zeilinger, S.; Illig, T.; Steer, C. Genetic variants of the fatty acid desaturase gene cluster predict amounts of red blood cell docosahexaenoic and other polyunsaturated fatty acids in pregnant women: Findings from the Avon Longitudinal Study of Parents and Children. Am. J. Clin. Nutr. 2011, 93, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Kwak, J.H.; Paik, J.K.; Kim, O.Y.; Jang, Y.; Lee, S.H.; Ordovas, J.M.; Lee, J.H. FADS gene polymorphisms in Koreans: Association with omega6 polyunsaturated fatty acids in serum phospholipids, lipid peroxides, and coronary artery disease. Atherosclerosis 2011, 214, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, S.; Hugenschmidt, C.E.; Rudock, M.E.; Ziegler, J.T.; Ivester, P.; Ainsworth, H.C.; Vaidya, D.; Case, L.D.; Langefeld, C.D.; Freedman, B.I.; et al. Differences in arachidonic acid levels and fatty acid desaturase (FADS) gene variants in African Americans and European Americans with diabetes or the metabolic syndrome. Br. J. Nutr. 2012, 107, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Steer, C.D.; Hibbeln, J.R.; Golding, J.; Davey, S.G. Polyunsaturated fatty acid levels in blood during pregnancy, at birth and at 7 years: Their associations with two common FADS2 polymorphisms. Hum. Mol. Genet. 2012, 21, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Porenta, S.R.; Ko, Y.A.; Gruber, S.B.; Mukherjee, B.; Baylin, A.; Ren, J.; Djuric, Z. Interaction of fatty acid genotype and diet on changes in colonic fatty acids in a Mediterranean diet intervention study. Cancer Prev. Res. 2013, 6, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.H.; Kwak, J.H.; Paik, J.K.; Chae, J.S.; Lee, J.H. Association of polymorphisms in FADS gene with age-related changes in serum phospholipid polyunsaturated fatty acids and oxidative stress markers in middle-aged nonobese men. Clin. Interv. Aging 2013, 8, 585–596. [Google Scholar] [PubMed]
- Harslof, L.B.; Larsen, L.H.; Ritz, C.; Hellgren, L.I.; Michaelsen, K.F.; Vogel, U.; Lauritzen, L. FADS genotype and diet are important determinants of DHA status: A cross-sectional study in Danish infants. Am. J. Clin. Nutr. 2013, 97, 1403–1410. [Google Scholar] [CrossRef] [PubMed]
- Li, S.W.; Lin, K.; Ma, P.; Zhang, Z.L.; Zhou, Y.D.; Lu, S.Y.; Zhou, X.; Liu, S.M. FADS gene polymorphisms confer the risk of coronary artery disease in a Chinese Han population through the altered desaturase activities: Based on high-resolution melting analysis. PLoS ONE 2013, 8, e55869. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, L.G.; Harding, S.V.; Rideout, T.C.; Yurkova, N.; Cunnane, S.C.; Eck, P.K.; Jones, P.J. Dietary oils and FADS1-FADS2 genetic variants modulate (13C)alpha-linolenic acid metabolism and plasma fatty acid composition. Am. J. Clin. Nutr. 2013, 97, 195–207. [Google Scholar] [CrossRef] [PubMed]
- Freemantle, E.; Lalovic, A.; Mechawar, N.; Turecki, G. Age and haplotype variations within FADS1 interact and associate with alterations in fatty acid composition in human male cortical brain tissue. PLoS ONE 2012, 7, e42696. [Google Scholar] [CrossRef] [PubMed]
- Lattka, E.; Koletzko, B.; Zeilinger, S.; Hibbeln, J.R.; Klopp, N.; Ring, S.M.; Steer, C.D. Umbilical cord PUFA are determined by maternal and child fatty acid desaturase (FADS) genetic variants in the Avon Longitudinal Study of Parents and Children (ALSPAC). Br. J. Nutr. 2013, 109, 1196–1210. [Google Scholar] [CrossRef] [PubMed]
- Tintle, N.L.; Pottala, J.V.; Lacey, S.; Ramachandran, V.; Westra, J.; Rogers, A.; Clark, J.; Olthoff, B.; Larson, M.; Harris, W.; et al. A genome-wide association study of saturated, mono- and polyunsaturated red blood cell fatty acids in the Framingham Heart Offspring Study. Prostaglandins Leukot. Essent. Fat. Acids 2015, 94, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Martinelli, N.; Girelli, D.; Malerba, G.; Guarini, P.; Illig, T.; Trabetti, E.; Sandri, M.; Friso, S.; Pizzolo, F.; Schaeffer, L.; et al. FADS genotypes and desaturase activity estimated by the ratio of arachidonic acid to linoleic acid are associated with inflammation and coronary artery disease. Am. J. Clin. Nutr. 2008, 88, 941–949. [Google Scholar] [PubMed]
- Ameur, A.; Enroth, S.; Johansson, A.; Zaboli, G.; Igl, W.; Johansson, A.C.; Rivas, M.A.; Daly, M.J.; Schmitz, G.; Hicks, A.A.; et al. Genetic adaptation of fatty-acid metabolism: A human-specific haplotype increasing the biosynthesis of long-chain omega-3 and omega-6 fatty acids. Am. J. Hum. Genet. 2012, 90, 809–820. [Google Scholar] [CrossRef] [PubMed]
- Amorim, C.E.; Nunes, K.; Meyer, D.; Comas, D.; Bortolini, M.C.; Salzano, F.M.; Hunemeier, T. Genetic signature of natural selection in first Americans. Proc. Natl. Acad. Sci. USA 2017, 114, 2195–2199. [Google Scholar] [CrossRef] [PubMed]
- Mathias, R.A.; Fu, W.Q.; Akey, J.M.; Ainsworth, H.C.; Torgerson, D.G.; Ruczinski, I.; Sergeant, S.; Barnes, K.C.; Chilton, F.H. Adaptive evolution of the FADS gene cluster within africa. PLoS ONE 2012, 7, e44926. [Google Scholar] [CrossRef] [PubMed]
- Dorajoo, R.; Sun, Y.; Han, Y.; Ke, T.; Burger, A.; Chang, X.; Low, H.Q.; Guan, W.; Lemaitre, R.N.; Khor, C.C.; et al. A genome-wide association study of n-3 and n-6 plasma fatty acids in a Singaporean Chinese population. Genes Nutr. 2015, 10, 53. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Steffen, B.T.; Lemaitre, R.N.; Wu, J.H.Y.; Tanaka, T.; Manichaikul, A.; Foy, M.; Rich, S.S.; Wang, L.; Nettleton, J.A.; et al. Genome-wide association study of plasma N6 polyunsaturated fatty acids within the cohorts for heart and aging research in genomic epidemiology consortium. Circ. Cardiovasc. Genet. 2014, 7, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, H.; Lu, L.; Manichaikul, A.; Zhu, J.; Chen, Y.D.; Sun, L.; Liang, S.; Siscovick, D.S.; Steffen, L.M.; et al. Genome-wide meta-analyses identify novel loci associated with n-3 and n-6 polyunsaturated fatty acid levels in Chinese and European-ancestry populations. Hum. Mol. Genet. 2016, 25, 1215–1224. [Google Scholar] [CrossRef] [PubMed]
- Lattka, E.; Illig, T.; Heinrich, J.; Koletzko, B. Do FADS genotypes enhance our knowledge about fatty acid related phenotypes? Clin. Nutr. 2010, 29, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.C.; Zhang, W.; Sehmi, J.; Li, X.; Wass, M.N.; Van der Harst, P.; Holm, H.; Sanna, S.; Kavousi, M.; Baumeister, S.E.; et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat. Genet. 2011, 43, 1131–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirkov, S.; Myers, J.L.; Ramirez, J.; Liu, W. SNPs affecting serum metabolomic traits may regulate gene transcription and lipid accumulation in the liver. Metabolism 2012, 61, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, J.; Langenberg, C.; Prokopenko, I.; Saxena, R.; Soranzo, N.; Jackson, A.U.; Wheeler, E.; Glazer, N.L.; Bouatia-Naji, N.; Gloyn, A.L.; et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat. Genet. 2010, 42, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burghardt, K.J.; Gardner, K.N.; Johnson, J.W.; Ellingrod, V.L. Fatty Acid desaturase gene polymorphisms and metabolic measures in schizophrenia and bipolar patients taking antipsychotics. Cardiovasc. Psychiatry Neurol. 2013, 2013, 596945. [Google Scholar] [CrossRef] [PubMed]
- Standl, M.; Lattka, E.; Stach, B.; Koletzko, S.; Bauer, C.P.; von, B.A.; Berdel, D.; Kramer, U.; Schaaf, B.; Roder, S.; et al. FADS1 FADS2 gene cluster, PUFA intake and blood lipids in children: Results from the GINIplus and LISAplus studies. PLoS ONE 2012, 7, e37780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fumagalli, M.; Moltke, I.; Grarup, N.; Racimo, F.; Bjerregaard, P.; Jorgensen, M.E.; Korneliussen, T.S.; Gerbault, P.; Skotte, L.; Linneberg, A.; et al. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science 2015, 349, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Harris, D.N.; Ruczinski, I.; Yanek, L.R.; Becker, L.C.; Guio, H.; Cui, T.; Chilton, F.H.; Mathias, R.A.; O’Conner, T. Evolution of hominim polyunsatruated fatty acid metabolism; from Africa to the New World. BioRxiv 2017. [Google Scholar] [CrossRef]
- Mathieson, I.; Lazaridis, I.; Rohland, N.; Mallick, S.; Patterson, N.; Roodenberg, S.A.; Harney, E.; Stewardson, K.; Fernandes, D.; Novak, M.; et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature 2015, 528, 499–503. [Google Scholar] [CrossRef] [PubMed]
- Kothapalli, K.S.; Ye, K.; Gadgil, M.S.; Carlson, S.E.; O’Brien, K.O.; Zhang, J.Y.; Park, H.G.; Ojukwu, K.; Zou, J.; Hyon, S.S.; et al. Positive selection on a regulatory insertion-deletion polymorphism in FADS2 influences apparent endogenous synthesis of arachidonic acid. Mol. Biol. Evol. 2016, 33, 1726–1739. [Google Scholar] [CrossRef] [PubMed]
- Buckley, M.T.; Racimo, F.; Allentoft, M.E.; Jensen, M.K.; Jonsson, A.; Huang, H.; Hormozdiari, F.; Sikora, M.; Marnetto, D.; Eskin, E.; et al. Selection in europeans on fatty acid desaturases associated with dietary changes. Mol. Biol. Evol. 2017, 34, 1307–1318. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Enattah, N.S.; Sahi, T.; Savilahti, E.; Terwilliger, J.D.; Peltonen, L.; Jarvela, I. Identification of a variant associated with adult-type hypolactasia. Nat. Genet. 2002, 30, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Tishkoff, S.A.; Reed, F.A.; Ranciaro, A.; Voight, B.F.; Babbitt, C.C.; Silverman, J.S.; Powell, K.; Mortensen, H.M.; Hirbo, J.B.; Osman, M.; et al. Convergent adaptation of human lactase persistence in Africa and Europe. Nat. Genet. 2007, 39, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Lettre, G.; Palmer, C.D.; Young, T.; Ejebe, K.G.; Allayee, H.; Benjamin, E.J.; Bennett, F.; Bowden, D.W.; Chakravarti, A.; Dreisbach, A.; et al. Genome-wide association study of coronary heart disease and its risk factors in 8,090 African Americans: The NHLBI CARe Project. PLoS Genet. 2011, 7, e1001300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kark, J.D.; Kaufmann, N.A.; Binka, F.; Goldberger, N.; Berry, E.M. Adipose tissue n-6 fatty acids and acute myocardial infarction in a population consuming a diet high in polyunsaturated fatty acids. Am. J. Clin. Nutr. 2003, 77, 796–802. [Google Scholar] [PubMed]
- Baylin, A.; Campos, H. Arachidonic acid in adipose tissue is associated with nonfatal acute myocardial infarction in the central valley of Costa Rica. J. Nutr. 2004, 134, 3095–3099. [Google Scholar] [PubMed]
- Hester, A.G.; Murphy, R.C.; Uhlson, C.J.; Ivester, P.; Lee, T.C.; Sergeant, S.; Miller, L.R.; Howard, T.D.; Mathias, R.A.; Chilton, F.H. Relationship between a common variant in the fatty acid desaturase (FADS) cluster and eicosanoid generation in humans. J. Biol. Chem. 2014, 289, 22482–22489. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Paik, J.K.; Kim, O.Y.; Chae, J.S.; Jang, Y.; Lee, J.H. Interactions between the APOA5-1131T>C and the FEN1 10154G>T polymorphisms on omega6 polyunsaturated fatty acids in serum phospholipids and coronary artery disease. J. Lipid Res. 2010, 51, 3281–3288. [Google Scholar] [CrossRef] [PubMed]
- Hellstrand, S.; Ericson, U.; Gullberg, B.; Hedblad, B.; Orho-Melander, M.; Sonestedt, E. Genetic variation in FADS1 has little effect on the association between dietary PUFA intake and cardiovascular disease. J. Nutr. 2014, 144, 1356–1363. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, C.M.; Minihane, A.M. The impact of fatty acid desaturase genotype on fatty acid status and cardiovascular health in adults. Proc. Nutr. Soc. 2017, 76, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.R.; Jorgensen, M.J.; Kaplan, J.R.; Seeds, M.C.; Rahbar, E.; Morgan, T.M.; Welborn, A.; Chilton, S.M.; Gillis, J.; Hester, A.; et al. Alterations in levels and ratios of n-3 and n-6 polyunsaturated fatty acids in the temporal cortex and liver of vervet monkeys from birth to early adulthood. Physiol. Behav. 2016, 156, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Kitson, A.P.; Stark, K.D.; Duncan, R.E. Enzymes in brain phospholipid docosahexaenoic acid accretion: A PL-ethora of potential PL-ayers. Prostaglandins Leukot. Essent. Fat. Acids 2012, 87, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kuipers, R.S.; Luxwolda, M.F.; Offringa, P.J.; Boersma, E.R.; Dijck-Brouwer, D.A.; Muskiet, F.A. Fetal intrauterine whole body linoleic, arachidonic and docosahexaenoic acid contents and accretion rates. Prostaglandins Leukot. Essent. Fat. Acids 2012, 86, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Cheatham, C.L.; Lupu, D.S.; Niculescu, M.D. Genetic and epigenetic transgenerational implications related to omega-3 fatty acids. Part II: Maternal FADS2 rs174575 genotype and DNA methylation predict toddler cognitive performance. Nutr. Res. 2015, 35, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Lupu, D.S.; Cheatham, C.L.; Corbin, K.D.; Niculescu, M.D. Genetic and epigenetic transgenerational implications related to omega-3 fatty acids. Part I: Maternal FADS2 genotype and DNA methylation correlate with polyunsaturated fatty acid status in toddlers: An exploratory analysis. Nutr. Res. 2015, 35, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Hoile, S.P.; Clarke-Harris, R.; Huang, R.C.; Calder, P.C.; Mori, T.A.; Beilin, L.J.; Lillycrop, K.A.; Burdge, G.C. Supplementation with N-3 long-chain polyunsaturated fatty acids or olive oil in men and women with renal disease induces differential changes in the DNA methylation of FADS2 and ELOVL5 in peripheral blood mononuclear cells. PLoS ONE 2014, 9, e109896. [Google Scholar] [CrossRef] [PubMed]
- Howard, T.D.; Mathias, R.A.; Seeds, M.C.; Herrington, D.M.; Hixson, J.E.; Shimmin, L.C.; Hawkins, G.A.; Sellers, M.; Ainsworth, H.C.; Sergeant, S.; et al. DNA methylation in an enhancer region of the FADS cluster is associated with fads activity in human liver. PLoS ONE 2014, 9, e97510. [Google Scholar] [CrossRef] [PubMed]
- Hoile, S.P.; Irvine, N.A.; Kelsall, C.J.; Sibbons, C.; Feunteun, A.; Collister, A.; Torrens, C.; Calder, P.C.; Hanson, M.A.; Lillycrop, K.A.; et al. Maternal fat intake in rats alters 20:4n-6 and 22:6n-3 status and the epigenetic regulation of FADS2 in offspring liver. J. Nutr. Biochem. 2013, 24, 1213–1220. [Google Scholar] [CrossRef] [PubMed]
- Bang, H.O.; Dyerberg, J.; Nielsen, A.B. Plasma lipid and lipoprotein pattern in Greenlandic West-coast Eskimos. Lancet 1971, 1, 1143–1145. [Google Scholar] [CrossRef]
- Dyerberg, J.; Bang, H.O. Haemostatic function and platelet polyunsaturated fatty acids in Eskimos. Lancet 1979, 2, 433–435. [Google Scholar] [CrossRef]
- Ander, B.P.; Dupasquier, C.M.; Prociuk, M.A.; Pierce, G.N. Polyunsaturated fatty acids and their effects on cardiovascular disease. Exp. Clin. Cardiol. 2003, 8, 164–172. [Google Scholar] [PubMed]
- Burr, M.L.; Fehily, A.M.; Gilbert, J.F.; Rogers, S.; Holliday, R.M.; Sweetnam, P.M.; Elwood, P.C.; Deadman, N.M. Effects of changes in fat, fish, and fibre intakes on death and myocardial reinfarction: Diet and reinfarction trial (DART). Lancet 1989, 2, 757–761. [Google Scholar] [CrossRef]
- Investigators, G.-P. Dietray supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: Results of the GISSIPrevenzione trial. Lancet 1999, 354, 447–455. [Google Scholar]
- Marchioli, R.; Barzi, F.; Bomba, E.; Chieffo, C.; Di Gregorio, D.; Di Mascio, R.; Franzosi, M.G.; Geraci, E.; Levantesi, G.; Maggioni, A.P.; et al. Early protection against sudden death by n-3 polyunsaturated fatty acids after myocardial infarction: Time-course analysis of the results of the Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto Miocardico (GISSI)-Prevenzione. Circulation 2002, 105, 1897–1903. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- Tavazzi, L.; Tognoni, G.; Franzosi, M.G.; Latini, R.; Maggioni, A.P.; Marchioli, R.; Nicolosi, G.L.; Porcu, M.; Investigators, G.-H. Rationale and design of the GISSI heart failure trial: A large trial to assess the effects of n-3 polyunsaturated fatty acids and rosuvastatin in symptomatic congestive heart failure. Eur. J. Heart Fail. 2004, 6, 635–641. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Song, Y.; Daviglus, M.L.; Liu, K.; Van Horn, L.; Dyer, A.R.; Greenland, P. Accumulated evidence on fish consumption and coronary heart disease mortality: A meta-analysis of cohort studies. Circulation 2004, 109, 2705–2711. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Warnakula, S.; Kunutsor, S.; Crowe, F.; Ward, H.A.; Johnson, L.; Franco, O.H.; Butterworth, A.S.; Forouhi, N.G.; Thompson, S.G.; et al. Association of dietary, circulating, and supplement fatty acids with coronary risk: A systematic review and meta-analysis. Ann. Intern. Med. 2014, 160, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Del Gobbo, L.C.; Imamura, F.; Aslibekyan, S.; Marklund, M.; Virtanen, J.K.; Wennberg, M.; Yakoob, M.Y.; Chiuve, S.E.; Dela Cruz, L.; Frazier-Wood, A.C.; et al. Omega-3 polyunsaturated fatty acid biomarkers and coronary heart disease: Pooling project of 19 cohort studies. JAMA Intern. Med. 2016, 176, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Kromhout, D.; Giltay, E.J.; Geleijnse, J.M.; Alpha Omega Trial, G. n-3 fatty acids and cardiovascular events after myocardial infarction. N. Engl. J. Med. 2010, 363, 2015–2026. [Google Scholar] [CrossRef] [PubMed]
- Rauch, B.; Schiele, R.; Schneider, S.; Diller, F.; Victor, N.; Gohlke, H.; Gottwik, M.; Steinbeck, G.; Del Castillo, U.; Sack, R.; et al. OMEGA, a randomized, placebo-controlled trial to test the effect of highly purified omega-3 fatty acids on top of modern guideline-adjusted therapy after myocardial infarction. Circulation 2010, 122, 2152–2159. [Google Scholar] [CrossRef] [PubMed]
- Galan, P.; Kesse-Guyot, E.; Czernichow, S.; Briancon, S.; Blacher, J.; Hercberg, S.; Group, S.F.O.C. Effects of B vitamins and omega 3 fatty acids on cardiovascular diseases: A randomised placebo controlled trial. BMJ 2010, 341, c6273. [Google Scholar] [CrossRef] [PubMed]
- Bosch, J.; Gerstein, H.C.; Dagenais, G.R.; Diaz, R.; Dyal, L.; Jung, H.; Maggiono, A.P.; Probstfield, J.; Ramachandran, A.; Riddle, M.C.; et al. n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N. Engl. J. Med. 2012, 367, 309–318. [Google Scholar] [PubMed]
- Roncaglioni, M.C.; Tombesi, M.; Avanzini, F.; Barlera, S.; Caimi, V.; Longoni, P.; Marzona, I.; Milani, V.; Silletta, M.G.; Tognoni, G.; et al. n-3 fatty acids in patients with multiple cardiovascular risk factors. N. Engl. J. Med. 2013, 368, 1800–1808. [Google Scholar] [PubMed]
- Bourre, J.M. Roles of unsaturated fatty acids (especially omega-3 fatty acids) in the brain at various ages and during ageing. J. Nutr. Health Aging 2004, 8, 163–174. [Google Scholar] [PubMed]
- Van Elst, K.; Bruining, H.; Birtoli, B.; Terreaux, C.; Buitelaar, J.K.; Kas, M.J. Food for thought: Dietary changes in essential fatty acid ratios and the increase in autism spectrum disorders. Neurosci. Biobehav. Rev. 2014, 45, 369–378. [Google Scholar] [CrossRef] [PubMed]
- James, S.; Montgomery, P.; Williams, K. Omega-3 fatty acids supplementation for autism spectrum disorders (ASD). Cochrane Database Syst. Rev. 2011. [Google Scholar] [CrossRef]
- Gow, R.V.; Hibbeln, J.R. Omega-3 fatty acid and nutrient deficits in adverse neurodevelopment and childhood behaviors. Child Adolesc. Psychiatr. Clin. N. Am. 2014, 23, 555–590. [Google Scholar] [CrossRef] [PubMed]
- Bondi, C.O.; Taha, A.Y.; Tock, J.L.; Totah, N.K.; Cheon, Y.; Torres, G.E.; Rapoport, S.I.; Moghaddam, B. Adolescent behavior and dopamine availability are uniquely sensitive to dietary omega-3 fatty acid deficiency. Biol. Psychiatry 2014, 75, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.W.; Schafer, M.R.; Klier, C.M.; Berk, M.; Rice, S.; Allott, K.; Bartholomeusz, C.F.; Whittle, S.L.; Pilioussis, E.; Pantelis, C.; et al. Relationship between membrane fatty acids and cognitive symptoms and information processing in individuals at ultra-high risk for psychosis. Schizophr. Res. 2014, 158, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.J.; Grenyer, B.F.; Crowe, T.; Owen, A.J.; Grigonis-Deane, E.M.; Howe, P.R. Improvement of major depression is associated with increased erythrocyte DHA. Lipids 2013, 48, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Gillies, D.; Sinn, J.; Lad, S.S.; Leach, M.J.; Ross, M.J. Polyunsaturated fatty acids (PUFA) for attention deficit hyperactivity disorder (ADHD) in children and adolescents. Cochrane Database Syst. Rev. 2012, 7. [Google Scholar]
- Lattka, E.; Klopp, N.; Demmelmair, H.; Klingler, M.; Heinrich, J.; Koletzko, B. Genetic variations in polyunsaturated fatty acid metabolism--implications for child health? Ann. Nutr. Metab. 2012, 60 (Suppl. 3), 8–17. [Google Scholar] [CrossRef] [PubMed]
- Horrobin, D.F.; Manku, M.S.; Hillman, H.; Iain, A.; Glen, M. Fatty acid levels in the brains of schizophrenics and normal controls. Biol. Psychiatry 1991, 30, 795–805. [Google Scholar] [CrossRef]
- Assies, J.; Lieverse, R.; Vreken, P.; Wanders, R.J.; Dingemans, P.M.; Linszen, D.H. Significantly reduced docosahexaenoic and docosapentaenoic acid concentrations in erythrocyte membranes from schizophrenic patients compared with a carefully matched control group. Biol. Psychiatry 2001, 49, 510–522. [Google Scholar] [CrossRef]
- McNamara, R.K.; Vannest, J.J.; Valentine, C.J. Role of perinatal long-chain omega-3 fatty acids in cortical circuit maturation: Mechanisms and implications for psychopathology. World J. Psychiatry 2015, 5, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.J.; Zentall, S.S.; Deck, J.L.; Abate, M.L.; Watkins, B.A.; Lipp, S.R.; Burgess, J.R. Essential fatty acid metabolism in boys with attention-deficit hyperactivity disorder. Am. J. Clin. Nutr. 1995, 62, 761–768. [Google Scholar] [PubMed]
- Wu, S.; Ding, Y.; Wu, F.; Li, R.; Hou, J.; Mao, P. Omega-3 fatty acids intake and risks of dementia and Alzheimer’s disease: A meta-analysis. Neurosci. Biobehav. Rev. 2015, 48, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Li, Q.; Chu, J.; Zeng, W.; Yang, M.; Zhu, S. Effect of n-3 PUFA supplementation on cognitive function throughout the life span from infancy to old age: A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2014, 100, 1422–1436. [Google Scholar] [CrossRef] [PubMed]
- Hawkey, E.; Nigg, J.T. Omega-3 fatty acid and ADHD: Blood level analysis and meta-analytic extension of supplementation trials. Clin. Psychol. Rev. 2014, 34, 496–505. [Google Scholar] [CrossRef] [PubMed]
- Bloch, M.H.; Hannestad, J. Omega-3 fatty acids for the treatment of depression: Systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 1272–1282. [Google Scholar] [CrossRef] [PubMed]
- Cooper, R.E.; Tye, C.; Kuntsi, J.; Vassos, E.; Asherson, P. Omega-3 polyunsaturated fatty acid supplementation and cognition: A systematic review and meta-analysis. J. Psychopharmacol. 2015, 29, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Gould, J.F.; Smithers, L.G.; Makrides, M. The effect of maternal omega-3 (n-3) LCPUFA supplementation during pregnancy on early childhood cognitive and visual development: A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2013, 97, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Shulkin, M.L.; Pimpin, L.; Bellinger, D.; Kranz, S.; Duggan, C.; Fawzi, W.; Mozaffarian, D. Effects of omega-3 supplementation during pregnancy and youth on neurodevelopment and cognition in childhood: A systematic review and meta-analysis. FASEB J. 2016, 30 (Suppl. 295), 5. [Google Scholar]
- Woods, R.K.; Thien, F.C.; Abramson, M.J. Dietary marine fatty acids (fish oil) for asthma in adults and children. Cochrane Database Syst. Rev. 2002. [Google Scholar] [CrossRef]
- Best, K.P.; Gold, M.; Kennedy, D.; Martin, J.; Makrides, M. Omega-3 long-chain PUFA intake during pregnancy and allergic disease outcomes in the offspring: A systematic review and meta-analysis of observational studies and randomized controlled trials. Am. J. Clin. Nutr. 2016, 103, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Fortin, P.R.; Lew, R.A.; Liang, M.H.; Wright, E.A.; Beckett, L.A.; Chalmers, T.C.; Sperling, R.I. Validation of a meta-analysis: The effects of fish oil in rheumatoid arthritis. J. Clin. Epidemiol. 1995, 48, 1379–1390. [Google Scholar] [CrossRef]
- Goldberg, R.J.; Katz, J. A meta-analysis of the analgesic effects of omega-3 polyunsaturated fatty acid supplementation for inflammatory joint pain. Pain 2007, 129, 210–223. [Google Scholar] [CrossRef] [PubMed]
- Leitzmann, M.F.; Stampfer, M.J.; Michaud, D.S.; Augustsson, K.; Colditz, G.C.; Willett, W.C.; Giovannucci, E.L. Dietary intake of n-3 and n-6 fatty acids and the risk of prostate cancer. Am. J. Clin. Nutr. 2004, 80, 204–216. [Google Scholar] [PubMed]
- Chavarro, J.E.; Stampfer, M.J.; Li, H.; Campos, H.; Kurth, T.; Ma, J. A prospective study of polyunsaturated fatty acid levels in blood and prostate cancer risk. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Sakai, M.; Kakutani, S.; Horikawa, C.; Tokuda, H.; Kawashima, H.; Shibata, H.; Okubo, H.; Sasaki, S. Arachidonic acid and cancer risk: A systematic review of observational studies. BMC Cancer 2012, 12, 606. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.; Thompson, R.L.; Harrison, R.A.; Summerbell, C.D.; Ness, A.R.; Moore, H.J.; Worthington, H.V.; Durrington, P.N.; Higgins, J.P.; Capps, N.E.; et al. Risks and benefits of omega 3 fats for mortality, cardiovascular disease, and cancer: Systematic review. BMJ 2006, 332, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Rice, H.B.; Bernasconi, A.; Maki, K.C.; Harris, W.S.; von Schacky, C.; Calder, P.C. Conducting omega-3 clinical trials with cardiovascular outcomes: Proceedings of a workshop held at ISSFAL 2014. Prostaglandins Leukot. Essent. Fat. Acids 2016, 107, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Superko, H.R.; Superko, A.R.; Lundberg, G.P.; Margolis, B.; Garrett, B.C.; Nasir, K.; Agatston, A.S. Omega-3 fatty acid blood levels clinical significance update. Curr. Cardiovasc. Risk Rep. 2014, 8, 407. [Google Scholar] [CrossRef] [PubMed]
- Draisma, H.H.M.; Pool, R.; Kobl, M.; Jansen, R.; Petersen, A.K.; Vaarhorst, A.A.M.; Yet, I.; Haller, T.; Demirkan, A.; Esko, T.; et al. Genome-wide association study identifies novel genetic variants contributing to variation in blood metabolite levels. Nat. Commun. 2015, 6, 7208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, S.Y.; Fauman, E.B.; Petersen, A.K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, T.; Hicks, M.; Yu, H.C.; Biggs, W.H.; Kirkness, E.F.; Menni, C.; Zierer, J.; Small, K.S.; Mangino, M.; Messier, H.; et al. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat. Genet. 2017, 49, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Raffler, J.; Pfeufer, A.; Suhre, K.; Kastenmuller, G. SNiPA: An interactive, genetic variant-centered annotation browser. Bioinformatics 2015, 31, 1334–1336. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SNPs | ||||
|---|---|---|---|---|
| n-3 PUFA/Metabolite | rs174547 p-Value | rs174537 p-Value | rs174548 p-Value | Data Source |
| 1-hexadecanoyl-2-docosapentaenoyl-GPC (16:0/22:5n3) | 2.97 × 10−95 | 3.83 × 10−93 | 9.17 × 10−88 | Draisma et al. [198] |
| 1-tetradecanoyl-2-docosapentaenoyl-GPC (14:0/22:5n3) | 2.76 × 10−58 | 1.94 × 10−57 | 5.08 × 10−54 | Draisma et al. [198] |
| 1-octadecanoyl-2-docosapentaenoyl-GPC (18:0/22:5n-3) | 2.23 × 10−42 | 4.61 × 10−41 | 2.53 × 10−40 | Draisma et al. [198] |
| 1-O-docosanyl-2-docosahexaenoyl-GPC (o-22:0/22:6n-3) | 1.67 × 10−40 | 6.03 × 10−40 | 3.15 × 10−37 | Draisma et al. [198] |
| 1-O-hexadecyl-2-docosahexaenoyl-GPC (o-16:0/22:6n-3) | 1.35 × 10−25 | 4.90 × 10−24 | 1.36 × 10−23 | Draisma et al. [198] |
| 1-eicosanoyl-2-docosahexaenoyl-GPC (20:0/22:6n-3) | 2.19 × 10−23 | Draisma et al. [198] | ||
| eicosapentaenoate (EPA; 20:5n3) | 1.12 × 10−21 | 2.55 × 10−21 | 3.71 × 10−22 | Shin et al. [199] |
| 1-octadecanoyl-2-docosahexaenoyl-GPC (18:0/22:6n-3) | 8.48 × 10−20 | 3.88 × 10−19 | 1.70 × 10−18 | Draisma et al. [198] |
| 1-tetradecanoyl-2-docosahexaenoyl-GPC (14:0/22:6n-3) | 9.86 × 10−18 | 2.72 × 10−17 | 1.38 × 10−15 | Draisma et al. [198] |
| 1-octadecanoyl-2-docosapentaenoyl-GPC (18:0/22:5n3) | 4.43 × 10−14 | 9.83 × 10−14 | 9.99 × 10−16 | Long et al. [200] |
| octadecatetraenoic acid (stearidonate) (18:4n3) | 1.63 × 10−15 | 1.07 × 10−15 | 1.97 × 10−13 | Shin et al. [199] |
| 1-O-octadecyl-2-docosahexaenoyl-GPC (o-18:0/22:6n-3) | 2.02 × 10−15 | 5.04 × 10−15 | 1.47 × 10−14 | Draisma et al. [198] |
| 1-hexadecanoyl-2-docosahexaenoyl-GPC 16:0/22:6 | 1.21 × 10−14 | 4.16 × 10−14 | 3.02 × 10−13 | Draisma et al. [198] |
| 1-eicosatrienoyl-GPC (ETA; 20:3n-3) | 1.30 × 10−14 | Shin et al. [199] | ||
| docosapentaenoate (DPA; 22:5n-3) | 2.93 × 10−13 | 5.07 × 10−13 | Shin et al. [199] | |
| 1-eicosapentaenoyl-GPC (20:5n-3) | 1.59 × 10−12 | Long et al. [200] | ||
| 1-octadecenoyl-2-eicosapentaenoyl-GPC (18:1/20:5n-3) | 1.78 × 10−12 | Long et al. [200] | ||
| 1-palmitoyl-2-eicosapentaenoyl-GPC (16:0/20:5n-3) | 1.01 × 10−11 | Long et al. [200] | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chilton, F.H.; Dutta, R.; Reynolds, L.M.; Sergeant, S.; Mathias, R.A.; Seeds, M.C. Precision Nutrition and Omega-3 Polyunsaturated Fatty Acids: A Case for Personalized Supplementation Approaches for the Prevention and Management of Human Diseases. Nutrients 2017, 9, 1165. https://doi.org/10.3390/nu9111165
Chilton FH, Dutta R, Reynolds LM, Sergeant S, Mathias RA, Seeds MC. Precision Nutrition and Omega-3 Polyunsaturated Fatty Acids: A Case for Personalized Supplementation Approaches for the Prevention and Management of Human Diseases. Nutrients. 2017; 9(11):1165. https://doi.org/10.3390/nu9111165
Chicago/Turabian StyleChilton, Floyd H., Rahul Dutta, Lindsay M. Reynolds, Susan Sergeant, Rasika A. Mathias, and Michael C. Seeds. 2017. "Precision Nutrition and Omega-3 Polyunsaturated Fatty Acids: A Case for Personalized Supplementation Approaches for the Prevention and Management of Human Diseases" Nutrients 9, no. 11: 1165. https://doi.org/10.3390/nu9111165